Introduction
Prostate cancer (PCa) is one of the most common
malignancies and one of the leading causes of cancer deaths in men
in the Western world. There are many therapeutic options against
localized prostate cancer including prostatectomy and radiation
therapy (1). However, in advanced
cancer, most tumors ultimately relapse after a period of initial
response to therapy and progress to metastatic cancer (2,3). It is
still a clinical challenge to deal with advanced prostate cancer in
patients. Unfortunately, many aspects remain unknown of the
cellular and molecular mechanisms for metastatic disease (4). As is known, epithelial-mesenchymal
transition (EMT) plays essential roles in development, invasion and
migration of prostate cancer. EMT is characterized by loss of
homotypic adhesion and cell polarity (5). The most familiar change that occurs
during EMT is the downregulation of surface E-cadherin expression
and increased expression of N-cadherin. Many signaling pathways,
including TGF-β, Wnt, notch, PI3K/AKT, and hedgehog, have been
intricately connected to the onset of EMT (6). A number of studies have reported that
EMT-inducing transcription factors, such as Snail, Slug, Twist, and
Zeb, are directly or indirectly involved in cancer cell metastasis
through different signaling cascades and pathways (7).
Nuclear factor κB (NF-κB) signaling has been
previously identified as an important pathway in the regulation of
EMT in tumor progression (8). The
NF-κB family is composed of five proteins, including Rel-A/p65,
Rel-B, C-Rel, p52, and p50. In unstimulated cells, uninduced NF-κB
dimers are restrained in the cytoplasm by complex formation with a
member of the IκB family (9). On
stimulation, IκB proteins are phosphorylated by the multisubunit
IκB kinase (IKK) complex, subsequently ubiquitinated and degraded
through the proteasomal pathway. Then, the liberated NF-κB
heterodimer rapidly translocates into the nucleus, where it binds
to the κB site and induces transcription of a wide variety of
target genes involved in cancer development and progression
(10). The IKK complex consists of
IKKα/IKK1, IKKβ/IKK2, and IKKγ/NEMO. Recently, two protein kinases
called IKKε/IKKi and TBK1 (TANK-banding kinase) were identified
that exhibit structural similarity to IKKα and IKKβ (11). However, it remains unclear what is
the regulation relationship between upstream NF-κB activator IKK
family members and EMT in prostate cancer.
Because IKK is a key molecular complex specifically
regulating IκB proteins and subsequently targeting NF-κB, we
speculated that IKK would be a potential therapeutic target for
prostate cancer. A potent small-molecule compound, BMS-345541, was
identified as a highly selective IKKα and IKKβ inhibitor to inhibit
kinase activity (12). To determine
whether IKK inhibitor manipulates the process of EMT and cell
death, we delivered the BMS-345541 drug to human prostate cancer
PC-3 cell in vitro. Here we investigated the detailed effect
of IKK inhibitor on EMT, apoptosis, and metastasis in prostate
cancer cells.
Materials and methods
Biological reagent
BMS-345541 (4(2′-aminoethyl) amino-1,
8-dimethylimidazo(1,2-α) quinoxaline) were obtained from Calbiochem
(San Diego, CA, USA). BMS-345541 was dissolved in DMSO to produce a
50-mmol/l stock solution for experiments. All phosphospecific or
total antibodies used in this study were purchased from Cell
Signaling Tech (Denver, MA, USA) and Santa Cruz Biotech (Santa
Cruz, CA, USA).
Cell culture
PC-3 and LNCaP cells were obtained directly from the
American Type Culture Collection (Manassas, VA, USA). All cell
lines were grown in RPMI-1640 medium (Gibco) both supplemented 10%
heat-inactivated FBS (fetal bovine serum), 100 IU/ml penicillin,
100 µg/ml streptomycin, 0.1 mM non-essential amino acids,
0.2 mM glutamine, and 1 mM pyruvate and incubated at 37°C in a 5%
CO2 incubator. Cells were treated with BMS-345541 in the
following experiments. Cell treated with DMSO were used as
controls. Each experiment was repeated three times.
Cell viability assay by MTT
The cells were seeded in a 96-well culture plate and
cultured overnight. The MTT assay was used to determine cell
viability. BMS-345541 was added to the cells in different time and
concentration. The MTT regent (5 mg/ml) was added and the cells
then incubated for a further 4 h. The reduced MTT crystals were
dissolved in DMSO and the absorbance was detected on BioTek ELISA
reader (Winooski, VT, USA) at 570 nm wavelength.
Cell invasion assay
The cell invasion assay was performed using Boyden
chambers with 8 µm porosity polyvinylpyrrolidone-free
polycarbonate filters coated with 50 µg/ml Matrigel
solution. The cells in 24-well plates at a concentration of
5×104/well were cultured for 24 h with DMSO and
BMS-345541, respectively. Normal culture medium was added to the
bottom chamber to induce the cancer cell lines. Pretreated cell
were seeded in the top chamber. The Matrigel invasion chamber was
incubated for 24 h in a humidified culture incubator, and after 24
h, the non-invasive cells were removed from the upper surface of
the separating membrane using a cotton swab. The invading cells
were then fixed in 100% methanol and stained with 0.1% crystal
violet solution. They were counted using a microscope
(magnification, ×200).
