Introduction
Experimental and clinical evidence has confirmed
that aldosterone induces deleterious structural and functional
changes in the heart (1–3). Clinical trials have demonstrated that
aldosterone antagonists improve the outcome of patients with heart
failure and left ventricular dysfunction after myocardial
infarction (MI) (4–6). Potential mechanisms for the
contribution of aldosterone in cardiovascular diseases may include
the induction of myocyte apoptosis and cardiovascular extracellular
matrix turnover, sympathoadrenergic stimulation, endothelial
dysfunction and/or myocardial structural and electrical remodeling
(7,8).
Additionally, previous studies provide evidence that
aldosterone-mediated myocyte apoptosis is an important component of
cardiovascular diseases (9,10).
Several mechanisms which may link aldosterone and apoptosis have
been suggested, including the activation of the membrane
receptor-mediated calcineurin-Bad, c-Jun N-terminal kinase (JNK)
and ERK1/2 pathways (11–13). However, whether the mechanism of
aldosterone-mediated apoptosis in cardiac myocytes involves the p38
mitogen-activated protein kinase (p38 MAPK) pathway remains
controversial.
The p38 MAPK pathway is important in numerous cell
processes, including myocyte apoptosis, hypertrophy and
inflammation, which may contribute to progressive left ventricular
remodeling post-MI and the transition to heart failure (14,15).
Pharmacological inhibitors of p38 MAPK have been shown to decrease
myocyte apoptosis and improve cardiac function and heart failure
in vitro and in vivo(16,17).
However, a number of studies have demonstrated that SB203580, a p38
MAPK inhibitor, has minimal effects on aldosterone-induced
apoptosis (18). Thus, whether the
inhibition of p38 MAPK improves aldosterone-mediated apoptosis
requires clarification.
The present study aimed to provide further
understanding of the mechanism of the association between
aldosterone and apoptosis. We assessed the direct effects of gene
silencing of the p38 MAPK signaling cascade to determine whether
this was able to attenuate aldosterone-mediated myocyte apoptosis.
We also explored a potential mechanism that may underlie these
effects.
Materials and methods
Neonatal rat cardiac cell cultures
Cardiac cells were isolated from 1 to 3-day-old
neonatal Sprague-Dawley rats and cultured, as described previously
(19). Myocytes were seeded in
Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 10%
horse serum and then transferred after 24 h to a defined medium,
which prevented the growth of proliferating cells.
Design and cloning of lentiviral short
hairpin RNA (ShRNA) vectors
The target ShRNAs against Rattus norvegicus
mRNA for p38 MAPK (GenBank GI no. 157890411; Genepharma Co., Ltd.,
Shanghai, China) were as follows: ShRNA1,
5′-GGACCTCCTTATAGACGAATG-3′; ShRNA2, 5′-GAAGCTGTCGAGACCGTTTCA-3′;
ShRNA3, 5′-GCCGAAGATGAACTTCGCAAA-3′; and the ShRNA control
sequence, 5′-GCGAGGGCTGAAGTATATACA-3′. These ShRNAs were cloned
into lentiviral vectors (PGLV; Genepharma Co., Ltd.) and a negative
control PGLV was also designed (PGLV-NC). The lentiviral vectors
used in this study were third generation, self-inactivating vectors
that contained green fluorescent protein (GFP) (20). HEK-293T cells were co-transfected
with appropriate amounts of the vector plasmids; helper construct,
envelope plasmid and PGLVs containing ShRNA. The culture medium was
collected over 48 h, concentrated by ultracentrifugation, aliquoted
and stored at −80ºC until it was used. The virus titer was
calculated as the number of cells expressing GFP multiplied by the
corresponding dilution and the titer of lentivirus was determined
by a hole-by-dilution titer assay. The final titer of recombinant
virus was 1×108 transducing units (TU)/ml. All
constructs were verified by sequence analysis and results of the
DNA sequencing were as expected.
Transduction of cultured myocytes and GFP
fluorescence observation
Dissociated myocytes were infected with lentivirus
vectors for 72 h. GFP fluorescence in the cells was monitored using
a fluorescence microscope (Leica, Solms, Germany) at 24, 48 and 72
h post-transduction. At 48 h after infection, the cells were
assayed for expression of the transgenes.
Administration of aldosterone and
PGLV-ShRNAs in vivo
Procedures involving animals and their care were
conducted in accordance with the guidelines approved by the China
Association of Laboratory Animal Care. Male adult normotensive
Sprague-Dawley rats (body weight, 280–300 g) were obtained from the
Animal Research Center of Zhongshan University (Zhongshan, China).
The rats were treated once with a hydrodynamic tail vein injection
containing 2×108 infectious units (IFU) of PGLV-ShRNA2,
-ShRNA3 or -NC (n=6). At 24 h after the hydrodynamic tail vein
injection, d-aldosterone (1 mg/kg; Sigma Chemicals, St. Louis, MO,
USA) was administered by gavage for 24 h. The control group
received vehicle only (5% ethanol; n=6 per group).
