Introduction
Thirty to 80% of cancer patients display a cachectic
state mostly attributed to involuntary weight loss greater than 5%
after 6 months of diagnosis (1,2). This
complication is responsible for a decreased quality of life, a
reduced tolerance to therapy (3)
and for more than 20% of death in cancer patients (4). Cancer cachexia is a complex clinical
syndrome with progressive development from pre-cachexia stage to
refractory cachexia (2).
Clinically, cancer cachexia leads to a severe loss of fat and
skeletal muscle mass usually associated with anorexia, anemia and a
pronounced asthenia with considerable alterations of lipid and
protein metabolism (2–4). Cancer cachexia is not fully explained
by caloric restriction. Although it can have a key role in muscle
loss, it remains elusive whether caloric restriction is responsible
for the genesis of cancer cachexia. Indeed, the mechanisms of
muscle loss and metabolic profiles are distinct between
tumor-bearing and long-term fasting subjects (5). Furthermore, nutritional
supplementation and pharmacological manipulation of appetite are
unable to restore loss of lean body mass (4,6). It
has been proposed that skeletal muscle is the major tissue involved
in cancer cachexia, accounting for 40% of total body weight loss
(3,6). However, other recent studies suggest
that other tissues such as brain, liver and heart could also be
implicated in the cachectic processes and directly impact muscle
loss (7). Hence, cancer cachexia
should be considered as a multifactorial syndrome.
Numerous studies managed to characterize mechanisms
underlying skeletal muscle atrophy during cancer. However, little
attention has been paid to the alteration of myocardial structure
and metabolic disorders that occur in the heart. It was proposed in
the 1970s that 7% of death during cancer could be attributed to
heart failure (8), an assumption
supported by the reduction in cardiac mass and function in patients
(9). Independently from cancer
itself, anticancer treatment including chemo- and/or radiotherapies
may have some deleterious effects on myocardium and can thus
exacerbate cardiac impairments that occur during cancer cachexia
(10,11). Patients being treated for a chronic
illness like cancer were often told by their doctor to rest and
reduce their physical activity. For a long time, exercise was
thought to cause pain, high heart rate, dyspnea and fatigue. In
contrast, randomized controlled trials showed a beneficial effect
of physical activity on physical fitness, quality of life, anxiety
or self-esteem, and exercise is now thought as a counter-measure
that could attenuate cardiac cachexia in patients (12).
Given the importance of cardiac function on health,
it is crucial to identify the molecular basis of cancer-induced
cardiac cachexia and to propose alternative tools for its
prevention. The present review aims to characterize cardiac
remodeling and to depict the potential benefits of physical
exercise on heart function during cancer cachexia.
Phenotype of cardiac remodeling during
cancer
Structural and functional aspects
Ectopic implantation of C26 colon carcinoma cells is
a widely used model of cancer cachexia in rodent. Myocardial
atrophy is a common feature observed in this model (13–15)
with a decrease in heart weight reaching ~20% in tumor-bearing mice
(Table I). This atrophy seems to be
greater in males compared to females (−22 vs. −10% in heart weight
27 days after tumor inoculation) (15). This striking difference in
myocardial atrophy could be explained by sexual hormones as
inhibition of estrogens receptor signaling, totally abolished this
protective mechanism (15). The
mode of administration can also influence the severity of atrophy,
a more pronounced decrease in heart weight being recorded after
intraperitoneal vs. subcutaneous injection of C26 tumor cells
(16). Importantly, cardiac atrophy
is not restricted to C26 model as it has been observed in several
animal models including cancer cell inoculation, genetic models and
chemically-induced tumorigenesis (Table
I). Moreover, reduced heart mass has been reported in cachectic
patients (gastrointestinal, pancreatic and non-small cell lung
cancer) compared with non-cachectic cancer patients and controls
(17).
 | Table I.Structural and functional alterations
of cardiac muscle during cancer cachexia. |
Table I.
Structural and functional alterations
of cardiac muscle during cancer cachexia.
| Model | Species | Duration (days) or
age | Structure | Function | Ref. |
|---|
| Apcmin/+
(small intestine/colon tumors) | M | 12 weeks old | HW −8% |
| (26) |
|
|
| 20 weeks old | HW −6% |
|
|
|
| M | 14 weeks old | HW −14% |
| (21) |
| Yoshida AH-130
hepatoma cells | R | 5 to 13 | HW −13 to 40% | LV EF −38% | (17) |
|
|
|
| Progressive loss of
LV mass | LV FS −40% |
|
|
|
|
| LV SV −89% |
|
|
|
|
|
| LV CO −86% |
|
|
| R | 11–14 | HW −34% | LV FS −37% | (27) |
|
|
|
|
| LV EF −28% |
|
|
|
|
|
| CO −46% |
|
|
| R | 11–14 | HW −33% | LV EF −23% | (28) |
|
|
|
| LV mass −25% | LV FS −40% |
|
|
|
| LV PWT −26% | CO −47% |
| C26 colon carcinoma
cells | M | 15 to 27 | HW −8 to −22% | Aortic velocity
−16% | (15) |
|
|
|
|
| Aortic pressure
−30% |
|
| M | 14 | HW −23% |
| (16) |
|
|
|
| HW −6% |
|
|
| M | 21 | HW −14% | FS −22% | (21) |
|
|
|
|
| EF −39% |
|
|
|
|
|
| SV −34% |
|
|
| M | 17 | HW −21% | FS −33% | (13,18) |
|
|
|
| PWT −30% | EF −21% |
|
|
|
|
|
| LVSD +28% |
|
|
| M | 17 | HW −27% | FS −27% | (19) |
|
|
|
| LV mass −24% | EF −16% |
|
|
|
|
| PWT −20% |
|
|
|
| M | 25 | HW −20% |
| (29) |
| Inhibin-α KO
(gonadal tumor) | M | 8–12 weeks old | HW −26 to −29% |
| (29) |
| Lewis lung
carcinoma cells | M | 21 | LV mass = | FS = |
|
|
|
|
| PWT = |
| (30) |
| Murine
adenocarcinoma cell 16 (colon) | M | 30 | HW −25% |
| (31) |
| MCA-induced
sarcoma | M | 11 | HW −15% |
| (20) |
| Walker-256 cells
(breast) | R | 5 and 10 | HW = |
| (32) |
|
|
|
| Right HW −30 and
−40% |
|
Echocardiographic measures confirm the loss of
cardiac mass since a decrease in left ventricle (LV) posterior wall
thickness was observed in many studies (Table I). Tian et al (18) also reported a 28% decrease in
interventricular septum along with a 30% decrease of LV posterior
wall thickness. Atrophy of cardiac wall is related to a reduction
of cardiomyocyte cross sectional area (14,19–21).
Moreover, electronic microscopy revealed disturbed alignment of
sarcomeric arrangement further demonstrating the impairment of
myocardium muscle structure with cancer (13). De novo expression of fetal
genes is commonly observed in studies underlying mechanisms of
cardiac remodeling including cancer cachexia. For example, strong
re-expression of myosin heavy chain (MyHC)-β was recorded in C26
tumor-bearing mice at the expense of MyHC-α (13,15,18).
In line with this, other contractile protein such as troponin I,
myosin light chain, α-actin are degraded (13,15).
Accordingly, this degradation of contractile protein could
deleteriously impact cardiac strength generation (22).
Besides, histological analyses show a potent
fibrosis as 50–65% of fibrosis was found in heart of C26 tumor
bearing mice (13,15). Fibrosis of cardiac tissue is usually
implicated in myocardial pathology and contributes to decrease
heart performance. This could be explained by the deposit of
collagen causing heart stiffness, hence the alteration of cardiac
function. In fact, fibrosis can induce heart failure (HF) through
the fading of diastolic function (23).
Altogether, these data unveil the potential
impairment of myocardium structure during cancer cachexia which is
essentially represented by a high fibrosis, an impaired sarcomeric
structure and a decrease in wall cardiac thickness. These
morphological abnormalities will negatively impact cardiac function
(Table I). For example, Cosper and
Leinwand (15) in 2011 reported a
30% reduction in aortic pressure and a 16% decrease in aortic
velocity in tumor-bearing mice compared to controls. Impaired
fraction ejection and fractional shortening have also been observed
in C26 and AH-130 tumor bearing animals (Table I). Moreover, MyHC-β is associated
with a lower ATPase activity compared to α isoform and
re-expression of the fetal MyHC-β is therefore associated with
altered LV function (24).