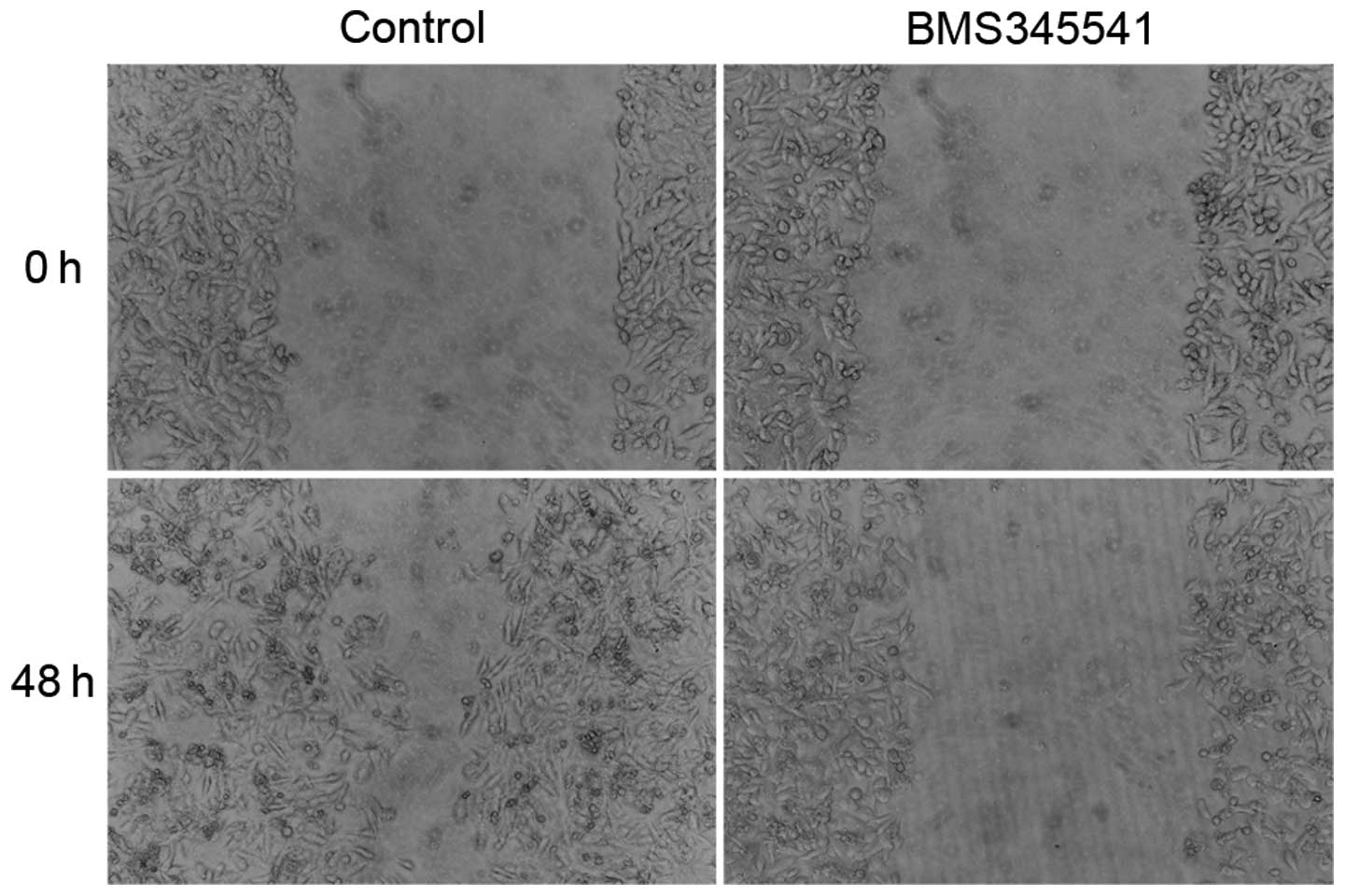
Wound healing assay
Cells were cultured to reach 100% confluency and
were pretreated with DMSO or IKK inhibitor (BMS-345541) for 12 h in
culture medium supplemented with 10% FBS. A scratch wound was
created on the cell surface using a micropipette tip. The wound
area was photographed by bright-field microscopy every 8 h for 48
h. The width of the wound was measured and the wound closure rate
was calculated.
Western blotting
For isolation of total protein, control and treated
cells were washed in ice cold PBS. Briefly, treated cells were
lysed in modified lysis buffer containing 50 mM Tris-HCl (pH 7.4),
150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM EDTA, 1 mM EGTA, 20 mM NaF,
1% sodium deoxycholate and protease inhibitor cocktail (Roche,
Mannheim, Germany). After lysis, the lysates were centrifuged for
10 min at 13,000 g at 4°C. Total protein samples (25–50 μg)
were transferred onto PVDF membrane after electrophoretic
separation in 12% SDS polyacrylamide gel. After blocked with 5%
non-fat milk in Tris-buffered saline for 1 h at room temperature,
the membranes were incubated overnight with the primary antibody at
4°C and washed three times in PBS containing 0.1% Tween-20, then
incubated with the horseradish peroxidase conjugated secondary
antibodies at room temperature for 1 h. The membranes were washed
three times in PBS, and then developed with a horseradish
peroxidase chemiluminescence detection reagent and exposed to X-ray
film.
