Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fifth
leading cause of cancer-related mortality with ~70,000 estimated
deaths each year and is predicted to become the fourth cause of
cancer-related mortality in both sexes in due course in the
European Union (1). This is in part
caused by the biology of this tumor type which is characterized by
early metastasis, poor response to chemotherapy and radiotherapy
and overexpression of transforming growth factor (TGF)-β (2). This growth factor elicits its
biological effects through type II and I transmembrane receptors
with serine-threonine kinase activity and signaling intermediates
from the Smad family of proteins, which transduce the TGF-β signal
to the nucleus to alter the activity of responsive genes (3). TGF-β can also signal through several
non-Smad pathways, e.g. PI3K/AKT, p38 mitogen-activated protein
kinase (MAPK), small G-proteins such as RAC and RHO, and reactive
oxygen species (ROS) (4,5). These Smad-independent TGF-β-induced
pathways are thought to drive late events in cancer progression
such as epithelial-mesenchymal transition (EMT), migration,
invasion and metastasis (6). A
better understanding of the signaling pathways involved in their
induction by TGF-β may be mandatory to prevent malignant
progression in PDAC.
Accumulating evidence suggests that ROS induce
genetic instability and that cancer cells use redox signaling
pathways to drive aberrant proliferation, survival and invasion, as
well as interactions with the tumor microenvironment (reviewed in
ref. 7), eventually resulting in
cancer progression (8,9). ROS are generated by cellular NADPH
oxidases (NOX proteins) that catalyze the production of superoxide
(O2−) which serves as a starting material for the
production of other reactive oxidants. The core enzyme from
phagocytes comprises 5 components: p40phox,
p47phox, p67phox, p22phox and
gp91phox (=NOX2). Upon exposure to appropriate stimuli,
the entire cytosolic complex (consisting of p40, p47 and p67)
migrates to the membrane, where it associates with flavocytochrome
b558 (consisting of gp91phox and p22phox) to
assemble the active oxidase (10).
Activation, in addition, requires the participation
of RAC proteins (11) which are
recruited to the membrane independently of
p47phox or p67phox. At the
membrane, RAC is converted to the GTP-bound active form via the
function of guanine nucleotide-releasing factors where it directly
interacts with the p67phox N-terminal region,
which is crucial for activation of gp91phox.
Binding of RAC is believed to induce a conformational change in
p67phox, which may allow the activation domain to
act on gp91phox/NOX2. Tumor cells harbor
non-phagocytic NADPH oxidases that differ from the phagocyte NADPH
oxidase in that they contain Rac1 rather than Rac2 as well as
low-activity NOX2 homologs with modest levels of ROS formation
(NOX1, NOX3-5, Duox1, Duox2) (10,12).
NOX-derived ROS appear to be particularly important for activation
of p38 and ERK MAPKs (13–15), a process which is thought to be
mediated through the inactivation of various protein phosphatases
(PP1, PP2A, MKP-1, PTP-1B) (16,17).
Our laboratory has previously demonstrated that
TGF-β1 induces EMT in PDAC cells (18) along with upregulation of various
mesenchymal markers such as the small leucine-rich proteoglycan
biglycan (19). Notably,
TGF-β1-induced biglycan expression was found to be dependent on
activation of p38 MAPK which in turn required integrin ligation
through adhesion (19) and both
TGF-β1-induced biglycan expression and p38 MAPK activation required
functional RAC1 and was sensitive to the NOX4/flavine oxidase
inhibitor diphenylene iodonium (DPI) (20). Since conversion to a mesenchymal
phenotype and integrin activation are considered prerequisites for
epithelial cells to adopt a motile phenotype, it is conceivable
that TGF-β-induced motility is coordinately controlled by RAC1, NOX
and p38 MAPK signaling. Indeed, a previous study in a pancreatic
cancer cell line showed that NOX4-derived ROS can transmit
TGF-β-triggered EMT signals through PTP-1B (17). However, the role of RAC1 and p38
MAPK signaling in mediating the NOX4-derived ROS actions in
response to TGF-β stimulation have not been analyzed. Specifically,
a direct involvement of p38 MAPK, and its possible activation by
RAC1 and NOX4, in TGF-β1-dependent random cell migration
(chemokinesis) in PDAC-derived cells remains to be studied. In
order to clarify this issue, several important questions must be
addressed: i) are NOX isoforms expressed in response to TGF-β1 in
PDAC-derived cells other than Panc1? ii) is the induction of
chemokinesis by TGF-β1, ROS, RAC1, NOX4 and p38 MAPK-dependent? and
iii) is p38 MAPK activation by TGF-β1, RAC1 and NOX4-dependent?
Materials and methods
Reagents
The Nox2 inhibitor apocynin and the chemical
antioxidant N-acetyl-L-cysteine (NAC) were purchased from
Sigma (Deisenhofen, Germany). DPI, the p38 MAPK inhibitor SB203580,
and the RAC1 inhibitor NSC23766 were obtained from Merck
(Darmstadt, Germany). Recombinant human (rh) TGF-β1 was obtained
from ReliaTech (Wolfenbüttel, Germany) and was used at a
concentration of 5 ng/ml in all experiments.
Ethical approval
The present research does not consist of any studies
using human participants or animals.
Cell culture and treatments
Human PDAC-derived Panc1 and Colo357 cells were
cultured as previously described (18). The generation of Panc1 cells stably
expressing a dominant-negative mutant of MKK6 (MKK6-Ala) was
previously described (19). For the
stimulation experiments, cells in normal growth medium [RPMI with
10% fetal calf serum (FCS)] were seeded into 6-well plates
(2×105/well), 12-well plates (0.8×105/well)
or 24-well plates (0.4×105/well) (all from Nunclon,
Thermo Fisher Scientific, Dreieich, Germany) for the migration
assays, protein analysis and qPCR, respectively. After cells had
reached 80% confluence, they were starved overnight in medium
containing 0.5% FCS. Cells were then treated with TGF-β1 for 24 h
in the same medium in the presence or absence of the various
inhibitors. These were administered to cells 30 min before the
addition of TGF-β1.
RNA isolation and RT-PCR
Total RNA was isolated using PeqGold reagent
(PeqGold, Erlangen, Germany) and further processed as recommended
by the manufacturer. Synthesis of cDNA was carried out with M-MLV
reverse transcriptase (Thermo Fisher Scientific). NOX protein
subunit RNAs were either amplified with Taq polymerase (Life
Technologies, Darmstadt, Germany) by standard endpoint PCR followed
by agarose gel electrophoresis and ethidium bromide staining or by
quantitative real-time RT-PCR (qPCR) on a CFX96 instrument
(Bio-Rad, München, Germany) using SYBR-Green as previously
described (18). The housekeeping
gene TATA box-binding protein (TBP) was used for normalization. The
human PCR primers used for amplification of NOX and PHOX mRNAs and
TBP mRNA are listed in Table I.
 | Table I.PCR-primers and siRNAs. |
Table I.