In summary, along with morphological and functional
abnormalities observed during cancer cachexia, all the data exposed
above provide evidence claiming that cancer cachexia will
ultimately lead to HF. In fact, HF would be a slow mechanism
related to cancer cachexia and could be prominent before death. In
turn, HF will accelerate cachexia (11). It is hypothesized that myocardial
energetic perturbation that occurs during HF is due to depletion of
ATP with impaired efficiency of mechanical work leading to
increased resting energy expenditure (25). These data underline the importance
of investigating myocardial energy balance during cancer-mediated
cardiac cachexia.
Metabolic aspects
It is well established that alteration of energy
balance contributes to cancer cachexia, increased energy
expenditure being the main cause of wasting associated with
cachexia (33). Indeed, a study
conducted on a group of 297 unselected cancer patients showed that
49% of patients developed a hyper metabolism (34) and similar conclusion was already
drawn earlier (35). Abnormalities
in carbohydrate and lipid metabolism and/or mitochondrial
dysfunction are major biochemical bases of elevated resting energy
expenditure. Mitochondria within the myocardium represent 30% of
the cardiac volume (36). In adult
mammalian myocardium, ATP production is essentially mediated by
mitochondrial oxidative phosphorylation as 70% of ATP is derived
from fatty acid oxidation while the rest is provided by glucose and
lactate oxidation along with other ketone body (37). The capacity of heart to oxidize
fatty acids is a key feature as an alteration of lipid metabolism
is classically associated to HF (38).
Early report evidenced a decrease in the total
mitochondrial volume and in citrate synthase activity (a marker of
mitochondrial content) within hearts of methylcholanthrene-induced
sarcoma mice (20). In addition,
Tian et al (13) showed
impaired mitochondrial structures in heart of tumor-bearing mice,
including broken membranes and disorganized cristae. These
alterations can result in a decreased capacity to oxidize energetic
substrates. Although they did not report significant alteration in
mitochondrial structure, Muhlfeld et al (30) observed an increase in triglyceride
content which is consistent with an impaired capacity of lipid
oxidation. However, others found no change (20) or even a decrease in lipid content
(39). Among the few data
available, results from Drott and coworkers show an increase in
cardiac oxygen consumption together with a decrease in glucose
uptake in non-cachectic tumor-bearing rats (39,40).
Glucose entry into cardiac cells is enhanced by
insulin which triggers glucose transporter type 4 (GLUT4)
translocation to the membrane. A decrease in insulin sensitivity
can thus explain the reduction in glucose uptake observed in these
rats. In addition, upregulation of GLUT1 expression concomitantly
to a decrease in GLUT4 expression has been observed in
tumor-bearing mice (18). GLUT1
expression is usually restricted to fetal tissue and is responsible
for a lower level of glucose transport compared to the GLUT4
isoform (41). Re-expression of
GLUT1 could be an adaptive response of the organism in order to
compensate hypoglycemia that can occur at the latest stage of
cancer, although the issue of heart metabolism during cancer
cachexia needs to be better characterized.
Overall, these alterations could explain the
morphological and functional abnormalities in myocardium of
tumor-bearing mice, especially the adenocarcinoma C26 model.
Regardless of tumor model, however, few studies have investigated
the extended myocardium atrophy including functional implications
and signaling pathways.
Mechanisms initiating cardiac cachexia
during cancer
Inflammation
Heart metabolism can be altered by chronic low grade
inflammation. In the C26 tumor model, pro-inflammatory cytokines
are released by the tumor and this process is believed to promote
cancer cachexia (42). The
pro-inflammatory interleukin-6 (IL-6) is strongly induced in heart
from cancer mice (13–15,18).
If acute cytokines release can protect tissues from insult, chronic
elevation in pro-inflammatory mediators fosters heart dysfunction.
Indeed, elevated IL-6 levels are associated with LV dysfunction
(43) and it has been proposed that
IL-6 contributes to the induction of fetal genes such as MyHC-β
(44).
Similarly to IL-6, tumor necrosis factor-α (TNF-α),
another key mediator of inflammatory response, is involved in the
physiopathology of diverse cardiomyopathies (45). TNF-α is notably a potent activator
of MuRF1, an E3 ligase involved in protein breakdown (cf. below),
and can thus promote muscle atrophy (46). Accordingly, an early study reported
that inhibition of TNF-α slowed down skeletal and cardiac
proteolysis in rats bearing the Yoshida AH-130 hepatoma (47).
Furthermore, Fn14, the cognate receptor of the
cytokine TNF-like weak inducer of apoptosis (TWEAK), is highly
expressed by C26 tumor cells and tumor-secreted Fn14 was
demonstrated to initiate cachexia (48). In fact, treatment with Fn14 antibody
but not genetic knockdown of Fn14 or TWEAK in host slowed tumor
growth and extended lifespan of tumor-bearing C26 mice.
Additionally, myocardium atrophy was prevented in cancer
cell-injected animals treated with anti-Fn14. Although TWEAK/Fn14
promotes inflammation via activation of the nuclear factor-kappa B
(NF-κB), it was not established whether the protective effects of
Fn14 inhibition were related to modulation of inflammatory
response.
Recently, seven tumor-secreted factors by C26 cells
have been identified to trigger cardiomyocytes atrophy and aberrant
lipid metabolism (21). Among these
so-called ‘cachexokines’, elevated serum Ataxin-10 is a common
feature of several colon tumor secretomes and is also highly
expressed in pancreatic cancer and in mouse model of obesity and
type II diabetes. Furthermore, serum Ataxin-10 expression
correlates with body wasting level. Altogether, these findings led
the authors to propose serum Ataxin-10 as a potential marker of
cardiac cachexia. Besides, deleterious effects of prolonged
inflammation are also related to the increase in oxidative
stress.
Oxidative stress
Signs of oxidative stress have been observed in
heart of rats inoculated either with Walker-256 tumors (32) or with AH-130 Yoshida hepatoma cells
(49). In this model, it was
demonstrated that proteins involved in muscle contraction and
metabolism are significantly more oxidized in heart of the
cachectic animals (14). Moreover,
administration of resveratrol, a potent antioxidant, prevented
cardiac atrophy in C26 tumor-bearing mice. Surprisingly, positive
effect of resveratrol on skeletal muscle mass was not retrieved in
LLC or AH-130 tumor-bearing animals, suggesting cancer-specific
effects (50).
Mitochondrion is a major source of reactive oxygen
species (ROS). In addition to inflammation, abnormality in
respiratory chain function can lead to enhanced oxidative stress. A
disruption in the balance between pro- and anti-oxidant systems may
also occur via impaired delivery of vitamins and antioxidants
resulting from anorexia, nausea and vomiting in cancer patients
(51). Lastly, anti-neoplastic
drugs such as cisplatin or doxorubicin could be responsible for ROS
accumulation, a condition known for promoting muscle atrophy
(52,53). In addition to mitochondria, xanthine
oxidase can generate ROS. Accordingly, inhibition of xanthine
oxidase reduces cardiac atrophy and maintains heart function in
rats inoculated with AH-130 Yoshida hepatoma cells (54). This further demonstrates the role of
ROS overproduction in cardiac cachexia.
ROS could alter calcium homeostasis by stimulating
calcium release and inhibiting calcium re-uptake by the
sarcoplasmic reticulum which result in calcium overload. Calcium
overload contributes to impair contractile function by promoting
release of the pro-apoptotic factor cytochrome c. In fact,
being located near the sites of calcium efflux, mitochondria will
capture excessive calcium load. This in turn results in the opening
of the mitochondrial permeability transition pore and the
subsequent release of cytochrome c which activates apoptosis
pathways. The unfavorable oxidative balance that occurs during
cancer also induces apoptosis through p38 MAPK-dependent
activation. Inhibitors of p38-MAPK have been shown to prevent
ROS-induced apoptosis, suggesting that targeting this signaling
pathway could attenuate cardiac dysfunction and decrease myocyte
apoptosis (55).
Insulin resistance
Cardiac mass and metabolism are also regulated by
insulin. Insulin activates protein synthesis and represses
proteolysis through binding to its receptor and subsequent
activation of the Akt/mammalian target of rapamycin complex 1
(mTORC1) pathway (see next section). In addition, insulin promotes
GLUT4-mediated glucose uptake into cardiac cells in an
Akt-dependent way. A defect in insulin sensitivity or secretion,
both observed in cancer (7,56), would thus impair both cardiac mass
and metabolism. Chronic inflammation is an identified cause of
insulin resistance in skeletal muscle but also in heart (57). Notably, insulin resistance and
mitochondrial dysfunction initiate heart failure in a model of
pressure overload (58),
underlining the importance of metabolic features in cardiac
function.
