1. Endoplasmic reticulum (ER) stress
The ER is a key organelle in cell survival and
normal cellular function. In the ER, nascent proteins are folded
with the assistance of molecular chaperones and folding enzymes.
Correctly folded proteins are subsequently transported to the Golgi
apparatus, whereas unfolded and misfolded protein are retained in
the ER or returned to the cytoplasm by the process of ER-associated
degradation (ERAD) and ultimately degraded by the proteasome.
Various factors have been reported to contribute to ER stress,
including: i) Disturbances in cellular redox regulation, as a
consequence of oxidants, hypoxia or reducing agents, which affect
the disulfide bonds of proteins in the ER lumen; ii) glucose
deprivation, which disrupts N-linked protein glycosylation in the
ER; iii) disruption of Ca2+, which impairs the functions
of Ca2+-dependent chaperones, including
glucose-regulated protein (GRP) 78, GRP94 and calreticulin; iv)
viral infections which result in the presence of virus-encoded
proteins; v) a high-fat diet and vi) protein mutations (1). ER stress activates a stress response
signaling network known as the unfolded protein response (UPR),
which is capable of inducing apoptosis and inflammatory reactions
(2). Of note, the ER also has a
role in lipid biosynthesis, including lipid membrane synthesis and
controlling the synthesis of cholesterol and other membrane lipid
components (3).
2. ER stress and the UPR
The UPR is mediated by at least three transmembrane
proteins, including inositol-requiring enzyme 1 (IRE1), protein
kinase R (PKR)-like ER kinase (PERK) and activating transcription
factor 6 (ATF6) (2,4). Under unstressed conditions, ER stress
transducers, predominantly IRE1, PERK and ATF6, are maintained in
an inactive state by binding to the major ER chaperone GRP78 at the
ER lumen periphery. Under conditions leading to ER stress, GRP78 is
displaced to interact with misfolded luminal proteins, resulting in
the release and activation of IRE1, PERK and ATF6. In ER stress,
PERK phosphorylates the α subunit of eukaryotic translation
initiation factor 2 (eIF2α), leading to a rapid reduction in the
initiation of mRNA translation and a reduction in the level of
newly synthesized proteins in the ER. However, eIF2α
phosphorylation by PERK enables the translation of ATF4. ATF4
promotes apoptosis by inducing the transcription of genes involved
in apoptosis, including tribbles homolog 3 and C/EBP homologous
protein (CHOP).
ER stress-induced IRE1 activation initiates
non-spliceosomal splicing of the mRNA of the transcription factor
X-box binding protein 1 (XBP1). XBP1 controls protective responses
to ER stress, for example by upregulating the transcription of
genes encoding ER chaperones and ERAD components. Using its
non-specific endoribonuclease activity, IRE1 induces the
degradation of ER-localized mRNAs, a process known as regulated
IRE1-dependent decay (5). This
process further reduces protein accumulation in the ER. Of note,
IRE1 also activates c-Jun N-terminal kinase (JNK) by recruiting the
scaffold protein tumor necrosis factor receptor-associated factor 2
(TRAF2), as well as apoptosis signal-regulating kinase (ASK1) and
caspase 12 (2,6,7).
Following translocation to the Golgi apparatus under
ER stress, ATF6 is cleaved by site 1 and site 2 proteases in the
process of regulated intramembrane proteolysis. The cytoplasmic
region of ATF6 is an active transcription factor responsible for
the transactivation of various genes encoding ER chaperones, ERAD
components and protein foldases. ATF6 and PERK also activate
nuclear factor κ-light-chain-enhancer of activated B cells (NFκB)
during ER stress (8–11). Increased levels of JNK and NFκB
promote the production of pro-inflammatory cytokines, which cause
further inflammation and ER stress (12).
3. ER stress in adipose tissue (AT)
Adipose tissue is increasingly recognized as a
tissue containing a molecular network that connects obesity,
adipokine secretion, chronic inflammation and insulin resistance
(IR). In AT, adipocytes have been found to constitute ≤50% of the
total number of cells, with other cell types including
preadipocytes, macrophages and vascular cells. Under conditions of
ER stress, AT secretes a number of protein signals and factors. A
previous study showed that UPR markers are overexpressed in the AT
of obese rodents (13). In
adult-derived human adipocyte stem cells (ADHASCs), ER stress has
been reported to increase the levels of ER stress genes and eIF2α
phosphorylation (14).
Furthermore, AT inflammation, which is observed in obesity and
diabetes, is associated with the infiltration of macrophages into
the AT. This may be triggered by adipocyte death, the secretion of
adipokines, including tumor necrosis factor-α (TNF-α) and
interleukin-6 (IL-6), and increased levels of adipocyte-associated
chemokines, including monocyte chemoattractant protein 1 (MCP-1).
Similar to other cells that demonstrate a high secretory capacity,
including mature B lymphocytes, liver cells and pancreatic β cells,
adipocytes have also been reported to adapt their ER capabilities
under conditions of stress, particularly in diabetes and obesity
(15). ER stress has been observed
in human AT in numerous studies, indicating that ER stress may play
a crucial role in AT disorders, including inflammation and
apoptosis (16–18). In obesity, the AT is poorly
oxygenated (19,20), which leads to AT hypoxia. This
interferes with disulfide bonding in the ER lumen, resulting in ER
stress, initiation of the UPR and increased levels of ER stress
markers in the AT, including CHOP and GRP78 (13,21–23).
Hosogai et al (22)
reported that, in 3T3-L1 adipocytes, hypoxia is associated with ER
stress and increased levels of GRP78 and CHOP. Furthermore, using
ATF4, GADD34 and ATF3 as markers of the apoptotic pathway, Sharma
et al (17) found that ER
stress induced apoptosis. Hypoxia does not only stimulate the
inflammatory response of macrophages (24,25),
but also induces apoptosis and G0/G1-phase
cell cycle arrest through AKT and JNK (26). Furthermore, Yin et al
(27) found that hypoxia induces
cell death by promoting the release of free fatty acids (FFAs) and
inhibiting glucose uptake in adipocytes via the inhibition of the
insulin signaling pathway.