PCR-primers and siRNAs.
| Name | Sequence
(5′→3′) |
|---|
| NOX1-forward |
acaaattccagtgtgcagaccac |
| NOX1-reverse |
agactggaatatcggtgacagca |
| NOX2-forward |
gggctgttcaatgcttgtggct |
| NOX2-reverse |
acatctttctcctcatcatggtgc |
| NOX3-forward |
atgaacacctctggggtcagctga |
| NOX3-reverse |
ggatcggagtcactcccttcgctg |
| NOX4-forward |
ctcagcggaatcaatcagctgtg |
| NOX4-reverse |
agaggaacacgacaatcagccttag |
| NOX5-forward |
atcaagcggccccctttttttcac |
| NOX5-reverse |
ctcattgtcacactcctcgacagc |
|
p22phox-forward |
gtgtttgtgtgcctgctggagt |
|
p22phox-reverse |
ctgggcggctgcttgatggt |
|
p47phox-forward |
gtacccagccagcactatgtgt |
|
p47phox-reverse |
aaagtagcctgtgacgtcgtct |
|
p67phox-forward |
ttcgagggaaccagctgataga |
|
p67phox-reverse |
gcatgggaacactgagcttcac |
| RAC1-forward |
accatgcaggccatcaagtgtgtgg |
| RAC1-reverse |
ttacaacagcaggcattttctcttc |
|
β-actin-forward |
gaccaggcccagagcaagag |
|
β-actin-reverse |
atctccttctgcttcctgtc |
| TBP-forward |
gctggcccatagtgatcttt |
| TBP-reverse |
cttcacacgccaagaaacag |
| NOX4 siRNA #1 |
acuaugauaucuucuggua |
| NOX4 siRNA #2 |
gaaauuaucccaagcugua |
| NOX4 siRNA #3 |
gggcuaggauugugucuaa |
| NOX4 siRNA #4 |
gaucacagccucuacauau |
Immunoblotting
Immunoblots were performed as described in detail
previously (18,20). The antibodies used were anti-NOX4
(#14347-1-AP, Acris Antibodies), phospho-p38 MAPK (Thr180/Tyr182)
(#4370), p38 MAPK (#3104), HSP90α/β (H-114, sc-7947) (all from Cell
Signaling Technology, Frankfurt, Germany) and β-actin (Sigma). In
some cases, a densitometric analysis of underexposed images was
performed using the NIH ImageJ program.
Transfection of small interfering RNAs
(siRNAs) and expression vectors
Panc1 cells were seeded in 12-well plates and
transfected on the next day serum-free for 4 h with Lipofectamine
RNAiMAX (Life Technologies) and 50 nM of either NOX4 (a mix of 4
premade siRNAs, ON-TARGETplus SMARTpool #L-010194-00-0005; Thermo
Scientific) (for sequences see Table
I), RAC1 siRNA (a mix of 4 premade siRNAs, siGENOME SMARTpool
reagent, #M-003560), or a siCONTROL nontargeting siRNA (both from
Dharmacon via Biomol, Hamburg, Germany). Following removal of the
transfection mixture, cells received standard culture medium and
were allowed to recover overnight. Twenty-four hours after the
first transfection, cells underwent a second round of transfection.
In some experiments which comprised only one round of transfection,
cells received Lipofectamine 2000 (Life Technologies) along with
expression vectors for RAC1-N17, MKK6-EE (19) or empty vector (pcDNA3), alone or in
combination. Forty-eight hours after the last transfection, cells
were subjected to chemokinesis assay, immunoblot analysis, or
qPCR.
Real-time cell analysis (RTCA)
migration assay
Real-time cell analysis (RTCA) migration assays were
performed with xCELLigence® technology (Acea
Biosciences, San Diego, CA, USA, distributed by OLS, Bremen,
Germany) as described elsewhere (18,21).
In brief, experiments were performed according to the
manufacturer's protocol with modified 16-well plates consisting of
an upper and a lower chamber separated by a porous membrane (8-µm
pores). The CIM-Plate 16 (OLS) is analogous to a Transwell plate,
except that the underside of the membrane is equipped with
microelectrodes for impedance-based detection of migrated cells.
Prior to assembly, the underside of the porous membrane was coated
with 30 µl of collagen I (Sigma) to facilitate adhesion of the
cells and thereby increase the likelihood of cell-electrode
contacts. To start an experiment, the lower chamber was filled with
165 µl of serum-reduced medium (containing 1% FCS plus
inhibitors/TGF-β1) and the CIM-Plate 16 was placed in the RTCA DP
device and incubated at 37°C in 5% CO2 for 1 h to
equilibrate the medium followed by a measurement step for recording
a background signal generated by cell-free media. Cells
(30,000–60,000, overnight serum-starved, per well) were mixed and
preincubated in 150 ml of culture medium containing 1% FCS and
inhibitors/TGF-β1 at the same concentrations as in the lower
chamber (chemokinesis setup). After 30 min, TGF-β1 was added and
cells were seeded in the wells of the upper chambers. After cell
addition, CIM-Plates 16 remained at room temperature in the laminar
flow hood for 30 min to allow cells to settle onto the membrane.
Phosphate-buffered saline (PBS) was added to the empty space
surrounding the wells in order to prevent interference from
evaporation. Each condition was performed in quadruplicate with a
programmed signal detection (RTCA software version 1.2.1.; OLS)
every 15 min for a total of 24–40 h, depending on the cell type and
conditions.
Measurement of ROS
The generation of intracellular ROS was detected
with H2-DCF-DA (2′,7′-dichlorodihydrofluorescein-diacetate;
Molecular Probes, Karlsruhe, Germany) by flow cytometry. H2-DCF-DA
is first deacetylated to the non membrane-permeable H2-DCF-DA and
then oxidized, emitting a fluorescent signal. For the ROS assay,
Panc1 cells were seeded in 6-well plates (2×105/well)
with RPMI containing 10% FCS. When 80% confluent, the cells were
incubated overnight in growth medium containing 0.5% FCS and on the
next day were loaded with 10 mM H2-DCF-DA for 30 min. Following
this incubation, the medium was replaced with fresh medium
containing TGF-β1 (5 ng/ml) and further incubated for various
times. Following trypsinization, cells were centrifuged and
resuspended in 500 µl PBS and fluorescence was measured by flow
cytometry (FACS; BD Biosciences, Heidelberg, Germany) at a
wavelength of 488 nm. For each sample, the specific fluorescence
from 10,000 cells was determined using Cell Quest Software from BD
Biosciences.
Statistical analysis
Statistical significance was calculated using the
non-parametric Mann-Whitney U test. Data were considered
significant at p<0.05.
Results
PDAC cells exhibit TGF-β1-inducible
NOX4 expression and ROS production in response to TGF-β1
stimulation
A study by Hiraga et al showed that
NOX4-derived ROS signaling contributes to TGF-β-induced EMT in
Panc1 cells (17). In order to test
which NOX isoforms are expressed in PDAC-derived cells, we
performed semi-quantitative RT-PCR analysis. Notably, Panc1 cells
expressed NOX2, NOX4, NOX5, p22phox, p47phox,
p67phox and RAC1, while NOX1 and NOX3 were not detected,
neither in the Panc1 cell line nor in freshly isolated peripheral
blood monocytes used as control (Fig.
1A, left). A quantification of NOX2, NOX4, and NOX5 in Panc1
and Colo357 cells by qPCR revealed that NOX4 exhibited by far the
strongest expression of all 3 isoforms with expression levels even
higher than those in peripheral blood monocytes (Fig. 1A, right). Panc1 cells have been
shown to respond to TGF-β1 with upregulation of NOX4 (17). To ascertain whether NOX4, NOX2 and
NOX5 are also induced in other PDAC-derived cells we treated
Colo357 cells for 24 h with rhTGF-β1 and determined their
expression by qPCR. These cells exhibited a strong upregulation of
NOX4 (6.1±3.2, p<0.05), a moderate induction of NOX2 (2.5±1.1,
p<0.05) (Fig. 1B), but no
induction of NOX5 (data not shown). Due to its higher expression
(see Fig. 1A) and the greater
response to TGF-β1 (Fig. 1B) and
the demonstration that it is a major source for ROS production in
Panc1 cells (17) we focussed on
NOX4 in all subsequent experiments. Upregulation of NOX4 by TGF-β1
was also noted at the protein level (Fig. 1C). To reveal whether the TGF-β1
effect on NOX4 temporarily correlates with the relatively rapid
induction of cell migration by this growth factor (compare Fig. 3), we measured NOX4 expression in
Panc1 and Colo357 cells after short stimulation periods. As shown
in Fig. 1D, NOX4 expression was
strongly induced already after 1 h of TGF-β1 treatment and remained
elevated until 8 h after TGF-β1 addition.
