1. Introduction
LXRs were originally considered as ‘orphan’ nuclear
receptors, since their natural ligands were unknown; however, these
receptors were ‘adopted’ following the discovery that metabolites
of cholestorol oxysterols bind to and activate these receptors at
physiological concentrations. More and more evidence indicates that
LXRs function as key modulators of cholesterol metabolism and play
an important role in inflammatory signaling. This review focuses on
recent advances concerning LXRs in cholesterol efflux and
inflammatory signaling in order to explore their use as potential
therapies for cardiovascular and inflammatory diseases.
Construction and transcription of
LXRs
LXRs have two subtypes: LXRα (nuclear receptor
subfamily 1, group H, member 3, NR1H3) and LXRβ (nuclear receptor
subfamily 1, group H, member 2, NR1H2). The two LXRs share
considerable sequence homology and appear to respond to the same
endogenous ligands. Similar to other members of the nuclear
receptor family, these proteins contain a zinc finger DNA-binding
domain that accommodates specific small lipophilic molecules.
Ligand binding triggers a conformational change that promotes
interaction with coactivator proteins and facilitates the
activation of specific target genes (1,2).
However, their tissue distribution differs. LXRα is highly
expressed in liver and at lower levels in adrenal glands,
intestine, adipose, macrophages, lung and kidney, whereas LXRβ is
expressed in all tissues examined. LXRs play a role in the
regulation of energy homeostasis; LXRα as well as LXRβ may play a
crucial role in the regulation of energy homeostasis. Both LXRα and
LXRβ are activated by physiological concentrations of sterol
metabolites. Natural ligands that activate LXRs include oxysterol
derivatives such as 25-hydroxycholesterol, 27-hydroxycholesterol,
22(R)-hydroxycholesterol, 24(S), 25-epoxycholesterol [24(S),25-EC],
and 5α,6α-epoxycholesterol [5,6-EC] (3–5).
LXRs bind to target DNA sequences in heterodimer complexes with
retinoid X receptor (RXR). The LXR/RXR is a so-called permissive
heterodimer; in that it can be activated by ligands for either LXR
or RXR. The heterodimer binding to LXR-responsive elements (LXREs)
in DNA consists of direct repeats (DRs) of the core sequence AGGTCA
separated by 4 nucleotides (DR-4). LXR/RXR heterodimer is bound to
LXREs in the promoters of target genes and in complex with
co-repressors such as silencing mediator of retinoic acid and
thyroid hormone receptor (SMRT) and nuclear receptor co-repressor
(N-COR). The function of nuclear proteins can be affected by their
sequestration in the nucleoli. Mutations in the activation
function-2 of LXR, an important protein-protein interaction site in
all nuclear receptors, was found to result in exclusion of LXRβ
from the nucleolus. In the absence of ligand these co-repressors
are maintained and the transcriptional activity of target genes is
repressed. Binding of ligand to LXR results in a conformational
change that facilitates an exchange of co-repressor-complex for
coactivator and transcription of target genes (6–8).
2. The role of LXRs in cholesterol
efflux
CYP7A1
Cholesterol 7α-hydroxylase (CYP7A1) is a member of
the cytochrome P450 family of enzymes and the rate-limiting enzyme
in the classical pathway of bile acid synthesis, and is the first
direct target of LXRs. CYP7A1 encodes for the rate-limiting step in
the conversion of cholesterol to bile acids in the liver. The
inability of LXRα−/− mice to induce hepatic CYP7A1
expression results in a diminished ability to metabolize
cholesterol to bile acids and the accumulation of cholesterol
esters (9). In response to acute
cholesterol feeding, CYP7A1 was up-regulated in mice via
stimulation of the liver X receptor α (LXRα). However, chronic
cholesterol feeding also results in activation of the
mitogen-activated protein (MAP) kinases, c-Jun N-terminal kinase
(JNK) and extracellular signal-regulated kinase (ERK), which leads
to suppression of CYP7A1 via activation of JNK and ERK signaling
pathways (10,11). The mRNA expression of CYP7A1 and
synthesis of bile acids was found to be low in embryonic stages,
suggesting that farnesoid X receptor (FXR) might be a key regulator
of CYP7A1 gene expression in the chicken embryo. In rabbits,
activation of FXR is dominant over activation LXRα in the
regulation of CYP7A1. The increased recruitment of LXRα, a CYP7A1
stimulatory pathway and decreased expression of FGF15 and
phosphorylated ERK1/2, a CYP7A1 repressive pathway, combined to
increase CYP7A1 expression during lactation (12). However, LXR agonists would not be
expected to promote bile acid syntheses in humans, since the LXR
response element is not conserved in the promoter of the human
CYP7A1 gene.
