Introduction
According to the amyloid cascade hypothesis, there
is an imbalance between the production and clearance of amyloid-β
(Aβ), leading to the formation of insoluble aggregates and soluble
monomers and larger assemblies of Aβ which drive the synaptic loss
and neuronal degeneration in the Alzheimer's disease (AD) process
(1). Data supporting the view that
life-long amyloid precursor protein (APP) overexpression triggers
Aβ deposition and neurodegeneration come from studies on Down's
syndrome patients and the finding of a duplication of the APP locus
in a certain familial form of AD (2,3).
Aβ is produced through the amyloidogenic pathway
from APP by proteolytic cleavage involving the two aspartyl
proteases β- and γ-secretase generating Aβ1-42 and C-terminal
truncated Aβ isoforms ranging from Aβ1-40 down to Aβ1-17 (4,5). In
another catabolic APP pathway, β-secretase cleavage in conjunction
with α-secretase cleavage result in the release of several short Aβ
isoforms (Aβ1-16 down to Aβ1-13) (5) of which Aβ1-16 previously have been
shown to be elevated in sporadic AD (SAD) (6) and familial AD (FAD) (7) compared to non-demented controls.
Moreover, this pathway is induced upon pharmacological γ-secretase
inhibitor treatment of cells, mice, dogs, rhesus monkeys and humans
(5,8–11).
The first identified mutation causing FAD was a
missense mutation in the APP gene (12). However, most FAD are caused by
mutations in the highly homologous genes of the presenilin enzymes
that constitute the active site γ-secretase, presenilin 1 (PSEN1)
and presenilin 2 (PSEN2) (13,14).
These mutations appear to accelerate Aβ plaque formation and have
been shown to increase the Aβ1-42/Aβ1-40 ratio in primary
fibroblasts and plasma of affected individuals (15–18).
Using immunoprecipitation in combination with mass spectrometry, we
recently showed that carriers of the FAD-associated PSEN1
A431E mutation have low CSF levels of Aβ1-37, Aβ1-38 and Aβ1-39
(7). Here we investigated the Aβ
isoform pattern in the CSF from carriers of the FAD-associated
PSEN1 M139T and L286P mutations.
Materials and methods
Study participants
Participants were recruited from the Genetic
Counseling Program for Familial Dementias (PICOGEN) at the Hospital
Clinic, Barcelona, Spain (19).
Eleven subjects from 3 families with 2 PSEN1 mutations
(L286P, M139T) (6 mutation carriers and 5 non-carriers) were
included in the study (20,21).
The L286P mutation causes familial early onset AD at a median age
of 40 years and cerebral hematomas in 42% of the affected subjects.
The neuropathological studies in this family reveal the typical
features of AD associated with severe amyloid angiopathy and
‘cotton-wool plaques’. The M139T mutation causes familial early
onset AD at a median age of 46 with typical clinical and
neuropathological characteristics of AD associated with intense
amyloid angiopathy but no ‘cotton-wool’ plaques or lobar hematomas.
Because of the relatively predictable age of onset within a family
we calculate for each participant the ‘adjusted age’ as the
subject's age relative to the median familial age of onset.
Subjects underwent clinical and cognitive evaluation and were
classified clinically as asymptomatic if they had normal cognitive
evaluation and the Clinical Dementia Rating (CDR) scale was equal
to 0; or symptomatic if cognitive performance was more than 1.5 SD
below the mean, with respect to age and education level, in any in
any cognitive test or CDR >0 (Table
I).
 | Table ISummary of the 11 subjects included
in the study. |
Table I
Summary of the 11 subjects included
in the study.