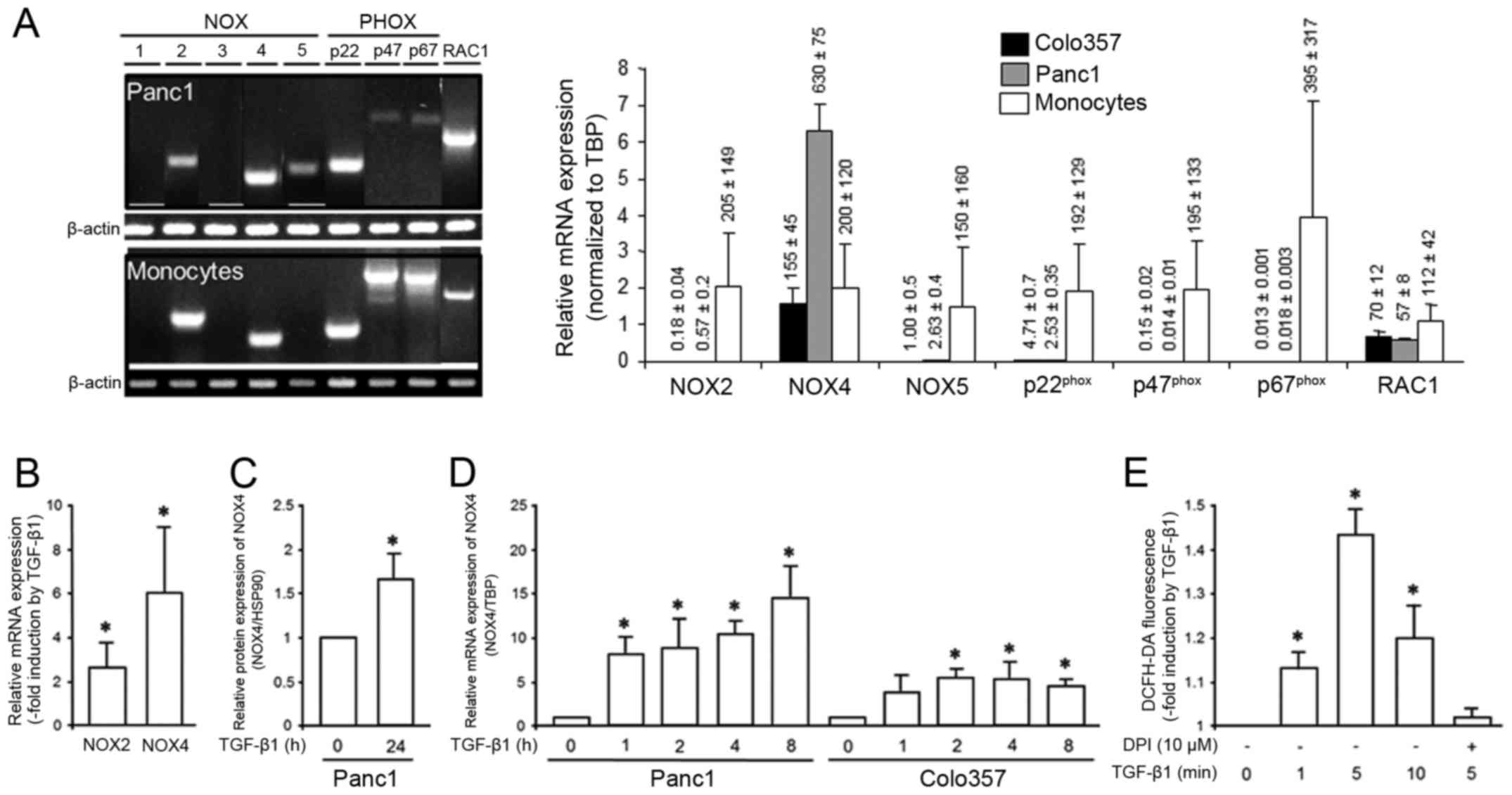 | Figure 1.PDAC-derived cells upregulate NOX4 in
response to TGF-β1 treatment and respond to TGF-β1 with
DPI-sensitive ROS production. (A) Panc1 cells and freshly isolated
peripheral blood monocytes were subjected to semi-quantitative
RT-PCR with primers specific for NOX1-5, p22phox,
p47phox, p67phox and RAC1. The resulting
amplification products were visualized by gel electrophoresis and
ethidium bromide staining (left-hand side). On the right-hand side,
results of qPCR analyses of NOX2, NOX4, NOX5, and RAC1 in Colo357
and Panc1 cells and in peripheral blood monocytes in relation to
the housekeeping gene TBP are shown. (B) Colo357 cells were treated
with TGF-β1 for 24 h and then subjected to qPCR for NOX2 and NOX4.
Data represent the mean ± SD of 3 assays and are expressed as -fold
induction by TGF-β1 over non-stimulated controls (n=3). (C) Panc1
cells were treated with TGF-β1 for 24 h and then subjected to
immunoblot analysis for NOX4 and HSP90 as loading control. The
graph shows results from densitometric analysis of band intensities
from underexposed blots (mean ± SD of 3 experiments). (D) As in C,
except that cells were treated with TGF-β1 for shorter time periods
(as indicated) and processed for qPCR rather than immunoblot
analysis. Data shown are the mean ± SD of triplicate samples and
are representative of 3 assays. (E) Panc1 cells were incubated with
H2-DCF-DA and remained untreated or were treated for the indicated
times with TGF-β1, alone or in combination with DPI, and
subsequently processed for detection of the oxidized dye by flow
cytometry. Data represent the mean ± SD of triplicate samples.