4. IR in brief
Prior to discussing the association between IR and
AT ER stress, an understanding of IR is required. IR is caused by
an impaired sensitivity of target organs, including AT, the liver
and muscle, to insulin. Insulin regulates glucose uptake in the
liver and muscle. Furthermore, insulin is a key regulator of
circulating FFA concentrations. In AT, insulin decreases lipolysis
and thereby reduces FFA efflux from adipocytes; in skeletal muscle,
insulin predominantly induces glucose uptake by stimulating the
translocation of glucose transporter type 4 (GLUT4) to the plasma
membrane. Furthermore, in the liver, insulin inhibits
gluconeogenesis by reducing the activity of key enzymes. Therefore
IR has been suggested to increase circulating FFA concentrations,
leading to ectopic fat deposition that impedes insulin-mediated
glucose uptake in skeletal muscle and increases glucose generation
in the liver (28). A combination
of IR and abnormalities in insulin secretion can lead to Type 2
diabetes mellitus (T2DM).
5. IR at the molecular level
In normal conditions, insulin is associated with
complex signaling cascades. Briefly, insulin receptor-mediated
tyrosine phosphorylation of insulin receptor substrates (IRSs)
induces the activation of at least two major pathways: The
phosphatidylinositol 3-kinase (PI3K)-AKT and mitogen-activated
protein kinase (MAPK) pathways. The PI3K-AKT pathway is primarily
responsible for the effect of insulin on glucose uptake and the
suppression of gluconeogenesis, whereas the MAPK pathway regulates
gene expression and interacts with the PI3K-AKT pathway to control
cell growth and differentiation.
While tyrosine phosphorylation activates, serine
phosphorylation of IRSs at specific serine residues inhibits
insulin signaling. IκB kinase-β (IKK-β), JNK 1 and MAPKs are
examples of serine kinases that phosphorylate IRS1 and consequently
inactivate its insulin signaling activity. Of note, these serine
kinases are also mediators of inflammatory signaling pathways,
demonstrating that an inhibitory crosstalk may exist between
inflammatory and insulin signaling at a molecular level. The
association between cytokine signaling and the inhibition of
insulin signaling has also been indicated by the presence of
molecular mediators, including suppressor of cytokine signaling
(SOCS) 1 and 3 and nitric oxide (NO). IL-6 has been shown to induce
the activation of SOCS proteins during inflammation, causing the
ubiquitinylation and degradation of IRS proteins. Endogenous NO
production by inducible NO synthase (iNOS), under the action of
numerous inflammatory cytokines, can limit IRS1 and iNOS activity,
resulting in reduced AKT activity; AKT is a key mediator of IRS
signaling (29).
6. Adipocyte ER stress and IR: Mechanisms
and significance
ER stress was first observed by Ozcan et al
(13) in the AT of obese mice and
proposed as a risk factor for IR. It has since been demonstrated
that stress-inducing conditions, such as obesity, are not only
associated with ER stress and the apoptosis of β-cells, hepatocytes
and adipocytes, but also with metabolic disorders, particularly IR.
In patients with IR, treatment with taurine-conjugated
ursodeoxycholic acid (TUDCA), a conjugated bile acid derivative
that inhibits ER stress-induced apoptosis, was observed to increase
insulin sensitivity (30).
Furthermore, the administration of chaperones, including 4-phenyl
butyric acid (PBA), trimethylamine N-oxide dihydrate, dimethyl
sulfoxide and 150 kDa oxygen-regulated protein, which protect cells
from ER stress by stabilizing protein conformation and improving ER
folding capacity, was found to increase insulin sensitivity in
obese diabetic mice (31,32). These data indicate that ER stress
in adipocytes, hepatocytes and β-cells may, at least in part,
initiate IR, as well as aggravate pre-existing IR, particularly in
the context of obesity. The mechanism by which ER stress interferes
with insulin receptor signaling is multifactorial, and the role of
adipose cells/AT in IR is discussed later in this review.
7. Role of the IRE1-JNK-IRS-1 signaling
pathway
It has been suggested that ER stress in adipocytes
may disrupt insulin signaling through the activation of IRE1. ER
stress induces insulin receptor signaling through increasing the
serine phosphorylation and decreasing the tyrosine phosphorylation
of IRS-1, leading to IR (33). As
mentioned previously, IRS-1 becomes inactivated upon
phosphorylation of specific serine residues (34). Several intracellular serine kinases
may mediate this IRS-1 phosphorylation, including IKK, JNK,
mammalian target of rapamycin (mTOR) and protein kinase C-θ
(PKC-θ). Insulin-resistant states, including obesity and T2DM, are
associated with the activation of JNK and/or IKK, which leads to
serine phosphorylation of IRS1 and thus the induction of IR
(35–39). In obesity, JNK is activated by IRE1
kinase activity as a consequence of AT ER. Furthermore, PKR, which
is activated by saturated FAs, ceramides and lipopolysaccharide, is
capable of inhibiting insulin signaling directly by phosphorylating
IRS1 on serine residues or indirectly by stimulating JNK activity
(40). The IRE1-JNK signaling
pathway has also been reported to directly inhibit cytoplasmic
insulin signaling in ob/ob mice due to JNK-mediated phosphorylation
of IRS-1 at serine-307 (13).