ABC transporters
The ABC (ATP binding cassette) transporters are
critical for the ability of LXRs to enhance efflux to cholesterol
acceptors. ABCA1, ABCG1, ABCG5, ABCG8 and ABCG4 of the ABC
transporter family are target genes of LXRs.
ABCA1 protein is critical for the efflux of excess
cellular cholesterol to Apo acceptors such as ApoA1, the first step
in reverse cholesterol transport. ATP-binding cassette protein A1
(ABCA1)-mediated cholesterol efflux is highly regulated at the
transcriptional level through the activity of the nuclear receptor
LXR. Cells from patients suffering from Tangier disease, which is
caused by a mutation in the ABCA1 gene, are defective in their
ability to efflux cholesterol. Expression of ABCA1 is strongly
induced by natural and synthetic LXR ligands which are attributed
to the presence of LXREs in the proximal promoter of the ABCA1
expression. LXRβ directly binds to the C-terminal region of ABCA1
to mediate its post-translational regulation. LXRβ can cause a
post-translational response by directly binding to ABCA1, as well
as a transcriptional response, to maintain cholesterol homeostasis
(13–15). However, Genvigir et al found
that lipid-lowering drugs down-regulate ABCA1 and ABCG1 mRNA
expression in individuals and exhibit differential effects on HepG2
cells. Moreover, they found that the ABCA1 and ABCG1 transcript
levels were not correlated directly to LXR mRNA expression in both
cell models treated with lipid-lowering drugs (16). LXR/RXR functions as a sensor of
cellular cholesterol concentration and mediates cholesterol efflux
by inducing the transcription of key cholesterol shuffling vehicles
namely, ABCA1 and ApoE. The LXR/RXR-induced up-regulation of ABCA1
and ApoE levels may be the molecular determinants of cholesterol
dyshomeostasis (17). ABCG1 has
recently been identified as a direct target of LXRs. It is also
strongly induced by cholesterol loading of macrophages. ABCG1 is
thought to function as a homodimer, although a functional
partnership with ABCG4 has also been suggested. Induction of ABCG1
may provide an additional pathway for cholesterol efflux from
macrophages or may act in concert with ABCA1. ABCG1 regulates
macrophage cholesterol efflux and hence plays a vital role in
macrophage foam cell formation. The sequential synergistic role of
ABCG1 in promoting cholesterol efflux involves phospholipid-rich
nascent HDL particles first generated by the lipidation of ApoA1 by
the ABCA1 transporter. The expression of ABCG1 induced by synthetic
or natural LXR ligands [TO901317, 22-(R)-hydroxycholesterol] was
attenuated by inhibitors of c-Jun N-terminal kinase and
phosphoinositide 3-kinase (PI3K). LXR agonists also induced the
binding of activator protein-1 (AP-1), a key transcription factor
family regulated by JNK, to recognition sequences present in the
regulatory regions of the ABCG1 gene (18–20).
In in vitro experiment assays, ABCG1 was demonstrated to
facilitate cholesterol efflux to HDL-2 and HDL-3 particles but not
to ApoA1. In in vitro experiments, macrophages lacking ABCG1
showed a diminished cholesterol efflux capacity to HDL, while
cholesterol efflux to ApoA1 which is mainly mediate by ABCA1 was
unchanged. Conjugated linoleic acid isomer trans-9, trans-11 (t9,
t11-CLA) is an agonist of LXRα in human macrophages and its effects
on macrophage lipid metabolism can be attributed to transcriptional
regulations associated with ABCG1 (21,22).
ABCG1 is known to be expressed in numerous cell
types and tissues, whereas ABCG4 expression is limited to the
central nervous system. ABCG4 is also modestly induced in
macrophages by cholesterol loading and LXR ligands and has been
reported to promote cholesterol efflux to HDL particles when
overexpressed in HEK293 cells (23,24).