Myocyte apoptosis in vivo and in
vitro
Cells were cultured in a normal growth medium for 72
h following infection. To evaluate the effects of p38 MAPK,
myocytes were pre-treated with PGLV-ShRNA1, -ShRNA2, -ShRNA3 or -NC
for 48 h and stimulated with 10−5 mol/l aldosterone for
24 h. Control myocytes were incubated in DMEM. Cultured rat myocyte
apoptosis was assessed by terminal deoxynucleotidyl
transferase-mediated deoxyuridine triphosphate nick end-labeling
(TUNEL). Positive (myocardial sections treated with DNase I) and
negative controls (omission of biotin-16-dUTP or TdT) were also
included. Interference contrast was used to exclude apoptotic
nuclei of a non-cardiomyocyte origin from cell counts.
Caspase-3 activity
Caspase-3 enzymatic activity in myocytes was
determined using a CPP32 assay kit (MBL), which detects the
production of the chromophore p-nitroanilide after it is cleaved
from the peptide substrate DEVD p-nitroanilide, as described
previously (21).
Real-time polymerase chain reaction (PCR)
analysis
Total RNA from cultured and rat myocytes was
extracted using an RNA isolation kit (Invitrogen, Carlsbad, CA,
USA), according to the manufacturer’s instructions. The reactions
were carried out on a real-time PCR system (iQ5 real-time PCR;
Bio-Rad, Hercules, CA, USA) under the following cycle conditions:
95ºC for 15 sec, 45 cycles at 95ºC for 5 sec and at 60ºC for 30
sec. A standard curve for p38 MAPK was generated using serially
diluted total RNA from myocytes and this was used to quantify
relative p38 MAPK mRNA levels. The sequences of the p38 MAPK gene
primers were as follows: forward, 5′-AAC CTGTCCCCGGTGGGCTCG-3′; and
reverse, 5′-CGATGT CCCGTCTTTGTATGA-3′. GAPDH was used as a control
for normalization. The primers of GAPDH were as follows: forward,
5′-GAAGGTGAAGGTCGGAGTC-3′; and reverse, 5′-GAAGATGGTGATGGGATTTC-3′.
The relative expression of mRNA was calculated using the
2−ΔΔCt method (Livak and Schmittgen).
Western blot analysis
Cultured and rat myocyte protein extracts (20 μg)
were resolved using 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and then transferred to polyvinylidene fluoride
membranes. Membranes were incubated with a p38 MAPK antibody
(1:2000 anti-p38 MAPK; Cell Signaling Technology, Inc., Danvers,
MA, USA), a caspase-3 antibody (1:1000 anti-cleaved caspase-3; Cell
Signaling Technology, Inc.) and β-actin (1:2000; Sigma). The
membranes were then washed with TBS containing 0.05% Tween-20,
incubated with an anti-mouse immunoglobulin G horseradish
peroxidase secondary antibody (1:2000; Sigma) and washed again.
Protein expression was analyzed using the Bandscan software and
normalized by the quantity of β-actin on the same membrane.
Statistical analysis
Results are expressed as mean ± SEM. Data sets were
analyzed using one-way analysis of variance (Kruskal-Wallis
analysis of variance by ranks) followed by Bonferroni’s post-hoc
test for comparisons between the groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
Lentiviral vector transduction in
cultured cardiac myocytes
The lentiviral vectors were transduced into cardiac
cells in vitro for 72 h. No significant change in the cell
morphology and no cell death were observed. The transduction
efficiency was measured by the frequency of GFP-positive cells. A
strong GFP signal was visible in cardiac cells 48 h after
transduction with PGLV-ShRNA, indicating that the recombinant
lentivirus led to the successful transduction of cardiac myocytes.
As demonstrated by the representative experiment shown, >70% of
cardiomyocytes were transduced successfully.
Myocyte apoptosis in vitro and in
vivo
The stimulation of myocytes with 10−5
mol/l aldosterone significantly increased the level of myocyte
apoptosis compared with the serum-deprived controls in vitro
(19.31±3.35 vs. 6.86±1.56%; P<0.01; Fig. 1A). PGLV-NC and PGLV-ShRNA1 had no
significant effect on apoptosis compared with the aldosterone group
in vitro. PGLV-ShRNA2 and -ShRNA3 decreased the level of
myocyte apoptosis (to 10.2 and 15.4%, respectively) compared with
the aldosterone group in vitro (P<0.05 and P<0.01,
respectively; Fig. 1A). The gavage
of d-aldosterone for 24 h significantly increased the level of
myocyte apoptosis compared with the vehicle group in normotensive
rats (15.20±2.18 vs. 5.83±1.42%; P<0.01; Fig. 1B). PGLV-NC had no significant
effect on apoptosis compared with the aldosterone group in
normotensive rats (16.65±2.24 vs. 15.20±2.18%; P>0.05).
Pre-treatment with PGLV-ShRNA2 and -ShRNA3 significantly decreased
the level of myocyte apoptosis (to 9.2 and 11.3%, respectively)
compared with the aldosterone group in vitro (P<0.05 and
P<0.01, respectively; Fig.
1B).
