Introduction
Vascular smooth muscle cells (VSMCs) shorten during
contraction, decreasing the internal diameter of blood vessels to
regulate blood flow and pressure (1). VSMC contraction and relaxation are
largely mediated by phosphorylation and dephosphorylation of the
20-kDa regulatory myosin light chain (MLC20) at
threonine-18 and serine-19 by myosin light chain kinase (MLCK) and
myosin light chain phosphatase (MLCP) (2). The initial phase of contraction is
mediated by a rise in intracellular calcium which results in
calmodulin-dependent activation of MLCK. The sustained phase of
vascular contraction is thought to involve Ca2+
sensitization mechanisms (3). The
major mechanism of Ca2+ sensitization of contraction is
mediated by inhibition of MLCP, leading to increased
MLC20 phosphorylation and VSMC contraction. The
RhoA/RhoA kinase (ROCK) pathway is hypothesized to be involved in
Ca2+ sensitization. The pathway is associated with
sustained vasoconstriction by phosphorylating and inhibiting MLCP,
subsequently increasing MLC20 phosphorylation (4,5). A
previous study demonstrated that the RhoA/ROCK pathway is important
for drug-induced VSMC contraction or relaxation through activation
or inhibition of the pathway itself. Additional studies (6–8) have
revealed that extracellular signal-regulated kinase (ERK) and p38
are involved in agonist-induced smooth muscle stimulation and
activation of these signaling pathways leads to phosphorylation of
caldesmon (CaD), thus increasing myosin ATPase activity and
promoting VSMC contraction. Glucocorticoids (GCs) are important in
the stress response and are currently utilized as anti-allergic,
anti-inflammatory and immunosuppressive agents. Previously, it was
widely assumed that GCs function solely through regulation of gene
expression and protein synthesis, a long-term response which takes
several hours or days to take biological effect. However, more
recently, GCs have also been identified to exert rapid non-genomic
effects on various tissues and cells. Previous studies by this
research group (9–12) identified that GCs inhibit
degranulation of mast cells and neutrophils and phagocytosis of
macrophages via a non-genomic mechanism, to exert
immunosuppressive, anti-allergic and anti-inflammatory effects. The
role of GCs on the circulatory system is largely mediated by
permissive regulation of VSMC contraction by catecholamine, leading
to enhanced maintenance of vascular tone and blood pressure
(13). In clinical practice, the
pressor effect of norepinephrine (NE) alone to lower blood pressure
is considered unsatisfactory, particularly during rescue therapy
for cases of septic shock. Administration of a small amount of
cortisol is known to significantly enhance the pressor effect of
NE. However, the mechanism by which GCs rapidly enhance NE-mediated
contraction of VSMCs remains unclear. A previous study using sepsis
models, hypothesized that the mechanism of vascular hyporeactivity
was associated with decreased Ca2+ sensitization of
VSMCs (14). Therefore, the aim of
the present study was to characterize the rapid effect of
dexamethasone (Dex) in NE-mediated contraction in vitro and
clarify the mechanism behind this clinically important
interaction.
Materials and methods
VSMC culture
Rat vascular smooth muscle cells (A7r5 cells) were
purchased from the Committee on Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China). Cells were incubated
with growth medium for 24 h and then replaced with serum-free
medium. After 18 h, cells were treated with various drugs. The
study was approved by the ethics committee of the Second Military
Medical University.
Protein extraction
Total protein was extracted from cells using RIPA
buffer, supplemented with protease and phosphatase inhibitors.
Protein concentration in the supernatant of cell lysate was
measured using BCA Protein Assay kit. Following this, the protein
was prepared with 5X sample buffer and stored at 80°C until
use.
Western blot analysis
Protein-matched samples were electrophoresed by
SDS-PAGE, transferred to PVDF membranes and blocked with 5% non-fat
milk. Membranes then were incubated with the appropriate primary
antibody, followed by the corresponding secondary antibody.
Membranes were developed with ECL reagents and bands were
visualized and quantified using Quantity One imaging software
(Bio-Rad, Hercules, CA, USA).
Solutions and materials
Solutions and materials included lipopolysaccharide
(LPS), NE and Dex (Sigma, St. Louis, MO, USA); Y-27632 (generously
provided by the Welfide Corp., Osaka, Japan); RIPA buffer, BCA
Protein Assay kit and 5X sample buffer (Beyotime Institute of
Biotechnology, Jiangsu, China); antibodies against MLC,
P-MLCSer19, MAPK, P-MAPK (Cell Signaling Technology,
Inc., Danvers, MA, USA) myosin phosphatase target subunit 1 (MYPT1)
and P-MYPT1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); PVDF
membrane (Millipore, Billerica, MA, USA); and ECL reagents (Pierce
Biotechnology, Inc., Rockford, IL, USA).