Administration of the LXR agonist T0901317 to
pregnant mice via their diet led to induced fetal hepatic
expression levels of the cholesterol transporter genes Abcg5/g8 and
Abca1. ABCG5 and ABCG8 perhaps play a prominent role in the
inhibition of intestinal absorption of cholesterol and plant-sterol
absorption and cholesterol efflux from hepatocytes into bile. The
ABCG5 and ABCG8 proteins form a functional heterodimer that resides
in the apical membrane of hepatocytes. ABCG8 has a more profound
effect upon biliary cholesterol secretion than sitosterol;
ABCG5/G8, unlike ABCA1, together with bile acids should participate
in sterol efflux on the apical surface of Caco-2 cells (25). The mutation in either ABCG5 or
ABCG8 causes the rare genetic disease sitosterolemia. Patients with
this disease exhibit hyperabsorption of cholesterol and plant
sterols and show diminished secretion of sterols into bile and
hypercholesterolemia and develop premature atherosclerosis
(26).
Apolipoproteins
A subset of apolipoproteins (APOs) such as ApoE,
ApoCI, ApoCII, ApoD and ApoCIV may contribute to LXR-driven reverse
cholesterol transport and may serve as cholesterol acceptors. ApoE
is a principal protein component of chylomicron remnants, very
low-density lipoproteins. Recognition of ApoE by LDL receptors
mediates hepatic uptake of these particles. Hepatic ApoE expression
is controlled by a distal enhancer known as the hepatic control
region, whereas expression in macrophages and adipocytes is
directed by a distinct flanking sequence termed the multiple
enhancer (ME) region. ApoE was the first gene shown to be regulated
by LXR/RXR heterodimers in a tissue-specific manner. LXR mediates
lipid inducible expression of the ApoE gene in adipose tissue and
macrophages but not in liver. This differential regulation
correlates with the presence of a critical LXR response element in
the ME region of the ApoE gene. LXR-regulated ApoE expression by
JNK and PI3K/AKT signaling in macrophages of several key genes has
been implicated in atherosclerosis (18,27).
Mice lacking ApoE exhibit greatly elevated plasma VLDL and
intermediate-density lipoprotein cholesterol levels. Interestingly,
Apos such as ApoE, ApoCI, ApoCII, ApoD and ApoCIV have been shown
to serve as acceptors in ABCA1-mediated cholesterol efflux. The
elaboration of these acceptors by macrophages within the arterial
wall would be expected to promote cholesterol efflux and reverse
cholesterol transport. LXR/RXR-induced up-regulation of ABCA1 and
ApoE levels may be the molecular determinants of cholesterol
dyshomeostasis (19,28,29).
Lipoprotein remodeling enzymes
LXR has been shown to influence the expression of
several enzymes that act on lipoproteins, including lipoprotein
lipase (LPL), cholesterol ester transfer protein (CETP) and the
phospholipid transfer protein (PLTP). The functions of these
enzymes are complex and likely to be context-dependent. LXR
activation induces expression of LPL, which plays a crucial role in
binding of modified lipoproteins and may promote the conversion of
triglyceride-rich lipoproteins to cholesterol-rich lipoproteins
such as LDL. CETP is secreted by the liver and circulates in plasma
principally bound to HDL. LXRα has an essential role in the
regulation of CETP expression and maintaintence of RCT. Synthetic
LXR agonist enhanced plasma CETP activity resulted in non-high
density lipoprotein (non-HDL) increase and HDL decrease in
cynomolgus monkeys and human CETP transgenic mice (30,31).
LXR-mediated induction of human CETP expression is switched on
during monocyte-to-macrophage differentiation, is magnified by
lipid loading and is selectively lost in inflammatory macrophages.
LXRs affect remodeling of ApoB-containing lipoproteins via
induction of hepatic expression of CETP, which facilitates the
transfer of CE from HDL to ApoB-containing lipoproteins. PLTP has
been identified as a modulator of HDL metabolism and may also be
involved in reverse cholesterol transport (32). PLTP can also mediate lipid transfer
between LDL particles to produce a small pre-β-HDL and a large
α-HDL. Expression of a human PLTP transgene in mice increased
production of pre-β-HDL and enhanced hepatic uptake and clearance
of the cholesterol ester. Increased PLTP expression in the artery
wall may serve to generate cholesterol acceptors and therefore
contribute to cholesterol efflux.
Lipogenesis
Earliest studies on LXR pointed to an important role
in the control of fatty acid as well as cholesterol metabolism.
LXRs have been proposed as a glucose sensor affecting LXR-dependent
gene expression and de novo lipogenesis (33,34).