Asterisks in B-E indicate significance. |
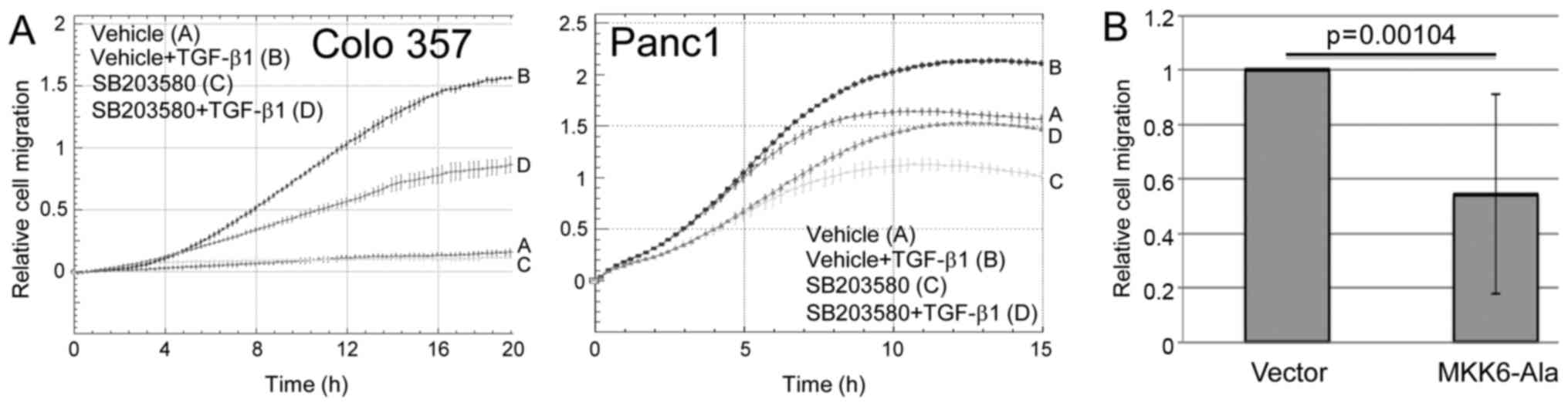
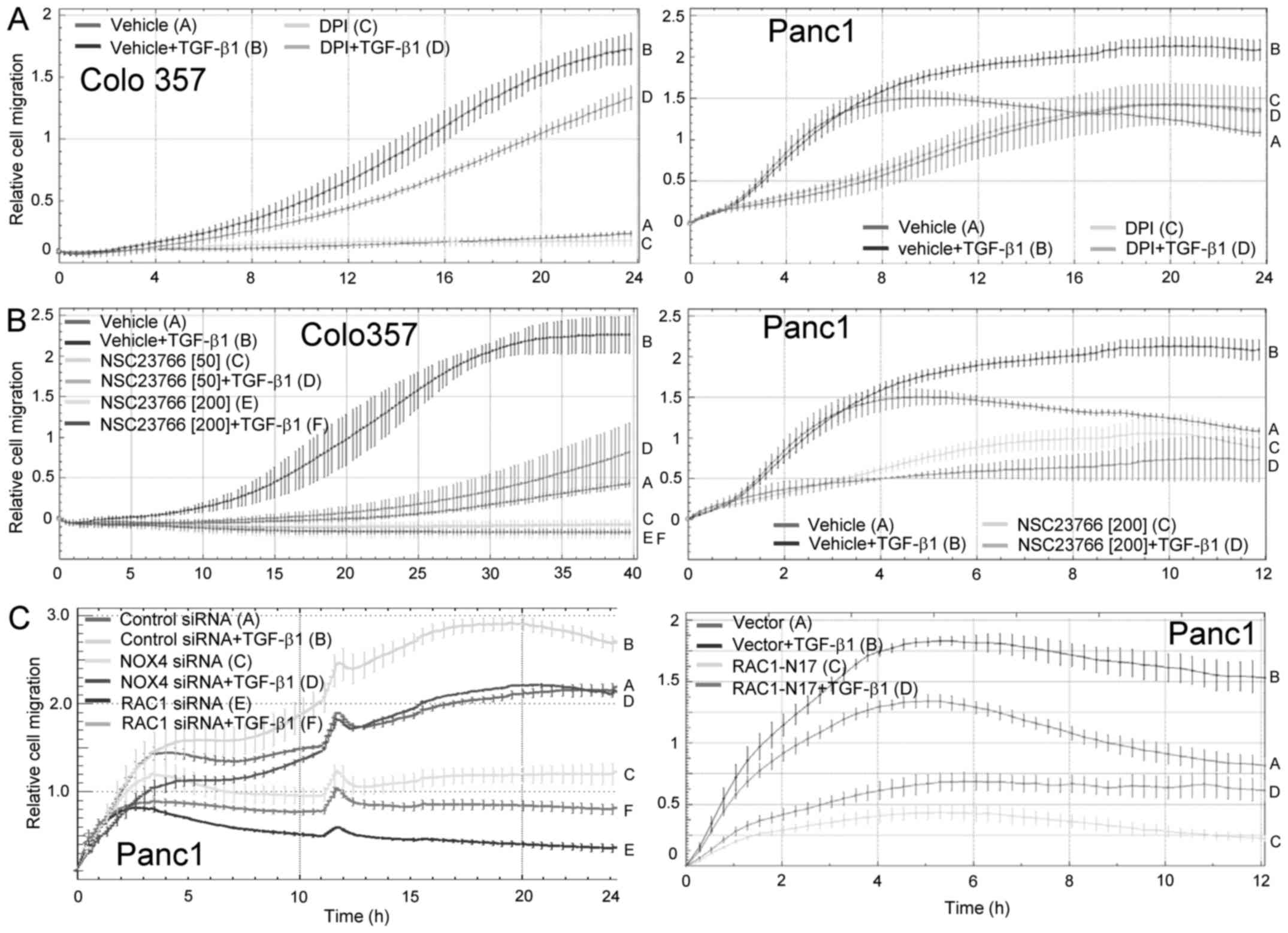 | Figure 3.Inhibition of NOX4 expression
suppresses TGF-β1-induced chemokinesis. Colo357 (A, left panel) and
Panc1 (A, right panel) cells were stimulated with TGF-β1 in the
absence or presence of the NOX4 inhibitor DPI followed by
chemokinesis assay. The inhibitory effect of DPI on
TGF-β1-dependent migration in Colo357 and Panc1 cells (tracing D
vs. B) was first significant at 12 h and 3 h, respectively, and all
later time points. As control, Colo357 (B, left panel) and Panc1
(B, right panel) cells were subjected to realtime chemokinesis
assay in the presence of the chemical RAC1 inhibitor NSC23766 or
vehicle as control. Data are depicted as relative cell migration
and are the mean ± SD of quadruplicate samples of a representative
experiment out of three experiments performed in total. For Colo357
cells, the inhibitory effect of TGF-β1 + NSC23766 (both 50 mM and
200 mM, tracing D and F, respectively) relative to TGF-β1 + vehicle
control cells (tracing B) was first significant at the 10 h time
point and remained so all later time points, while for Panc1 cells
significance of the inhibitory effect for 200 mM NSC23766 + TGF-β1
was first noted at the 2 h time point (tracing D vs. B). (C, left
panel) Panc1 were transfected twice by lipofection with irrelevant
negative control siRNA, or siRNA to either NOX4, or siRNA to RAC1
as positive control for inhibition of TGF-β-dependent chemokinesis
(see Fig. 5 for effectiveness of
the NOX4 and RAC1 siRNAs in reducing expression of the respective
proteins). Forty-eight hours after the second round of
transfection, cells were subjected to realtime cell migration assay
in the absence or presence of 5 ng/ml TGF-β1. The inhibitory effect
of Nox4 siRNA and RAC1 siRNA on TGF-β1-dependent chemokinesis when
compared to that of control siRNA transfected cells (NOX4: tracing
D vs. B; RAC1: tracing F vs. B) was significant at 3 h and all
later time points. (C, right panel) As an additional positive
control, Panc1 cells were transiently transfected with RAC1-N17, an
activation-deficient RAC1 mutant, or empty vector as negative
control. Forty-eight hours after the start of transfection, cells
were analyzed by realtime cell migration assay. Data are depicted
as relative cell migration and are the mean ± SD of quadruplicate
samples from a representative experiment out of three experiments
performed in total. The inhibitory effect of TGF-β1-treated
RAC1-N17 expressing cells relative to TGF-β1-treated empty vector
control cells (tracing D vs. B) was first significant at 1 h and
remained so at all later time points. Data in (A)-(C) are displayed
as relative cell migration and are the mean ± SD of quadruplicate
samples. In each panel, one representative experiment is shown out
of three experiments performed in total. |
Since PDAC cells appear to express the necessary
components of the nonphagocytic enzyme, we tested more directly in
Panc1 cells whether TGF-β1 was able to generate ROS. Intracellular
ROS formation was monitored over a period of 20 min using
H2-DCF-DA, a redox-sensitive dye that readily diffuses into the
cell where it is freed from the acetate groups by cellular
esterases and converted into the highly fluorescent
2′,7′-dichlorofluorescein in the presence of ROS. We observed a
rapid, although moderate and transient increase in ROS peaking at 5
min (Fig. 1E). In the presence of
the NOX4/flavine oxidase inhibitor DPI, TGF-β1-stimulated ROS
production was suppressed (Fig.
1E). Although DPI is not specific for NOX4, high expression of
this isoform and low or absent expression of the other NOX isoforms
suggests that the observed effect was due to inhibition of NOX4.
The data showed that TGF-β1 can induce ROS generation in
PDAC-derived cells and, based on its high expression and its
responsivity to TGF-β1 suggest that NOX4 accounted for
TGF-β1-stimulated ROS production.