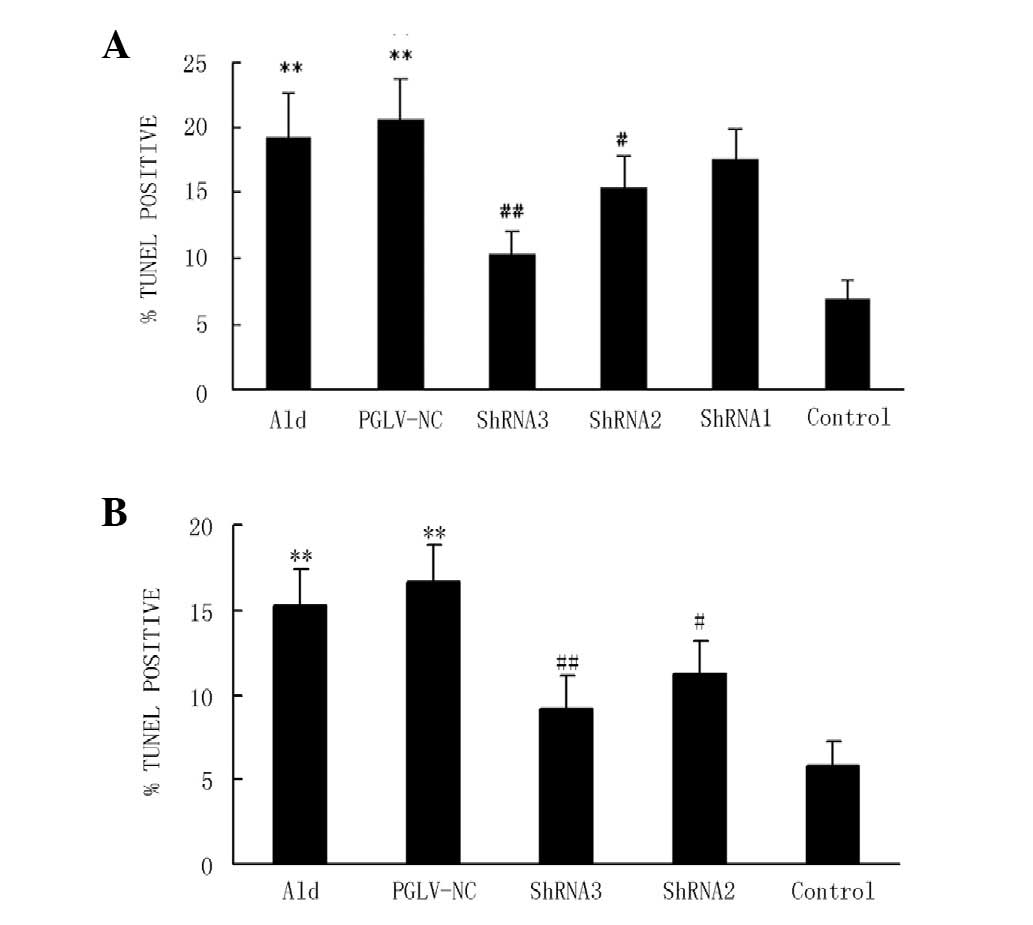 | Figure 1Myocyte apoptosis induced by
aldosterone in vitro and in vivo. (A) A graph showing
the quantification of apoptotic cells in six groups in vitro
(n=6 per group). Myocytes were pre-treated with PGLV-ShRNA1,
-ShRNA2, -ShRNA3 or -NC for 48 h and then stimulated with
10−5 mol/l aldosterone for 24 h. Control myocytes were
incubated with DMEM. Groups were as follows: Ald, aldosterone group
(myocytes were stimulated with aldosterone); PGLV-NC, negative
control PGLV. *P<0.05 vs. the control group,
#P<0.05 vs. the Ald group. (B) A graph showing the
quantification of apoptotic cells in five groups in vivo
(n=6 per group). At 24 h after the hydrodynamic tail vein
injection, d-aldosterone (1 mg/kg) was administered by gavage for
24 h. Groups were as follows: Ald, aldosterone group; PGLV-NC,
negative control PGLV; Control, tail vein injection contained
vehicle only. *P<0.05 vs. the control group,
#P<0.05 vs. the Ald group. PGLV, lentiviral vectors;
ShRNA, short hairpin RNA; DMEM, Dulbecco’s modified Eagle’s
medium. |
Caspase-3 expression in vitro and in
vivo
The stimulation of myocytes with 10−5
mol/l aldosterone significantly increased the level of caspase-3
expression (to 41%) compared with the serum-deprived controls in
vitro (P<0.01; Fig. 2A).
PGLV-NC and -ShRNA1 had no significant effect on caspase-3 compared
with the aldosterone group in vitro. PGLV-ShRNA2 and -ShRNA3
decreased the level of caspase-3 (to 10.6% and 17.7%, respectively)
compared with the aldosterone group in vitro (P<0.05 and
P<0.01, respectively; Fig. 2A).
The gavage of d-aldosterone for 24 h significantly increased the
level of caspase-3 expression compared with the vehicle group in
normotensive rats (P<0.01; Fig.
2B). PGLV-NC had no significant effect on caspase-3 expression
compared with the aldosterone group in normotensive rats.
Pre-treatment with PGLV-ShRNA2 and -ShRNA3 significantly decreased
‘sthe level of caspase-3 expression (to 35.0% and 46.5%,
respectively) compared with the aldosterone group in vivo
(P<0.05 and P<0.01, respectively; Fig. 2B).