However, nuclear factor erythroid-2-related factor-2 (Nrf2)
activation promoted deacetylation of farnesoid X receptor (FXR) by
competing for p300, leading to FXR-dependent induction of the small
heterodimer partner (SHP), which was responsible for the repression
of LXRα-dependent gene transcription and inhibits LXRα-dependent
hepatic lipogenesis (35). Mice
lacking LXR were noted to be deficient in the expression of sterol
regulatory element binding protein 1c (SREBP-1c), fatty acid
synthase (FAS), steroyl coenzyme A desaturase 1 (SCD-1) and
acylcoenzyme A carboxylase (ACC). LXRs play an important role in
cholesterol synthesis and uptake and the effect of LXR agonists on
cholesterol synthesis plays only a minor role in the regulation of
cellular sterol homeostasis (36).
On one hand, the expression of SREBP-1C and FAS induced by LXR
activation promotes the esterification of free cholesterol to fatty
acid which is an important mechanism for buffering free cholesterol
levels; on the other hand, it is also likely to cause the elevation
of plasma and hepatic triglyceride levels.
3. The role of LXRs in inflammatory
signaling
LXRs and innate immunity
Recent studies have uncovered a common mechanism by
which different microbial pathogens might contribute to foam cell
formation and accelerate lesion development and interference with
LXR-dependent cholesterol metabolism. The innate immune system
recognizes conserved motifs found in microbes through so-called
pattern recognition receptors that include the TLR (Toll-like
receptor) family of proteins. Activation of TLR3 or TLR4 during
viral bacterial infections in macrophages severely compromises the
expression of ABCA1, ABCG1, ApoE and other LXR target genes both
in vitro and in vivo. A synthetic liver X receptor
(LXR) ligand, TO-901317, was found to restore cholesterol efflux
from HIV-infected T lymphocytes and macrophages. TO-901317 potently
suppressed HIV-1 replication in both cell types and inhibited HIV-1
replication in ex vivo cultured lymphoid tissue and in
RAG-hu mice infected in vivo (37). Activation of LXR represents a novel
lipid-signaling paradigm that alters the inflammatory response of
human dendritic cells (DCs), and LXR-positive DCs are present in
reactive lymph nodes in vivo. Administration of LXR-specific
natural or synthetic activators induced target gene expression
accompanied by increased expression of DC maturation markers
(38). T cell responses were
strongly affected in LXRα−/−LXRβ−/− mice,
Treatment of WT mice with the LXR agonists TO901317 and GW3965
resulted in a decrease in the pulmonary bacterial burden and a
comparable increase of Th1/Th17 function in the lungs. The
dependence of LXR signaling on the neutrophil IL-17 axis may be a
novel function for these nuclear receptors in resistance to M.
tuberculosis infection (39).
Consistent with these effects on LXR-dependent gene expression, LXR
activation was found to increase reactive oxygen species generation
by enhancing the expression of NADPH oxidase subunits. Activation
of TLR3 or TLR4 potently inhibits cholesterol efflux from
macrophages. TLR3/4-dependent inhibition of LXR is accomplished
through activation of the viral response transcription factor IFN
regulatory factor 3 (40),
however, the mechanism by which this factor blocks LXR action
remains to be determined. LXR-TLR crosstalk provides a potential
mechanism to explain how microbial infections may interfere with
cholesterol metabolism.
LXRs and inflammation
Atherosclerosis is now recognized to be a chronic
inflammatory disease as well as a disorder of lipid metabolism.
Activation of inflammatory signaling pathways and release of
inflammatory mediators are fundamental to the diverse immune
functions of macrophages. The microenvironment within the
atherosclerotic lesion is pro-inflammatory and results in
activation of these same pathways. Studies have demonstrated that
excessive inflammation is a risk factor for the promotion of
atherogenesis.