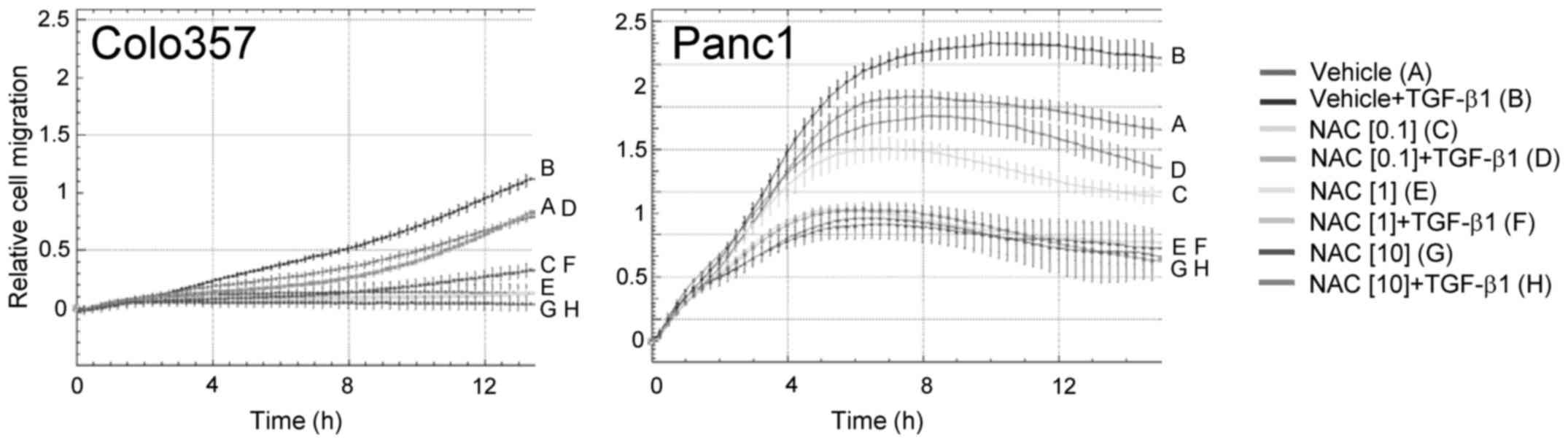
NAC reduces TGF-β1-induced
chemokinesis
We previously showed in a series of studies that
TGF-β1 is a powerful driver of random cell migration in
PDAC-derived cells (18,21). To evaluate the possibility that
TGF-β acts through intermediate production of ROS to control this
response, we employed the antioxidant N-acetyl-cysteine
(NAC). This drug which has been previously shown to be a specific
inhibitor of TGF-β signaling (22)
at 0.1–10 mM final concentrations strongly and in a dose-dependent
manner downregulated the migratory response to TGF-β1 in both Panc1
and Colo357 cells (Fig. 2).
Notably, in Panc1 cells, NAC also inhibited the early and strong
rise in migratory activity that was TGF-β1-independent up to ~4 h
when the TGF-β effect initially became statistically significant
(Fig. 2).
Inhibition of NOX4 suppresses
TGF-β1-induced chemokinesis
Above we showed that Panc1 cells produced ROS in
response to TGF-β1 and that the ROS scavenger NAC can blunt
TGF-β1-mediated chemokinesis. TGF-β is known to produce ROS through
the activation of RAC1 and we previously demonstrated that RAC1 is
activated rapidly (within 5 min) by TGF-β1 in Panc1 cells (19). However, the above data led us to
hypothesize that other components of the NADPH oxidase
multi-subunit complex, e.g. NOX proteins are involved in mediating
the TGF-β1 effect on random cell migration in PDAC cells. Above, we
showed that expression of NOX4 was rapidly induced by TGF-β1
(within 1 h) in both Panc1 and Colo357 cells and was closely
correlated temporarily with the TGF-β1 effect on cell migration. To
verify the involvement of NOX proteins, in particular NOX4, we
treated cells with DPI, which at 10 µM strongly inhibited
TGF-β1-induced chemokinesis in Colo357 (Fig. 3A, left panel) and Panc1 (Fig. 3A, right panel) cells. Notably, the
effect of DPI was generally more pronounced in Panc1 cells compared
to Colo357 cells (Fig. 3A). As
control, we employed the small molecule NSC23766 which blocks
activation of RAC1 by the guanine nucleotide exchange factors TRIO
and TIAM1. NSC23766 efficiently reduced TGF-β1-dependent
chemokinesis in both Colo357 (Fig.
3B, left panel) and Panc1 (Fig.
3B, right panel) cells.
We complemented these data with a genetic approach
using an siRNA to NOX4. Notably, NOX4 silencing in Panc1
cells by siRNA transfection led to a markedly reduced chemokinetic
response to TGF-β1 (Fig. 3C, left
panel). However, the effect was not as strong as that resulting
from siRNA-mediated depletion of RAC1 which was performed as an
internal control (Fig. 3C, left
panel), or from ectopic expression of RAC1-N17, a RAC1 mutant that
is unable to bind GTP and upon ectopic expression can block RAC1
activation in a dominant-negative fashion (Fig. 3C, right panel). As observed above
for NAC, NSC23766 (Fig. 3B, right
panel), DPI (Fig. 3A, right panel),
RAC1-N17 (Fig. 3C, right panel) and
NOX4 siRNA (Fig. 3C, left panel)
also effectively inhibited the early, TGF-β1-independent phase of
strongly increasing migratory activity (here, 0–7 h) in Panc1
cells. TGF-β1-induced chemokinesis was also inhibited by apocynin,
a selective inhibitor of NOX2 (23)
(data not shown). Together, these data clearly showed that both
RAC1 and NOX proteins, particularly NOX4, play an essential role in
the regulation of random cell migration by TGF-β1 and are in
keeping with our assumption that RAC1 cooperates with NOX proteins
to control TGF-β1-driven chemokinesis.
Inhibition of p38 MAPK suppresses
TGF-β1-induced chemokinesis
The above results suggest that RAC1 and NOX4 are
required for TGF-β1-induced chemokinesis of PDAC cells. Both RAC1
and ROS can activate p38 MAPK; however, whether p38 MAPK is also
involved in TGF-β1-induced chemokinesis has not yet been
demonstrated. To analyze this, we studied the effect of the p38
MAPK inhibitor SB203580 on TGF-β1-induced random cell migration of
Colo357 and Panc1 cells in real-time cell migration assays. To this
end, treatment of both cell types with SB203580 (10 µM) moderately
and strongly, respectively, suppressed the chemokinetic response to
TGF-β1 stimulation (Fig. 4A).
Likewise, the migratory activity of Panc1 cells stably expressing
MKK6-Ala, a dominant-negative inhibitor of MKK6-p38 MAPK signaling
was reduced by 45% (p=0.00104) compared to the empty
vector-expressing control cells (Fig.
4B). These data showed that the activation of p38 MAPK is
crucial for TGF-β1-mediated chemokinesis and identified p38 MAPK as
a downstream target of RAC1 and NOX4 in this process.