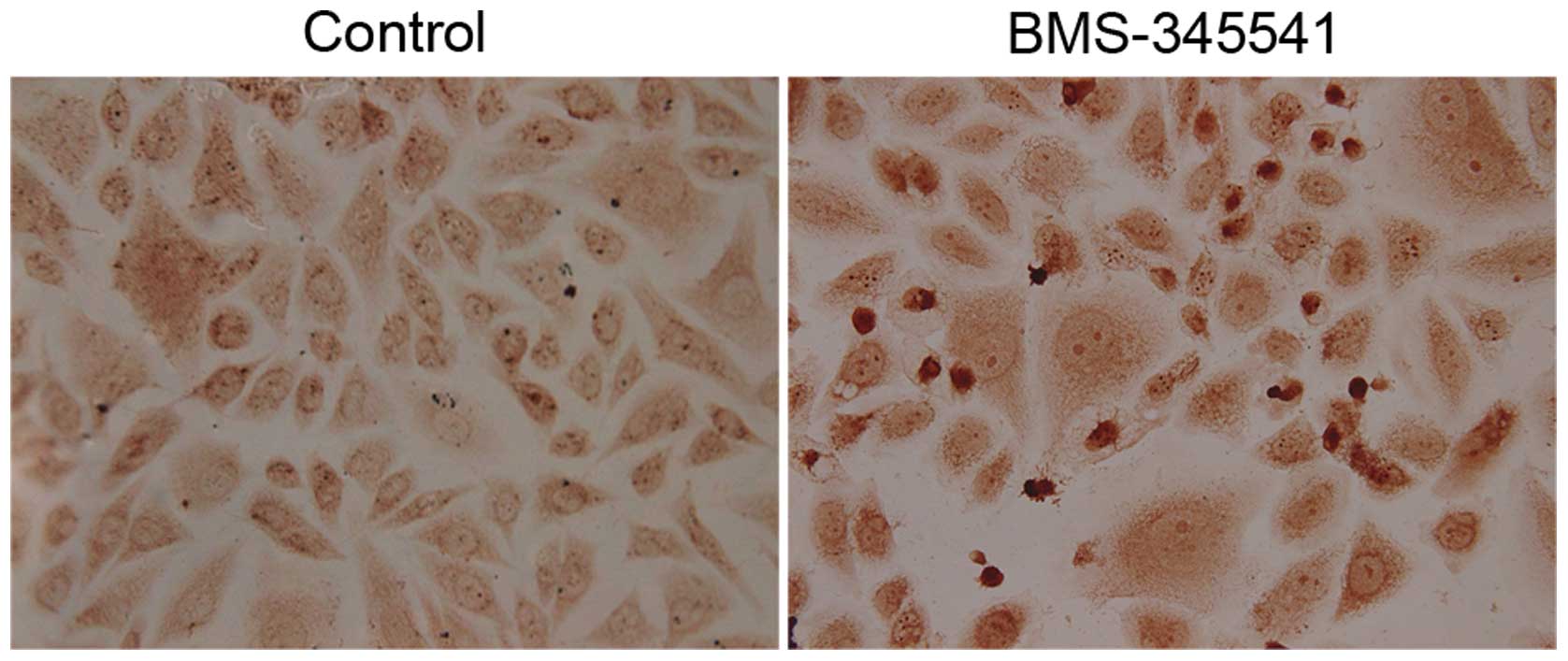
TUNEL apoptosis assay
A quantitative evaluation method was applied by
using terminal deoxynucleotidyl transferase- mediated deoxyuridine
triphosphate (TdT) nick-end labelling (TUNEL) kit to examine
apoptotic cells. Briefly, coverslips with adherent cells treated
with BMS-345541 (5 µmol/l) for 72 h were fixed in 4%
paraformaldehyde at room temperature for 30 min. Then they were
rinsed in distilled water and incubated in PBS containing 0.2%
Triton X-100. DNA fragments were labeled with TUNEL-enzyme
(Boehringer Mannheim). The kit was used according to the
manufacturer's instructions, with the addition of incubation in TdT
reaction buffer for 10 min before TUNEL reaction. The coverslips
were then incubated in TdT reaction mixture for 60 min at 37°C in
humidified chamber, rinsed in stop wash buffer for 10 min and
washed by PBS for 3 times. The reaction was detected by incubating
coverslips with streptavidin-HRP in PBS for 30 min at room
temperature. Then washed by PBS 3 times, and the sections were
incubated with DAB solution for 10 min.
Assessment of the apoptotic index
Positive signal was defined as the presence of a
dark brown staining on the nuclei of the neoplastic cells or on
apoptotic bodies as morphologically defined. Cells were defined as
apoptotic if the whole nuclear area of cells labeled positively.
Apoptotic bodies were defined as small, positively labeled,
globular bodies in the cytoplasm of the tumor cells that could be
found either singly or in groups. The apoptotic index was
determined by the percentage of apoptotic cells divided by the
number of tumor cells in ×400 magnification. A total of ≥1,000
neoplastic nuclei were counted based on 10 randomly chosen fields
at ×400 magnification. Apoptotic cells were identified by TUNEL in
conjunction with characteristic morphological changes, such as cell
shrinkage, membrane blebbing, and chromatin condensation.
Statistical analysis
Data are expressed as mean ± standard deviation (SD)
from three independent experiments. All statistical analyses were
performed using the SPSS 19.0 for Windows software system. Data
with two groups were analyzed by Student's t-tests, and data with
multiple groups were analyzed by one-way ANOVA. A significant
difference was considered when the P-value from a two-tailed test
was <0.05.