Statistical analysis
Statistical analysis was performed using SPSS
software (v17.0) using raw data. All values are expressed as mean ±
SE. Data for BP were analyzed by paired and unpaired Student's
t-tests. Additional results were analyzed using one-way ANOVA.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Dex rapidly enhances NE-induced
MLC20 phosphorylation in VSMCs
Mice treated with LPS are widely accepted as an
acute septic shock model. NE was oxidized and deactivated by ONOO-1
and this deactivation induced the hyporeactivity of
vasoconstriction to NE in septic shock (15). Phosphorylation of MLC20
is a key event in the activation of Ca2+-induced
contraction and Ca2+ sensitization in smooth muscle
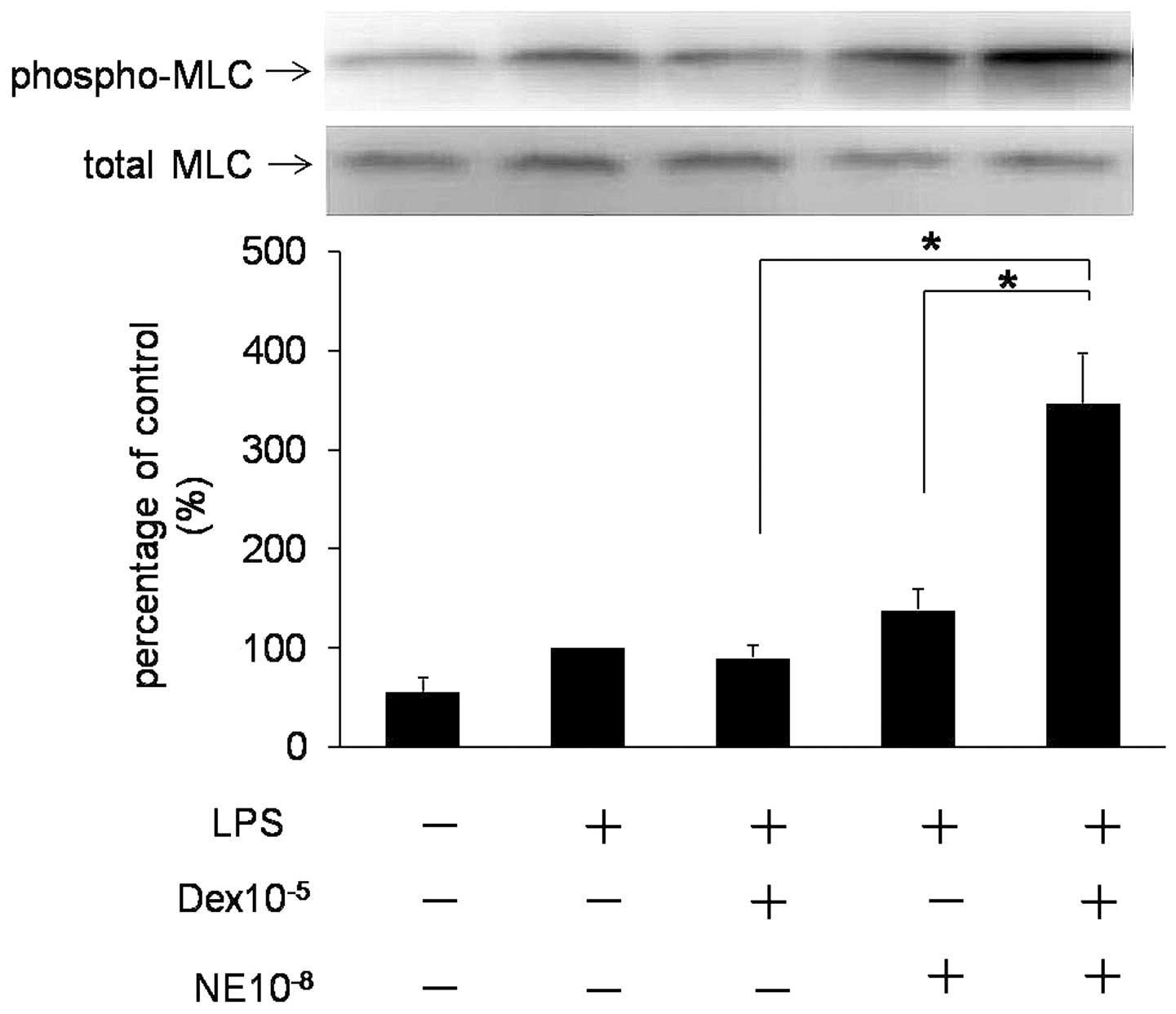
(16). Fig. 1 demonstrates phosphorylation of
MLC20 in VSMCs with various treatments. LPS enhanced
phosphorylation of MLC20 as compared with the control
group, while NE and Dex alone caused a reduced increase in the
phosphorylation of MLC20 compared with the LPS group.
However, preincubation with Dex enhanced phosphorylation of
MLC20 in cells treated with NE (P<0.05; n=3).
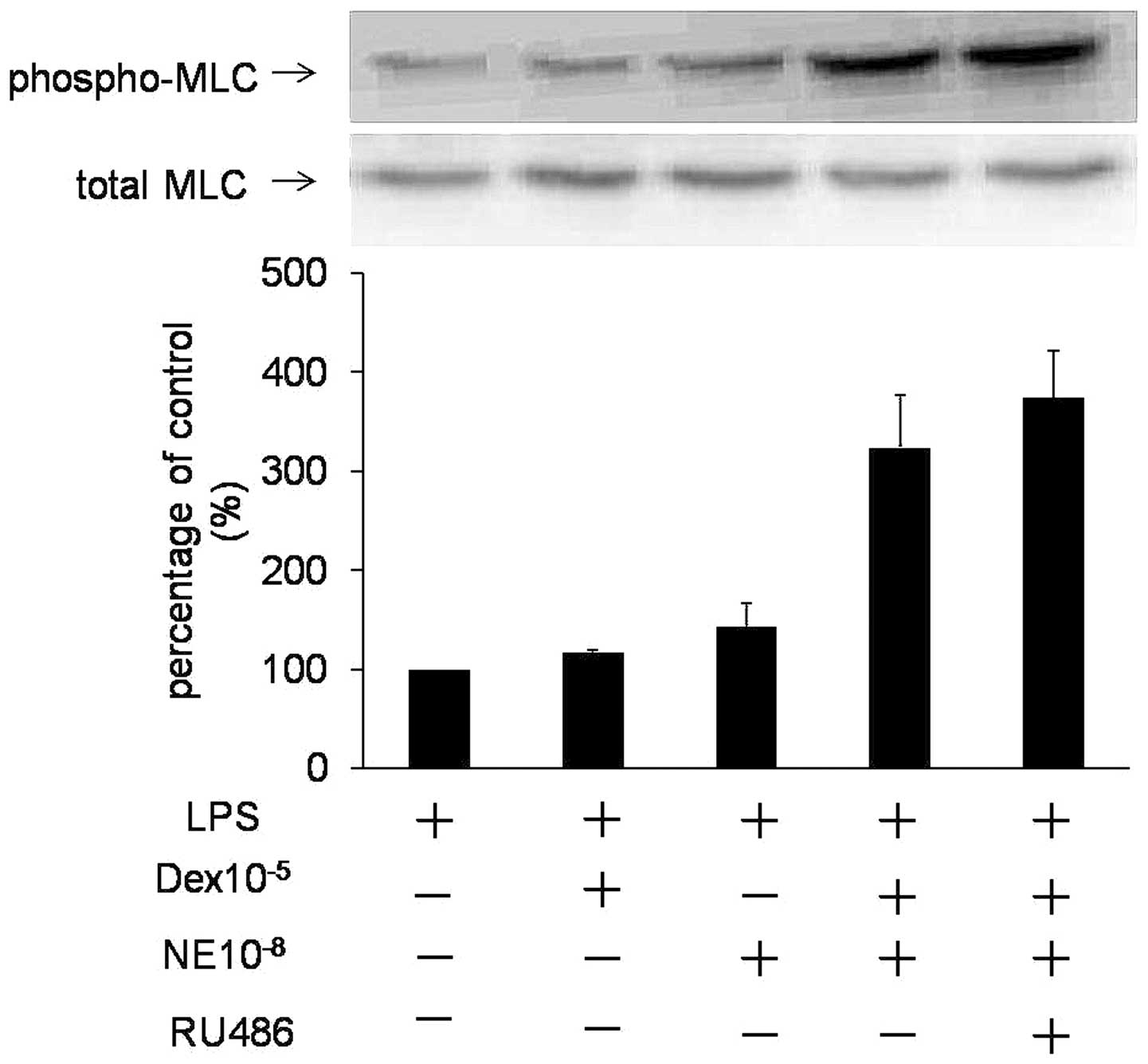
Fig. 2 demonstrates that RU486, a
GC nuclear receptor antagonist, did not block this rapid action.