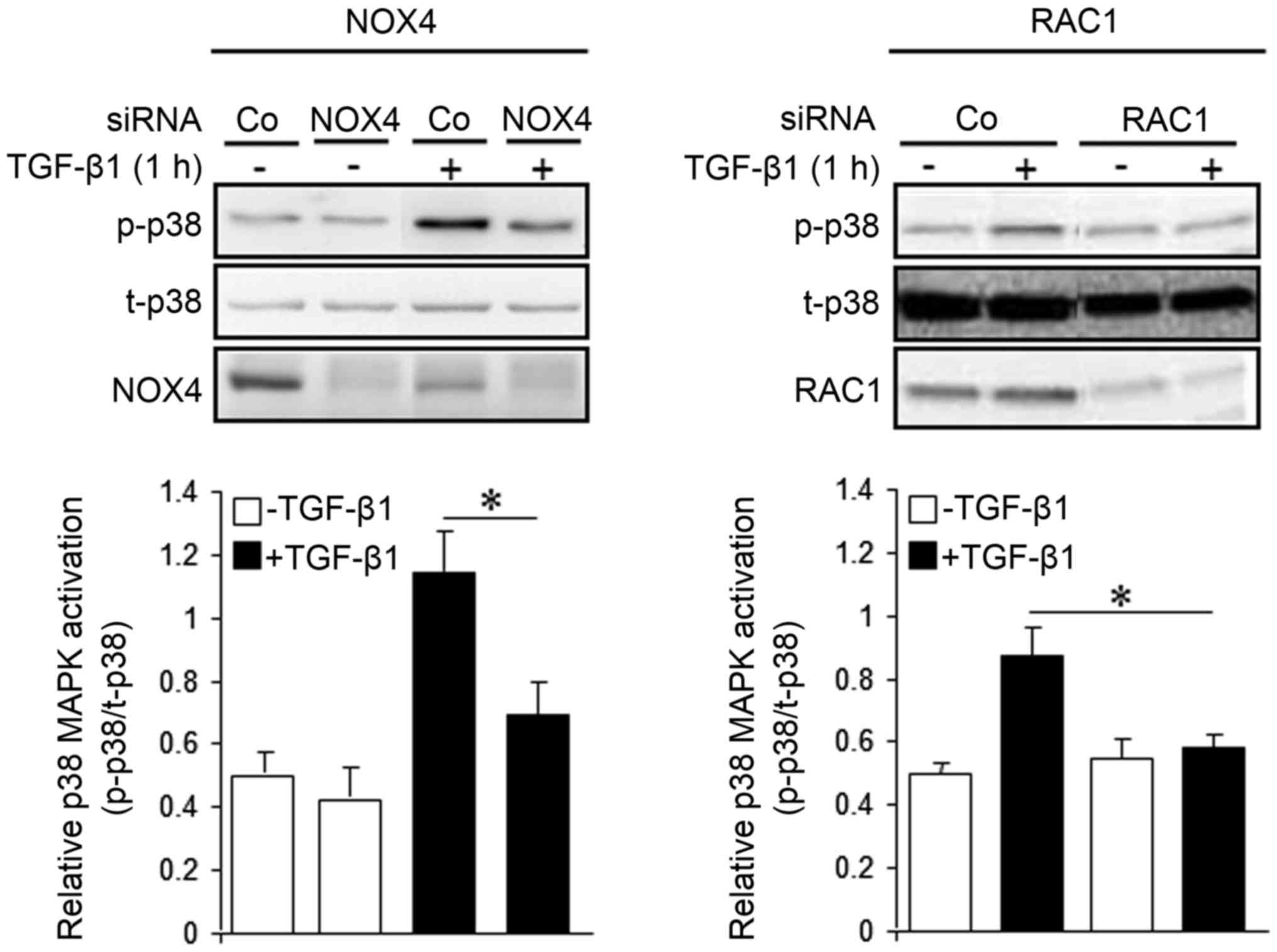
Inhibition of NOX4 expression impairs
TGF-β1-dependent activation of p38 MAPK
To analyze whether inhibition of NOX4
expression/function impacts TGF-β1-induced p38 MAPK activation, we
performed phospho-immunoblotting with antibodies associated with
p38 MAPK activation (Thr180/Tyr182). As shown in Fig. 5, transient transfection of NOX4
siRNA (Fig. 5, left panel), or RAC1
siRNA as control (Fig. 5, right
panel), prevented the phosphorylation of these sites by TGF-β1.
However, the expression of NOX4 was not affected by transfection of
RAC1 siRNA (data not shown). These data showed that TGF-β1-driven
activation of p38 MAPK is dependent on functional NOX4 and RAC1
protein. In addition, it is suggested that although NOX4 appears to
function independently of RAC1, both proteins cooperate to induce
p38 MAPK activation in order to mediate the TGF-β1 effect on
chemokinesis.
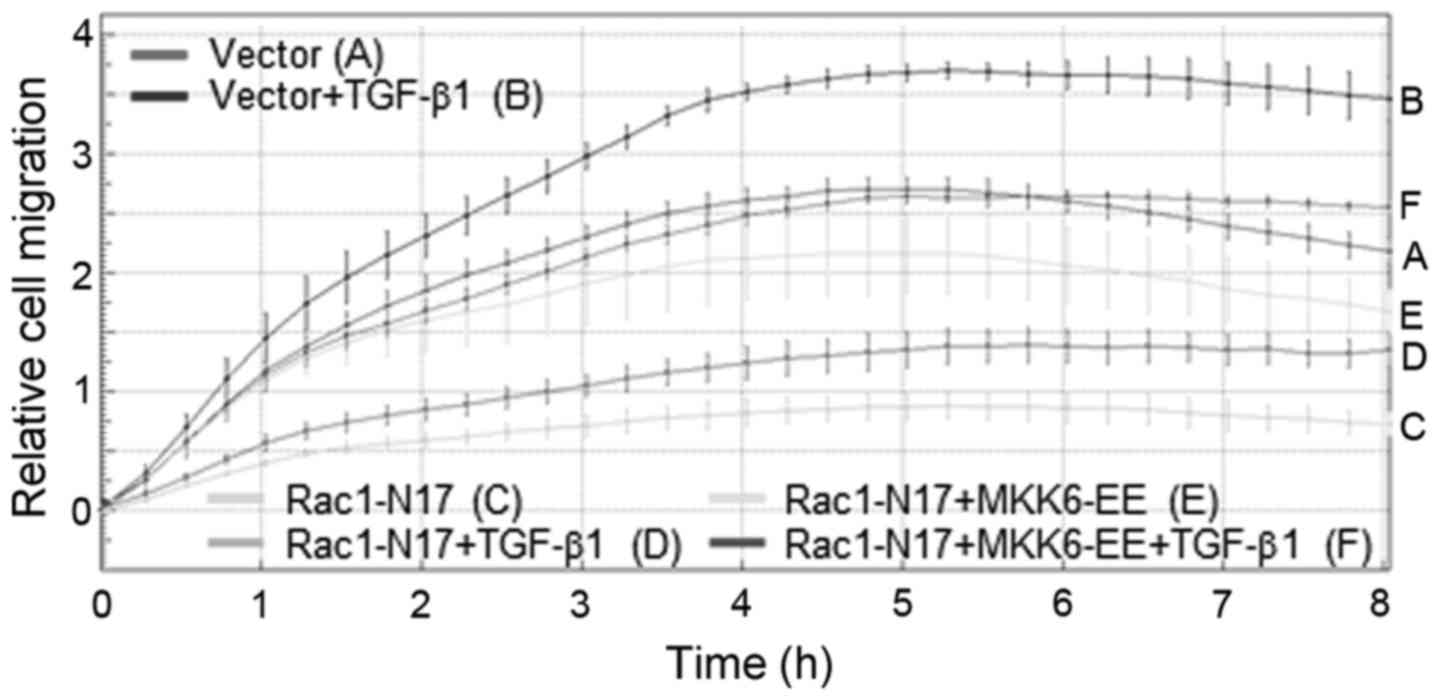
Ectopic expression of constitutively
active MKK6 rescues cells from the RAC1-N17-mediated decline in
TGF-β1-induced chemokinesis
The data to date suggest that RAC1 and NOX4 act
upstream of p38 MAPK in promoting cell migration. To prove that
more directly, we ascertained whether activating p38 MAPK
signaling, e.g. by ectopic expression of a kinase-active mutant of
MKK6 (MKK6-EE), the upstream activator of p38, overcomes the
antimigratory effect of Rac1 inhibition on TGF-β1-induced
chemokinesis. To this end, while RAC1-N17 potently inhibited basal
and TGF-β1-driven chemokinesis in Panc1 cells, co-transfection of
MKK6-EE was able to partially rescue the cells from the decline in
migratory activity (Fig. 6). These
data provide additional evidence for our contention that p38 MAPK
acts downstream of RAC1 and NOX4 in promoting TGF-β1-dependent
chemokinesis.