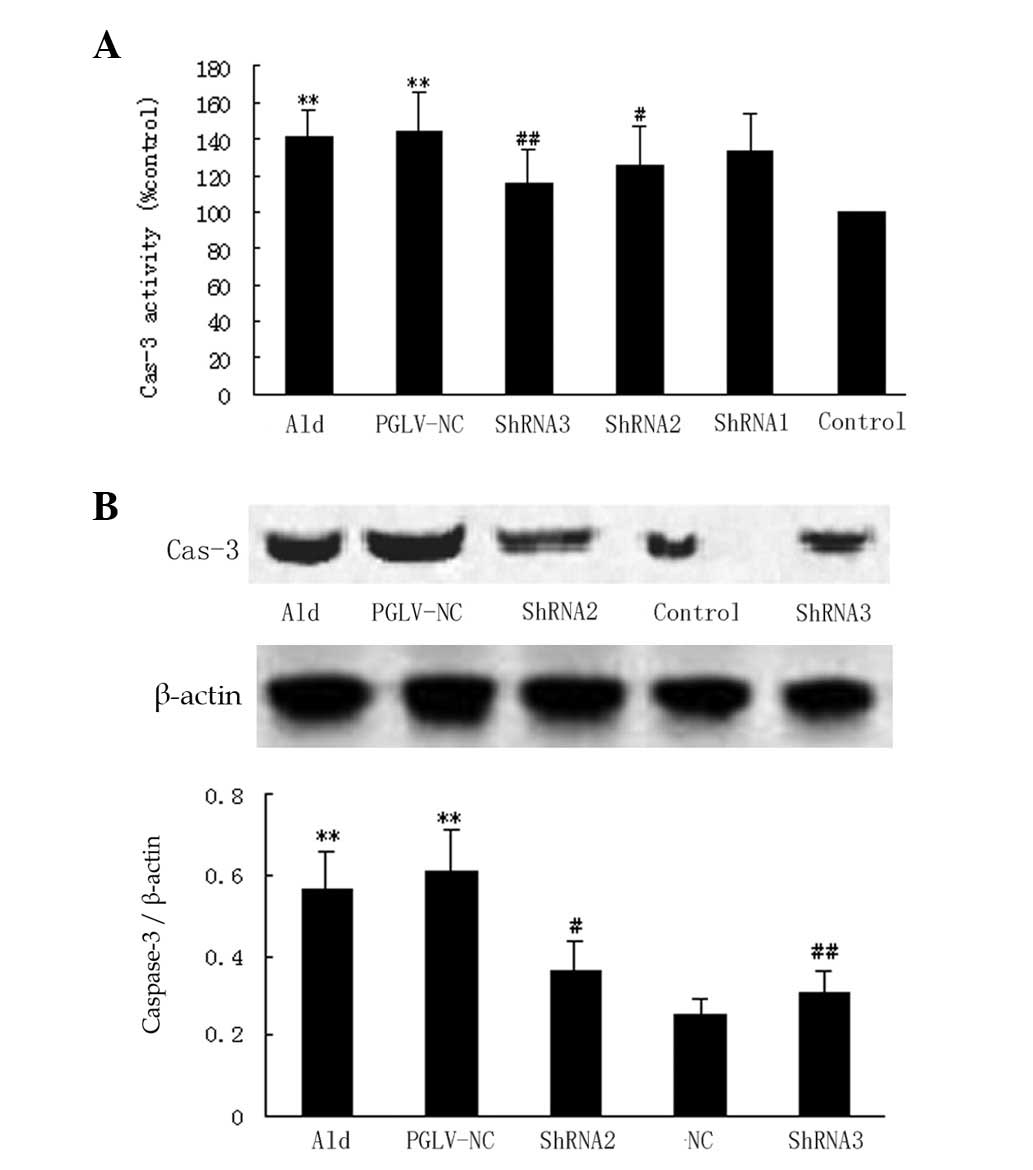 | Figure 2Caspase-3 expression in vitro
and in vivo. (A) A graph showing the quantification of
caspase-3 expression in six groups in vitro (n=6 per group).
Myocytes were pre-treated with PGLV-ShRNA1, -ShRNA2, -ShRNA3 or -NC
for 48 h and then stimulated with 10−5 mol/l aldosterone
for 24 h. Control myocytes were incubated with DMEM. Groups were as
follows: Ald, aldosterone group (myocytes were stimulated with
aldosterone); PGLV-NC, negative control PGLV. *P<0.05
vs. the control group, #P<0.05 vs. the Ald group.(B)
Western blotting and a graph showing western blot analysis of
caspase-3 expression in five groups in vivo (n=6 per group).
At 24 h after the hydrodynamic tail vein injection, d-aldosterone
(1 mg/kg) was administered by gavage for 24 h. Groups were as
follows: Ald, aldosterone group; PGLV-NC, negative control PGLV;
Control, tail vein injection contained vehicle only.