Results
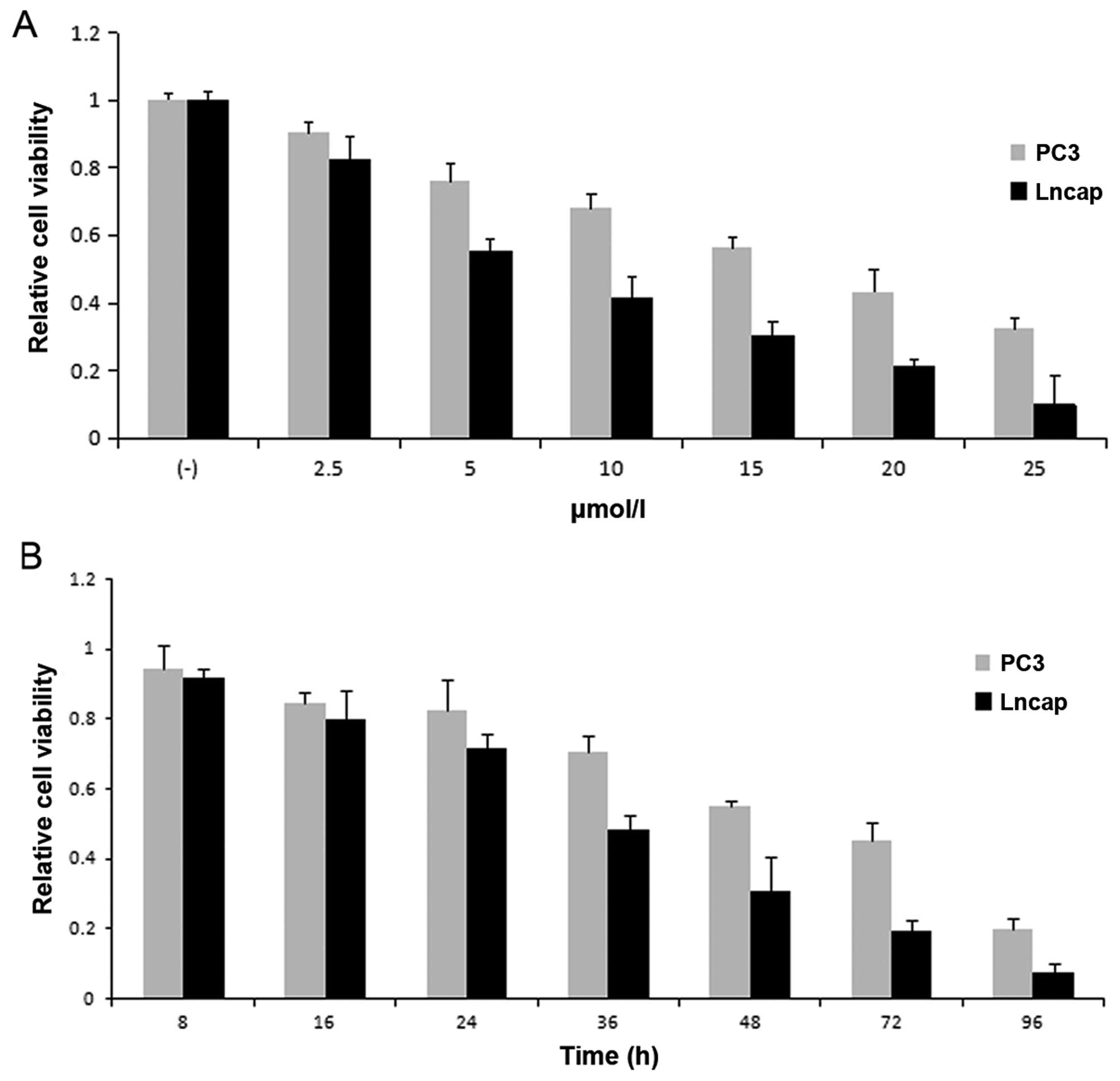
BMS-345541 inhibits the growth of
prostate cancer cells
Since inappropriate regulation of IKK/NF-κB
correlates with prostate caner progression, IKK inhibitor might be
a potential therapeutic agent (13). Hereby, we first assessed the effects
of BMS-345541 on cell viability in PC-3 and LNCaP prostate cancer
cells, respectively. PC-3 (1×103) and LNCaP
(5×103) cell lines were cultured in medium with
BMS-345541 at 0, 2.5, 5, 10, 15, 20 and 25 µmol/l
concentrations. We then measured cell viability at different time
points (8–96 h). From the results of MTT assay, the inhibition rate
of prostate cancer cells treated with BMS-345541 showed a
dose-dependent and time-dependent increase (Fig. 1).
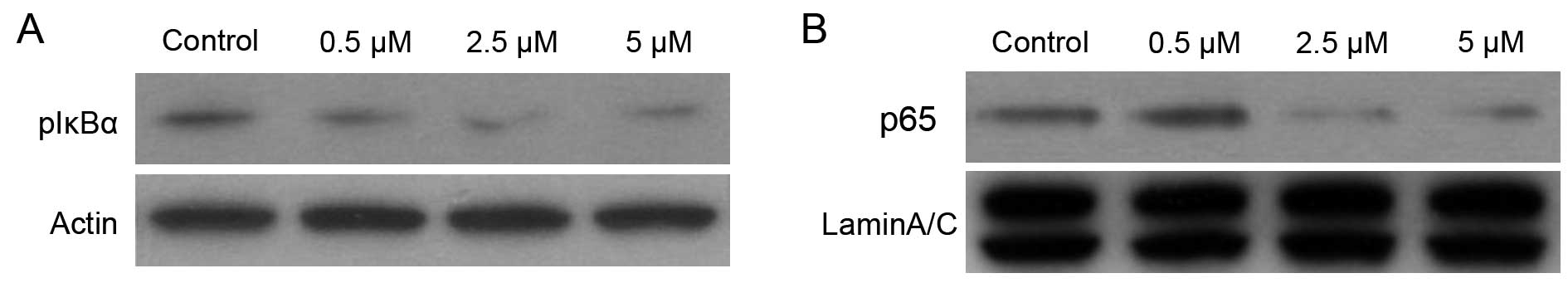
BMS-345541 inhibited IκBα phosphorylation
and nuclear level of NF-κB/p65 in PC-3 cells
BMS-345541 was identified as a selective inhibitor
of the catalytic subunits of IKK (IKKβ IC50=0.3 micron,
IKKα IC50=4 micron). It bands to similar allosteric
sites on IKKα and IKKβ, which then affects the active sites of
subunits differently (14). The
IκBα is constitutively phosphorylated in PC-3 cells by IKK
(15). To evaluate the effect of
the IKK inhibitor BMS-345541 on phosphorylation of IκBα, we
performed western blotting using cytoplasmic extracts from PC-3
cells treated with DMSO or BMS-345541 (final concentration, 0.5,
2.5 and 5 µM) in DMSO for 12 h. As shown in Fig. 2, treatment of PC-3 cells with 0.5,
2.5 and 5 µM doses of BMS-345541 for 12 h resulted in a
significant decrease in p-IκBα level in a dose-dependent manner.