These results indicate that Dex rapidly enhances NE-induced
MLC20 activation in VSMCs by a non-genomic
mechanism.
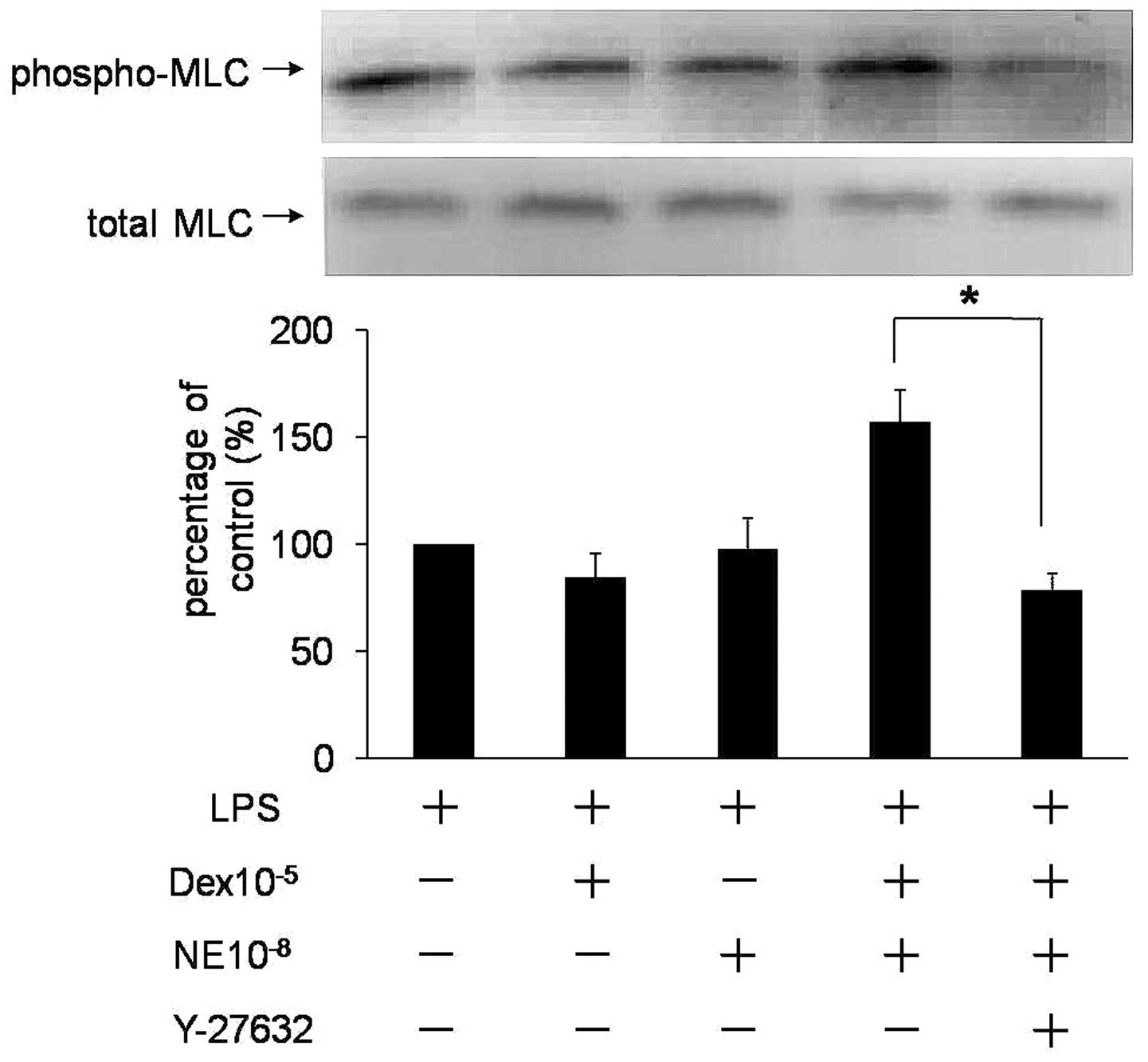
Inhibition of Rho kinase activity
reverses rapid Dex-induced promotion of NE-mediated
MLC20 phosphorylation
The RhoA/ROCK pathway participates in sustained
vasoconstriction and has been proposed to be important for
Ca2+ sensitization. Y-27632, a selective inhibitor of
Rho kinase, was used to determine the role of Rho kinase in rapid
regulation of NE-mediated VSMC contraction by Dex. Fig. 3 reveals that the rapid function of
Dex for NE-mediated VSMC contraction (P<0.05, n=3) was
eliminated by Y-27632, indicating that the RhoA/ROCK pathway is
involved in Dex-induced rapid promotion of NE-mediated VSMC
contraction.