*P<0.05 vs. the control group, #P<0.05
vs. the Ald group. PGLV, lentiviral vectors; ShRNA, short hairpin
RNA; DMEM, Dulbecco’s modified Eagle’s medium. |
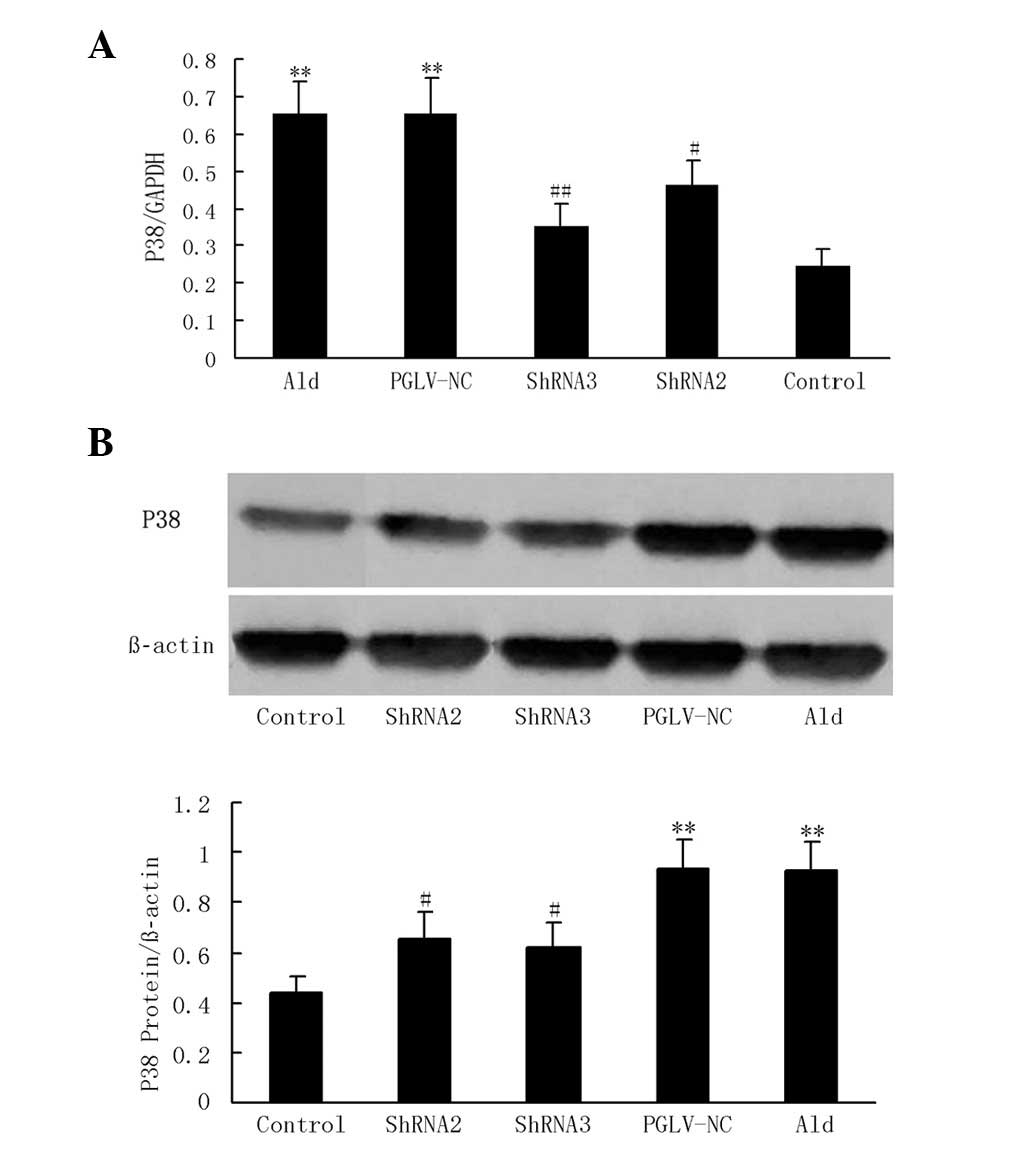
p38 MAPK expression in myocytes
With aldosterone treatment, P38 MAPK mRNA and
protein expression levels were increased 2.7- and 2.6-fold,
respectively, compared with the serum-deprived controls in
vitro (P<0.01 and P<0.01, respectively; Fig. 3). PGLV-NC and -ShRNA1 had no
significant effect on p38 MAPK mRNA or protein expression compared
with the aldosterone group in vitro. PGLV-ShRNA2
transduction resulted in an ~39.9 and 49.9% reduction of p38 MAPK
mRNA and protein expression levels, respectively, compared with the
aldosterone group in vitro (P<0.05 and P<0.05,
respectively). PGLV-ShRNA3 transduction resulted in an ~50.1 and
56.7% reduction of p38 MAPK mRNA and protein expression levels,
respectively, compared with the aldosterone group in vitro
(P<0.01 and P<0.01, respectively; Fig. 3).
 | Figure 3P38 MAPK expression in vitro.
(A) A graph showing real-time PCR analysis of p38 MAPK mRNA levels
in six groups in vitro (n=6 per group). (B) Western blotting
and a graph showing western blot analysis of p38 MAPK protein
levels in six groups in vitro (n=6 per group). Myocytes were
pre-treated with PGLV-ShRNA1, -ShRNA2, -ShRNA3 or -NC for 48 h and
then stimulated with 10−5 mol/l aldosterone for 24 h.