Additionally, the inflammatory cytokines secreted by adipocytes and
macrophages in AT in obese patients can activate JNK pathways
(41). Furthermore, inflammatory
cytokines can impair insulin signaling by interfering with
IRS-1-insulin receptor interactions and promoting IRS-1 degradation
(42). However, the effect of ER
stress on insulin signaling requires further investigation in order
to elucidate the specific mechanisms underlying the development of
IR in AT in obese patients. In a study on AT from obese volunteers,
approximately a two-fold upregulation of phosphorylated JNK-1 and
an upregulation of the spliced form of XBP1, which is part of the
IRE-1/XBP1 proximal ER stress sensor, was observed in the AT
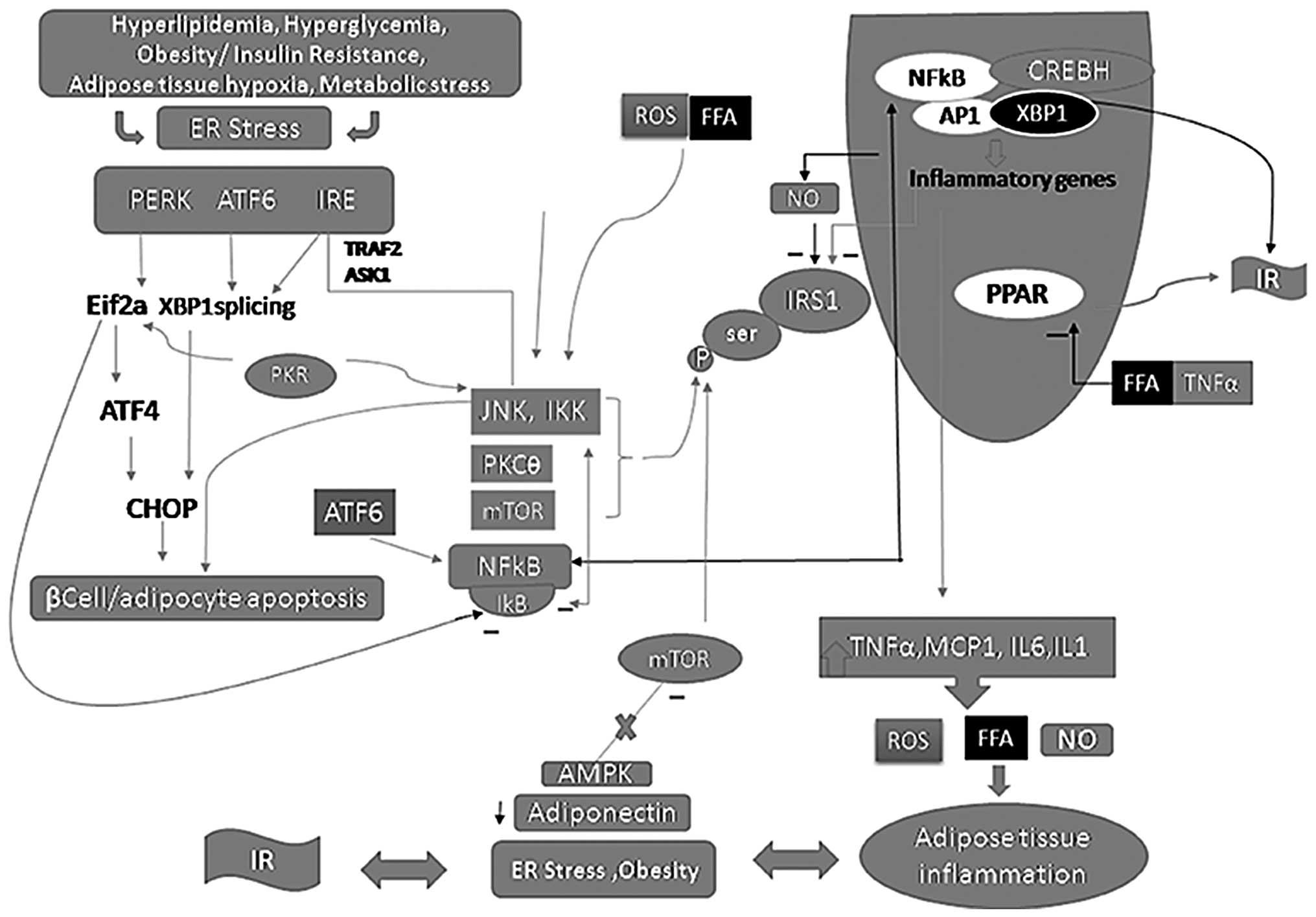
(43) (Fig. 1).
8. Role of adipokines
Several investigations in rodents and humans have
found an association between IR and ER stress in AT, as well as
increased lipolysis and altered adipokine production (13,16–18,44).
In ER stress-associated conditions, including obesity, a decrease
in leptin and adiponectin secretion and an increase in IL-6
secretion may be observed (44).
Furthermore, studies have shown that adiponectin folding and
multimerization are impaired in obese states due to a decreased
expression of ER disulfide-bond A oxidoreductase-like protein
(DsbA-L), which leads to ER stress (45,46).
However, CHOP is upregulated under ER stress, which impairs
resistin transcription in adipocytes and alters its secretion
(47). Of note, a number of
studies have shown that a decreased expression of the cytokine
adiponectin may promote IR in obesity (48–50).
ER stress has been reported to decrease levels of
high-molecular-weight fractions as well as total adiponectin in
human adipocytes, which may have role in insulin sensitivity and
metabolic syndrome (51,52). Decreased levels of adiponectin are
observed in conditions associated with excessive nutrient intake,
including obesity. Adiponectin signaling targets adenosine
monophosphate-activated protein kinase, which is a negative
regulator of mTOR. High levels of mTOR in turn induce serine
phosphorylation of IRS1. Therefore, when adiponectin levels are
low, the inhibition of IRS1 causes IR through the activation of the
mTOR signaling pathway. An investigation using tunicamycin and
thapsigargin as ER stress inducers in ADHASCs found that ER stress
decreased adiponectin and increased TNF-α mRNA expression, in
addition to decreasing levels of inhibitor of NFκB-α (IκB) protein
(14).
Resistin is an adipokine secreted by preadipocytes
of human AT (53) and has been
associated with IR. ER stress has been found to reduce resistin
mRNA expression in 3T3-L1 adipocytes in a time- and dose-dependent
manner, indicating that resistin is regulated by ER stress
(47). Furthermore, in adipocytes,
ER stress affects IR through resistin. However, further
investigations are required as resistin was observed to be
positively correlated with IR in a study involving individuals who
were obese and diabetic (54),
while no correlation was observed in other studies involving
individuals with a normal weight and children who were obese
(55–57).
9. Role of inflammatory cytokines during ER
stress with IR
In a study by Hotamisligil (58), inflammation was observed to induce
IR through the inhibition of IRS in the insulin signaling pathway.
Furthermore, inflammation and ER stress in AT are known to exhibit
a dual relationship. Inflammation can cause ER stress in AT, while
ER stress, either due to obesity or obesity-associated
consequences, such as IR and increased FFA levels, can increase the
activity of anti- or pro-inflammatory proteins. Studies have shown
that obese patients exhibit increased plasma levels of C-reactive
protein, inflammatory cytokines, including TNF-α, IL-6, MCP-1 and
IL-8, and the multifunctional proteins leptin (59) and osteopontin (60,61),
indicating a continuous low-grade inflammation in the obese
state.