Evidence indicates that LXRs not only induce genes
involved in cholesterol efflux, but also repress a set of
inflammatory genes after bacterial, LPS, TNF-α or IL-1β
stimulation. The inflammatory genes include those involved in the
generation of bioactive molecules such as iNOS and COX2, IL-6 and
IL-1β, the chemokine monocyte chemoattractant protein-1 (MCP-1) and
MCP-3 and MMP-9 (38,41). In mature DCs, LXR activation
increased the production of inflammatory cytokines IL-12, TNF-α,
IL-6 and IL-8 and resulted in an increased capacity to activate
CD4+ T cell proliferation upon ligation with TLR4 or
TLR3 ligands (42). LXRs centrally
control reverse cholesterol transport, but also negatively modulate
TLR-mediated inflammatory pathways. LXR ligands repress these genes
in macrophages derived from LXRα−/− or
LXRβ−/− mice but are unable to do so in macrophages from
LXRαβ−/− mice, indicating both LXR isoforms possess
anti-inflammatory activity. LXR ligands also exhibit a similar
repression of tissue factor (TF) and osteopontin, both of which are
associated with the development of atherosclerosis. LXR null mice
exhibit enhanced responses to inflammatory stimuli, while the LXR
ligands can significantly reduce inflammation in a murine model of
contact dermatitis. Astrocytic LXRα activation and subsequent
release of ApoE by astrocytes is critical for the ability of
microglia to remove fibrillar Aβ in response to treatment with
TO901317 (43). In addition,
treatment of APOE−/− mice with LXR agonists was found to
reduce the expression of the inflammatory mediator MMP-9 and tissue
factor in atherosclerotic aortas while inducing expression of
ABCA1. LXRαβ−/− mice, challenged with LPS, exhibit an
exacerbated systemic inflammatory response and increased hepatic
expression of iNOS, TNF-α or IL-1β.
The mechanisms underlying the repression of
inflammatory genes by LXRs are poorly understood. LXREs have not
been identified in the proximal promoters of the repressed genes,
thus may be an indirect mechanism. In addition to the possible
competition for transcriptional co-activators, evidence suggests
that inhibition of the NF-κB pathway is involved. This inhibition,
referred to as transrepression, is thought to underlie
anti-inflammatory actions of nuclear receptors such as LXRs.
Transrepression of NF-κB by LXR involves a nuclear event. In a
recent study of transrepression of the iNOS promoter by peroxisome
proliferator activator receptor γ (PPARγ), sumoylation of PPARγ was
identified as a possible mechanism involved in this process.
Sumoylated PPARγ was suggested to prevent the LPS-dependent
exchange of co-repressor for co-activators, thus maintaining the
iNOS promoter in a repressed state.
4. Conclusion
On the whole, it is clear that LXRs play an
important role in cholesterol metabolism and inflammatory signaling
as summarized in Fig. 1. LXR
agonists show promise as potential therapeutics, given their
anti-atherogenic and anti-inflammatory properties. Future research
should continue to define the roles of LXRs in cholesterol
metabolism and the inflammatory response in order to identify new
compounds which can be designed to avoid the side effects of