Molecular mechanisms underlying cardiac
cachexia during cancer
Atrophic pathways
Cardiac muscle mass is finely controlled by the
balance between protein synthesis and breakdown. The Akt/mTORC1
pathway controls the protein metabolism through the concomitant
stimulation of protein translation and inhibition of proteolytic
pathways. Akt is activated by insulin or insulin-like growth
factor-1 (IGF-1). A decrease in Akt/mTORC1 pathway activation will
thus result in muscle atrophy because of reduced protein synthesis
and enhanced protein breakdown. Accordingly, Manne et al
(26) reported a decrease in
cardiac protein synthesis together with a reduction of mTORC1
activity in adenomatous polyposis coli muted mice
(ApcMin/+), a model of colorectal cancer. Consistent
with these findings, tumor-bearing AH-130 rats display a marked
reduction in IGF-1 receptor and reduced phosphorylation of key
downstream targets, including Akt and GSK3β (17). In contrast, Akt/mTORC1 pathway was
overactivated in N-Methyl-N-nitrosourea (MNU)-induced breast cancer
rats (59) and although mTORC1
activity was reduced, Akt phosphorylation was indeed increased in
ApcMin/+ mice (26). If
sustained activity of the Akt/mTORC1 pathway may spare cardiac mass
under cancer cachexia, excessive activation can lead to
pathological hypertrophy associated with fibrosis (59). This points to a complex
dysregulation of the Akt/mTORC1 pathway in heart with cancer. In
contrast to IGF-1, myostatin has negative effects on Akt/mTORC1
activation (60). Mammary
tumorigenesis induced by administration of MNU leads to enhanced
myostatin expression in the myocardium (59). Higher levels of phosphorylated
Smad-3, the down-stream effector of myostatin, were also observed
(59). Similar results were found
in myocardium of AH-130 tumor-bearing rats (17). Notably, pharmacological blockage of
the myostatin receptor reversed both skeletal and cardiac muscle
wasting during cancer cachexia (29). Old myostatin KO mice display lower
fibrosis and improved cardiac function (61) further underlining the potential role
of myostatin in the pathogenesis of HF. However, cardiac-specific
myostatin deletion did not restore either heart mass or function in
a model of HF in mice (62).
Therefore, the effect of myostatin inhibition on heart function
during cancer remains to be ascertained.
Importantly, the Akt/mTORC1 signaling inhibits the
two major pathways responsible for proteolysis: the
ubiquitin/proteasome system and the autophagy/lysosome process.
Proteasomal degradation requires the labelling of targeted proteins
through the successive activity of the so-called
ubiquitin-activating (E1), -conjugating (E2) and -ligase (E3)
enzymes. Polyubiquitinylated proteins are then addressed to and
hydrolyzed by the 26S proteasome. Two muscle-specific E3 enzymes,
Atrogin-1 (also called MAFbx or Fbox32) and muscle ring finger 1
(MuRF1), have been widely studied because of their crucial role in
the regulation of both skeletal and cardiac muscle mass. Indeed,
MuRF1 KO mice are protected against cardiac atrophy induced by
glucocorticoids or by the reversal of transaortic constriction
(63) and MuRF1 transgenic mice
have thinner LV wall thickness (64). Atrogin-1 has been shown to inhibit
cardiac hypertrophy, notably through the degradation of calcineurin
(65) while its adenoviral
overexpression reduces cardiomyocytes size (66). Consistently, increased cardiac
expression of MuRF1 and Atrogin-1 has been reported in mice
inoculated with colon-26 adenocarcinoma cells (16,18,19) or
colon adenocarcinoma cell lines 16 (31). Interestingly, Atrogin-1 expression
is also increased in response to doxorubicin, an effective
antitumor agent (66). MuRF1
targets structural proteins such as MyHC or troponin 1 (reviewed in
ref. 67). However, recent advances
point out the role of MuRF1 in energetic metabolism, notably
through the degradation of the creatine kinase. There are few data
on the role of other E3 ligases (e.g. F-box and leucine-rich repeat
protein 22, murine double minute 2 or c-terminus of heat shock
protein 70-interacting protein) in cancer. Therefore, one may
suppose that the knowledge about the implication of the
ubiquitin/proteasome system in cardiac remodeling during cancer
cachexia is still fragmented.
Autophagy (ATG) is a process that ensures the
degradation of long-lived proteins, macromolecules and organelles
including lipids, glycogen, mitochondria or ribosomes by lysosomal
vesicles. In skeletal or cardiac muscle, basal ATG is important to
recycle damaged-mitochondria and to provide amino acids for protein
synthesis. Excessive activation or inhibition of ATG flux
contributes to muscle atrophy (68)
and to the development of heart disease (69). An increase in the expression of LC3,
which controls a key step in ATG initiation, and of cathepsin L, a
lysosomal protease, has been observed in the hearts of C26
tumor-bearing mice (15).
Importantly, these animals displayed strong increase in the protein
content of activated LC3 and atrophic hearts have increased
autolysosomes and autophagosomes number. Manne et al
(26) also observed an increase in
Beclin protein expression, another marker of ATG activation.
Recently, restoration of cardiac mass and function in tumor-bearing
rats by appetite stimulation was associated with the normalization
of ATG markers (70). This emerging
literature reinforces the idea that ATG contributes to cardiac
muscle atrophy with cancer.
ATG and ubiquitin/proteasome systems can be
transcriptionally activated by the forkhead family of transcription
factors FOXO1, FOXO3A and FOXO4 in skeletal muscle (71). FOXO is directly phosphorylated by
Akt, leading to its cytosolic sequestration. FOXOs inhibition
partly prevented skeletal muscle atrophy following C26 cells
inoculation (72). In heart, FOXO3A
promotes MuRF1 and Atrogin-1 expression while it inhibits
cardiomyocyte hypertrophy and reduces cardiac cell size (73). Therefore, FOXOs could be considered
as a key regulator of cardiac atrophy in tumor-bearing mice.
However, while there is strong presumption about the implication of
FOXOs in heart atrophy during cancer cachexia, further work remains
to be done in order to characterize its role.
NF-κB is another central transcription factor
involved in cardiac cachexia. NF-κB exists as a heterodimer of two
subunits (p65 and p50), which are sequestered in an inactive form
in the cytoplasm by the repressor IκBα. Inhibition of NF-κB
prevents skeletal muscle loss under denervation and cancer cachexia
(74). NF-κB acts notably through
MuRF1 to promote muscle atrophy (74,75).
NF-κB expression is upregulated in the heart of tumor-bearing
animals (14,59). Resveratrol inhibits IκB
kinase-mediated NF-κB activation, and promotes SIRT1-dependent p65
deacetylation, resulting in the repression of NF-κB transcriptional
activity (14). Resveratrol also
blunted MuRF1 expression (14) and
prevented cardiomyocyte atrophy as well as functional impairment in
mice implanted with C26 cancer cells (19). Importantly these beneficial effects
occurred without alteration of tumor-derived cytokines release
underlining the direct effect of NF-κB on cardiac cells.
Oxidative metabolism
Cardiac ATP production is highly dependent on lipid
metabolism. Lipid oxidation is transcriptionally promoted by the
peroxisome-proliferator activated receptor α (PPARα). Among the
genes upregulated by PPARα, CPT1 (carnitine palmitoyltransferase)
is a critical step of fatty acid β oxidation as it allows the entry
of fatty acid in mitochondria. At a refractory stage, cardiac PPARα
and CPT1 mRNA expressions were significantly reduced in C26
tumor-bearing mice (18). Similar
results were observed using unloaded hearts that exhibit
cardiomyocyte atrophy (76). As
stated before, this metabolic reshuffle from lipid to glucose is
harmful to cardiac metabolism as it has been associated to imminent
HF occurrence (77). A greater
glucose oxidation can nevertheless reflect a positive adaptation.
Indeed, glucose oxidation provides more energy (ATP) per molecule
of oxygen consumed compared to fatty acid (higher P/O ratio).