Control myocytes were incubated with DMEM. Groups were as follows:
Ald, aldosterone group (myocytes were stimulated with aldosterone);
PGLV-NC, negative control PGLV. *P<0.05 vs. the
control group, #P<0.05 vs. the Ald group. p38 MAPK,
p38 mitogen-activated protein kinase; PCR, polymerase chain
reaction; PGLV, lentiviral vectors; ShRNA, short hairpin RNA; DMEM,
Dulbecco’s modified Eagle’s medium. |
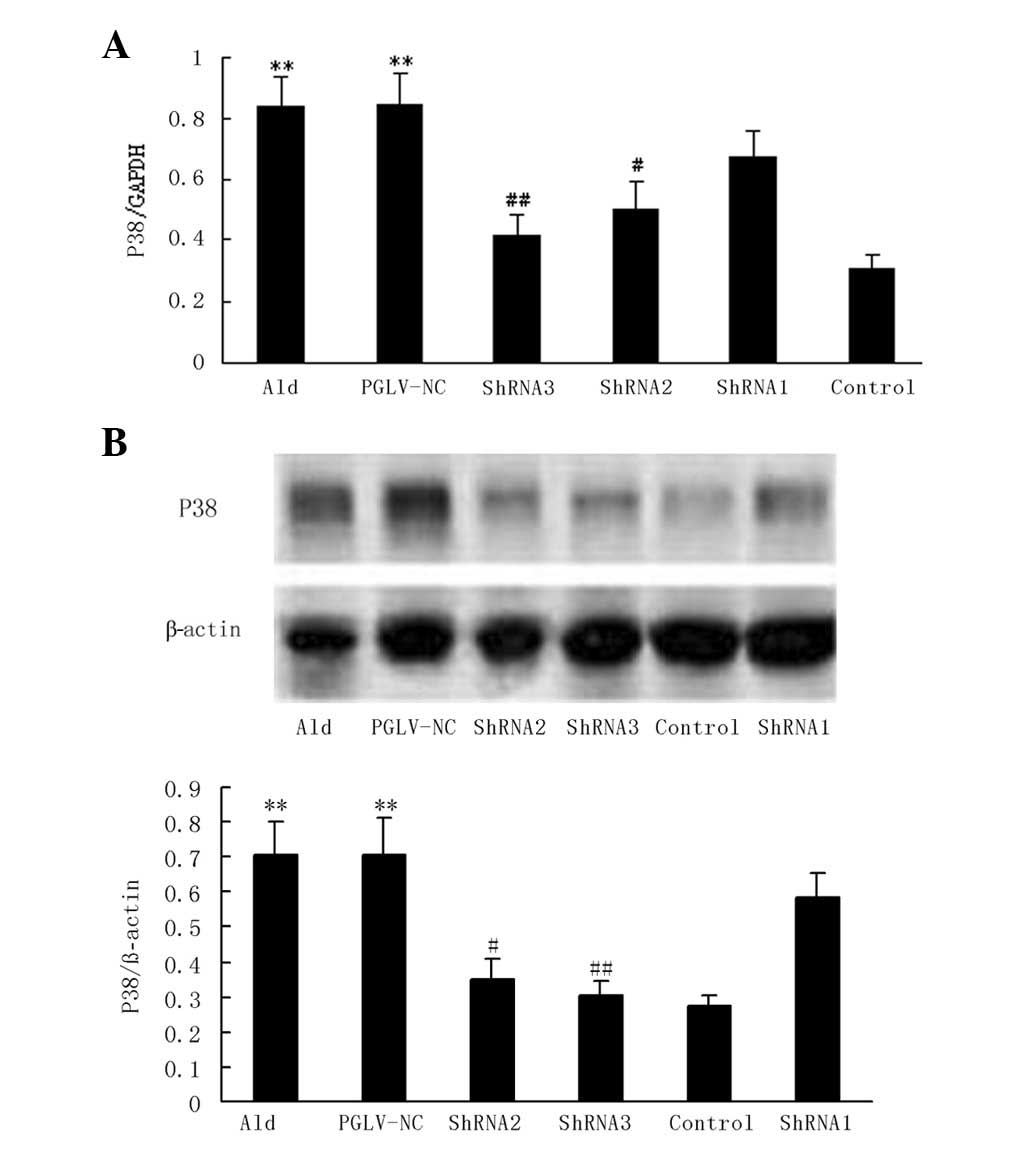
P38 MAPK expression in normotensive
rats
With aldosterone treatment, P38 MAPK mRNA and
protein expression levels were increased 2.2- and 2.6-fold,
respectively, compared with the vehicle group in normotensive rats
(P<0.01 and P<0.01, respectively; Fig. 4). PGLV-NC had no significant
effects on p38 MAPK mRNA or protein expression compared with the
aldosterone group in vivo. PGLV-ShRNA2 transduction resulted
in an ~29.3 and 29.4% reduction in p38 MAPK mRNA and protein
expression levels, respectively, compared with the aldosterone
group in vivo (P<0.05 and P<0.05, respectively).
PGLV-ShRNA3 transduction resulted an ~46 and 33.6% reduction in p38
MAPK mRNA and protein expression levels, respectively, compared
with the aldosterone group in vivo (P<0.01 and P<0.05,
respectively; Fig. 4).
Discussion
The present study demonstrated the direct adverse
effect of aldosterone on cardiomyocyte p38 MAPK expression and
apoptosis. Previous studies have demonstrated that aldosterone
directly induces myocyte apoptosis in a dose- and time-dependent
manner (11,12). Our results showed that aldosterone
(10−5 mol/l for 24 h) significantly increased the level
of myocyte apoptosis. A previous study also demonstrated that
d-aldosterone (1 mg/kg, infused for 24 h) was able to stimulate
cardiac myocyte apoptosis in adult normotensive rats (22,23).