Compared with DMSO-treated control, the level of inhibition was 50%
at 5 µM doses for 12 h.
In addition, p50/p65 dimer is considered to be the
most important of NF-κB proteins, and nuclear translocation of
p50/p65 is required for their transactivation potential (16). Western blot analysis of nuclear
protein from PC-3 cells treated with BMS-345541 showed a
dose-dependent inhibition of the NF-κB/p65 level (Fig. 2). Compared with DMSO-treated
control, a 60% inhibition in NF-κB/p65 protein expression in the
nucleus was observed 10 µM dose of BMS-345541 for 12 h.
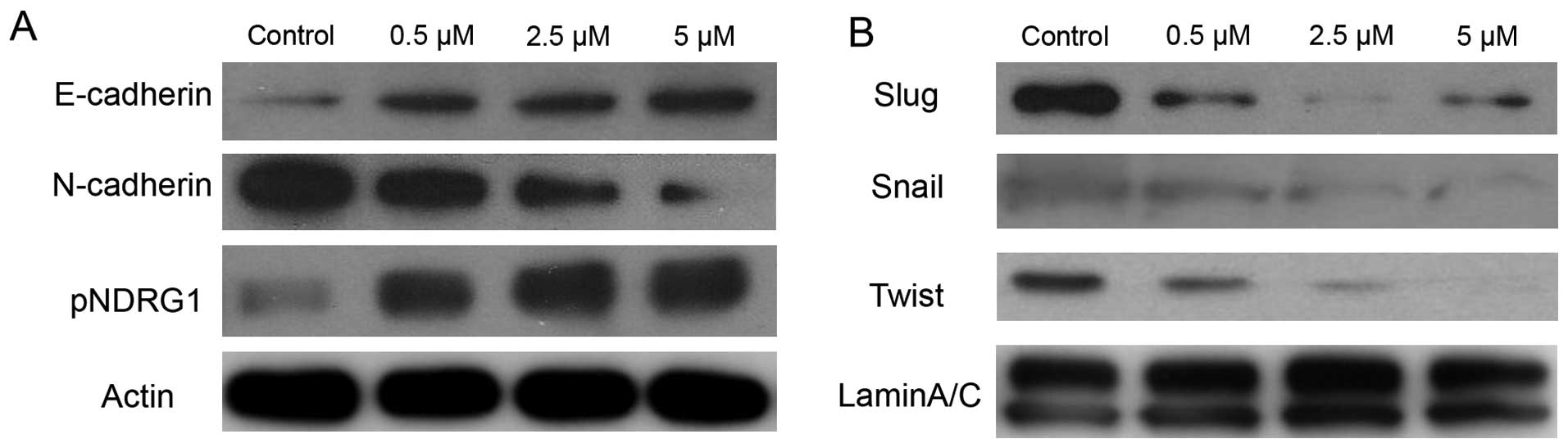
BMS-345541 reverses EMT in PCa cells
Accumulated evidence suggests that prostate cancer
cells can activate the process of EMT, and epithelial cells undergo
multiple biochemical changes including expression of mesenchymal
biomarkers, induction of angiogenesis and resistance to apoptosis
(17–19). To characterize the effect of IKK
inhibitor on EMT, we used BMS-345541 with varying concentration in
PC-3 cells. As shown in Fig. 3,
BMS-345541 induced upregulation of the epithelial marker E-cadherin
at protein level. Nevertheless, we observed downregulation of the
N-cadherin, Snail, Slug and Twist proteins in a dose-dependent
manner.
N-myc downstream-regulated gene 1 (NDRG1) is a
potent metastasis suppressor that has been demonstrated to inhibit
the TGF-β induced EMT in prostate cancer cells (20). To elucidate the molecular role of
NDRG1 in EMT and the IKK inhibitor effect on NDRG1, immunoblot
analysis was used to measure the level of phosphorylation of NDRG1
in PC-3 treated by BMS-345541. There was a significant increase in
phosphorylated NDRG1 by blocking IKK in a dose-dependent manner
(Fig. 3).