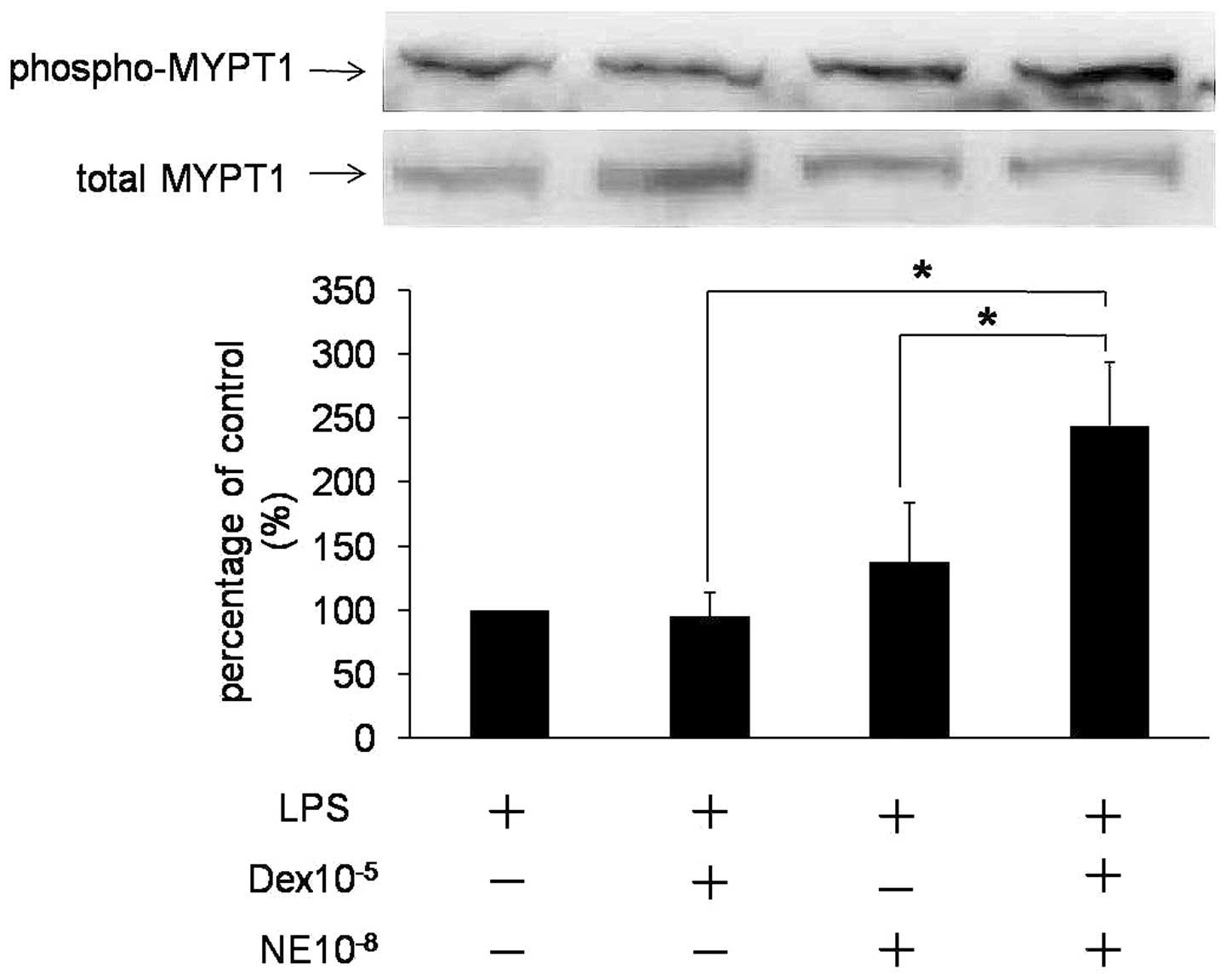
Dex enhances NE-mediated phosphorylation
of MYPT1
GTP-RhoA activates ROCK, which in turn
phosphorylates MYPT1 to inactivate MLCP activity (17). Therefore, we examined whether
MYPT1Thr853 phosphorylation accounted for NE-mediated
contraction in VSMCs. Fig. 4
indicates that MYPT1 was activated by NE or Dex alone, resulting in
an increase in P-MYPT1. However, when NE and Dex were administered
together, phosphorylation of MYPT1 was identified to be
significantly upregulated compared with NE or Dex alone (P<0.05;
n=3; Fig. 4), indicating that Dex
may enhance NE-mediated phosphorylation of MYPT1. Therefore,
activation of the RhoA/ROCK pathway is involved in Dex-induced
rapid promotion of NE-mediated VSMC contraction.