Our results showed that exposure to 10−5 mol/l
aldosterone treatment for 24 h significantly increased myocyte
apoptosis 2.8-fold compared with the serum-deprived control in
vitro. The gavage of d-aldosterone for 24 h significantly
increased myocyte apoptosis 2.6-fold compared with the vehicle
group in normotensive rats.
Apoptosis is important in cardiovascular disease and
may contribute to the development and progression of cardiac
dysfunction and heart failure (8–10).
In vitro and in vivo studies have suggested a number
of potential mechanisms of these adverse effects, including the
theory that aldosterone induces apoptosis in myocytes via
activation of the calcineurin, p38 MAPK, JNK and ERK1/2 pathways.
Ma et al(16) and See et
al(17) reported that
treatment with the p38 MAPK inhibitor SB203580 decreased myocyte
apoptosis and improved postischemic cardiac function. The
cardioprotective effects of SB203580 were shown to be closely
associated with its ability to inhibit p38 MAPK. However, another
study showed that SB203580 had minimal effects on
aldosterone-mediated apoptosis (18). Of the several possible mechanisms
which may underly the effects of aldosterone on cardiomyocyte
apoptosis, our results suggest a key role for p38 MAPK activation
that is known to be associated with cardiomyocyte apoptosis.
Despite observations that p38 MAPK inhibitors have a
cardioprotective role in isolated ventricular myocytes and in
vivo, p38 MAPK inhibitors may also affect other signaling
pathways, including the JNK and ERK1/2 pathways. Whether p38 MAPK
gene silencing ameliorates aldosterone-mediated cardiomyocyte
apoptosis has rarely been investigated. Compared with gene knockout
techniques, RNAi-based gene silencing is more rapid and
cost-effective (24). Various
delivery methods for the expression of ShRNA include direct
application of naked siRNA and the use of lipid-based delivery
vehicles. However, these methods are limited, due to low
transduction efficiencies, weak control of gene expression and the
short duration of effects (25).
It is possible to achieve targeted ShRNA against p38 MAPK to
cardiac myocytes by lentiviral vectors and this may be exploited as
a novel approach to refine our understanding of the mechanisms of
myocyte apoptosis.
In the present study, three individual strands of
p38 MAPK ShRNA were constructed, delivered by lentiviral vectors,
to investigate their effects on aldosterone-mediated myocyte
apoptosis in vitro and in vivo. PGLV-ShRNA3
transduction significantly decreased the levels of myocyte
apoptosis and caspase-3 expression, which were associated with
significant reductions in p38 MAPK mRNA and protein expression
levels. Thus, the PGLV-ShRNA3 sequence was a specific ShRNA target.
The present study also demonstrated that p38 MAPK PGLV-ShRNA3
protected against aldosterone-mediated peri-infarct myocyte
apoptosis and attenuated pathological cardiac remodeling and LV
dysfunction in post-MI aldosterone overload rats (unpublished
data). Based on these findings, we conclude that aldosterone
induced cardiac dysfunction and apoptosis by activating p38 MAPK.
Gene silencing of the p38 MAPK signaling cascade was able to
attenuate aldosterone-mediated myocyte apoptosis.
Acknowledgements
This study was supported, in part, by grants from
the Science and Technology Foundation of Hubei Province
(2010CDB06806) and the Nature Science Foundation of Guangdong
Province (Grant 7001020).
Abbreviations:
|
DMEM
|
Dulbecco’s modified Eagle’s medium
|
|
ERK
|
extracellular regulated protein
kinases
|
|
GFP
|
green fluorescent protein
|
|
IFU
|
infectious unit
|
|
JNKs
|
Jun N-terminal kinases
|
|
MI
|
myocardial infarction
|
|
p38 MAPK
|
p38 mitogen-activated protein
kinase
|
|
ShRNA
|
short hairpin RNA
|
|
TU
|
transducing units
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated deoxyuridine triphosphate nick
end-labeling
|
References
|
1
|
Fraccarollo D, Berger S, Galuppo P, et al:
Deletion of cardiomyocyte mineralocorticoid receptor ameliorates
adverse remodeling after myocardial infarction. Circulation.
123:400–408. 2011. View Article : Google Scholar
|
|
2
|
Messaoudi S, Azibani F, Delcayre C and
Jaisser F: Aldosterone, mineralocorticoid receptor, and heart
failure. Mol Cell Endocrinol. 350:266–272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Güder G, Bauersachs J, Frantz S, et al:
Complementary and incremental mortality risk prediction by cortisol
and aldosterone in chronic heart failure. Circulation.
115:1754–1761. 2007.PubMed/NCBI
|
|
4
|
Iraqi W, Rossignol P, Angioi M, et al:
Extracellular cardiac matrix biomarkers in patients with acute
myocardial infarction complicated by left ventricular dysfunction
and heart failure: insights from the Eplerenone Post-Acute
Myocardial Infarction Heart Failure Efficacy and Survival Study
(EPHESUS) study. Circulation. 119:2471–2479. 2009.
|
|
5
|
Rossignol P, Ménard J, Fay R, Gustafsson
F, Pitt B and Zannad F: Eplerenone survival benefits in heart
failure patients post-myocardial infarction are independent from
its diuretic and potassium-sparing effects. Insights from an
EPHESUS (Eplerenone Post-Acute Myocardial Infarction Heart Failure
Efficacy and Survival Study) substudy. J Am Coll Cardiol.