However, if insufficiently compensated, the disturbance of energy
balance capacity will impede heart function. In physiological
conditions, AMPK plays a crucial role in protecting cardiac cells
from energy homeostasis perturbations via activation of catabolic
pathways to generate ATP. ROS can reduce AMPK activation by
oxidation of cysteine residues (78), leading to decreased phosphorylation
of ACC (acetyl-CoA carboxylase), an AMPK downstream target. The
lack of ACC inhibition will then result in the impairment of
CPT1-dependent fatty acid entry into the mitochondria.
A TNF-α/NF-κB pathway has been proposed to increase
glucose oxidation at the expense of lipids, by the inhibition of
PPARγ coactivator 1-α (PGC-1α) (79). In addition, although its use has
been restricted due to cardiac complications, the PPARγ agonist
rosiglitazone has been shown to improve the phenotype of AH-130
tumor-bearing rats (28). Indeed,
treatment restored several markers of cardiac function, increased
septum thickness together with the abolition of proteasome
enzymatic activity upregulation and enhanced locomotor activity.
Rosiglitazone has anti-diabetic effects via the improvement of
insulin sensitivity, and PPARγ activation has also
anti-inflammatory actions on cardiovascular system notably by the
inhibition of NF-κB (reviewed in ref. 80). However, these results should be
interpreted with cautious because of differences in PPARγ agonists
effects between mouse models and human (80). Cardiomyocytes cultured with
conditioned medium from C26 cells display aberrant fatty acid
oxidation (21), suggesting that
tumor released-factors will not systematically inhibit lipid
oxidation as previously suggested by Drott and coworkers.
Altogether this points to the importance of better understanding
lipid metabolism regulation in heart during cancer. Interestingly,
PPARs and AMPK could be major targets for an effective strategy to
offset the altered cardiac metabolism.
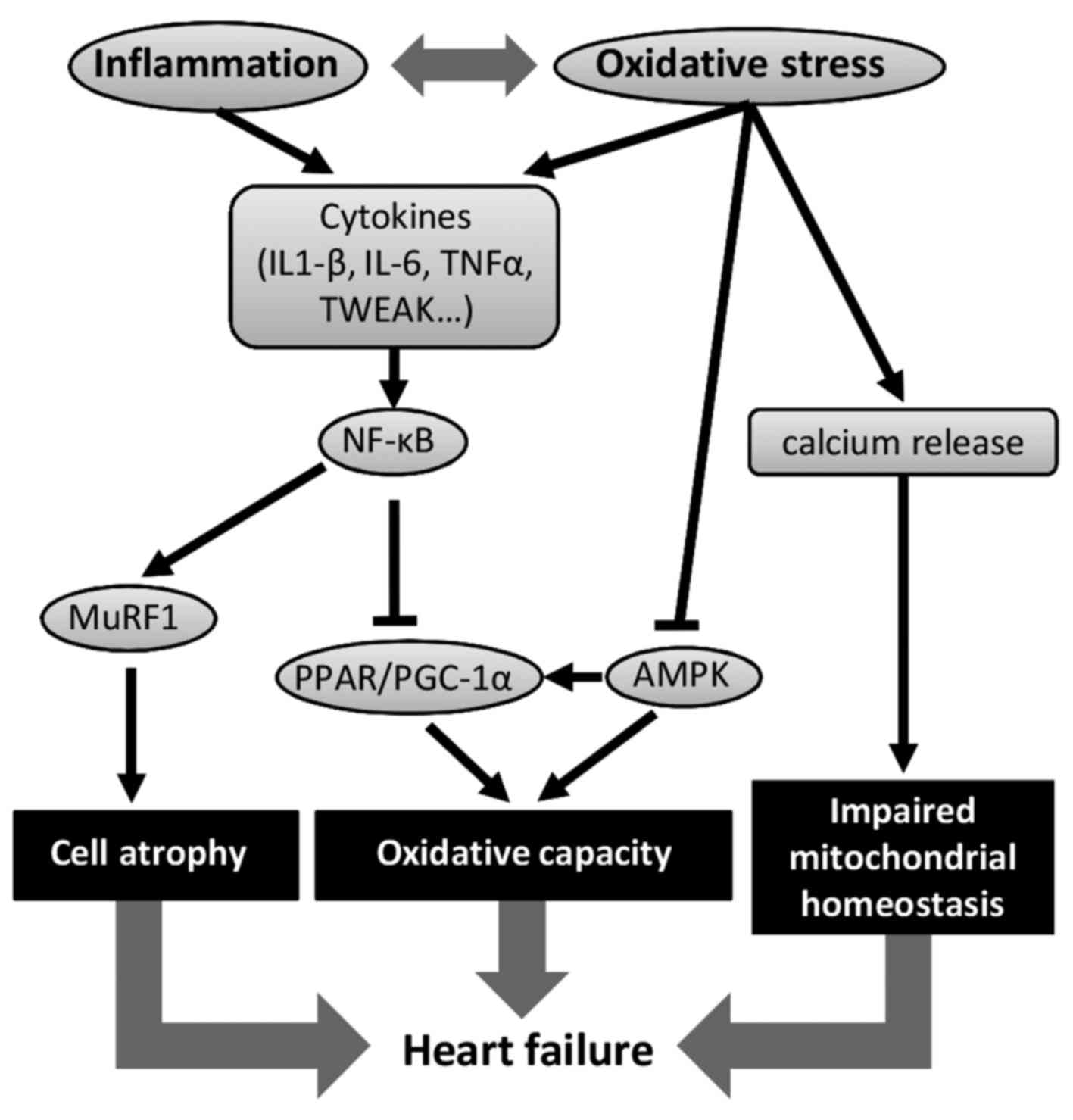
All the mechanisms described above highlight the
importance of decreasing inflammation, oxidative stress and insulin
resistance in order to maintain: i) cardiac muscle mass; ii)
mitochondrial homeostasis; and iii) oxidative capacity (Fig. 1).
Positive effects of physical exercise on
cardiac cachexia
A number of studies confirm the beneficial effects
of regular aerobic exercise on heart function in pathological
conditions (reviewed in ref. 81).
For instance, hypertensive rats displayed significant reduction of
cardiac fibrosis (40%) upon aerobic training and this was
associated with improved diastolic function (82). Similarly, endurance training reduced
myocardial infarction-mediated fibrosis (83). Endurance training is also
responsible for physiological hypertrophy in cardiac myocytes and
improves contractile function as attested by higher calcium
sensitivity and enhanced calcium handling in female rats compared
to sedentary (84). In
physiological conditions, aerobic exercise increases MyHC-α protein
expression while β isoform content remains unchanged (85,86).
This mechanism also occurs in pathological conditions since
exercise prevented the increase in MyHC-β to MyHC-α mRNA ratio
after myocardial infarction (83).
In cancer, aerobic training restores the tolerance to physical
activity allowing therefore, a better quality of life (87). Importantly, regular physical
activity seems to slow down tumor growth (88). This finding is confirmed by Goh
et al (89), which unveiled
the potent role of exercise training as a preventive measure for
tumor progression. In fact, results of this study show that the
longer distance tumor- bearing mice browse, the smaller their tumor
is. Furthermore, it has been shown that aerobic training enhances
life span of rats bearing Walker 256 tumor (90).
Exercise and systemic inflammation
Aerobic training has been shown to reduce chronical
inflammation (87), although acute
endurance exercise increases serum IL-6 and myocardial IL-6
receptor protein content (91).
Noteworthy, cytokines response to exercise differs from the ones
that occur in chronic diseases characterized by an inflammatory
state (92). Indeed, plasmatic
TNF-α and IL-1β (two pro-inflammatory cytokines) do not increase in
response to exercise. Exercise is also associated with high levels
of anti-inflammatory cytokines like IL-10 and pro-inflammatory
cytokines inhibitors such as IL-1ra (antagonist receptor for IL-1),
sTNFα-r1 and sTNFα-r2 (antagonist receptors for TNF-α).