current drugs, including hypertriglyceridemia and steatosis while
conferring beneficial effects on cholesterol metabolism and
inflammation. Investigation of LXRs can offer additional potential
therapies for cardiovascular and inflammatory diseases.
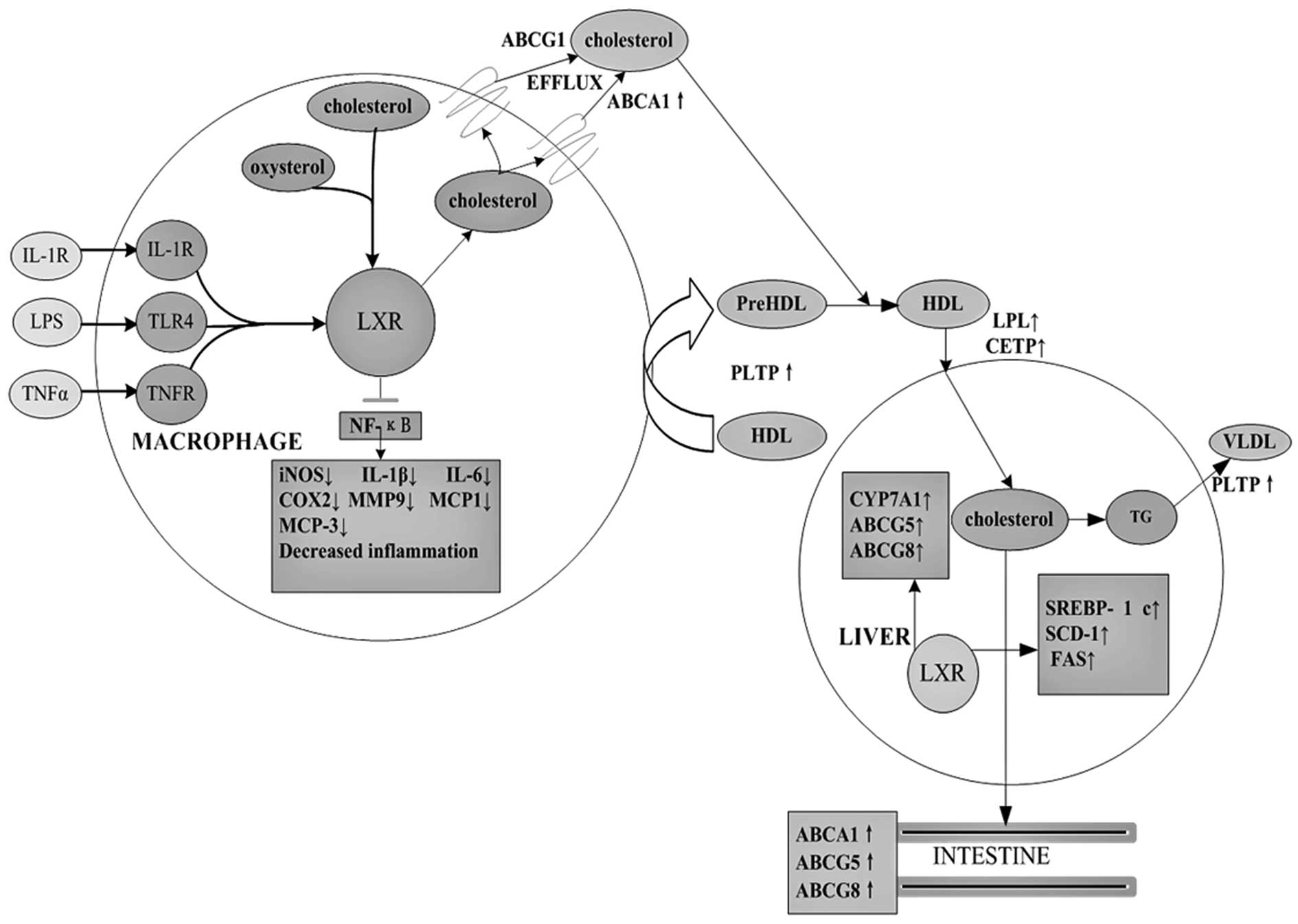 | Figure 1The role of LXR target genes in
cholesterol metabolism and inflammatory signaling. As
cholesterol-sensing nuclear receptors, LXRs promote cholesterol
efflux via regulation of CPY7A1, ABCA1, ABCG1, ABCG4, ABCG5
apolipoproteins, lipoprotein remodeling enzymes and lipogenesis.
Following their ligand-induced activation, LXRs inhibit expression
of inflammatory genes such as iNOS and COX2, IL-1β, IL-6, IL-8,
IL-12, MCP-1, MCP-3, MMP-9 and TNF-α. |
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 30772098, 81070374).
References
|
1
|
V LegryD CottelJ FerrièresAssociation
between liver X receptor α gene polymorphisms and risk of metabolic
syndrome in French populationsInt J Obes (Lond)324214282008
|
|
2
|
BM StensonM RydénN VenteclefLiver X
receptor (LXR) regulates human adipocyte lipolysisJ Biol
Chem286370379201110.1074/jbc.M110.17949921030586
|
|
3
|
TJ BerrodinQ ShenEM QuinetIdentification
of 5α, 6α-epoxycholesterol as a novel modulator of liver X receptor
activityMol Pharmacol78104610582010
|
|
4
|
M Korach-AndréA ArcherRP BarrosBoth
liver-X receptor (LXR) isoforms control energy expenditure by
regulating brown adipose tissue activityProc Natl Acad Sci
USA108403408201121173252
|
|
5
|
K MotoshimaT Noguchi-YachideK
SugitaSeparation of α-glucosidase-inhibitory and liver X
receptor-antagonistic activities of phenethylphenyl phthalimide
analogs and generation of LXRα-selective antagonistsBioorg Med
Chem17500150142009
|
|
6
|
T YasudaD GrillotJT
BillheimerTissue-specific liver X receptor activation promotes
macrophage reverse cholesterol transport in vivoArterioscler Thromb
Vasc Biol30781786201010.1161/ATVBAHA.109.19569320110577
|
|
7
|
Q ShenY BaiKC ChangLiver X
receptor-retinoid X receptor (LXR-RXR) heterodimer cistrome reveals
coordination of LXR and AP1 signaling in keratinocytesJ Biol
Chem2861455414563201110.1074/jbc.M110.16570421349840
|
|
8
|
R LallS KuruvillaK PrüferLiver X receptor
beta with mutations in the activation function-2 region is excluded
from the nucleolusCell Biol
Int33447452200910.1016/j.cellbi.2009.01.01519356704
|
|
9
|
M NoshiroE UsuiT KawamotoMultiple
mechanisms regulate circadian expression of the gene for
cholesterol 7α-hydroxylase (Cyp7a), a key enzyme in hepatic bile
acid biosynthesisJ Biol Rhythms22299311200717660447
|
|
10
|
AS HenkelKA AndersonAM DeweyA chronic
high-cholesterol diet paradoxically suppresses hepatic CYP7A1
expression in FVB/NJ miceJ Lipid
Res52289298201110.1194/jlr.M01278121097822
|
|
11
|
M SatoK SatoM FuruseChange in hepatic and
plasma bile acid contents and its regulatory gene expression in the
chicken embryoComp Biochem Physiol B Biochem Mol
Biol150344347200810.1016/j.cbpb.2008.04.00318499494
|
|
12
|
CR Wooton-KeeDJ CoyAT AthippozhyMechanisms
for increased expression of cholesterol 7α-hydroxylase (Cyp7a1) in