58:1958–1966. 2011.
|
|
6
|
Zannad F, McMurray JJV, Krum H, et al;
EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart
failure and mild symptoms. N Engl J Med. 364:11–21. 2011.
View Article : Google Scholar
|
|
7
|
Kuster GM, Kotlyar E, Rude MK, et al:
Mineralocorticoid receptor inhibition ameliorates the transition to
myocardial failure and decreases oxidative stress and inflammation
in mice with chronic pressure overload. Circulation. 111:420–427.
2005. View Article : Google Scholar
|
|
8
|
Usher MG, Duan SZ, Ivaschenko CY, et al:
Myeloid mineralocorticoid receptor controls macrophage polarization
and cardiovascular hypertrophy and remodeling in mice. J Clin
Invest. 120:3350–3364. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun HY, Wang NP, Halkos M, et al:
Postconditioning attenuates cardiomyocyte apoptosis via inhibition
of JNK and p38 mitogen-activated protein kinase signaling pathways.
Apoptosis. 11:1583–1593. 2006. View Article : Google Scholar
|
|
10
|
Kinoshita H, Kuwahara K, Takano M, et al:
T-type Ca2+ channel blockade prevents sudden death in
mice with heart failure. Circulation. 120:743–752. 2009.
|
|
11
|
Mano A, Tatsumi T, Shiraishi J, et al:
Aldosterone directly induces myocyte apoptosis through
calcineurin-dependent pathways. Circulation. 110:317–323. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferron L, Ruchon Y, Renaud JF and Capuano
V: T-type Ca2+ signalling regulates aldosterone-induced
CREB activation and cell death through PP2A activation in neonatal
cardiomyocytes. Cardiovasc Res. 90:105–112. 2011.
|
|
13
|
Dhingra S, Sharma AK, Singla DK and Singal
PK: p38 and ERK1/2 MAPKs mediate the interplay of TNF-alpha and
IL-10 in regulating oxidative stress and cardiac myocyte apoptosis.
Am J Physiol Heart Circ Physiol. 293:H3524–H3531. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yeh CC, Li H, Malhotra D, et al:
Distinctive ERK and p38 signaling in remote and infarcted
myocardium during post-MI remodeling in the mouse. J Cell Biochem.
109:1185–1191. 2010.PubMed/NCBI
|
|
15
|
Cheriyan J, Webb AJ, Sarov-Blat L, et al:
Inhibition of p38 mitogen-activated protein kinase improves nitric
oxide-mediated vasodilatation and reduces inflammation in
hypercholesterolemia. Circulation. 123:515–523. 2011. View Article : Google Scholar
|
|
16
|
Ma XL, Kumar S, Gao F, et al: Inhibition
of p38 mitogen-activated protein kinase decreases cardiomyocyte
apoptosis and improves cardiac function after myocardial ischemia
and reperfusion. Circulation. 99:1685–1691. 1999. View Article : Google Scholar
|
|
17
|
See F, Thomas W, Way K, et al: p38
mitogen-activated protein kinase inhibition improves cardiac
function and attenuates left ventricular remodeling following
myocardial infarction in the rat. J Am Coll Cardiol. 44:1679–1689.
2004. View Article : Google Scholar
|
|
18
|
De Silva DS, Wilson RM, Hutchinson C, et
al: Fenofibrate inhibits aldosterone-induced apoptosis in adult rat
ventricular myocytes via stress-activated kinase-dependent
mechanisms. Am J Physiol Heart Circ Physiol. 296:H1983–H1993.
2009.PubMed/NCBI
|
|
19
|
Thomas WG, Brandenburger Y, Autelitano DJ,
Pham T, Qian H and Hannan RD: Adenoviral-directed expression of the
type 1A angiotensin receptor promotes cardiomyocyte hypertrophy via
transactivation of the epidermal growth factor receptor. Circ Res.
90:135–142. 2002. View Article : Google Scholar
|
|
20
|
Coleman JE, Huentelman MJ, Kasparov S, et
al: Efficient large-scale production and concentration of
HIV-1-based lentiviral vectors for use in vivo. Physiol Genomics.
12:221–228. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Casciola-Rosen L, Nicholson DW, Chong T,
et al: Apopain/CPP32 cleaves proteins that are essential for
cellular repair: a fundamental principle of apoptotic death. J Exp
Med. 183:1957–1964. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Z, Ma JY, Kerr I, Chakravarty S, Dugar
S, Schreiner G and Protter AA: Selective inhibition of p38alpha
MAPK improves cardiac function and reduces myocardial apoptosis in
rat model of myocardial injury. Am J Physiol Heart Circ Physiol.
291:H1972–H1977. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Angelis N, Fiordaliso F, Latini R, et
al: Appraisal of the role of angiotensin II and aldosterone in
ventricular myocyte apoptosis in adult normotensive rat. J Mol Cell
Cardiol. 34:1655–1665. 2002.PubMed/NCBI
|
|
24
|
Aouadi M, Tesz GJ, Nicoloro SM, et al:
Orally delivered siRNA targeting macrophage Map4k4 suppresses
systemic inflammation. Nature. 458:1180–1184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Whitehead KA, Langer R and Anderson DG:
Knocking down barriers: advances in siRNA delivery. Nat Rev Drug
Discov. 8:129–138. 2009. View
Article : Google Scholar : PubMed/NCBI
|