| Subject no. | Carrier | Mutation | CDRsum | CDRtot | MMSE | Adjusted age |
|---|
| 1 | 1 | L286P | 2 | 0.5 | 28 | −2.7 |
| 2 | 1 | L286P | 2.5 | 0.5 | 24 | 2.6 |
| 3 | 1 | L286P | 5.5 | 1 | 24 | 4.7 |
| 4 | 0 | L286P | 0 | 0 | 30 | −5.2 |
| 5 | 0 | L286P | 0 | 0 | 29 | −1.2 |
| 6 | 0 | L286P | 0 | 0 | 29 | 3.8 |
| 7 | 1 | M139T | 0 | 0 | 30 | −21.8 |
| 8 | 1 | M139T | 0 | 0 | 30 | −13.1 |
| 9 | 1 | M139T | 0 | 0 | 28 | −12.5 |
| 10 | 0 | M139T | 0 | 0 | 29 | −20.9 |
| 11 | 0 | M139T | 0 | 0 | 29 | −10.6 |
CSF analysis
Ten milligrams of CSF was obtained in the morning
using a 22 gauge Sprotte needle. The CSF was then centrifuged,
aliquoted into siliconized polypropylene Eppendorf tubes and frozen
at −80˚C within 2 h of being obtained.
The study was approved by the Hospital Clinic Ethics
Committee, and all participants gave informed consent to
participate in the study, which was conducted according to the
provisions of the Helsinki Declaration.
Immunoprecipitation (IP)-mass
spectrometry (MS)
IP and MS analyses were conducted as previously
described with the monoclonal antibody 6E10 (epitope 4–9, Signet
Laboratories Inc., Dedham, MA, USA) and matrix-assisted laser
desorption/ionization time-of-flight mass spectrometry
(MALDI-TOFMS, Autoflex, Bruker Daltonics, Bremen, Germany)
operating in reflector mode (22).
The peak areas were normalized to the sum of the integrated peaks