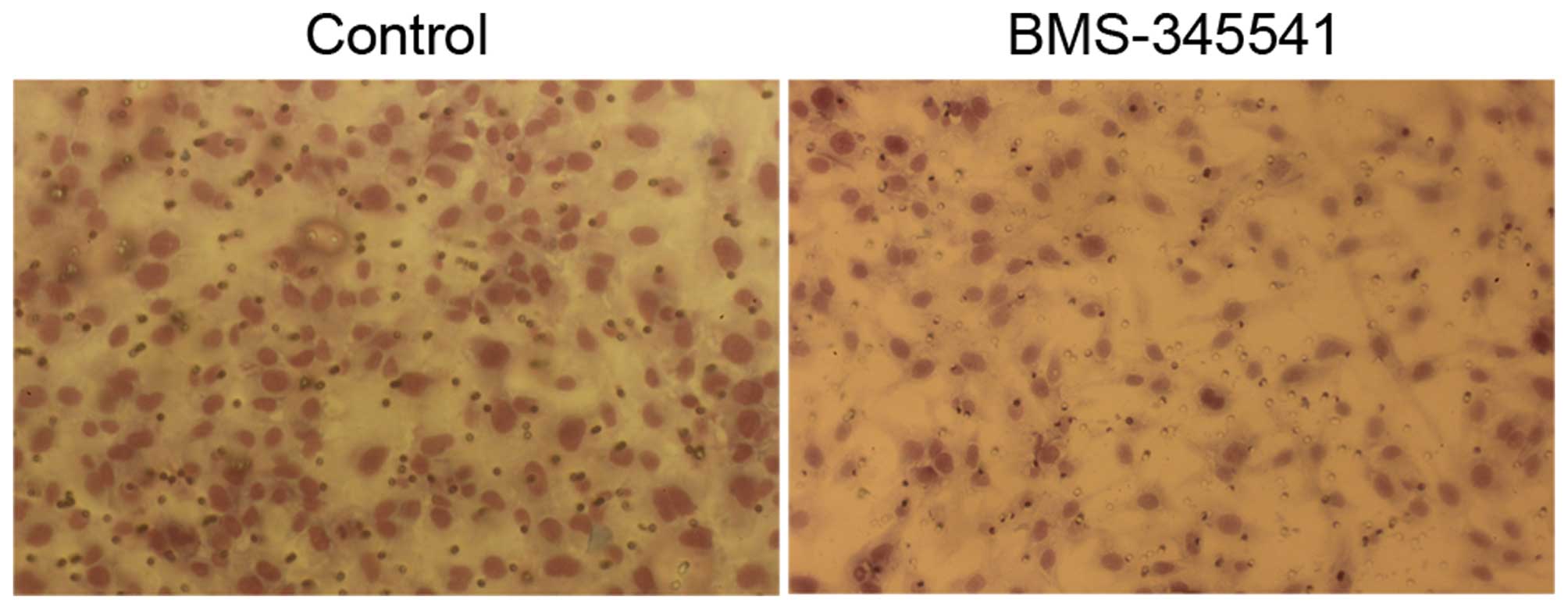
BMS-345541 decreases invasion and
metastasis of PC-3 cells in vitro
We investigated whether BMS-345541 has an impact on
invasion and migration of PC-3 cells, which is usually associated
with the propensity to metastasis. The invasion ability of PC-3
cells was assessed by transwell invasion assay, and BMS-345541
significantly decreased the cell invasion through matrigel-coated
filters by 40% at 24 h (Fig. 4). We
further examined the cell migration by wounding cells plated on the
cell culture plates. The result of wound healing assay showed that
PC-3 cells treated by BMS-345541 were unable to recolonize the
denuded zone as fast as the control cells did at 48 h after removal
(Fig. 5).
The above results demonstrate that IKK inhibitor
exerts a control on the invasion and migration capacities of
prostate cancer cells and suggests that, besides its effect on the
cell proliferative rate, it could also contribute to both tumor
aggressiveness and metastasis.
BMS-345541 induces cell apoptosis of PC-3
cells
PC-3 cells after exposure to BMS-345541 for 72 h
were examined by TUNEL assay. As shown in Fig. 6, apoptotic nuclei and fragmented DNA
were stained dark brown in treated cells, but not in the control
cells. The apoptotic index is significantly higher in the
BMS-345541 treated cells than in the control (Table I).
 | Table IEffect of BMS-345541 on apoptosis in
prostate cancer PC-3 cells (TUNEL assay). |
Table I
Effect of BMS-345541 on apoptosis in
prostate cancer PC-3 cells (TUNEL assay).
| Treatment | Apoptotic index
(%) | P-value |
|---|
| Control | 2.8±0.4 | |
| BMS-345541 | 30.5±1.2 | <0.01 |
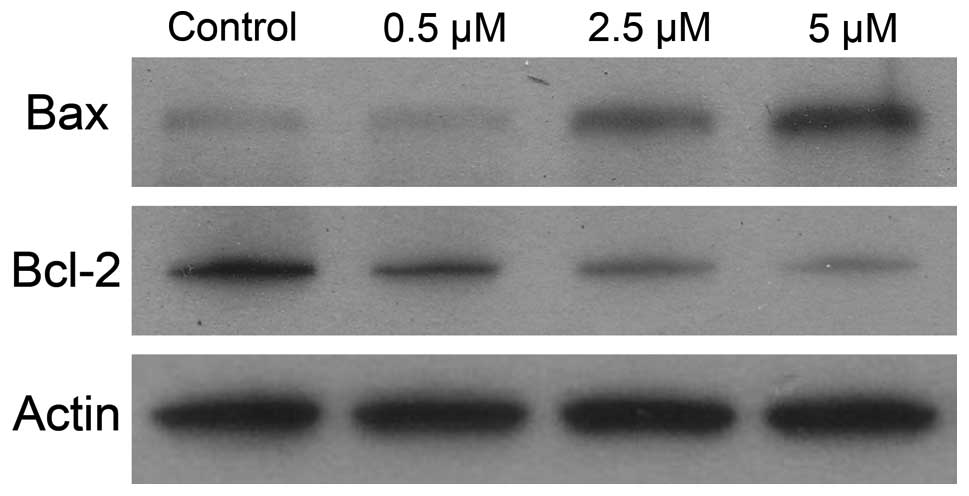
To gain better understanding of the mechanism
leading to cell death, we measured the combined effect of IKK
inhibitor on the expression of Bcl-2 and Bax proteins. As shown in
Fig. 7, the decreases in Bcl-2
expression were significantly greater in samples treated with
BMS-345541 than those in control samples. Moreover, IKK inhibitor
resulted in greater increases in Bax protein expression than
untreated control.
Discussion
Several lines of evidence suggest that IKK/NF-κB
pathway activation is a key event in the acquisition of invasive
and metastatic capacities in prostate cancer (21). Furthermore, NF-κB expression is
upregulated in patients with castration-resistant prostate cancer
(CRPC) who progress more rapidly (13). The kinase subunits of IKK complex
have previously been shown to be involved either directly or
indirectly in the regulation of cellular proliferation (22). Hence, both IKKα and IKKβ are
considered to be therapeutic targets for development of anticancer
agents (23,24). However, the precise mechanisms how
it is achieved in prostate cancer are only partly understood. In
this study, we characterized a highly selective IKK small-molecule
kinase inhibitor-BMS-345541, and demonstrated a key role of IKK in
both EMT and apoptosis of prostate cancer. Unlike other reported
IKK inhibitors, BMS-345541 was found to bind to an unidentified
allosteric site of the catalytic subunits, and so behaves as an
ATP-non-competitive inhibitor. The high selectivity of BMS-345541
for IKKα and IKKβ suggests that the allosteric site is unique to
the IKKs, although it cannot be excluded that the site may also be
present within other kinases not yet tested for selectivity
(14). Especially, BMS-345541 is a
potent and selective inhibitor of IKKα and IKKβ. It displays
10-fold greater selectivity towards IKKβ than IKKα with no activity
towards IKKε or other protein kinases, even at concentrations as
high as 100 µM.