lactating ratsHepatology512772852010
|
|
13
|
M Hozoji-InadaY MunehiraK NagaoLXRβ
directly interacts with ABCA1 to promote HDL formation during acute
cholesterol accumulationJ Biol Chem28620117201242011
|
|
14
|
JJ DonkinS StukasV
Hirsch-ReinshagenATP-binding cassette transporter A1 mediates the
beneficial effects of the liver X receptor agonist GW3965 on object
recognition memory and amyloid burden in amyloid precursor
protein/presenilin 1 miceJ Biol
Chem2853414434154201010.1074/jbc.M110.108100
|
|
15
|
A TsezouD IliopoulosKN MalizosImpaired
expression of genes regulating cholesterol efflux in human
osteoarthritic chondrocytesJ Orthop Res2810331039201020108316
|
|
16
|
FD GenvigirAC RodriguesA CerdaEffects of
lipid-lowering drugs on reverse cholesterol transport gene
expressions in peripheral blood mononuclear and HepG2
cellsPharmacogenomics1112351246201010.2217/pgs.10.9320860464
|
|
17
|
A AkramJ SchmeidlerP KatselIncreased
expression of RXRα in dementia: an early harbinger for the
cholesterol dyshomeostasis?Mol Neurodegener5362010
|
|
18
|
EA HuwaitKR GreenowNN SinghA novel role
for c-Jun N-terminal kinase and phosphoinositide 3-kinase in the
liver X receptor-mediated induction of macrophage gene
expressionCell
Signal23542549201110.1016/j.cellsig.2010.11.00221070853
|
|
19
|
JP MauldinMH NagelinAJ WojcikReduced
expression of ATP-binding cassette transporter G1 increases
cholesterol accumulation in macrophages of patients with type 2
diabetes
mellitusCirculation11727852792200810.1161/CIRCULATIONAHA.107.741314
|
|
20
|
CJ DelvecchioP BilanP NairLXR-induced
reverse cholesterol transport in human airway smooth muscle is
mediated exclusively by ABCA1Am J Physiol Lung Cell Mol
Physiol295L949L957200810.1152/ajplung.90394.200818820007
|
|
21
|
H WiersmaN NijstadJF de BoerLack of Abcg1
results in decreased plasma HDL cholesterol levels and increased
biliary cholesterol secretion in mice fed a high cholesterol
dietAtherosclerosis206141147200910.1016/j.atherosclerosis.2009.02.02219339012
|
|
22
|
J EckerG LiebischW PatschThe conjugated
linoleic acid isomer trans-9,trans-11 is a dietary occurring
agonist of liver X receptor αBiochem Biophys Res
Commun388660666200919682978
|
|
23
|
DD BojanicPT TarrGD GaleDifferential
expression and function of ABCG1 and ABCG4 during development and
agingJ Lipid Res51169181201010.1194/M900250-JLR20019633360
|
|
24
|
N WangL Yvan-CharvetD LütjohannATP-binding
cassette transporters G1 and G4 mediate cholesterol and desmosterol
efflux to HDL and regulate sterol accumulation in the brainFASEB
J2210731082200810.1096/fj.07-9944com18039927
|
|
25
|
EM Van StratenNC HuijkmanJF
BallerPharmacological activation of LXR in utero directly
influences ABC transporter expression and function in mice but does
not affect adult cholesterol metabolismAm J Physiol Endocrinol
Metab295E1341E1348200818840761
|
|
26
|
H KusuharaY SugiyamaATP-binding cassette,
subfamily G (ABCG family)Pflugers
Arch453735744200710.1007/s00424-006-0134-x16983557
|
|
27
|
NP HessvikMV BoekschotenMA BaltzersenLXRβ
is the dominant LXR subtype in skeletal muscle regulating
lipogenesis and cholesterol effluxAm J Physiol Endocrinol