Discussion
Previous studies from our group have shown a crucial
role for RAC1 in TGF-β/Smad and non-Smad-mediated cellular
responses. For instance, functional RAC1 was necessary for
TGF-β-dependent Smad2 C-terminal phosphorylation and induction of
biglycan gene expression as demonstrated by ectopic expression of
RAC1-N17 in Panc1 cells (18). In
the present study, we showed that activated RAC1 was crucial for
mediating the pro-migratory effect of TGF-β1 since both the small
molecule Rac1 inhibitor NSC23766 and RAC1-specific siRNA, and
RAC1-N17 were able to inhibit TGF-β1-driven random cell
migration.
Since RAC1 constitutes a subunit of the NOX
multienzyme complex, we investigated the possibility that
TGF-β1-activated RAC1 acts through NOX4-mediated production of ROS
to drive chemokinesis. A combination of semi-quantitative RT-PCR
and qPCR analyses revealed that although PDAC-derived Colo357 and
Panc1 cells expressed several NOX isoforms, only NOX4 displayed
expression levels in the range of those in peripheral blood
monocytes. Moreover, NOX4 mRNA was potently induced by a 24-h
treatment with TGF-β1 in Colo357 cells (6.1±3.2-fold, p<0.05),
together suggesting NOX4 as the NOX isoform required for
TGF-β1-dependent cell migration. In line with this, Panc1 cells
responded to TGF-β1 treatment with a rapid and time-dependent
generation of ROS that was sensitive to the NOX4 inhibitor DPI. The
fast kinetics of ROS production in response to TGF-β1 stimulation
which parallels that of NADPH oxidase activity (14) is in good agreement with the rapid
activation of RAC1 in these cells (<5 min) (20) and its rapid translocation (within 5
min) from the cytoplasm to the membrane (14). Both events precede the activation of
p38 MAPK (20) which is first
detectable at 10–15 min and peaks after 1 h of TGF-β1 treatment
(14,24). The activated p38 MAPK then drives
cell migration as the time course of p38 MAPK activation parallels
that of the migratory response (19). In line with a role of ROS in
TGF-β1-dependent cell motility, treatment of Panc1 and Colo357
cells with the radical scavenger NAC suppressed TGF-β1-mediated
chemokinesis in a dose-dependent manner.
The hypothesis that NOX4 drives TGF-β1-dependent
chemokinesis through ROS was given further credit by the results
from inhibition experiments with DPI and an NOX4-specific siRNA
both of which were able to inhibit TGF-β1-driven random cell
migration. Hiraga et al previously showed that NOX4
expression was induced by TGF-β1, was upregulated in tumors from
PDAC patients and was required for TGF-β1-induced ROS production.
Using Boyden chamber assays in combination with a chemotaxis setup,
these authors showed that NOX4 was involved in TGF-β regulation of
directed cell migration/chemotaxis (17). We extended these data by
demonstrating that NOX4 is also involved in random cell
migration/chemokinesis induced by TGF-β1. Moreover, by employing
real-time rather than endpoint measurements, we revealed that in
Panc1 cells NAC or DPI treatment, or NOX4 or RAC1 depletion also
inhibited the initial sharp rise in migratory activity that was
independent of TGF-β1. This rapid anti-migratory effect was likely
mediated by inhibition of SRC as we have shown previously that the
initial, TGF-β-independent rise in migration was SRC-dependent
(25). In line with this,
TGF-β1-initated c-SRC-Y416 activation is known to be ROS-dependent
(26). These findings remained
undiscovered by Hiraga et al since their Boyden chamber
assays only allowed for endpoint measurements. It should be
mentioned that NOX4 has also been implicated in TGF-β-driven EMT
and migration of breast epithelial cells (27), and in the stimulation of breast
cancer cells by mammary cells of stromal origin (28).
Vaquero et al were the first to demonstrate
that a nonmitochondrial NADPH oxidase is a major source of growth
factor-induced ROS in pancreatic cancer cells (29), and that these cells express various
subunits and NOX isoforms of the phagocytic NADPH oxidase including
phagocytic NOX2. Notably, we found that NOX2 inhibition with the
prodrug apocynin in Panc1 and Colo357 cells reduced the TGF-β
effect on chemokinesis (data not shown). It may be interesting to
test the idea that NOX2 acts in concert with NOX4 which could help
to explain why inhibiting NOX4 was not as effective as inhibiting
RAC1 in suppressing TGF-β1-induced chemokinesis (Fig. 3C). However, definite conclusions
should not be based on the use of these inhibitors alone as their
specificity has been questioned (30).
We further evaluated the possibility that
TGF-β1-induced NOX4 protein drives random migration through p38
MAPK signaling. This assumption was supported in the present study
by the demonstration that blocking p38 MAPK by pharmacological
inhibition with SB203580 abrogated TGF-β1-induced chemokinesis.
Moreover, treatment with the NOX4 inhibitor DPI (20) or NOX4 depletion by RNAi (Fig. 5) prevented p38 MAPK activation by
TGF-β1 in Panc1 cells, identifying p38 MAPK as a mediator of
NOX4-dependent TGF-β1-driven chemokinesis. Again, NOX4 functionally
cooperates with RAC1 through ROS production since both
siRNA-mediated depletion of RAC1 (the present study, Fig. 5) and stable ectopic expression of
RAC1-N17 (ref. 20 and the present
study, Fig. 3C, right panel) was
able to mimic the NOX4 effect on TGF-β1-induced p38 MAPK activation
(Fig. 5) and chemokinesis (Fig. 3). However, NOX4 does not appear to
be functionally linked to RAC1 since depleting RAC1 protein from
Panc1 cells did not affect the expression of NOX4, an important
observation that is in line with data from other studies (31–33,
reviewed in ref. 34).
Enhanced ROS production as observed in inflammation
and cancer can cause severe tissue damage. TGF-β1 via RAC1, NOX4,
ROS intermediates and p38 MAPK promote not only cell
migration/invasion and metastasis but also the synthesis and
accumulation of extracellular matrix proteins such as biglycan.
Thereby, TGF-β1 favors the development of fibrosis and desmoplasia,
the latter being a hallmark feature of PDAC. Antioxidants, radical
scavengers, or RAC1/NOX(4)/p38 MAPK inhibitors that can abrogate
TGF-β signaling may thus be promising tools with which to prevent
desmoplasia, migration, invasion and metastasis.
Acknowledgements
We thank H. Albrecht und S. Grammerstorf-Rosche for
excellent technical assistance, Dr F. Gieseler (Lübeck, Germany)
for providing °CIM-Plates 16 and Dr A. Sotnikova for assisting with
the flow cytometric analysis.