All three subdivisions of ER stress associated with
the UPR contribute to the low-grade inflammation associated with
obesity. Of note, IRE1 activates JNK and NFκB through TRAF2 and
ASK1. In addition, PERK inhibits IκB through eIF2α phosphorylation.
PKR also phosphorylates eIF2α and activates JNK and IKK, while the
ATF6 branch of the pathway activates NFκB. Finally, ER stress
activates cyclic adenosine monophosphate (cAMP) responsive
element-binding protein, hepatocyte-specific, which, together with
NFκB, augments the transcription of genes involved in inflammation
(38,62). The role of IκB and IKK in
ER-induced inflammation has been investigated in ADHASCs. The IκB
family of proteins controls the activation of NFκB, whose entry
into the nucleus can initiate inflammation. Therefore, IκB
regulates NFκB by sequestering it in the cytoplasm. In turn, IκB
can be degraded by IKK, which results in entry of the NFκB dimers
into the nucleus and inflammation. Therefore, ER stress may induce
inflammation through a decrease in IκB levels as a consequence of
increased IKK activity (58).
IL-6 and TNF-α, levels of which are increased in
obesity and IR, have been proposed to induce ER stress and
consequently promote IR in a positive feedback manner (63). In ATs, TNF-α inhibits lipogenesis
and adiponectin expression via the inhibition of peroxisome
proliferator-activated receptor-γ (PPAR-γ)-mediated mechanisms
(64–66). It has been suggested that ER stress
may activate TNF-α, which is then involved in inflammatory
processes and the PPAR-γ-mediated effects on adipocytes (67). Investigations using PPAR activators
such as thiazolidinediones have verified that the transcriptional
activity of PPAR-γ is required for the maintenance of insulin
sensitivity and lipid metabolism (26). A previous study investigated TNF-α
in the AT of lean and obese subjects using reverse
transcription-polymerase chain reaction methodology (43). Neutralizing TNF-α in rats and
knocking out the TNF-α or TNF-α receptor 1 genes in mice have also
shown protective effects against IR induced by diet and genetic
obesity (68,69). Furthermore, TNF-α activates several
IR-related pathways, including IKK-β and SOCS3, in cultured murine
adipocytes (70,71). TNF-α has been suggested to affect
insulin sensitivity by modifying the expression of IRS1, GLUT4,
adiponectin and PPAR-α (66,72).
Of note, TNF-α also induces reactive oxygen species (ROS)
generation through the activation of the nicotinamide adenine
dinucleotide phosphate-oxidase (NADPH oxidase) (73,74).
In addition, another inflammatory cytokine, IL-1β, plays an
important role in inducing ER stress in adipocytes in obese
individuals by increasing the levels of iNOS, which generates NO.
In turn, NO inhibits the activity of the ER Ca2+ pump,
resulting in a depletion of ER Ca2+ stores, and thus
inducing ER stress (75,76).
Obesity-induced ER stress was recently investigated
in AT using the 12/15-lipoxygenase (12/15-LO) enzyme, which is
involved in a unique inflammatory pathway that regulates the ER
stress response in key cells, tissues and organs, including
adipocytes, pancreatic islets and the liver (77). The addition of the 12/15-LO
cleavage products, 12-hydroxyeicosatetraenoic acid and
12-hydroperoxyeicosatetraenoic acid, to differentiated 3T3-L1
adipocytes was observed to induce the expression and activation of
ER stress markers, including GRP78, XBP1, phosphorylated-PERK and
phosphorylated-IRE1α. The study also found that 12/15-LO
upregulated IL-12 expression. These findings may represent a novel
therapeutic strategy for alleviating ER-stress associated
inflammation, β-cell dysfunction and IR, thereby reducing metabolic
complications associated with visceral adiposity by inhibiting
12/15-LO activation or downstream IL-12 signaling (77).
10. Role of DsbA-L
DsbA-L is a protein that has been proposed to have a
role in the downregulation of adiponectin during ER stress-induced
autophagy in adipocytes (78). It
was revealed that ER stress-induced autophagy plays an important
role in obesity-induced adiponectin downregulation in adipocytes,
and that increasing the expression of DsbA-L may increase
adiponectin levels and lead to enhanced insulin sensitivity in
vitro and in vivo. Therefore, increasing DsbA-L
expression levels could be a novel approach to protect cells from
obesity-induced ER stress and improve insulin sensitivity. Of note,
cellular DsbA-L levels were stimulated by the PPAR-γ agonist
rosiglitazone, an insulin-sensitizing drug (78).
11. A direct role for ER stress in insulin
signaling in AT
Xu et al (44) demonstrated a direct role for ER
stress in insulin signaling in adipose cells. ER stress inducers
were observed to decrease insulin signaling in 3T3-L1 adipocytes
without affecting insulin-stimulated glucose uptake (79), in a manner independent of the
IRE1/JNK pathway (44). Notably,
ER stress has also been revealed to directly increase lipolysis by
downregulating the expression of the lipid droplet-associated
protein perilipin A (80,81).
12. Role of FFAs
The roles of hypoxia and inflammatory mediators in
AT ER stress have been discussed previously in this review. A
number of studies have demonstrated that elevated FFA levels may
also have an important role in the induction of ER stress in
various cells, including adipocytes (62,82);
however, further investigation is required. This theory is
supported by the fact that numerous obese individuals exhibit
elevated FFA plasma levels (83,84).
Hotamisligil (58) showed that
inflammation may lead to IR by inhibiting IRSs in the insulin
signaling pathway, and it is well established that inflammation
inhibits the action of insulin by increasing the levels of FFAs and
decreasing those of adiponectin in the blood. Therefore, it is
possible that both inflammatory cytokines and FFAs may target IRSs,
leading to IR (85,86). High FFA levels have also been shown
to downregulate PPAR-γ protein and mRNA expression, further
enhancing IR (87). In AT, ER
stress promotes FFA efflux from adipocytes, and high levels of
circulating FFA have been suggested to be the cellular basis of
lipotoxicity, dyslipidemia and IR (13,17,88–90).