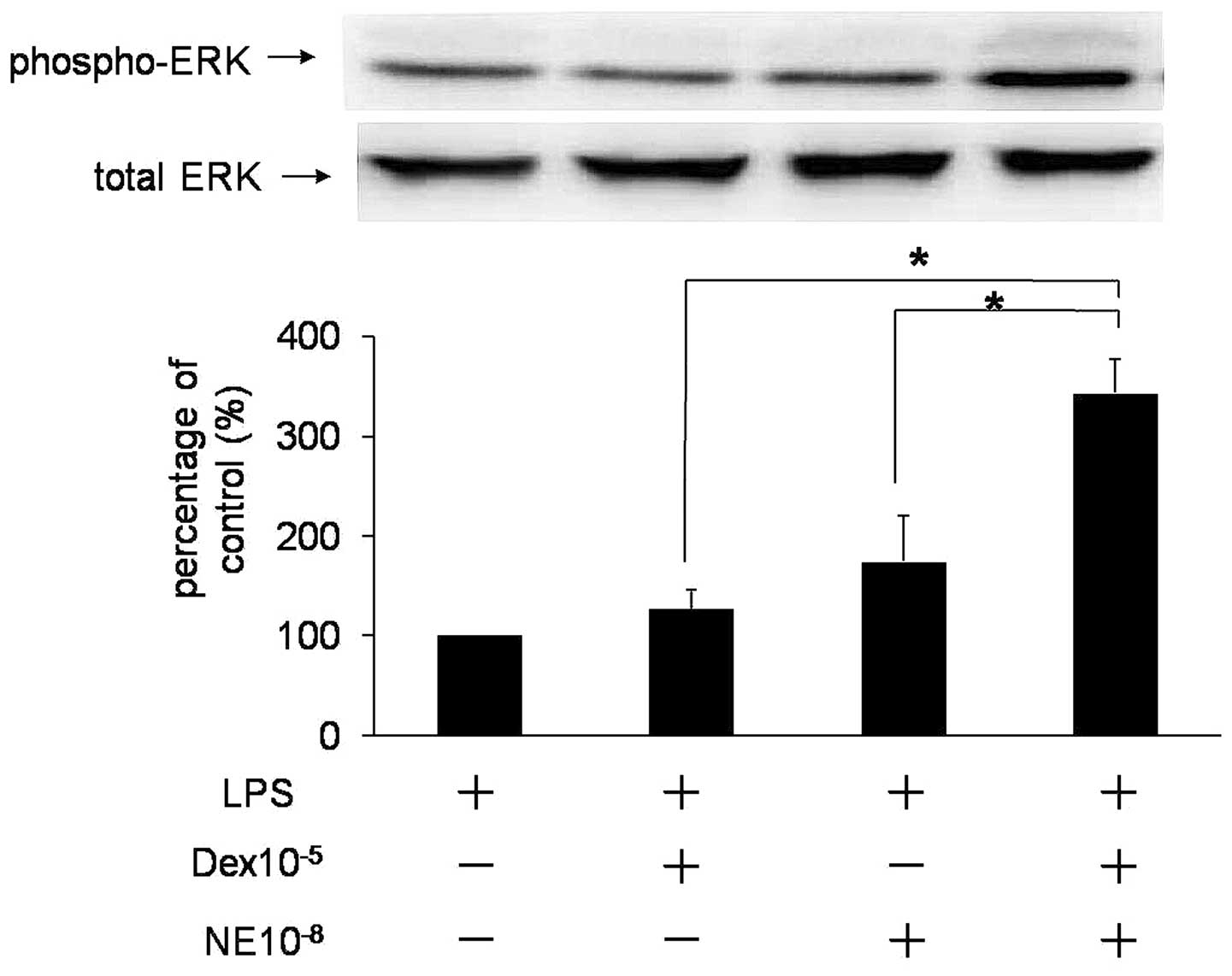
Dex increases NE-mediated activation of
ERK
Phosphorylation of CaD was previously hypothesized
to regulate smooth muscle contraction. Activation of ERK leads to
phosphorylation of CaD (6). To
determine whether ERK is involved in the rapid effect of Dex on
NE-mediated VSMC contraction, activation of ERK was examined.
Fig. 5 demonstrates that NE and
Dex enhanced activation of ERK. However, when NE and Dex were
administered together, the activation level of ERK was identified
as significantly upregulated compared with NE or Dex alone,
indicating that ERK activation occurred upstream when NE and Dex
were administered together, i.e., ERK was involved in the rapid
effect of Dex-induced promotion of NE-mediated VSMC
contraction.
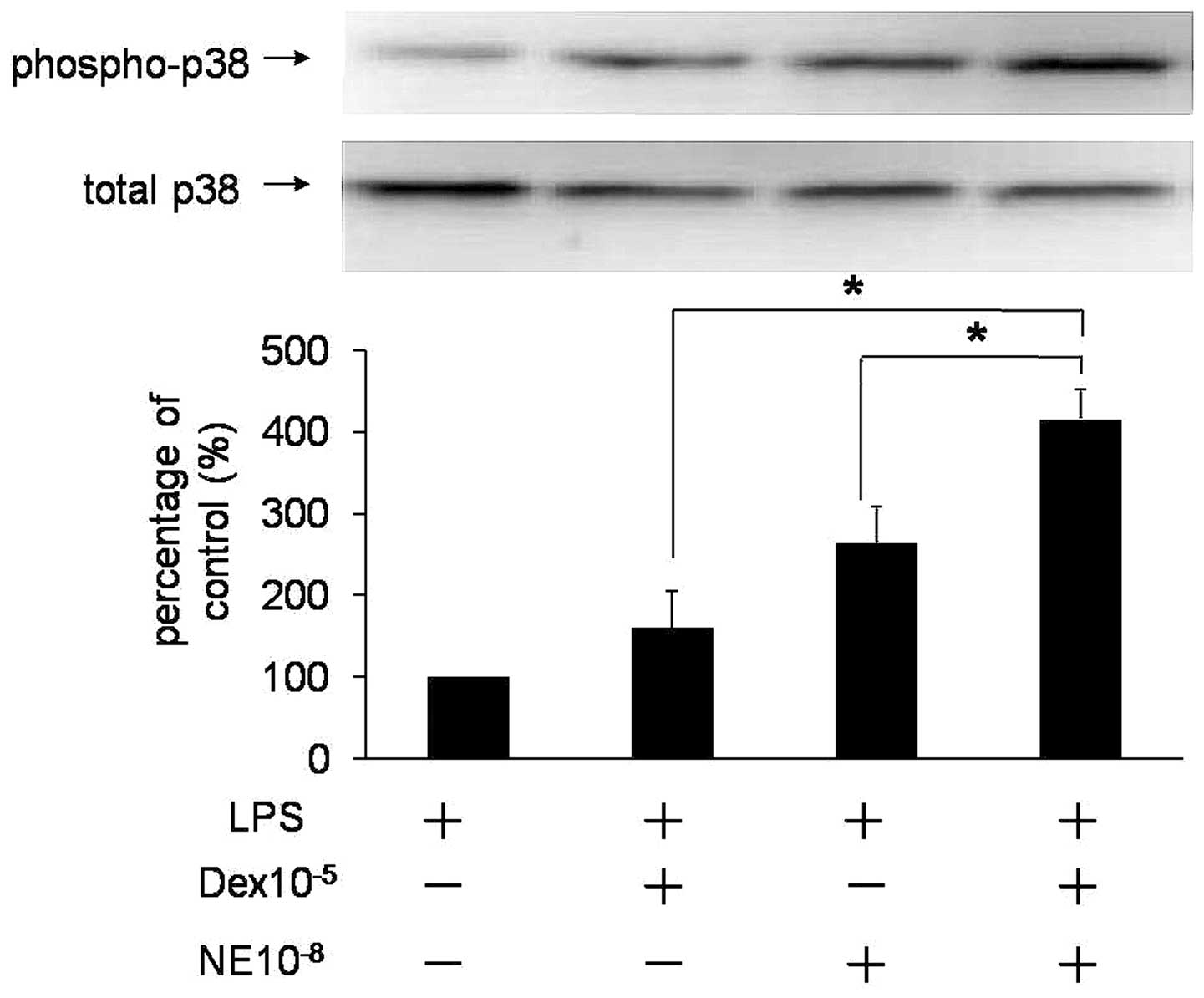
Dex increases NE-mediated activation of
p38
Activated p38 phosphorylates CaD and increases
phosphorylation and activation of heat-shock protein 27 (HSP27).