Accordingly, it has recently been shown that endurance exercise
blunts TNF-α-mediated NF-κB activation in skeletal muscle (93) and regular exercise limits TNF-α,
IL-6 and IL-1β induction in heart after myocardial infarction
(83). During multiple pathologies,
the stress is chronic with low and constant level of
pro-inflammatory cytokines as observed for IL-6 during C26-induced
cardiac cachexia (15). Exercise
will modulate this low grade of systemic inflammation by
establishing an anti-inflammatory environment via anti-inflammatory
cytokines release (IL-1ra and IL-10) and cytokines inhibitors
(sTNFα-r1 and sTNFα-r2). This releasing process, initiated within
local tissues and progresses to peripheral ones, is essentially
mediated by the myokine IL-6 underlining its anti-inflammatory
effect when induced by exercise (92). In agreement, a recent report showed
that 35 weeks of aerobic training prevented the increase in TWEAK
expression (in serum and heart) as well as NF-κB activation but did
not alter myostatin expression in C26 tumor- bearing mice (59). This result suggests that moderate
exercise training could modulate cancer-induced cardiac remodeling
through the TWEAK/NF-κB signaling.
Oxidative stress and exercise
The beneficial effect of aerobic training on redox
status is well documented (94) and
the protective effects of physical activity against prostate cancer
have been proposed to be related to enhancement of antioxidant
system (95). In heart, regular
physical activity at a low intensity enhances the expression of
anti-oxidant enzymes such as Mn-superoxide dismutase (SOD) and
Cu/Zn-SOD, glutathione peroxidase 1 and catalase (55). Importantly, Mn-SOD overexpression is
essential to integral heart protection against
ischemia/reperfusion, a condition known for inducing ROS. In
agreement, doxorubicin-mediated impairment in LV function and LV
lipid peroxidation, a sign of oxidative stress, were attenuated
with exercise (96).
Exercise also induces the expression of heat shock
protein (HSP) 60 and 72 that may contribute to myocardial
protection from ROS. The putative mechanisms by which HSP can
protect cardiomyocytes include control of protein folding,
prevention of denaturation and aggregation of intracellular
proteins, and acceleration of the breakdown of damaged proteins.
HSP72 seems to be involved in cellular protection against a variety
of stresses (97). Importantly,
exercise reduces cell death via modulation of pro- and
anti-apoptotic gene expression (81) and HSP are thought to protect against
apoptosis. For instance, the HSP70 overexpression protected against
ischemia-induced tissue damage and would be associated to enhanced
cell survival (98). Altogether,
these data are encouraging enough to consider aerobic exercise as a
promoting therapeutic target to prevent and counteract ROS-induced
cardiac dysfunction (55).
Oxidative metabolism
Endurance capacity is significantly reduced during
cancer cachexia. This functional impairment has been associated to
mitochondrial dysfunction in skeletal muscle and myocardium
(99). In physiological conditions,
aerobic training is able to stimulate oxidative metabolism and
insulin sensitivity (87). Indeed,
6 weeks of endurance training enhanced mitochondrial protein
content from 50 to 100% in hearts (100), an observation corroborated by
microarray analysis (85).
Mitochondrial biogenesis is notably driven by exercise-induced
PGC-1α upregulation (100,101). Activation of the PGC1-α-PPAR
complex with endurance training logically results in enhanced fatty
acid oxidation and could be associated to oxidative profile switch
(100,102). AMPK activation also results in
facilitation of fatty acid oxidation (cf. above) and exercise is a
potent activator of AMPK activity in cardiac tissue (103). Furthermore, GLUT4 translocation
and expression is enhanced ensuing aerobic training in myocardium
of sedentary subjects (104).
Therefore, aerobic training would prevent or restore cardiac lipid
oxidation during cancer cachexia through activation of AMPK and
PPAR/PGC-1α signaling.
IGF-1/Akt signaling and heart mass with
exercise
IGF-1/PI3K/Akt signaling pathway is involved during
physiological hypertrophy of cardiomyocytes in response to aerobic
training. For example, six weeks of aerobic training induce
physiological cardiac hypertrophy (85) and this response was blunted in KO
Akt mice after a 3 weeks swimming program (105). Importantly, hearts overexpressing
IGF-1 receptor displayed PI3K-dependent cardiac hypertrophy with no
evidence of histopathology (106).
Activation of the IGF-1/PI3K pathway seems to be beneficial in
pathogical models. Indeed, overexpressing PI3K in load-pressure
model mice reduced interstitial fibrosis and attenuated
pathological growth (107).
Cardiac-specific IGF-1 overexpression enhanced myocardial function
and cell reparation after cardiac infarction (108). Altogether, these data support the
idea that aerobic training could delay the onset and/or prevent the
progression of cardiac dysfunction during cancer via enhanced
IGF-1/PI3K activity.
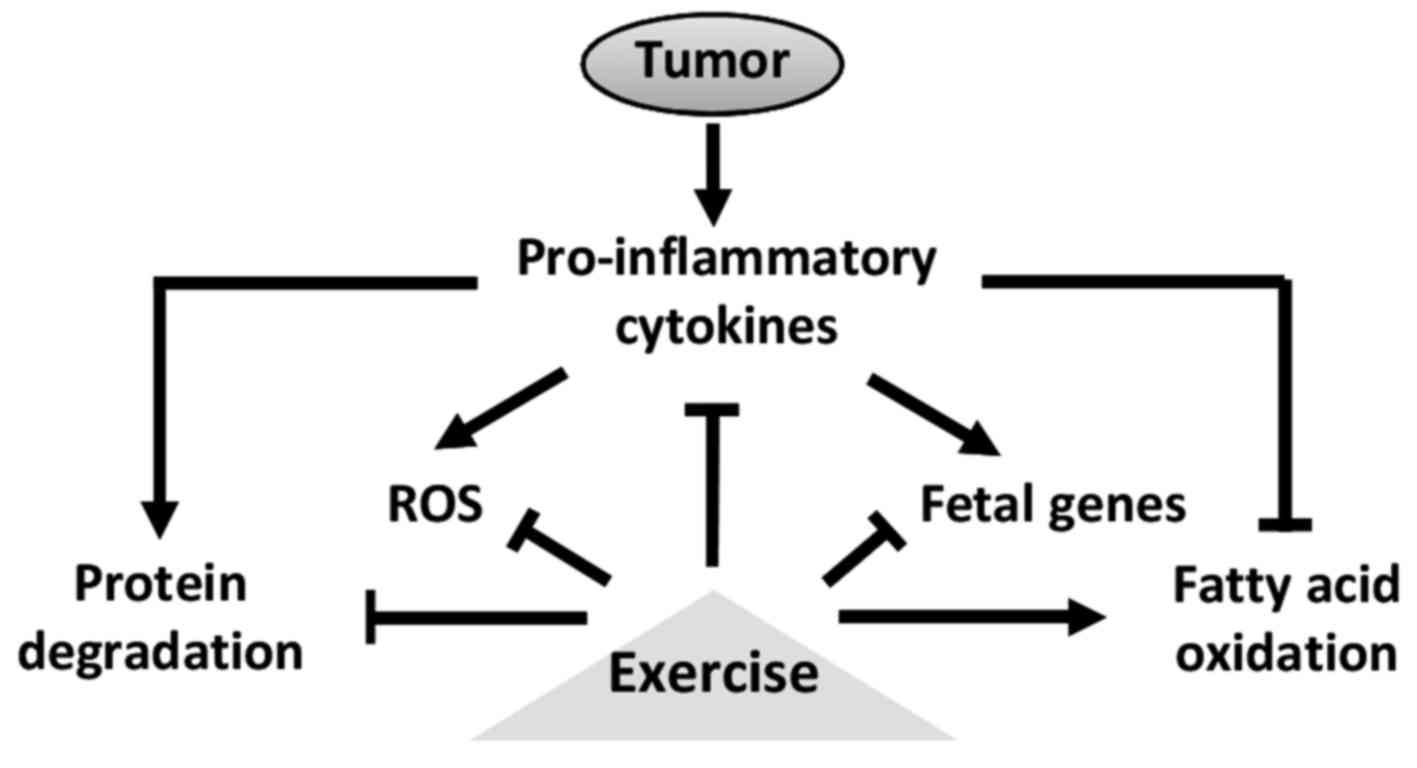
Conclusion
Understanding cancer physiopathology is a huge
challenge, notably because of the multiple types of cancers.
However, cachexia is a common feature of the disease at advanced
stage. This devastating syndrome induces HF, which in turn
exacerbates cachexia and worsens vital prognosis. The exact
sequence of events leading to cancer-related HF still remains to be
established. Cardiac muscle catabolism results from an imbalance
between energy expenditure and capacity production, and this
perturbation is related to inflammation and oxidative stress.
Exercise is a powerful antidote against such mechanisms and can
therefore protect myocardium from cancer itself and therapies side
effects (Fig. 2). However, the
modalities of exercise have to be adapted to fragile population.