which result in a relative abundance rather than an absolute
concentration pattern (6).
Results
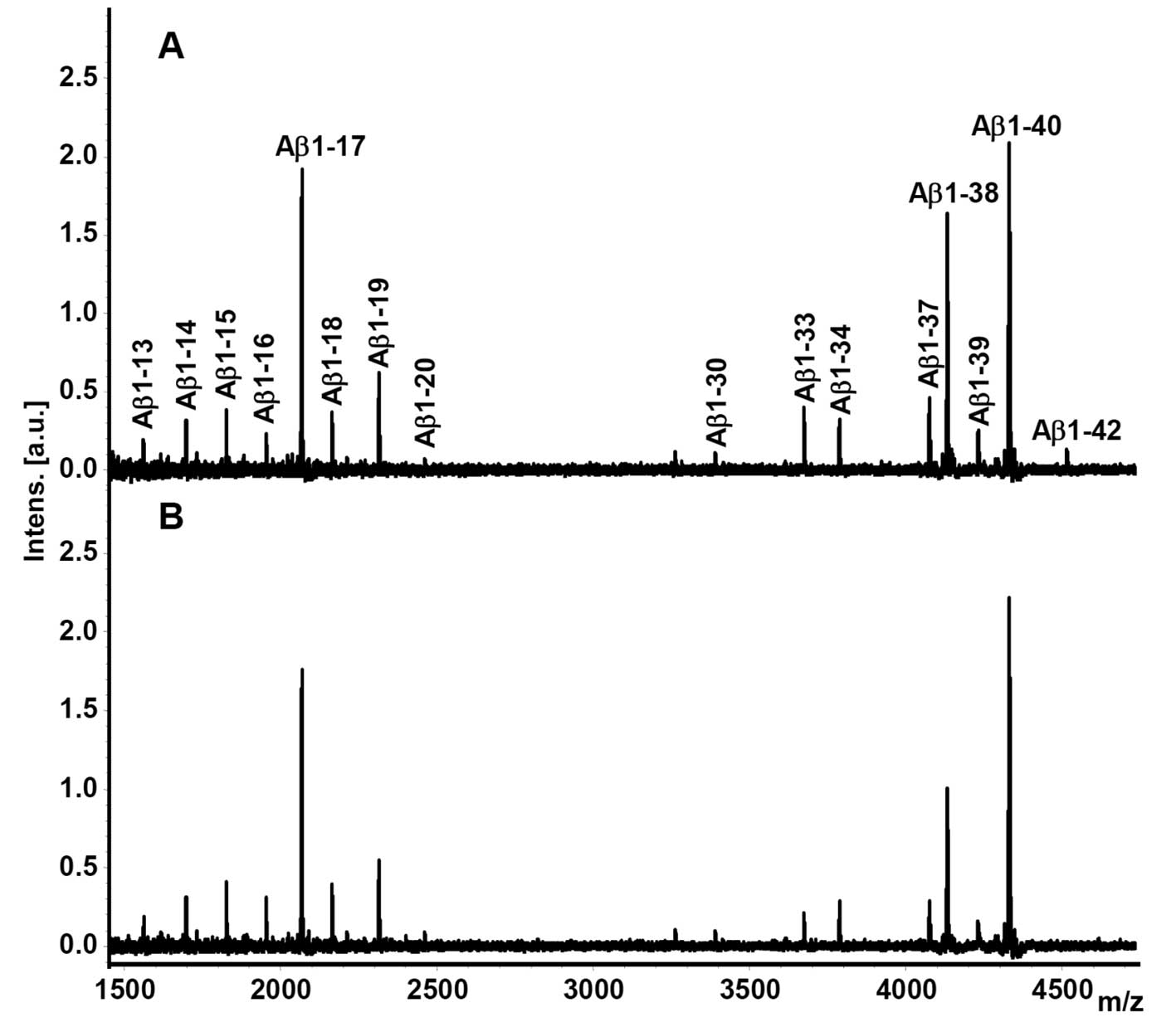
Representative CSF Aβ isoform mass spectra from a
PSEN1 L286P FAD mutation carrier and a non-carrier are shown
in Fig. 1. All isoforms (Aβ1-13,
Aβ1-14, Aβ1-15, Aβ1-16, Aβ1-17, Aβ1-18, Aβ1-19, Aβ1-20, Aβ1-30,
Aβ1-33, Aβ1-34, Aβ1-37, Aβ1-38, Aβ1-39, Aβ1-40 and Aβ1-42) were
reproducibly detected from all subjects and quantified in respect
to their relative abundance.
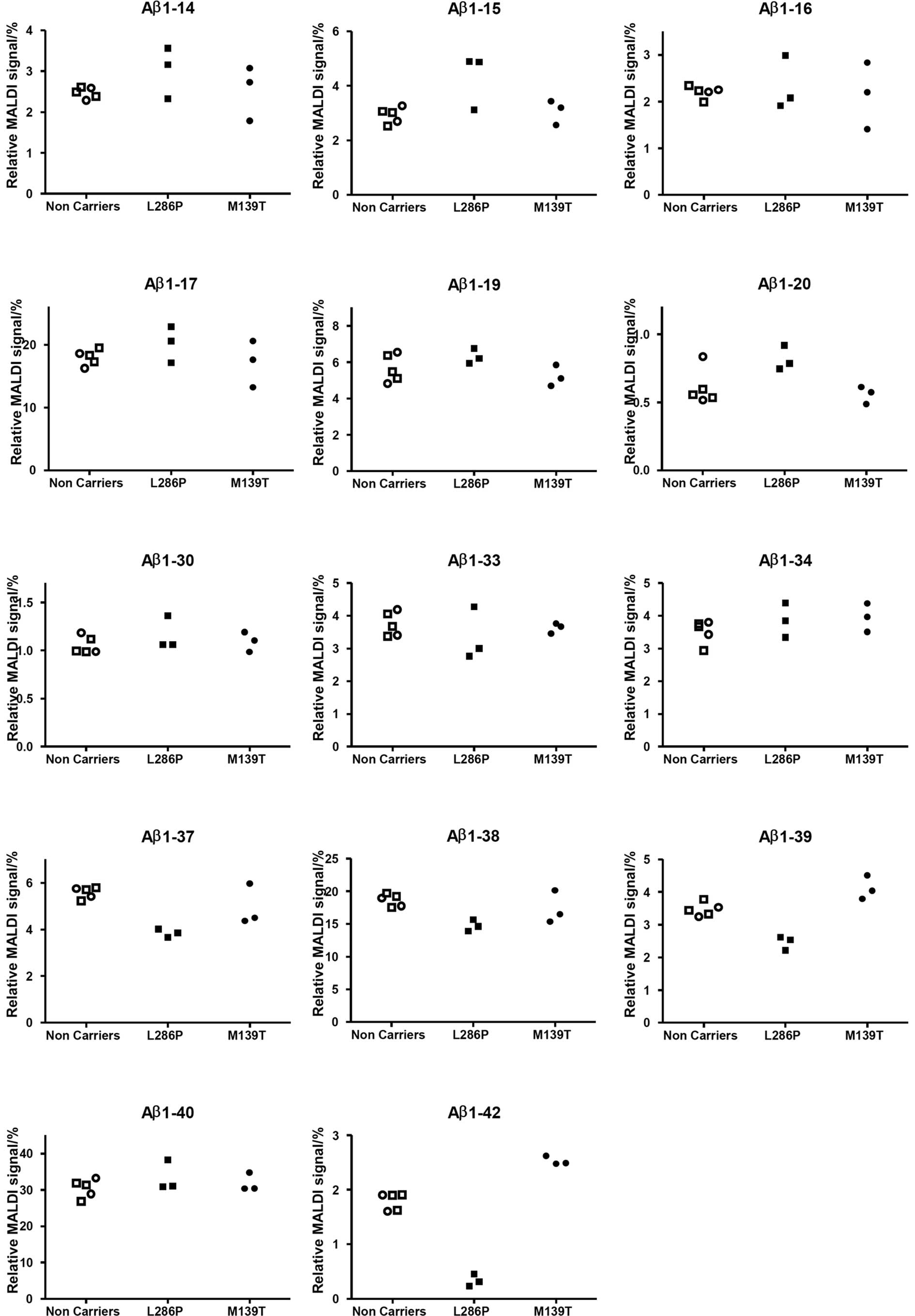
The MS peaks at mass-to-charge ratio 4073.0, 4130.0,
4229.1 and 4512.2, corresponding to Aβ1-37, Aβ1-38, Aβ1-39 and
Aβ1-42, respectively, were lower in the PSEN1 L286P FAD
mutation carriers (Fig. 2). The
relative level of Aβ1-37 was reduced by 31%, Aβ1-38 by 21%, Aβ1-39
by 29% and Aβ1-42 by 81%. In contrast Aβ1-15 and Aβ1-20 were
increased by 47 and 34%, respectively, while the other isoforms
detected were more or less unaffected.
In contrast, PSEN1 M139T carriers displayed
similar levels as non-carriers of all Aβ isoforms reproducibly
detected except Aβ1-42 that was present at 41% higher levels in the
group harboring the mutation as compared to non-carriers (Fig. 2).
Discussion
The reduction in Aβ1-42 in the CSF from AD patients
is a well-replicated finding thought to reflect the AD pathology
with plaques in the brain acting as sinks trapping Aβ1-42 (23). The data presented here on L286P