Metab298E602E6132010
|
|
28
|
JM TaylorF BorthwickC
BartholomewOverexpression of steroidogenic acute regulatory protein
increases macrophage cholesterol efflux to apolipoprotein
AICardiovasc Res86526534201010.1093/cvr/cvq015
|
|
29
|
S ZhongAL MagnoloM SundaramNonsynonymous
mutations within APOB in human familial hypobetalipoproteinemia:
evidence for feedback inhibition of lipogenesis and postendoplasmic
reticulum degradation of apolipoprotein BJ Biol
Chem28564536464201010.1074/jbc.M109.060467
|
|
30
|
S HonzumiA ShimaA HiroshimaLXRα regulates
human CETP expression in vitro and in transgenic
miceAtherosclerosis2121391452010
|
|
31
|
F BriandM TréguierA AndréLiver X receptor
activation promotes macrophage-to-feces reverse cholesterol
transport in a dyslipidemic hamster modelJ Lipid
Res51763770201010.1194/jlr.M00155219965597
|
|
32
|
D LakomyC RébéAL SbernaLiver X
receptor-mediated induction of cholesteryl ester transfer protein
expression is selectively impaired in inflammatory
macrophagesArterioscler Thromb Vasc
Biol2919231929200910.1161/ATVBAHA.109.193201
|
|
33
|
M Korach-AndréA ArcherC GabbiLiver-X
Receptors regulate de novo lipogenesis in a tissue specific manner
in C57Bl/6 female miceAm J Physiol Endocrinol
Metab301E210E222201121521718
|
|
34
|
I HongHS RhoDH KimActivation of LXRα
induces lipogenesis in HaCaT cellsArch Pharm Res33144314492010
|
|
35
|
HY KayWD KimSJ HwangNrf2 inhibits
LXRα-dependent hepatic lipogenesis by competing with FXR for
acetylase bindingAntioxid Redox Signal15213521462011
|
|
36
|
K SatoT KamadaRegulation of bile acid,
cholesterol, and fatty acid synthesis in chicken primary
hepatocytes by different concentrations of T0901317, an agonist of
liver X receptorsComp Biochem Physiol A Mol Integr
Physiol158201206201110.1016/j.cbpa.2010.10.02821056113
|
|
37
|
MP MorrowA GrantZ MujawarStimulation of
the liver X receptor pathway inhibits HIV-1 replication via
induction of ATP-binding cassette transporter A1Mol
Pharmacol78215225201010.1124/mol.110.06502920479131
|
|
38
|
D TöröcsikM BaráthS BenkoActivation of
liver X receptor sensitizes human dendritic cells to inflammatory
stimuliJ Immunol18454565465201020410489
|
|
39
|
H KorfS Vander BekenM RomanoLiver X
receptors contribute to the protective immune response against
Mycobacterium tuberculosis in miceJ Clin
Invest11916261637200910.1172/JCI3528819436111
|
|
40
|
C FontaineE RigamontiA NoharaLiver X
receptor activation potentiates the lipopolysaccharide response in
human macrophagesCirc
Res1014049200710.1161/CIRCRESAHA.106.13581417540978
|
|
41
|
W HuangS GhislettiK SaijoCoronin 2A
mediates actin-dependent de-repression of inflammatory response
genesNature470414418201110.1038/nature0970321331046
|
|
42
|
S ChenR SorrentinoK ShimadaChlamydia
pneumoniae-induced foam cell formation requires MyD88-dependent
and independent signaling and is reciprocally modulated by liver X
receptor activationJ
Immunol18171867193200810.4049/jimmunol.181.10.7186
|
|
43
|
D TerwelKR SteffensenPB VergheseCritical
role of astroglial apolipoprotein E and liver X receptor-α
expression for microglial Aβ phagocytosisJ
Neurosci3170497059201121562267
|