In our study, we showed that BMS-345541 treatment
results in a concentration-dependent suppression of prostate cancer
cell survival in vitro. In addition, BMS-345541 could induce
cell apoptosis and cause inhibition of tumor cell migration and
invasion in PC-3 cells. Thus, we demonstrate the significant
inhibitory effect on CRPC of IKK inhibitor. This phenomenon may
involve several biologic properties such as EMT and programmed cell
death existing in tumor cells. We found that BMS-345541 treatment
resulted in inhibition of cytoplasmic pIκBα and reduction of NF-κB
p65 nuclear translocation. Thus, significant blockade of NF-κB
pathway was achieved by IKK inhibitor. Based on the complexity of
signaling networks that regulate induction of EMT, and the
plasticity of these transitions, it is important to focus on the
most promising methods toward safe and effective reversal of EMT.
IKK/NF-κB appears to be a potential pathway in the regulation of
EMT. Our results showed that IKK inhibitor reduced the critical EMT
markers and transcription factors including N-cadherin, Snail, and
Slug in PC-3 cells. Nevertheless, the level of E-cadherin protein
increased in a dose-dependent manner with BMS-345541 treatment.
N-myc downregulated gene 1 (NDRG1) is a known
metastasis suppressor in multiple cancers, being also involved in
cell growth and differentiation, apoptosis, stress responses,
angiogenesis and EMT (25,26). However, the relationship of IKKs,
EMT and NDRG1 is unclear. Here, we first confirmed the effects of
IKK inhibitor on NDRG1 protein in prostate cancer cells. The
results showed BMS-345541 induced upregulation of membrane pNDRG1
in a dose-dependent manner. It has been reported that NDRG1
modulated EMT through upregulation of the E-cadherin expression,
but downregulation of the N-cadherin, Snail, Slug, and Vimentin
(27). We predict that there is a
complex signaling network among IKK, NDRG1 and EMT which needs
further studies in the future.
Several lines of evidence demonstrated that NF-κB
activation and NDRG1 downregulation can maintain tumor cell
viability, and regulation targeting these factors is sufficient to
induce apoptosis (28,29). In our research, the TUNEL assay
revealed IKK inhibitor regulated both NF-κB and NDRG1, and resulted
in a dramatic induction of prostate cancer cell apoptosis.
Molecules belonging to the B-cell lymphoma leukaemia-2 (Bcl-2)/Bax
system play a crucial role in the regulation of the apoptotic
process. In particular, Bcl-2 is an intracellular protein that
inhibits apoptosis while Bax counteracts the anti-apoptotic
function of Bcl-2 by binding to this molecule. Furthermore, we
conformed that the cell apoptosis was mediated through Bcl-2
downregulation and Bax overexpression. The Bax/Bcl-2 ratio appears
more important than the individual Bax or Bcl-2 level in
determining cell apoptosis, and high Bax/Bcl-2 ratio leads to
greater apoptotic activity.
In conclusion, the IKK inhibitor BMS-345541,
significantly suppresses the growth, invasion, migration of
prostate cancer cells in vitro, as well as induces cell
apoptosis. The mechanism may involve in the blockade of IKK/NF-κB
pathway and EMT, and NDRG1 plays an important role in this process.
Our studies provide a rationale and molecular basis for IKK
inhibitor in the clinical treatment of prostate cancer. IKK
inhibitors have the potential as novel therapeutic agents to deal
with the advanced prostate cancer in the future.
Acknowledgments
This study was supported by the Beijing Municipal
Administration of Hospitals Incubating Program (code:
PX2016050).
References
|
1
|
Bayne CE, Williams SB, Cooperberg MR,
Gleave ME, Graefen M, Montorsi F, Novara G, Smaldone MC,
Sooriakumaran P, Wiklund PN, et al: Treatment of the primary tumor
in metastatic prostate cancer: Current concepts and future
perspectives. Eur Urol. May 20–2015.Epub ahead of print.
S0302–2838. (15): 00378-4. PubMed/NCBI
|
|
2
|
Miyake H and Fujisawa M: Prognostic
prediction following radical prostatectomy for prostate cancer
using conventional as well as molecular biological approaches. Int
J Urol. 20:301–311. 2013. View Article : Google Scholar
|
|
3
|
Lei JH, Liu LR, Wei Q, Song TR, Yang L,
Meng Y and Han P: Androgen-deprivation therapy alone versus
combined with radiation therapy or chemotherapy for nonlocalized
prostate cancer: A systematic review and meta-analysis. Asian J
Androl. 18:102–107. 2016. View Article : Google Scholar :
|
|
4
|
Nandana S and Chung LW: Prostate cancer
progression and metastasis: Potential regulatory pathways for
therapeutic targeting. Am J Clin Exp Urol. 2:92–101.
2014.PubMed/NCBI
|
|
5
|
Clucas J and Valderrama F: ERM proteins in
cancer progression. J Cell Sci. 128:12532015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu J, Lamouille S and Derynck R:
TGF-β-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McConkey DJ, Choi W, Marquis L, Martin F,
Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, et al: Role
of epithelial-to-mesenchymal transition (EMT) in drug sensitivity
and metastasis in bladder cancer. Cancer Metastasis Rev.