mutation carriers suggests that this specific PSEN1 mutation
modulates γ-secretase function, which is reflected by decreased
levels of Aβ1-37, Aβ1-38 and Aβ1-39, in a similar manner as the
PSEN1 A431E mutation. It has been speculated that the
decrease in Aβ1-37, Aβ1-38, Aβ1-39, due to the PSEN1 A431E
mutation, indicates that the γ-secretase cleavage site preference
has been changed in a disease-promoting manner (7). Similar changes in the levels of the
isoforms have also been reported from cells expressing the
PSEN1 Δ9 or L166P mutation, or the PSEN2 N141I
mutation (15). Thus, modulating
the γ-secretase function to boost cleavages at Gly37, Gly38 and
Val39 may be an attractive approach to prevent aggregation of Aβ
and consequently the formation of plaques.
The increase in Aβ1-20 in L286P mutation carriers is
a finding which previously has been described for the PSEN1
A431E mutation (7). Two candidate
enzymes which possibly can cleave at this position are
insulin-degrading enzyme and BACE 2 (24). However, the underlying molecular
mechanism needs to be further investigated.
We could not detect an increase in Aβ1-16 for L286P
mutation carriers as previously described for both sporadic and
familial AD patients (6,7). On the other hand, an increase by 47%
was observed for Aβ1-15. Whether this is a specific finding for
this particular mutation needs to be confirmed in a larger patient
sample.