Furthermore, accumulating evidence suggests that saturated
long-chain FFAs, primarily palmitate, and, to a lesser extent,
unsaturated long chain FFAs may induce ER stress and mediate β-cell
apoptosis, ultimately leading to IR and T2DM (91–96).
In a recent study, a unique lipolysis pathway was reported that
occurred in response to ER stress in adipocytes. This pathway
occurred independently of hormone-sensitive lipolysis, but was
associated with elevated cAMP production and PKA activity (81). Chemically induced ER stress was
revealed to activate cAMP/PKA and extracellular signal-regulated
kinase 1/2 (ERK1/2) and regulate lipolysis in ER-stressed
adipocytes, with PKA being an acute regulator and ERK1/2 a chronic
regulator (81). Notably, it has
been suggested that JNK (97) and
PKC (98) may also modulate
lipolysis and that ERK1/2 and JNK are activated during ER
stress.
13. Role of ROS and ER stress in
adipocytes
ROS have been reported to play an important role in
the ER stress response in adipocytes, which then directly or
indirectly contributes to IR (21). Increased ROS generation has also
been observed in response to high levels of FFAs in the AT of obese
mice. Furthermore, TNF-α has been shown to induce ROS generation
through the activation of NADPH oxidase (74,75,99).
As a consequence of their oxidizing effects on nascent proteins and
their action on Ca2+ channels, ROS lower Ca2+
availability and increase the number of misfolded and unfolded
proteins in the ER, which further increases ER stress (39,100). This FFA-mediated ROS generation
model provides another mechanism by which ER stress may be induced
in AT and subsequently lead to IR.
14. Conclusion and future directions
In conclusion, this review demonstrates the
existence of a strong association between adipocyte ER stress and
IR that is complex and multifactorial. Therefore, the inhibition of
ER stress may lead to the discovery of novel therapeutics for the
treatment of metabolic diseases, including T2DM; investigations are
currently underway in this area. To date, at least two chaperones,
PBA and TUDCA, have been approved by the Food and Drug
Administration and have been shown to relieve ER stress-mediated
pathologies, including IR, in hepatocytes, adipocytes and β-cells
(31,32,94).
However, further investigations are required before these
strategies can be applied in patients for the treatment of
metabolic and nutritional disorders, including obesity and IR.
Abbreviations:
|
ASK1
|
apoptosis signal-regulating kinase
1
|
|
AT
|
adipose tissue
|
|
ATF
|
activating transcription factor
|
|
ADHASC
|
adult-derived human adipocyte stem
cell
|
|
CHOP
|
C/EBP homologous protein
|
|
DsbA-L
|
disulfide-bond A oxidoreductase-like
protein
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
eIF2α
|
eukaryotic translation initiation
factor
|
|
ER
|
endoplasmic reticulum
|
|
ERAD
|
ER-associated degradation
|
|
FFAs
|
free fatty acids
|
|
IKK-β
|
IκB kinase-β
|
|
IRE
|
inositol-requiring enzyme
|
|
IRS
|
insulin receptor substrate
|
|
JNK
|
c-jun N-terminal kinase
|
|
MCP
|
monocyte chemo-attractant protein
|
|
MAPK
|
mitogen-activated protein kinases
|
|
NFκB
|
nuclear factor κ-light-chain-enhancer
of activated B cells
|
|
PKR
|
protein kinase R
|
|
PERK
|
protein kinase R-like ER-regulated
kinase
|
|
PKA
|
protein kinase A
|
|
ROS
|
reactive oxygen species
|
|
SOCS
|
suppressor of cytokine signaling
|
|
TUDCA
|
taurine-conjugated ursodeoxycholic
acid
|
|
TRAF2
|
tumor necrosis factor
receptor-associated factor 2
|
|
UPR
|
unfolded protein response
|
|
XBP1
|
X-box binding protein 1
|
References
|
1
|
Schröder M: Endoplasmic reticulum stress
responses. Cell Mol Life Sci. 65:862–894. 2008.
|
|
2
|
Winnay JN and Kahn CR: PI 3-kinase
regulatory subunits as regulators of the unfolded protein response.
Methods Enzymol. 490:147–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van der Kallen CJ, van Greevenbroek MM,
Stehouwer CD and Schalkwijk CG: Endoplasmic reticulum
stress-induced apoptosis in the development of diabetes: is there a
role for adipose tissue and liver? Apoptosis. 14:1424–1434.
2009.
|
|
4
|
Schröder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
|
|
5
|
Hollien J and Weissman JS: Decay of
endoplasmic reticulum-localized mRNAs during the unfolded protein
response. Science. 313:104–107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu P, Han Z, Couvillon AD, Kaufman RJ and
Exton JH: Autocrine tumor necrosis factor alpha links endoplasmic
reticulum stress to the membrane death receptor pathway through
IRE1alpha-mediated NF-kappaB activation and down-regulation of
TRAF2 expression. Mol Cell Biol. 26:3071–3084. 2006. View Article : Google Scholar
|
|
7
|
Urano F, Wang X, Bertolotti A, et al:
Coupling of stress in the ER to activation of JNK protein kinases
by transmembrane protein kinase IRE1. Science. 287:664–666. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deng J, Lu PD, Zhang Y, et al:
Translational repression mediates activation of nuclear factor
kappa B by phosphorylated translation initiation factor 2. Mol Cell
Biol. 24:10161–10168. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang HY, Wek SA, McGrath BC, et al:
Phosphorylation of the alpha subunit of eukaryotic initiation
factor 2 is required for activation of NF-kappaB in response to
diverse cellular stresses. Mol Cell Biol. 23:5651–5663. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu S, Tan M, Hu Y, Wang JL, Scheuner D and
Kaufman RJ: Ultraviolet light activates NFkappaB through
translational inhibition of IkappaBalpha synthesis. J Biol Chem.
279:34898–34902. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamazaki H, Hiramatsu N, Hayakawa K, et
al: Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin
through the ATF6 branch of the unfolded protein response. J
Immunol. 183:1480–1487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang K, Shen X, Wu J, et al: Endoplasmic
reticulum stress activates cleavage of CREBH to induce a systemic
inflammatory response. Cell. 124:587–599. 2006. View Article : Google Scholar
|
|
13
|
Ozcan U, Cao Q, Yilmaz E, et al:
Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mondal AK, Das SK, Varma V, et al: Effect
of endoplasmic reticulum stress on inflammation and adiponectin
regulation in human adipocytes. Metab Syndr Relat Disord.