Glossary
Abbreviations
Abbreviations:
|
DPI
|
diphenylene iodonium
|
|
EMT
|
epithelial-mesenchymal transition
|
|
MAPK
|
mitogen-activated protein kinase
|
|
NAC
|
N-acetyl-L-cysteine
|
|
qPCR
|
quantitative real-time RT-PCR
|
|
ROS
|
reactive oxygen species
|
|
TGF-β
|
transforming growth factor-β
|
References
|
1
|
Seufferlein T, Bachet JB, Van Cutsem E and
Rougier P: ESMO Guidelines Working Group: Pancreatic
adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for
diagnosis, treatment and follow-up. Ann Oncol. 23 Suppl
7:vii33–vii40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fokas E, O'Neill E, Gordon-Weeks A,
Mukherjee S, McKenna WG and Muschel RJ: Pancreatic ductal
adenocarcinoma: From genetics to biology to radiobiology to
oncoimmunology and all the way back to the clinic. Biochim Biophys
Acta. 1855:61–82. 2015.PubMed/NCBI
|
|
3
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-β family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang Z, Seo JY, Ha H, Lee EA, Kim YS, Han
DC, Uh ST, Park CS and Lee HB: Reactive oxygen species mediate
TGF-β1-induced plasminogen activator inhibitor-1 upregulation in
mesangial cells. Biochem Biophys Res Commun. 309:961–966. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li WQ, Qureshi HY, Liacini A, Dehnade F
and Zafarullah M: Transforming growth factor Beta1 induction of
tissue inhibitor of metalloproteinases 3 in articular chondrocytes
is mediated by reactive oxygen species. Free Radic Biol Med.
37:196–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mu Y, Gudey SK and Landström M: Non-Smad
signaling pathways. Cell Tissue Res. 347:11–20. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Díaz B and Courtneidge SA: Redox signaling
at invasive microdomains in cancer cells. Free Radic Biol Med.
52:247–256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chiera F, Meccia E, Degan P, Aquilina G,
Pietraforte D, Minetti M, Lambeth D and Bignami M: Overexpression
of human NOX1 complex induces genome instability in mammalian
cells. Free Radic Biol Med. 44:332–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu JH and Kim H: Oxidative stress and
cytokines in the pathogenesis of pancreatic cancer. J Cancer Prev.
19:97–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bokoch GM and Knaus UG: NADPH oxidases:
Not just for leukocytes anymore! Trends Biochem Sci. 28:502–508.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bokoch GM and Diebold BA: Current
molecular models for NADPH oxidase regulation by Rac GTPase. Blood.
100:2692–2696. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Geiszt M and Leto TL: The Nox family of
NAD(P)H oxidases: Host defense and beyond. J Biol Chem.
279:51715–51718. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Herrera B, Fernández M, Roncero C, Ventura
JJ, Porras A, Valladares A, Benito M and Fabregat I: Activation of
p38MAPK by TGF-beta in fetal rat hepatocytes requires radical
oxygen production, but is dispensable for cell death. FEBS Lett.
499:225–229. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiu C, Maddock DA, Zhang Q, Souza KP,
Townsend AR and Wan Y: TGF-β-induced p38 activation is mediated by
Rac1-regulated generation of reactive oxygen species in cultured
human keratinocytes. Int J Mol Med. 8:251–255. 2001.PubMed/NCBI
|
|
15
|
Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh
ST and Lee HB: Role of reactive oxygen species in TGF-β1-induced
mitogen-activated protein kinase activation and
epithelial-mesenchymal transition in renal tubular epithelial
cells. J Am Soc Nephrol. 16:667–675. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korbecki J, Baranowska-Bosiacka I,
Gutowska I and Chlubek D: The effect of reactive oxygen species on
the synthesis of prostanoids from arachidonic acid. J Physiol
Pharmacol. 64:409–421. 2013.PubMed/NCBI
|
|
17
|
Hiraga R, Kato M, Miyagawa S and Kamata T:
Nox4-derived ROS signaling contributes to TGF-β-induced
epithelial-mesenchymal transition in pancreatic cancer cells.
Anticancer Res. 33:4431–4438. 2013.PubMed/NCBI
|
|
18
|
Ungefroren H, Groth S, Sebens S, Lehnert
H, Gieseler F and Fändrich F: Differential roles of Smad2 and Smad3
in the regulation of TGF-β1-mediated growth inhibition and cell
migration in pancreatic ductal adenocarcinoma cells: Control by
Rac1. Mol Cancer. 10:672011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ungefroren H, Lenschow W, Chen WB,
Faendrich F and Kalthoff H: Regulation of biglycan gene expression
by transforming growth factor-beta requires MKK6-p38
mitogen-activated protein kinase signaling downstream of Smad
signaling. J Biol Chem. 278:11041–11049. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Groth S, Schulze M, Kalthoff H, Fändrich F
and Ungefroren H: Adhesion and Rac1-dependent regulation of
biglycan gene expression by transforming growth factor-beta.
Evidence for oxidative signaling through NADPH oxidase. J Biol
Chem. 280:33190–33199. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Carl C, Flindt A, Hartmann J, Dahlke M,
Rades D, Dunst J, Lehnert H, Gieseler F and Ungefroren H: Ionizing
radiation induces a motile phenotype in human carcinoma cells in
vitro through hyperactivation of the TGF-beta signaling pathway.
Cell Mol Life Sci. 73:427–443. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meurer SK, Lahme B, Tihaa L, Weiskirchen R
and Gressner AM: N-acetyl-L-cysteine suppresses TGF-β
signaling at distinct molecular steps: The biochemical and
biological efficacy of a multifunctional, antifibrotic drug.
Biochem Pharmacol. 70:1026–1034. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
't Hart BA, Copray S and Philippens I:
Apocynin, a low molecular oral treatment for neurodegenerative
disease. Biomed Res Int. 2014:2980202014.PubMed/NCBI
|
|
24
|
Takekawa M, Tatebayashi K, Itoh F, Adachi
M, Imai K and Saito H: Smad-dependent GADD45beta expression
mediates delayed activation of p38 MAP kinase by TGF-beta. EMBO J.
21:6473–6482. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ungefroren H, Sebens S, Groth S, Gieseler
F and Fändrich F: Differential roles of Src in transforming growth
factor-β regulation of growth arrest, epithelial-to-mesenchymal
transition and cell migration in pancreatic ductal adenocarcinoma
cells. Int J Oncol. 38:797–805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Samarakoon R, Overstreet JM and Higgins
PJ: TGF-β signaling in tissue fibrosis: Redox controls, target
genes and therapeutic opportunities. Cell Signal. 25:264–268. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boudreau HE, Casterline BW, Rada B,
Korzeniowska A and Leto TL: Nox4 involvement in TGF-β and
SMAD3-driven induction of the epithelial-to-mesenchymal transition
and migration of breast epithelial cells. Free Radic Biol Med.
53:1489–1499. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tobar N, Guerrero J, Smith PC and Martínez
J: NOX4-dependent ROS production by stromal mammary cells modulates
epithelial MCF-7 cell migration. Br J Cancer. 103:1040–1047. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vaquero EC, Edderkaoui M, Pandol SJ,
Gukovsky I and Gukovskaya AS: Reactive oxygen species produced by
NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J
Biol Chem. 279:34643–34654. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aldieri E, Riganti C, Polimeni M, Gazzano
E, Lussiana C, Campia I and Ghigo D: Classical inhibitors of NOX
NAD(P)H oxidases are not specific. Curr Drug Metab. 9:686–696.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Manickam N, Patel M, Griendling KK, Gorin
Y and Barnes JL: RhoA/Rho kinase mediates TGF-β1-induced kidney
myofibroblast activation through Poldip2/Nox4-derived reactive
oxygen species. Am J Physiol Renal Physiol. 307:F159–F171. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meng D, Lv DD and Fang J: Insulin-like
growth factor-I induces reactive oxygen species production and cell
migration through Nox4 and Rac1 in vascular smooth muscle cells.
Cardiovasc Res. 80:299–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang B, Liu Z and Hu X: Inhibiting cancer
metastasis via targeting NAPDH oxidase 4. Biochem Pharmacol.
86:253–266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hordijk PL: Regulation of NADPH oxidases:
The role of Rac proteins. Circ Res. 98:453–462. 2006. View Article : Google Scholar : PubMed/NCBI
|