HSP27 is a known regulator of actin polymerization, an important
event in the mechanism of force maintenance during smooth muscle
contraction (8). To elucidate
whether p38 was involved in NE-mediated VSMC contraction stimulated
by Dex, phosphorylated p38 levels were analyzed. Results are
presented in Fig. 6. A higher
level of p38 activation was observed when NE and Dex were
administered together. Therefore, p38, as with ERK, was identified
as an additional molecule affected by Dex in NE-mediated VSMC
contraction.
Discussion
Septic shock usually results in a cardiac
dysfunction and a marked fall in systemic vascular resistance
(18). Impaired synthesis of
corticosteroids is commonly observed in the plasma of septic shock
patients (19). Specific patients
with sepsis and impaired adrenal function demonstrate a significant
decrease in pressor sensitivity to NE, which may be improved by
administration of hydrocortisone (20). In the clinic, the pressor effect of
NE alone for blood pressure elevation, particularly during rescue
therapy in septic shock, is considered unsatisfactory. When a small
amount of cortisol is administered, the pressor effect of NE is
significantly enhanced. However, the mechanism by which GCs rapidly
enhance NE-mediated contraction remains unclear. In the present
study, we observed the rapid effect of Dex on NE-mediated
contraction in vitro and investigated the mechanism of this
clinically important interaction.
Contractile activity in VSMCs is primarily
determined by phosphorylation of MLC20. MLC20
phosphorylation may induce VSMC contraction directly (17). In the present study, we demonstrate
a complex intracellular interaction between NE and Dex in VSMC
contraction. Treatment with NE alone induced MLC20
phosphorylation and enhanced the contraction of VSMCs. Dex alone
also caused low levels of MLC20 phosphorylation.
However, preincubation with Dex prior to addition of NE was
identified to significantly improve the sensitivity of VSMCs to NE
and enhance MLC20 phosphorylation, thus increasing the
contractile effect of VSMCs. In addition, the rapid effect of Dex
was not blocked by RU486, a GC nuclear receptor antagonist. The
present results indicate that Dex may rapidly enhance NE-mediated
contraction of VSMCs by a non-genomic mechanism.
Activation of MLCK leads to phosphorylation of
MLC20(17). In addition
to MLCK, the state of MLC20 phosphorylation is regulated
by MLCP, which removes the high-energy phosphate from the
MLC20 to cause VSMC relaxation. As this process does not
rely on [Ca2+]i, it is termed Ca2+
sensitization (21). The
Ca2+ sensitivity of contraction is affected by
variations in the ratio of MLCK/MLCP activity. A decrease in MLCP
activity is likely to alter the balance in favor of MLCK, resulting
in a greater degree of MLC20 phosphorylation and
contraction (22).
RhoA is a well-known member of the Rho protein
family (23). ROCK is the first
RhoA effector. RhoA and its downstream target ROCK are important
for Ca2+ sensitization (21). The activated RhoA-GTP activates
ROCK, which subsequently combines with the MYPT1 subunit of MLCP to
phosphorylate Thr853 and 696 sites and inhibit MLCP activity. These
phosphorylation events promote the phosphorylated state of
MLC20 and prolong VSMC contraction (17). The effect of ROCK is blocked by
Y-27632 (24). The
RhoA/ROCK-mediated pathway is critical for signal transduction
initiated by a number of agonists, including NE, angiotensin II,
serotonin, endothelin-1 and platelet-derived growth factor.
Previous studies have indicated that the RhoA/ROCK pathway is
important for numerous cellular functions, not only VSMC
contraction but also actin cytoskeleton organization, cytokinesis,
cell migration, proliferation and differentiation and gene
expression, all of which may participate in the pathogenesis of
cardiovascular disorders, including atherosclerosis, restenosis,
hypertension and cardiac hypertrophy (23–27).
The RhoA/ROCK pathway has also been associated with drug-induced
VSMC contraction or relaxation (28,29).
A previous study (28)
demonstrated that RhoA/ROCK is involved in
Ca2+-independent contractions induced by
phorbol-12,13-dibutyrate (PDBu). PDBu phosphorylates MYPT1 by
activating the RhoA/ROCK pathway, thus inactivating MLCP and
causing smooth muscle contraction. An additional study (29) revealed that the RhoA/ROCK signaling
pathway was involved in the regulation of vascular reactivity
following hemorrhagic shock. Chiba et al(30) demonstrated that GCs inhibit airway
hyperresponsiveness in allergic bronchial asthma. The mechanism of
this effect involves the reduction of augmented bronchial smooth
muscle contraction by GCs through inhibition of RhoA upregulation.
NE stimulates α1-adrenoreceptors to produce
inositol-1,4,5-triphosphate, which then releases Ca2+
that may promote the increase of [Ca2+]i.
Following this, Ca2+/CaM activates MLCK and
phosphorylates MLC20, leading to VSMC contraction. In
addition, α1-adrenoreceptors activate the smooth muscle
RhoA/ROCK pathway and upregulate Ca2+ sensitivity of the
contractile response (31). Using
the sepsis model, a previous study (14) identified that decreased
Ca2+ sensitization of VSMCs was the mechanism
responsible for vascular hyporeactivity. The present study
demonstrated that the RhoA/ROCK pathway was involved in enhancement
of Dex-induced rapid promotion of NE-mediated VSMC contraction.