10:297–306. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu J and Kaufman RJ: From acute ER stress
to physiological roles of the Unfolded Protein Response. Cell Death
Differ. 13:374–384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boden G, Duan X, Homko C, et al: Increase
in endoplasmic reticulum stress-related proteins and genes in
adipose tissue of obese, insulin-resistant individuals. Diabetes.
57:2438–2444. 2008. View Article : Google Scholar
|
|
17
|
Sharma NK, Das SK, Mondal AK, et al:
Endoplasmic reticulum stress markers are associated with obesity in
nondiabetic subjects. J Clin Endocrinol Metab. 93:4532–4541. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gregor MF, Yang L, Fabbrini E, et al:
Endoplasmic reticulum stress is reduced in tissues of obese
subjects after weight loss. Diabetes. 58:693–700. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fleischmann E, Kurz A, Niedermayr M, et
al: Tissue oxygenation in obese and non-obese patients during
laparoscopy. Obes Surg. 15:813–819. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Virtanen KA, Lönnroth P, Parkkola R, et
al: Glucose uptake and perfusion in subcutaneous and visceral
adipose tissue during insulin stimulation in nonobese and obese
humans. J Clin Endocrinol Metab. 87:3902–3910. 2002. View Article : Google Scholar
|
|
21
|
Furukawa S, Fujita T, Shimabukuro M, et
al: Increased oxidative stress in obesity and its impact on
metabolic syndrome. J Clin Invest. 114:1752–1761. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hosogai N, Fukuhara A, Oshima K, et al:
Adipose tissue hypoxia in obesity and its impact on adipocytokine
dysregulation. Diabetes. 56:901–911. 2007. View Article : Google Scholar
|
|
23
|
Lazar MA: How obesity causes diabetes: not
a tall tale. Science. 307:373–375. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Heilbronn LK and Campbell LV: Adipose
tissue macrophages, low grade inflammation and insulin resistance
in human obesity. Curr Pharm Des. 14:1225–1230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lewis JS, Lee JA, Underwood JC, Harris AL
and Lewis CE: Macrophage responses to hypoxia: relevance to disease
mechanisms. J Leukoc Biol. 66:889–900. 1999.PubMed/NCBI
|
|
26
|
Spiegelman BM: PPAR-gamma: adipogenic
regulator and thiazolidinedione receptor. Diabetes. 47:507–514.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yin J, Gao Z, He Q, Zhou D, Guo Z and Ye
J: Role of hypoxia in obesity-induced disorders of glucose and
lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab.
296:E333–E342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Samuel VT and Shulman GI: Mechanisms for
insulin resistance: common threads and missing links. Cell.
148:852–871. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeyda M and Stulnig TM: Obesity,
inflammation, and insulin resistance - a mini-review. Gerontology.
55:379–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kars M, Yang L, Gregor MF, et al:
Tauroursodeoxycholic Acid may improve liver and muscle but not
adipose tissue insulin sensitivity in obese men and women.
Diabetes. 59:1899–1905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakatani Y, Kaneto H, Kawamori D, et al:
Involvement of endoplasmic reticulum stress in insulin resistance
and diabetes. J Biol Chem. 280:847–851. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ozcan U, Yilmaz E, Ozcan L, et al:
Chemical chaperones reduce ER stress and restore glucose
homeostasis in a mouse model of type 2 diabetes. Science.
313:1137–1140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim DS, Jeong SK, Kim HR, Kim DS, Chae SW
and Chae HJ: Effects of triglyceride on ER stress and insulin
resistance. Biochem Biophys Res Commun. 363:140–145. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gual P, Le Marchand-Brustel Y and Tanti
JF: Positive and negative regulation of insulin signaling through
IRS-1 phosphorylation. Biochimie. 87:99–109. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan M, Konstantopoulos N, Lee J, et al:
Reversal of obesity- and diet-induced insulin resistance with
salicylates or targeted disruption of Ikkbeta. Science.
293:1673–1677. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Itani SI, Ruderman NB, Schmieder F and
Boden G: Lipid-induced insulin resistance in human muscle is
associated with changes in diacylglycerol, protein kinase C, and
IkappaB-alpha. Diabetes. 51:2005–2011. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Arkan MC, Hevener AL, Greten FR, et al:
IKK-beta links inflammation to obesity-induced insulin resistance.
Nat Med. 11:191–198. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hirosumi J, Tuncman G, Chang L, et al: A
central role for JNK in obesity and insulin resistance. Nature.
420:333–336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang K and Kaufman RJ: From
endoplasmic-reticulum stress to the inflammatory response. Nature.
454:455–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakamura T, Furuhashi M, Li P, et al:
Double-stranded RNA-dependent protein kinase links pathogen sensing
with stress and metabolic homeostasis. Cell. 140:338–348. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI
|
|
42
|
Shoelson SE, Lee J and Goldfine AB:
Inflammation and insulin resistance. J Clin Invest. 116:1793–1801.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Boden G and Merali S: Measurement of the
increase in endoplasmic reticulum stress-related proteins and genes
in adipose tissue of obese, insulin-resistant individuals. Methods
Enzymol. 489:67–82. 2011. View Article : Google Scholar
|
|
44
|
Xu L, Spinas GA and Niessen M: ER stress
in adipocytes inhibits insulin signaling, represses lipolysis, and
alters the secretion of adipokines without inhibiting glucose
transport. Horm Metab Res. 42:643–651. 2010. View Article : Google Scholar
|
|
45
|
Liu M, Zhou L, Xu A, et al: A
disulfide-bond A oxidoreductase-like protein (DsbA-L) regulates
adiponectin multimerization. Proc Natl Acad Sci USA.
105:18302–18307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhou L, Liu M, Zhang J, Chen H, Dong LQ
and Liu F: DsbA-L alleviates endoplasmic reticulum stress-induced
adiponectin downregulation. Diabetes. 59:2809–2816. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lefterova MI, Mullican SE, Tomaru T,
Qatanani M, Schupp M and Lazar MA: Endoplasmic reticulum stress
regulates adipocyte resistin expression. Diabetes. 58:1879–1886.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hu E, Liang P and Spiegelman BM: AdipoQ is
a novel adipose-specific gene dysregulated in obesity. J Biol Chem.