The median age at onset for the PSEN1 L286P
mutation is 40 years. The patients included in this group had a
mean age of 41.5 thus very close to the critical age and they all
displayed cognitive decline. In contrast, the mean age in the
patient group carrying the PSEN1 M139T mutation was 30.2
years, which is almost 16 years younger than the expected age of
onset (Table I). Furthermore,
L286P seems to be a more aggressive mutation than M139T with
cotton-wool plaques noted in the neuropath vs. no cotton-wool
present in the M139T mutation. The A431E mutation also presents a
median age of onset of 40 and cotton-wool plaques, suggesting
similarities in the biological effects of L286P and A431E different
from M139T.
The slightly increased relative levels of Aβ1-42 in
the CSF of M139T mutation carriers suggest that this particular
mutation starts with an over-production of Aβ very early in life or
that these subjects present normal values of Aβ1-42 while L286P
mutation carriers present very low levels. Furthermore, the
patients harboring the PSEN1 M139T mutation were all
cognitively healthy (Table I).
It should be noted that due to the low number of
individuals in the groups analyzed, no statistical analysis was
performed. Moreover, there are also several non-quantitative
aspects of IP-MS, which should be considered, as discussed
elsewhere (25). However, the
results presented here with decreased CSF levels of Aβ1-37, Aβ1-38
and Aβ1-39 in patients with a PSEN1 L286P mutation are in
agreement with previous findings in PSEN1 A431E mutation
carriers and suggest that the pathogenic effect of some
PSEN1 mutations may be related to impaired ability of the
γ-secretase complex to produce C-terminally truncated Aβ peptides
that may inhibit Aβ1-42 oligomerization.
In conclusion, the data suggest that IP-MS analysis
of the CSF Aβ isoform pattern may function as an additional
screening tool to detect certain forms of PSEN1 mutations in
patients with a family history indicating FAD. This hypothesis will
be further tested on subjects having other PSEN1
mutations.
Acknowledgements
This study was supported by grants from the Swedish
Research Council (projects 2006–6227, 2006–2740 and 2006–3505), the
Alzheimer's Association (NIRG-08-90356), The Torsten and Ragnar
Söderberg Foundation, Demensförbundet, cNEUPRO, the Royal Swedish
Academy of Sciences, the Sahlgrenska University Hospital, the
Inga-Britt and Arne Lundberg Research Foundation, the Göteborg
Medical Society, the Swedish Medical Society, Swedish Brain Power,
Stiftelsen Gamla Tjänarinnor, Gun och Bertil Stohnes stiftelse,
Åhlén-stiftelsen, Alzheimer Foundation, Sweden and the Spanish
Ministry of Science and Technology (FIS080036).
References
|
1
|
JA HardyGA HigginsAlzheimer's disease: the
amyloid cascade hypothesisScience2561841851992
|
|
2
|
CA LemereJK BlusztajnH YamaguchiT
WisniewskiTC SaidoDJ SelkoeSequence of deposition of heterogeneous
amyloid beta-peptides and APO E in Down syndrome: implications for
initial events in amyloid plaque formationNeurobiol
Dis31632199610.1006/nbdi.1996.00039173910
|
|
3
|
A Rovelet-LecruxD HannequinG RauxAPP locus
duplication causes autosomal dominant early-onset Alzheimer disease
with cerebral amyloid angiopathyNat
Genet382426200610.1038/ng171816369530
|
|
4
|
D BeherJD WrigleyAP OwensMS
ShearmanGeneration of C-terminally truncated amyloid-beta peptides
is dependent on gamma-secretase activityJ
Neurochem82563575200210.1046/j.1471-4159.2002.00985.x12153480
|
|
5
|
E PorteliusE PriceG BrinkmalmA novel
pathway for amyloid precursor protein processingNeurobiol
Aging3210901098201110.1016/j.neurobiolaging.2009.06.00219604603
|
|
6
|
E PorteliusH ZetterbergU AndreassonAn
Alzheimer's disease-specific beta-amyloid fragment signature in