28:335–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Min C, Eddy SF, Sherr DH and Sonenshein
GE: NF-kappaB and epithelial to mesenchymal transition of cancer. J
Cell Biochem. 104:733–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Napetschnig J and Wu H: Molecular basis of
NF-κB signaling. Annu Rev Biophys. 42:443–468. 2013. View Article : Google Scholar :
|
|
10
|
Kim SW, Schifano M, Oleksyn D, Jordan CT,
Ryan D, Insel R, Zhao J and Chen L: Protein kinase C-associated
kinase regulates NF-κB activation through inducing IKK activation.
Int J Oncol. 45:1707–1714. 2014.PubMed/NCBI
|
|
11
|
Courtois G and Israël A: IKK regulation
and human genetics. Curr Top Microbiol Immunol. 349:73–95.
2011.
|
|
12
|
Yang J, Amiri KI, Burke JR, Schmid JA and
Richmond A: BMS-345541 targets inhibitor of kappaB kinase and
induces apoptosis in melanoma: Involvement of nuclear factor kappaB
and mitochondria pathways. Clin Cancer Res. 12:950–960. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nguyen DP, Li J, Yadav SS and Tewari AK:
Recent insights into NF-κB signalling pathways and the link between
inflammation and prostate cancer. BJU Int. 114:168–176. 2014.
View Article : Google Scholar
|
|
14
|
Burke JR, Pattoli MA, Gregor KR, Brassil
PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad
J, et al: BMS-345541 is a highly selective inhibitor of I kappa B
kinase that binds at an allosteric site of the enzyme and blocks
NF-kappa B-dependent transcription in mice. J Biol Chem.
278:1450–1456. 2003. View Article : Google Scholar
|
|
15
|
Gasparian AV, Yao YJ, Kowalczyk D, Lyakh
LA, Karseladze A, Slaga TJ and Budunova IV: The role of IKK in
constitutive activation of NF-kappaB transcription factor in
prostate carcinoma cells. J Cell Sci. 115:141–151. 2002.PubMed/NCBI
|
|
16
|
Dyson HJ and Komives EA: Role of disorder
in IκB-NFκB interaction. IUBMB Life. 64:499–505. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luo Y, Cui X, Zhao J, Han Y, Li M, Lin Y,
Jiang Y and Lan L: Cells susceptible to epithelial-mesenchymal
transition are enriched in stem-like side population cells from
prostate cancer. Oncol Rep. 31:874–884. 2014.
|
|
18
|
Behnsawy HM, Miyake H, Harada K and
Fujisawa M: Expression patterns of epithelial-mesenchymal
transition markers in localized prostate cancer: Significance in
clinicopathological outcomes following radical prostatectomy. BJU
Int. 111:30–37. 2013. View Article : Google Scholar
|
|
19
|
Thiery JP, Acloque H, Huang RYJ and Nieto
MA: Epithelial- mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen Z, Zhang D, Yue F, Zheng M, Kovacevic
Z and Richardson DR: The iron chelators Dp44mT and DFO inhibit
TGF-β-induced epithelial-mesenchymal transition via upregulation of
N-Myc downstream-regulated gene 1 (NDRG1). J Biol Chem.
287:17016–17028. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jain G, Voogdt C, Tobias A, Spindler KD,
Möller P, Cronauer MV and Marienfeld RB: IκB kinases modulate the
activity of the androgen receptor in prostate carcinoma cell lines.
Neoplasia. 14:178–189. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bradford JW and Baldwin AS: IKK/nuclear
factor-kappaB and oncogenesis: Roles in tumor-initiating cells and
in the tumor microenvironment. Adv Cancer Res. 121:125–145. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tada Y, Kokabu S, Sugiyama G, Nakatomi C,
Aoki K, Fukushima H, Osawa K, Sugamori Y, Ohya K, Okamoto M, et al:
The novel IκB kinase β inhibitor IMD-0560 prevents bone invasion by
oral squamous cell carcinoma. Oncotarget. 5:12317–12330. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi J, Chen J, Serradji N, Xu X, Zhou H,
Ma Y, Sun Z, Jiang P, Du Y, Yang J, et al: PMS1077 sensitizes TNF-α
induced apoptosis in human prostate cancer cells by blocking NF-κB
signaling pathway. PLoS One. 8:e611322013. View Article : Google Scholar
|
|
25
|
Fang BA, Kovačević Ž, Park KC, Kalinowski
DS, Jansson PJ, Lane DJR, Sahni S and Richardson DR: Molecular
functions of the iron-regulated metastasis suppressor, NDRG1, and
its potential as a molecular target for cancer therapy. Biochim
Biophys Acta. 1845:1–19. 2014.
|
|
26
|
Sánchez-Tilló E, Liu Y, de Barrios O,
Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A
and Postigo A: EMT-activating transcription factors in cancer:
Beyond EMT and tumor invasiveness. Cell Mol Life Sci. 69:3429–3456.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JC, Chung LC, Chen YJ, Feng TH and
Juang HH: N-myc downstream-regulated gene 1 downregulates cell
proliferation, invasiveness, and tumorigenesis in human oral
squamous cell carcinoma. Cancer Lett. 355:242–252. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McCall P, Bennett L, Ahmad I, Mackenzie
LM, Forbes IWG, Leung HY, Sansom OJ, Orange C, Seywright M,
Underwood MA, et al: NFκB signalling is upregulated in a subset of
castrate- resistant prostate cancer patients and correlates with
disease progression. Br J Cancer. 107:1554–1563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kovacevic Z and Richardson DR: The
metastasis suppressor, Ndrg-1: A new ally in the fight against
cancer. Carcinogenesis. 27:2355–2366. 2006. View Article : Google Scholar : PubMed/NCBI
|