271:10697–10703. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Berg AH, Combs TP and Scherer PE:
ACRP30/adiponectin: an adipokine regulating glucose and lipid
metabolism. Trends Endocrinol Metab. 13:84–89. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Menzaghi C, Ercolino T, Di Paola R, et al:
A haplotype at the adiponectin locus is associated with obesity and
other features of the insulin resistance syndrome. Diabetes.
51:2306–2312. 2002. View Article : Google Scholar
|
|
51
|
Kern PA, Di Gregorio GB, Lu T, Rassouli N
and Ranganathan G: Adiponectin expression from human adipose
tissue: relation to obesity, insulin resistance, and tumor necrosis
factor-alpha expression. Diabetes. 52:1779–1785. 2003. View Article : Google Scholar
|
|
52
|
Lara-Castro C, Luo N, Wallace P, Klein RL
and Garvey WT: Adiponectin multimeric complexes and the metabolic
syndrome trait cluster. Diabetes. 55:249–259. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kaser S, Kaser A, Sandhofer A, Ebenbichler
CF, Tilg H and Patsch JR: Resistin messenger-RNA expression is
increased by proinflammatory cytokines in vitro. Biochem Biophys
Res Commun. 309:286–290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Silha JV, Krsek M, Skrha JV, Sucharda P,
Nyomba BL and Murphy LJ: Plasma resistin, adiponectin and leptin
levels in lean and obese subjects: correlations with insulin
resistance. Eur J Endocrinol. 149:331–335. 2003. View Article : Google Scholar
|
|
55
|
Vozarova de Courten B, Degawa-Yamauchi M,
Considine RV and Tataranni PA: High serum resistin is associated
with an increase in adiposity but not a worsening of insulin
resistance in Pima Indians. Diabetes. 53:1279–1284. 2004.PubMed/NCBI
|
|
56
|
Lee JH, Chan JL, Yiannakouris N, et al:
Circulating resistin levels are not associated with obesity or
insulin resistance in humans and are not regulated by fasting or
leptin administration: cross-sectional and interventional studies
in normal, insulin-resistant, and diabetic subjects. J Clin
Endocrinol Metab. 88:4848–4856. 2003. View Article : Google Scholar
|
|
57
|
Gherlan I, Vladoiu S, Alexiu F, et al:
Adipocytokine profile and insulin resistance in childhood obesity.
Maedica (Buchar). 7:205–213. 2012.PubMed/NCBI
|
|
58
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Friedman JM and Halaas JL: Leptin and the
regulation of body weight in mammals. Nature. 395:763–770. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kiefer FW, Zeyda M, Todoric J, et al:
Osteopontin expression in human and murine obesity: extensive local
up-regulation in adipose tissue but minimal systemic alterations.
Endocrinology. 149:1350–1357. 2008. View Article : Google Scholar
|
|
61
|
Kahn BB and Flier JS: Obesity and insulin
resistance. J Clin Invest. 106:473–481. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jiao P, Ma J, Feng B, et al: FFA-induced
adipocyte inflammation and insulin resistance: involvement of ER
stress and IKKβ pathways. Obesity (Silver Spring). 19:483–491.
2011.PubMed/NCBI
|
|
63
|
Bruun JM, Verdich C, Toubro S, Astrup A
and Richelsen B: Association between measures of insulin
sensitivity and circulating levels of interleukin-8, interleukin-6
and tumor necrosis factor-alpha. Effect of weight loss in obese
men. Eur J Endocrinol. 148:535–542. 2003. View Article : Google Scholar
|
|
64
|
Sung HY, Hong CG, Suh YS, et al: Role of
(−)-epigallocatechin-3-gallate in cell viability, lipogenesis, and
retinol-binding protein 4 expression in adipocytes. Naunyn
Schmiedebergs Arch Pharmacol. 382:303–310. 2010.
|
|
65
|
Meng L, Zhou J, Sasano H, Suzuki T,
Zeitoun KM and Bulun SE: Tumor necrosis factor alpha and
interleukin 11 secreted by malignant breast epithelial cells
inhibit adipocyte differentiation by selectively down-regulating
CCAAT/enhancer binding protein alpha and peroxisome
proliferator-activated receptor gamma: mechanism of desmoplastic
reaction. Cancer Res. 61:2250–2255. 2001.
|
|
66
|
Ruan H, Pownall HJ and Lodish HF:
Troglitazone antagonizes tumor necrosis factor-alpha-induced
reprogramming of adipocyte gene expression by inhibiting the
transcriptional regulatory functions of NF-kappaB. J Biol Chem.
278:28181–28192. 2003. View Article : Google Scholar
|
|
67
|
Ritchie SA, Ewart MA, Perry CG, Connell JM
and Salt IP: The role of insulin and the adipocytokines in
regulation of vascular endothelial function. Clin Sci (Lond).
107:519–532. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hotamisligil GS, Shargill NS and
Spiegelman BM: Adipose expression of tumor necrosis factor-alpha:
direct role in obesity-linked insulin resistance. Science.
259:87–91. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Uysal KT, Wiesbrock SM, Marino MW and
Hotamisligil GS: Protection from obesity-induced insulin resistance
in mice lacking TNF-alpha function. Nature. 389:610–614. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hotamisligil GS, Peraldi P, Budavari A,
Ellis R, White MF and Spiegelman BM: IRS-1-mediated inhibition of
insulin receptor tyrosine kinase activity in TNF-alpha- and
obesity-induced insulin resistance. Science. 271:665–668. 1996.
View Article : Google Scholar
|
|
71
|
Shi H, Tzameli I, Bjørbaek C and Flier JS:
Suppressor of cytokine signaling 3 is a physiological regulator of
adipocyte insulin signaling. J Biol Chem. 279:34733–34740. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jager J, Grémeaux T, Cormont M, Le
Marchand-Brustel Y and Tanti JF: Interleukin-1beta-induced insulin
resistance in adipocytes through down-regulation of insulin
receptor substrate-1 expression. Endocrinology. 148:241–251. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Griendling KK, Sorescu D and Ushio-Fukai
M: NAD(P)H oxidase: role in cardiovascular biology and disease.