Physical exercise is an interesting therapy as it can have both
protective but also preventive effects, so it is probably best to
start training as soon as possible.
References
|
1
|
Inui A: Cancer anorexia-cachexia syndrome:
Current issues in research and management. CA Cancer J Clin.
52:72–91. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fearon K, Strasser F, Anker SD, Bosaeus I,
Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N,
Mantovani G, et al: Definition and classification of cancer
cachexia: An international consensus. Lancet Oncol. 12:489–495.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Argilés JM, Moore-Carrasco R, Fuster G,
Busquets S and López-Soriano FJ: Cancer cachexia: The molecular
mechanisms. Int J Biochem Cell Biol. 35:405–409. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tisdale MJ: Cachexia in cancer patients.
Nat Rev Cancer. 2:862–871. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Costelli P and Baccino FM: Cancer
cachexia: From experimental models to patient management. Curr Opin
Clin Nutr Metab Care. 3:177–181. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tisdale MJ: Mechanisms of cancer cachexia.
Physiol Rev. 89:381–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Argilés JM, Busquets S, Stemmler B and
López-Soriano FJ: Cancer cachexia: Understanding the molecular
basis. Nat Rev Cancer. 14:754–762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ambrus JL, Ambrus CM, Mink IB and Pickren
JW: Causes of death in cancer patients. J Med. 6:61–64.
1975.PubMed/NCBI
|
|
9
|
Burch GE, Phillips JH and Ansari A: The
cachetic heart. A clinico-pathologic, electrocardiographic and
roentgenographic entity. Dis Chest. 54:403–409. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ewer MS and Ewer SM: Cardiotoxicity of
anticancer treatments. Nat Rev Cardiol. 12:547–558. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kazemi-Bajestani SM, Becher H, Fassbender
K, Chu Q and Baracos VE: Concurrent evolution of cancer cachexia
and heart failure: Bilateral effects exist. J Cachexia Sarcopenia
Muscle. 5:95–104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Murphy KT: The pathogenesis and treatment
of cardiac atrophy in cancer cachexia. Am J Physiol Heart Circ
Physiol. 310:H466–H477. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tian M, Nishijima Y, Asp ML, Stout MB,
Reiser PJ and Belury MA: Cardiac alterations in cancer-induced
cachexia in mice. Int J Oncol. 37:347–353. 2010.PubMed/NCBI
|
|
14
|
Shadfar S, Couch ME, McKinney KA,
Weinstein LJ, Yin X, Rodríguez JE, Guttridge DC and Willis M: Oral
resveratrol therapy inhibits cancer-induced skeletal muscle and
cardiac atrophy in vivo. Nutr Cancer. 63:749–762. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cosper PF and Leinwand LA: Cancer causes
cardiac atrophy and autophagy in a sexually dimorphic manner.
Cancer Res. 71:1710–1720. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsuyama T, Ishikawa T, Okayama T, Oka K,
Adachi S, Mizushima K, Kimura R, Okajima M, Sakai H, Sakamoto N, et
al: Tumor inoculation site affects the development of cancer
cachexia and muscle wasting. Int J Cancer. 137:2558–2565. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Springer J, Tschirner A, Haghikia A, von
Haehling S, Lal H, Grzesiak A, Kaschina E, Palus S, Pötsch M, von
Websky K, et al: Prevention of liver cancer cachexia-induced
cardiac wasting and heart failure. Eur Heart J. 35:932–941. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tian M, Asp ML, Nishijima Y and Belury MA:
Evidence for cardiac atrophic remodeling in cancer-induced cachexia
in mice. Int J Oncol. 39:1321–1326. 2011.PubMed/NCBI
|
|
19
|
Wysong A, Couch M, Shadfar S, Li L,
Rodriguez JE, Asher S, Yin X, Gore M, Baldwin A, Patterson C, et
al: NF-κB inhibition protects against tumor-induced cardiac atrophy
in vivo. Am J Pathol. 178:1059–1068. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sjöström M, Wretling ML, Karlberg I, Edén
E and Lundholm K: Ultrastructural changes and enzyme activities for
energy production in hearts concomitant with tumor-associated
malnutrition. J Surg Res. 42:304–313. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schäfer M, Oeing CU, Rohm M, Baysal-Temel
E, Lehmann LH, Bauer R, Volz HC, Boutros M, Sohn D, Sticht C, et
al: Ataxin-10 is part of a cachexokine cocktail triggering cardiac
metabolic dysfunction in cancer cachexia. Mol Metab. 5:67–78. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van der Velden J, Merkus D, Klarenbeek BR,
James AT, Boontje NM, Dekkers DH, Stienen GJ, Lamers JM and Duncker
DJ: Alterations in myofilament function contribute to left
ventricular dysfunction in pigs early after myocardial infarction.
Circ Res. 95:e85–e95. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bonne G, Carrier L, Richard P, Hainque B,
Tesson F, Komajda M and Schwartz K: Génétique des cardiomyopathies
hypertrophiques. Med Sci. 14:1054–1066. 1998.
|
|
24
|
Korte FS, Herron TJ, Rovetto MJ and
McDonald KS: Power output is linearly related to MyHC content in
rat skinned myocytes and isolated working hearts. Am J Physiol
Heart Circ Physiol. 289:H801–H812. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ashrafian H, Frenneaux MP and Opie LH:
Metabolic mechanisms in heart failure. Circulation. 116:434–448.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Manne ND, Lima M, Enos RT, Wehner P,
Carson JA and Blough E: Altered cardiac muscle mTOR regulation
during the progression of cancer cachexia in the
ApcMin/+ mouse. Int J Oncol. 42:2134–2140.
2013.PubMed/NCBI
|
|
27
|
Palus S, von Haehling S, Flach VC,
Tschirner A, Doehner W, Anker SD and Springer J: Simvastatin
reduces wasting and improves cardiac function as well as outcome in
experimental cancer cachexia. Int J Cardiol. 168:3412–3418. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Trobec K, Palus S, Tschirner A, von
Haehling S, Doehner W, Lainscak M, Anker SD and Springer J:
Rosiglitazone reduces body wasting and improves survival in a rat
model of cancer cachexia. Nutrition. 30:1069–1075. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou X, Wang JL, Lu J, Song Y, Kwak KS,
Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, et al: Reversal
of cancer cachexia and muscle wasting by ActRIIB antagonism leads
to prolonged survival. Cell. 142:531–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mühlfeld C, Das SK, Heinzel FR, Schmidt A,
Post H, Schauer S, Papadakis T, Kummer W and Hoefler G: Cancer
induces cardiomyocyte remodeling and hypoinnervation in the left
ventricle of the mouse heart. PLoS One. 6:e204242011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hinch EC, Sullivan-Gunn MJ, Vaughan VC,
McGlynn MA and Lewandowski PA: Disruption of pro-oxidant and
antioxidant systems with elevated expression of the ubiquitin
proteosome system in the cachectic heart muscle of nude mice. J
Cachexia Sarcopenia Muscle. 4:287–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Borges FH, Marinello PC, Cecchini AL,
Blegniski FP, Guarnier FA and Cecchini R: Oxidative and proteolytic
profiles of the right and left heart in a model of cancer-induced
cardiac cachexia. Pathophysiology. 21:257–265. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Argilés JM, Fontes-Oliveira CC, Toledo M,
López-Soriano FJ and Busquets S: Cachexia: A problem of energetic
inefficiency. J Cachexia Sarcopenia Muscle. 5:279–286. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bosaeus I, Daneryd P, Svanberg E and
Lundholm K: Dietary intake and resting energy expenditure in
relation to weight loss in unselected cancer patients. Int J
Cancer. 93:380–383. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lindmark L, Bennegård K, Edén E, Ekman L,
Scherstén T, Svaninger G and Lundholm K: Resting energy expenditure
in malnourished patients with and without cancer. Gastroenterology.