Addition of Y-27632 was observed to significantly lowered the
effect of Dex-induced promotion of NE-mediated MLC20
phosphorylation, indicating that the effect of Dex on the rapid
enhancement of NE-mediated contraction of VSMCs may be mediated by
increased phosphorylation of MLC20 by promoting
NE-mediated activation of the RhoA/ROCK pathway. In addition, the
mechanism by which ROCK inhibits MLCP is through phosphorylation of
the MYPT1. In the present study, activation of MYPT1 was analyzed
to reveal that NE-mediated phosphorylation of MYPT1 was also
increased by Dex, indicating that Dex may upregulate activation of
the RhoA/ROCK pathway and promote NE-mediated VSMC contraction by
enhanced activity of MYPT1.
Previous studies have identified additional
mechanisms of VSMC contraction, including filament rearrangement
and the ERK, p38 and protein kinase C (PKC) pathways. Firstly,
actin and myosin interactions result in the initial development of
force, a process similar to the function of these filaments in
adhesion to attachment sites where they form a cytoskeletal
scaffold that maintains tension in the absence of additional
cross-bridge cycling (32).
Secondly, a number of studies have identified that activated ERK
and p38 are involved in agonist-induced smooth muscle stimulation.
Their activation leads to CaD phosphorylation, which increases
myosin ATPase activity and promotes VSMC contraction. In addition,
p38 increases phosphorylation and activation of HSP27, a regulator
of actin polymerization (6–8).
Finally, PKC phosphorylates calponin (CaP) and CaD, subsequently
increasing myosin ATPase activity and contractile response in VSMCs
(33).
To determine whether ERK and p38 are involved in
rapid enhancement of NE-mediated VSMC contraction by Dex, we
examined phosphorylation of ERK and p38. The results demonstrated
that co-treatment with Dex and NE upregulated phosphorylation of
ERK and p38, consistent with the hypothesis that ERK and p38 are
involved in the effect of Dex on NE-mediated VSMC contraction. The
association of the PKC pathway in this mechanism was not examined
in the present study.
Numerous research groups (34,35)
have previously reported cross talk between the RhoA/ROCK and ERK
pathways or that they are associated with the same pathway.
Activation of ERK is involved in angiotensin II-induced contraction
of pressurized mesenteric arteries. This effect is blocked by the
ROCK inhibitor Y-27632, indicating that ROCK is upstream of ERK
activation (34). However, another
study maintained that α2-adrenoceptor-mediated vascular
contraction in the porcine palmar lateral vein involved RhoA/ROCK
and ERK activation, although these were separate pathways (35). Further studies are required to
clarify the correlation between RhoA/ROCK and ERK or p38
pathways.
In conclusion, the present study demonstrated that
Dex rapidly reversed the hyporeactivities of vasoconstriction to NE
in vitro and this effect may be mediated by non-genomic
mechanisms by increasing activation of the RhoA/ROCK signaling
pathway. In addition, we identified that ERK and p38 pathways were
important for Dex-induced promotion of NE-mediated contraction in
VSMCs. These results may provide insight into the mechanism of
rapid enhancement of NE-mediated VSMC contraction by Dex and aid
development of clinical therapies against septic shock.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 30971083), International
Cooperation Fund of Shanghai (no. 11410706800) and the Military
Twelfth Five-Year Plan (no. BMS11J016). The authors thank
co-workers at the Department of Nautical Medicine and Laboratory of
Stress Medicine of the Second Military Medical University.
References
|
1
|
Somlyo AP and Somlyo AV: Signal
transduction and regulation in smooth muscle. Nature. 372:231–236.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamm KE and Stull JT: The function of
myosin and myosin light chain kinase phosphorylation in smooth
muscle. Annu Rev Pharmacol Toxicol. 25:593–620. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wickman G, Lan C and Vollrath B:
Functional roles of the rho/rho kinase pathway and protein kinase C
in the relevance regulation of cerebrovascular constriction
mediated by hemoglobin: relevance to subarachnoid hemorrhage and
vasospasm. Circ Res. 92:809–816. 2003. View Article : Google Scholar
|
|
4
|
Ito M, Nakano T, Erdodi F and Hartshorne
DJ: Myosin phosphatase: structure, regulation and function. Mol
Cell Biochem. 259:197–209. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Somlyo AP and Somlyo AV: Signal
transduction by G-proteins, rho-kinase and protein phosphatase to
smooth muscle and non-muscle myosin II. J Physiol. 522:177–185.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hedges JC, Oxhorn BC, Carty M, Adam LP,
Yamboliev IA and Gerthoffer WT: Phosphorylation of caldesmon by ERK
MAP kinases in smooth muscle. Am J Physiol Cell Physiol.
278:718–726. 2000.PubMed/NCBI
|
|
7
|
Yamboliev IA, Hedges JC, Mutnick JL, Adam
LP and Gerthoffer WT: Evidence for modulation of smooth muscle
force by the p38 MAP kinase/HSP27 pathway. Am J Physiol Heart Circ
Physiol. 278:1899–1907. 2000.PubMed/NCBI
|
|
8
|
Gorenne I, Su X and Moreland RS: Caldesmon
phosphorylation is catalyzed by two kinases in permeabilized and
intact vascular smooth muscle. J Cell Physiol. 198:461–469. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou J, Li M, Liu L, Sheng CQ, Li Z, Wang
Y, Zhou JR, Jiang ZP, Chen YZ and Jiang CL: A novel strategy for
development of glucocorticoids through non-genomic mechanism. Cell
Mol Life Sci. 9:1–10. 2010.
|
|
10
|
Zhou J, Liu DF, Liu C, Kang ZM, Shen XH,
Chen YZ, Xu T and Jiang C: Glucocorticoids inhibit degranulation of
mast cells in allergic asthma via nongenomic mechanism. Allergy.