Circ Res. 86:494–501. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xue X, Piao JH, Nakajima A, et al: Tumor
necrosis factor alpha (TNFalpha) induces the unfolded protein
response (UPR) in a reactive oxygen species (ROS)-dependent
fashion, and the UPR counteracts ROS accumulation by TNFalpha. J
Biol Chem. 280:33917–33925. 2005. View Article : Google Scholar
|
|
75
|
Li WW, Alexandre S, Cao X and Lee AS:
Transactivation of the grp78 promoter by Ca2+ depletion.
A comparative analysis with A23187 and the endoplasmic reticulum
Ca(2+)-ATPase inhibitor thapsigargin. J Biol Chem.
268:12003–12009. 1993.PubMed/NCBI
|
|
76
|
Tsujino M, Hirata Y, Imai T, et al:
Induction of nitric oxide synthase gene by interleukin-1 beta in
cultured rat cardiocytes. Circulation. 90:375–383. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Cole BK, Kuhn NS, Green-Mitchell SM, et
al: 12/15-Lipoxygenase signaling in the endoplasmic reticulum
stress response. Am J Physiol Endocrinol Metab. 302:E654–E665.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhou L and Liu F: Autophagy: roles in
obesity-induced ER stress and adiponectin downregulation in
adipocytes. Autophagy. 6:1196–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Pfaffenbach KT, Gentile CL, Nivala AM,
Wang D, Wei Y and Pagliassotti MJ: Linking endoplasmic reticulum
stress to cell death in hepatocytes: roles of C/EBP homologous
protein and chemical chaperones in palmitate-mediated cell death.
Am J Physiol Endocrinol Metab. 298:E1027–E1035. 2010. View Article : Google Scholar
|
|
80
|
Zhou QG, Zhou M, Hou FF and Peng X:
Asymmetrical dimethylarginine triggers lipolysis and inflammatory
response via induction of endoplasmic reticulum stress in cultured
adipocytes. Am J Physiol Endocrinol Metab. 296:E869–E878. 2009.
View Article : Google Scholar
|
|
81
|
Deng J, Liu S, Zou L, Xu C, Geng B and Xu
G: Lipolysis response to endoplasmic reticulum stress in adipose
cells. J Biol Chem. 287:6240–6249. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Cnop M, Foufelle F and Velloso LA:
Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med.
18:59–68. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Boden G, Lebed B, Schatz M, Homko C and
Lemieux S: Effects of acute changes of plasma free fatty acids on
intramyocellular fat content and insulin resistance in healthy
subjects. Diabetes. 50:1612–1617. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Reaven GM, Hollenbeck C, Jeng CY, Wu MS
and Chen YD: Measurement of plasma glucose, free fatty acid,
lactate, and insulin for 24 h in patients with NIDDM. Diabetes.
37:1020–1024. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Nguyen MT, Satoh H, Favelyukis S, et al:
JNK and tumor necrosis factor-alpha mediate free fatty acid-induced
insulin resistance in 3T3-L1 adipocytes. J Biol Chem.
280:35361–35371. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Gao Z, Hwang D, Bataille F, et al: Serine
phosphorylation of insulin receptor substrate 1 by inhibitor kappa
B kinase complex. J Biol Chem. 277:48115–48121. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Haukeland JW, Dahl TB, Yndestad A, et al:
Fetuin A in nonalcoholic fatty liver disease: in vivo and in vitro
studies. Eur J Endocrinol. 166:503–510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Arner P: Insulin resistance in type 2
diabetes: role of fatty acids. Diabetes Metab Res Rev. 18(Suppl 2):
S5–S9. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Jensen MD: Adipose tissue metabolism - an
aspect we should not neglect? Horm Metab Res. 39:722–725. 2007.
View Article : Google Scholar
|
|
90
|
Gregor MF and Hotamisligil GS: Thematic
review series: Adipocyte Biology. Adipocyte stress: the endoplasmic
reticulum and metabolic disease. J Lipid Res. 48:1905–1914. 2007.
View Article : Google Scholar
|
|
91
|
Bollheimer LC, Skelly RH, Chester MW,
McGarry JD and Rhodes CJ: Chronic exposure to free fatty acid
reduces pancreatic beta cell insulin content by increasing basal
insulin secretion that is not compensated for by a corresponding
increase in proinsulin biosynthesis translation. J Clin Invest.
101:1094–1101. 1998. View
Article : Google Scholar
|
|
92
|
Liu YQ, Jetton TL and Leahy JL: beta-Cell
adaptation to insulin resistance. Increased pyruvate carboxylase
and malate-pyruvate shuttle activity in islets of nondiabetic
Zucker fatty rats. J Biol Chem. 277:39163–39168. 2002. View Article : Google Scholar
|
|
93
|
Prentki M and Nolan CJ: Islet beta cell
failure in type 2 diabetes. J Clin Invest. 116:1802–1812. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
McGarry JD and Dobbins RL: Fatty acids,
lipotoxicity and insulin secretion. Diabetologia. 42:128–138. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kharroubi I, Ladrière L, Cardozo AK,
Dogusan Z, Cnop M and Eizirik DL: Free fatty acids and cytokines
induce pancreatic beta-cell apoptosis by different mechanisms: role
of nuclear factor-kappaB and endoplasmic reticulum stress.
Endocrinology. 145:5087–5096. 2004. View Article : Google Scholar
|
|
96
|
Cunha DA, Hekerman P, Ladrière L, et al:
Initiation and execution of lipotoxic ER stress in pancreatic
beta-cells. J Cell Sci. 121:2308–2318. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ryden M, Dicker A, van Harmelen V, et al:
Mapping of early signaling events in tumor necrosis factor-alpha
-mediated lipolysis in human fat cells. J Biol Chem. 277:1085–1091.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Fricke K, Heitland A and Maronde E:
Cooperative activation of lipolysis by protein kinase A and protein
kinase C pathways in 3T3-L1 adipocytes. Endocrinology.
145:4940–4947. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kawasaki N, Asada R, Saito A, Kanemoto S
and Imaizumi K: Obesity-induced endoplasmic reticulum stress causes
chronic inflammation in adipose tissue. Sci Rep. 2:7992012.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: a vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|