87:402–408. 1984.PubMed/NCBI
|
|
36
|
Aon MA and Cortassa S: Mitochondrial
network energetics in the heart. Wiley Interdiscip Rev Syst Biol
Med. 4:599–613. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal
JS and Stanley WC: Myocardial fatty acid metabolism in health and
disease. Physiol Rev. 90:207–258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Madrazo JA and Kelly DP: The PPAR trio:
Regulators of myocardial energy metabolism in health and disease. J
Mol Cell Cardiol. 44:968–975. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Drott C, Waldenström A and Lundholm K:
Cardiac sensitivity and responsiveness to beta-adrenergic
stimulation in experimental cancer and undernutrition. J Mol Cell
Cardiol. 19:675–683. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Drott C and Lundholm K: Glucose uptake and
amino acid metabolism in perfused hearts from tumor-bearing rats. J
Surg Res. 49:62–68. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Montel-Hagen A, Blanc L, Boyer-Clavel M,
Jacquet C, Vidal M, Sitbon M and Taylor N: The Glut1 and Glut4
glucose transporters are differentially expressed during perinatal
and postnatal erythropoiesis. Blood. 112:4729–4738. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yasumoto K, Mukaida N, Harada A, Kuno K,
Akiyama M, Nakashima E, Fujioka N, Mai M, Kasahara T,
Fujimoto-Ouchi K, et al: Molecular analysis of the cytokine network
involved in cachexia in colon 26 adenocarcinoma-bearing mice.
Cancer Res. 55:921–927. 1995.PubMed/NCBI
|
|
43
|
Kanda T and Takahashi T: Interleukin-6 and
cardiovascular diseases. Jpn Heart J. 45:183–193. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saito S, Aikawa R, Shiojima I, Nagai R,
Yazaki Y and Komuro I: Endothelin-1 induces expression of fetal
genes through the interleukin-6 family of cytokines in cardiac
myocytes. FEBS Lett. 456:103–107. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pajak B, Orzechowska S, Pijet B, Pijet M,
Pogorzelska A, Gajkowska B and Orzechowski A: Crossroads of
cytokine signaling - the chase to stop muscle cachexia. J Physiol
Pharmacol. 59:(Suppl 9). 251–264. 2008.PubMed/NCBI
|
|
46
|
Bodine SC, Latres E, Baumhueter S, Lai VK,
Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K,
et al: Identification of ubiquitin ligases required for skeletal
muscle atrophy. Science. 294:1704–1708. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Costelli P, Carbó N, Tessitore L, Bagby
GJ, Lopez-Soriano FJ, Argilés JM and Baccino FM: Tumor necrosis
factor-alpha mediates changes in tissue protein turnover in a rat
cancer cachexia model. J Clin Invest. 92:2783–2789. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Johnston AJ, Murphy KT, Jenkinson L, Laine
D, Emmrich K, Faou P, Weston R, Jayatilleke KM, Schloegel J, Talbo
G, et al: Targeting of Fn14 prevents cancer-induced cachexia and
prolongs survival. Cell. 162:1365–1378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Marin-Corral J, Fontes CC, Pascual-Guardia
S, Sanchez F, Olivan M, Argilés JM, Busquets S, López-Soriano FJ
and Barreiro E: Redox balance and carbonylated proteins in limb and
heart muscles of cachectic rats. Antioxid Redox Signal. 12:365–380.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Busquets S, Fuster G, Ametller E, Olivan
M, Figueras M, Costelli P, Carbó N, Argilés JM and López-Soriano
FJ: Resveratrol does not ameliorate muscle wasting in different
types of cancer cachexia models. Clin Nutr. 26:239–244. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gould DW, Lahart I, Carmichael AR,
Koutedakis Y and Metsios GS: Cancer cachexia prevention via
physical exercise: Molecular mechanisms. J Cachexia Sarcopenia
Muscle. 4:111–124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mantovani G, Madeddu C, Macciò A,
Gramignano G, Lusso MR, Massa E, Astara G and Serpe R:
Cancer-related anorexia/cachexia syndrome and oxidative stress: An
innovative approach beyond current treatment. Cancer Epidemiol
Biomarkers Prev. 13:1651–1659. 2004.PubMed/NCBI
|
|
53
|
Min K, Kwon OS, Smuder AJ, Wiggs MP,
Sollanek KJ, Christou DD, Yoo JK, Hwang MH, Szeto HH, Kavazis AN,
et al: Increased mitochondrial emission of reactive oxygen species
and calpain activation are required for doxorubicin-induced cardiac
and skeletal muscle myopathy. J Physiol. 593:2017–2036. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Springer J, Tschirner A, Hartman K, von
Haehling S, Anker SD and Doehner W: The xanthine oxidase inhibitor
oxypurinol reduces cancer cachexia-induced cardiomyopathy. Int J
Cardiol. 168:3527–3531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Scott JM, Khakoo A, Mackey JR, Haykowsky
MJ, Douglas PS and Jones LW: Modulation of anthracycline-induced
cardiotoxicity by aerobic exercise in breast cancer: Current
evidence and underlying mechanisms. Circulation. 124:642–650. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Asp ML, Tian M, Wendel AA and Belury MA:
Evidence for the contribution of insulin resistance to the
development of cachexia in tumor-bearing mice. Int J Cancer.
126:756–763. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gray S and Kim JK: New insights into
insulin resistance in the diabetic heart. Trends Endocrinol Metab.
22:394–403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang L, Jaswal JS, Ussher JR,
Sankaralingam S, Wagg C, Zaugg M and Lopaschuk GD: Cardiac
insulin-resistance and decreased mitochondrial energy production
precede the development of systolic heart failure after
pressure-overload hypertrophy. Circ Heart Fail. 6:1039–1048. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Padrão AI, Moreira-Gonçalves D, Oliveira
PA, Teixeira C, Faustino-Rocha AI, Helguero L, Vitorino R, Santos
LL, Amado F, Duarte JA, et al: Endurance training prevents TWEAK
but not myostatin-mediated cardiac remodelling in cancer cachexia.
Arch Biochem Biophys. 567:13–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Amirouche A, Durieux AC, Banzet S,
Koulmann N, Bonnefoy R, Mouret C, Bigard X, Peinnequin A and
Freyssenet D: Down-regulation of Akt/mammalian target of rapamycin
signaling pathway in response to myostatin overexpression in
skeletal muscle. Endocrinology. 150:286–294. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Morissette MR, Stricker JC, Rosenberg MA,
Buranasombati C, Levitan EB, Mittleman MA and Rosenzweig A: Effects
of myostatin deletion in aging mice. Aging Cell. 8:573–583. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Heineke J, Auger-Messier M, Xu J, Sargent
M, York A, Welle S and Molkentin JD: Genetic deletion of myostatin
from the heart prevents skeletal muscle atrophy in heart failure.
Circulation. 121:419–425. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Willis MS, Schisler JC, Li L, Rodríguez
JE, Hilliard EG, Charles PC and Patterson C: Cardiac muscle ring
finger-1 increases susceptibility to heart failure in vivo. Circ
Res. 105:80–88. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Willis MS, Rojas M, Li L, Selzman CH, Tang
RH, Stansfield WE, Rodriguez JE, Glass DJ and Patterson C: Muscle
ring finger 1 mediates cardiac atrophy in vivo. Am J Physiol Heart
Circ Physiol. 296:H997–H1006. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li HH, Kedar V, Zhang C, McDonough H, Arya
R, Wang DZ and Patterson C: Atrogin-1/muscle atrophy F-box inhibits
calcineurin-dependent cardiac hypertrophy by participating in an
SCF ubiquitin ligase complex. J Clin Invest. 114:1058–1071. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yamamoto Y, Hoshino Y, Ito T, Nariai T,
Mohri T, Obana M, Hayata N, Uozumi Y, Maeda M, Fujio Y, et al:
Atrogin-1 ubiquitin ligase is upregulated by doxorubicin via
p38-MAP kinase in cardiac myocytes. Cardiovasc Res. 79:89–96. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Willis MS, Bevilacqua A, Pulinilkunnil T,
Kienesberger P, Tannu M and Patterson C: The role of ubiquitin
ligases in cardiac disease. J Mol Cell Cardiol. 71:43–53. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Masiero E and Sandri M: Autophagy
inhibition induces atrophy and myopathy in adult skeletal muscles.
Autophagy. 6:307–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Musolino V, Palus S, Tschirner A, Drescher
C, Gliozzi M, Carresi C, Vitale C, Muscoli C, Doehner W, von
Haehling S, et al: Megestrol acetate improves cardiac function in a
model of cancer cachexia-induced cardiomyopathy by autophagic
modulation. J Cachexia Sarcopenia Muscle. 7:555–566. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Milan G, Romanello V, Pescatore F, Armani
A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, et
al: Regulation of autophagy and the ubiquitin-proteasome system by
the FoxO transcriptional network during muscle atrophy. Nat Commun.