cerebrospinal fluidNeurosci Lett4092152192006
|
|
7
|
E PorteliusU AndreassonJM RingmanDistinct
cerebrospinal fluid amyloid beta peptide signatures in sporadic and
PSEN1 A431E-associated familial Alzheimer's diseaseMol
Neurodegener52201010.1186/1750-1326-5-220145736
|
|
8
|
E PorteliusB Van BroeckU AndreassonAcute
effect on the Abeta isoform pattern in CSF in response to
γ-secretase modulator and inhibitor treatment in dogsJ Alzheimers
Dis2110051012201020634579
|
|
9
|
E PorteliusB ZhangMK GustavssonEffects of
gamma-secretase inhibition on the amyloid beta isoform pattern in a
mouse model of Alzheimer's diseaseNeurodegener
Dis6258262200910.1159/00026463919955704
|
|
10
|
E PorteliusRA DeanMK GustavssonA novel
Abeta isoform pattern in CSF reflects gamma-secretase inhibition in
Alzheimer diseaseAlzheimers Res
Ther27201010.1186/alzrt3020350302
|
|
11
|
JJ CookKR WildsmithDB GilbertoAcute
gamma-secretase inhibition of nonhuman primate CNS shifts amyloid
precursor protein (APP) metabolism from amyloid-beta production to
alternative APP fragments without amyloid-beta reboundJ
Neurosci3067436750201010.1523/JNEUROSCI.1381-10.201020463236
|
|
12
|
A GoateMC Chartier-HarlinM
MullanSegregation of a missense mutation in the amyloid precursor
protein gene with familial Alzheimer's
diseaseNature349704706199110.1038/349704a0
|
|
13
|
E Levy-LahadW WascoP PoorkajCandidate gene
for the chromosome 1 familial Alzheimer's disease
locusScience269973977199510.1126/science.7638622
|
|
14
|
R SherringtonEI RogaevY LiangCloning of a
gene bearing missense mutations in early-onset familial Alzheimer's
diseaseNature375754760199510.1038/375754a0
|
|
15
|
M BentahirO NyabiJ VerhammePresenilin
clinical mutations can affect gamma-secretase activity by different
mechanismsJ
Neurochem96732742200610.1111/j.1471-4159.2005.03578.x16405513
|
|
16
|
M CitronD WestawayW XiaMutant presenilins
of Alzheimer's disease increase production of 42-residue amyloid
beta-protein in both transfected cells and transgenic miceNat
Med367721997
|
|
17
|
S Kumar-SinghJ TheunsB Van BroeckMean
age-of-onset of familial alzheimer disease caused by presenilin
mutations correlates with both increased Abeta42 and decreased
Abeta40Hum Mutat27686695200610.1002/humu.20336
|
|
18
|
JM RingmanSG YounkinD PraticoBiochemical
markers in persons with preclinical familial Alzheimer
diseaseNeurology718592200810.1212/01.wnl.0000303973.71803.8118509095
|
|
19
|
J ForteaA LladoJ ClarimonPICOGEN: Five
years experience with a genetic counselling program for
dementiaNeurologia26143149201121163230
|
|
20
|
R Sanchez-ValleA LladoM EzquerraMJ ReyL
RamiJL MolinuevoA novel mutation in the PSEN1 gene (L286P)
associated with familial early-onset dementia of Alzheimer type and
lobar haematomasEur J
Neurol1414091412200710.1111/j.1468-1331.2007.01988.x18028191
|
|
21
|
J ForteaA LladoB BoschCerebrospinal fluid
biomarkers in Alzheimer's disease families with PSEN1
mutationsNeurodegener Dis8202207201110.1159/000322229
|
|
22
|
E PorteliusAJ TranU
AndreassonCharacterization of amyloid beta peptides in
cerebrospinal fluid by an automated immunoprecipitation procedure
followed by mass spectrometryJ Proteome
Res644334439200710.1021/pr070362717927230
|
|
23
|
K BlennowH HampelM WeinerH
ZetterbergCerebrospinal fluid and plasma biomarkers in Alzheimer
diseaseNat Rev Neurol6131144201010.1038/nrneurol.2010.420157306
|
|
24
|
U AndreassonE PorteliusME AnderssonK
BlennowH ZetterbergAspects of beta-amyloid as a biomarker for
Alzheimer's diseaseBiomark Med15978200710.2217/17520363.1.1.59
|
|
25
|
E PorteliusN BogdanovicMK GustavssonMass
spectrometric characterization of brain amyloid beta isoform
signatures in familial and sporadic Alzheimer's diseaseActa
Neuropathol120185193201010.1007/s00401-010-0690-120419305
|