63:1177–1185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun HW, Miao CY, Liu L, Zhou J, Su DF,
Wang YX and Jiang CL: Rapid inhibitory effect of glucocorticoids on
airway smooth muscle contractions in guinea pigs. Steriods.
71:154–159. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Wang YX, Zhou J, Long F, Sun HW,
Liu Y, Chen YZ and Jiang CL: Rapid nongenomic inhibitory effects of
glucocorticoids on human neutrophil degranulation. Inflam Res.
54:37–41. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang S and Zhang L: Glucocorticoids and
vascular reactivity. Curr Vasc Pharmacol. 2:1–12. 2004. View Article : Google Scholar
|
|
14
|
Mansart A, Bollaert PE, Giummelly P,
Capdeville-Atkinson C and Atkinson J: Effects of dexamethasone and
L-canavanine on the intracellular calcium-contraction relation of
the rat tail artery during septic shock. Am J Physiol Heart Circ
Physiol. 291:H1177–H1182. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takakura K, Xiaohong W, Takeuchi K, Yasuda
Y and Fukuda S: Deactivation of norepinephrine by peroxynitrite as
a new pathogenesis in the hypotension of septic shock.
Anesthesiology. 98:928–934. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jeon SB, Jin F, Kim JI, Kim SH, Suk K,
Chae SC, Jun JE, Park WH and Kim IK: A role for rho kinase in
vascular contraction evoked by sodium fluoride. Biochem Biophys Res
Commun. 343:27–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Somlyo AP and Somlyo AV: Ca2+
sensitivity of smooth muscle and nonmuscle myosin: modulated by G
proteins, kinases andmyosin phosphatase. Physiol Rev. 83:1325–1358.
2003.
|
|
18
|
Mansart A, Bollaert PE, Seguin C, Levy B,
Longrois D and Mallie JP: Hemodynamic effects of early versus late
glucocorticoid administration in experimental septic shock. Shock.
19:38–44. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zaloga GP and Marik P:
Hypothalamic-pituitary-adrenal insufficiency. Crit Care Clin.
17:25–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Annane D, Bellissant E, Sebille V, Lesieur
O, Mathieu B, Raphael JC and Gajdos P: Impaired pressor sensitivity
to noradrenaline in septic shock patients with and without impaired
adrenal function reserve. Br J Clin Pharmacol. 46:589–597. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hilgers RH and Webb RC: Molecular aspects
of arterial smooth muscle contraction: focus on Rho. Exp Biol Med.
230:829–835. 2005.PubMed/NCBI
|
|
22
|
Ihara E, Moffat L, Ostrander J, Walsh MP
and Macdonald JA: Characterization of protein kinase pathways
responsible for Ca2+ sensitization in rat ileal
longitudinal smooth muscle. Am J Physiol Gastrointest Liver
Physiol. 293:699–710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Loirand G, Guerin P and Pacaud P: Rho
kinases in cardiovascular physiology and pathophysiology. Circ Res.
98:322–334. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Noma K, Oyama N and Liao JK: Physiological
role of ROCKs in the cardiovascular system. Am J Physiol Cell
Physiol. 290:C661–C668. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shimokawa H and Takeshita A: Rho-kinase is
an important therapeutic target in cardiovascular medicine.
Arterioscler Thromb Vasc Biol. 25:1767–1775. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hirokazu T, Hiroki C, Tadashi I, Jun K,
Haruhiko M, Yoshisato T, Miyako T, Kazuhiko Y, Masaki N and Hisashi
S: Diverse activation states of RhoA in human lung cancer cells:
contribution of G protein coupled receptors. Int J Oncol.
30:709–715. 2007.PubMed/NCBI
|
|
27
|
Bellizzi A, Mangia A, Chiriatti A, Petroni
S, Quaranta M, Schittulli F, Malfettone A, Cardone RA, Paradiso A
and Reshkin SJ: RhoA protein expression in primary breast cancers
and matched lymphocytes is associated with progression of the
disease. Int J Mol Med. 22:25–31. 2008.PubMed/NCBI
|
|
28
|
Baek I, Jeon SB, Kim J, Seok YM, Song MJ,
Chae SC, Jun JE, Park WH and Kim IK: A role for Rho-kinase in
Ca2+ independent contractions induced by
phorbol-12,13-dibutyrate. Clin Exp Pharmacol Physiol. 36:256–261.
2009.
|
|
29
|
Li T, Liu L, Liu J, Ming J, Xu J, Yang G
and Zhang Y: Mechnisms of Rho kinase regulation of vascular
reactivity following hemorrhagic shock in rats. Shock. 29:65–70.
2008.PubMed/NCBI
|
|
30
|
Chiba Y, Goto K, Hirahara M, Sakai H and
Misawa M: Glucocorticoids ameliorate antigen-induced bronchial
smooth muscle hyperresponsiveness by inhibiting upregulation of
RhoA in rats. J Pharmacol Sci. 106:615–625. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Berridge MJ: Smooth muscle cell calcium
activation mechanisms. J Physiol. 586:5047–5061. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tseng S, Kim R, Kim T, Morgan KG and Hai
CM: F-actin disruption attenuates agonist-induced
[Ca2+], myosin phosphorylation and force in smooth
muscle. Am J Physiol. 41:1960–1967. 1997.PubMed/NCBI
|
|
33
|
Tang DC, Xiang JZ and Lu WT: Calponin: a
new regulatory protein for smooth muscle contraction. Prog Biochem
Biophys. 23:325–329. 1996.
|
|
34
|
Matrougui K, Tanko LB, Loufrani L, Gorny
D, Levy BI, Tedgui A and Henrion D: Involvement of rho-kinase and
the actin filament network in angiotensin II-induced contraction
and extracellular signal-regulated kinase activity in intact rat
mesenteric resistance arteries. Arterioscler Thromb Vasc Biol.
21:1288–1293. 2001. View Article : Google Scholar
|
|
35
|
Roberts RE: The role of Rho kinase and
extracellular regulated kinase-mitogen-activated protein kinase in
α2-adrenoceptor-mediaed vasoconstriction in the porcine
palmar lateral vein. J Pharmacol Exp Ther. 311:742–747. 2004.
|