6:66702015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Judge SM, Wu CL, Beharry AW, Roberts BM,
Ferreira LF, Kandarian SC and Judge AR: Genome-wide identification
of FoxO-dependent gene networks in skeletal muscle during C26
cancer cachexia. BMC Cancer. 14:9972014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Skurk C, Izumiya Y, Maatz H, Razeghi P,
Shiojima I, Sandri M, Sato K, Zeng L, Schiekofer S, Pimentel D, et
al: The FOXO3a transcription factor regulates cardiac myocyte size
downstream of AKT signaling. J Biol Chem. 280:20814–20823. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cai D, Frantz JD, Tawa NE Jr, Melendez PA,
Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, et
al: IKKbeta/NF-kappaB activation causes severe muscle wasting in
mice. Cell. 119:285–298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Li H, Malhotra S and Kumar A: Nuclear
factor-kappa B signaling in skeletal muscle atrophy. J Mol Med
(Berl). 86:1113–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Razeghi P, Wang ME, Youker KA, Golfman L,
Stepkowski S and Taegtmeyer H: Lack of NF-kappaB1 (p105/p50)
attenuates unloading-induced downregulation of PPARalpha and
PPARalpha-regulated gene expression in rodent heart. Cardiovasc
Res. 74:133–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Sack MN, Rader TA, Park S, Bastin J,
McCune SA and Kelly DP: Fatty acid oxidation enzyme gene expression
is downregulated in the failing heart. Circulation. 94:2837–2842.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Shao D, Oka S, Liu T, Zhai P, Ago T,
Sciarretta S, Li H and Sadoshima J: A redox-dependent mechanism for
regulation of AMPK activation by Thioredoxin1 during energy
starvation. Cell Metab. 19:232–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Li YY, Chen D, Watkins SC and Feldman AM:
Mitochondrial abnormalities in tumor necrosis factor-alpha-induced
heart failure are associated with impaired DNA repair activity.
Circulation. 104:2492–2497. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hamblin M, Chang L, Fan Y, Zhang J and
Chen YE: PPARs and the cardiovascular system. Antioxid Redox
Signal. 11:1415–1452. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Gielen S, Schuler G and Adams V:
Cardiovascular effects of exercise training: Molecular mechanisms.
Circulation. 122:1221–1238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Holloway TM, Bloemberg D, da Silva ML,
Simpson JA, Quadrilatero J and Spriet LL: High intensity interval
and endurance training have opposing effects on markers of heart
failure and cardiac remodeling in hypertensive rats. PLoS One.
10:e01211382015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Puhl SL, Müller A, Wagner M, Devaux Y,
Böhm M, Wagner DR and Maack C: Exercise attenuates inflammation and
limits scar thinning after myocardial infarction in mice. Am J
Physiol Heart Circ Physiol. 309:H345–H359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wisløff U, Loennechen JP, Falck G, Beisvag
V, Currie S, Smith G and Ellingsen O: Increased contractility and
calcium sensitivity in cardiac myocytes isolated from endurance
trained rats. Cardiovasc Res. 50:495–508. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Burniston JG: Adaptation of the rat
cardiac proteome in response to intensity-controlled endurance
exercise. Proteomics. 9:106–115. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Rafalski K, Abdourahman A and Edwards JG:
Early adaptations to training: Upregulation of alpha-myosin heavy
chain gene expression. Med Sci Sports Exerc. 39:75–82. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Alves CR, da Cunha TF, da Paixão NA and
Brum PC: Aerobic exercise training as therapy for cardiac and
cancer cachexia. Life Sci. 125:9–14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Gueritat J, Lefeuvre-Orfila L, Vincent S,
Cretual A, Ravanat JL, Gratas-Delamarche A, Rannou-Bekono F and
Rebillard A: Exercise training combined with antioxidant
supplementation prevents the antiproliferative activity of their
single treatment in prostate cancer through inhibition of redox
adaptation. Free Radic Biol Med. 77:95–105. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Goh J, Tsai J, Bammler TK, Farin FM,
Endicott E and Ladiges WC: Exercise training in transgenic mice is
associated with attenuation of early breast cancer growth in a
dose-dependent manner. PLoS One. 8:e801232013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Deuster PA, Morrison SD and Ahrens RA:
Endurance exercise modifies cachexia of tumor growth in rats. Med
Sci Sports Exerc. 17:385–392. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
McGinnis GR, Ballmann C, Peters B,
Nanayakkara G, Roberts M, Amin R and Quindry JC: Interleukin-6
mediates exercise preconditioning against myocardial ischemia
reperfusion injury. Am J Physiol Heart Circ Physiol.
308:H1423–H1433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Petersen AM and Pedersen BK: The
anti-inflammatory effect of exercise. J Appl Physiol 1985.
98:1154–1162. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Rodriguez J, Fernández-Verdejo R, Pierre
N, Priem F and Francaux M: Endurance training attenuates catabolic
signals induced by TNF-α in muscle of mice. Med Sci Sports Exerc.
48:227–234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Gomes EC, Silva AN and de Oliveira MR:
Oxidants, antioxidants, and the beneficial roles of
exercise-induced production of reactive species. Oxid Med Cell
Longev. 2012:7561322012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Rebillard A, Lefeuvre-Orfila L, Gueritat J
and Cillard J: Prostate cancer and physical activity: Adaptive
response to oxidative stress. Free Radic Biol Med. 60:115–124.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Chicco AJ, Schneider CM and Hayward R:
Voluntary exercise protects against acute doxorubicin
cardiotoxicity in the isolated perfused rat heart. Am J Physiol
Regul Integr Comp Physiol. 289:R424–R431. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Powers SK, Morton AB, Ahn B and Smuder AJ:
Redox control of skeletal muscle atrophy. Free Radic Biol Med.
98:208–217. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ellison GM, Waring CD, Vicinanza C and
Torella D: Physiological cardiac remodelling in response to
endurance exercise training: Cellular and molecular mechanisms.
Heart. 98:5–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Constantinou C, de Fontes Oliveira CC,
Mintzopoulos D, Busquets S, He J, Kesarwani M, Mindrinos M, Rahme
LG, Argilés JM and Tzika AA: Nuclear magnetic resonance in
conjunction with functional genomics suggests mitochondrial
dysfunction in a murine model of cancer cachexia. Int J Mol Med.
27:15–24. 2011.PubMed/NCBI
|
|
100
|
Coffey VG and Hawley JA: The molecular
bases of training adaptation. Sports Med. 37:737–763. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Scarpulla RC: Transcriptional activators
and coactivators in the nuclear control of mitochondrial function
in mammalian cells. Gene. 286:81–89. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Iemitsu M, Miyauchi T, Maeda S, Tanabe T,
Takanashi M, Irukayama-Tomobe Y, Sakai S, Ohmori H, Matsuda M and
Yamaguchi I: Aging-induced decrease in the PPAR-alpha level in
hearts is improved by exercise training. Am J Physiol Heart Circ
Physiol. 283:H1750–H1760. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Coven DL, Hu X, Cong L, Bergeron R,
Shulman GI, Hardie DG and Young LH: Physiological role of
AMP-activated protein kinase in the heart: Graded activation during
exercise. Am J Physiol Endocrinol Metab. 285:E629–E636. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Kraniou GN, Cameron-Smith D and Hargreaves
M: Acute exercise and GLUT4 expression in human skeletal muscle:
Influence of exercise intensity. J Appl Physiol 1985. 101:934–937.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
DeBosch B, Treskov I, Lupu TS, Weinheimer
C, Kovacs A, Courtois M and Muslin AJ: Akt1 is required for
physiological cardiac growth. Circulation. 113:2097–2104. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
McMullen JR: Role of insulin-like growth
factor 1 and phosphoinositide 3-kinase in a setting of heart
disease. Clin Exp Pharmacol Physiol. 35:349–354. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
McMullen JR, Amirahmadi F, Woodcock EA,
Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang
Y, Shioi T, et al: Protective effects of exercise and
phosphoinositide 3-kinase(p110alpha) signaling in dilated and
hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 104:612–617.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Santini MP, Tsao L, Monassier L,
Theodoropoulos C, Carter J, Lara-Pezzi E, Slonimsky E, Salimova E,
Delafontaine P, Song YH, et al: Enhancing repair of the mammalian
heart. Circ Res. 100:1732–1740. 2007. View Article : Google Scholar : PubMed/NCBI
|