Introduction
Genistein (4′,5,7-trihydroxyisoflavone), a naturally
occurring soybean isoflavone glycoside with a heterocyclic
diphenolic structure similar to estrogen (1), is considered to be a potent
anticancer agent (2,3). The potential importance of genistein
was highlighted by a previous study that reported an increased
consumption of soy in Asia resulting in increased levels of
isoflavone in serum, which is closely associated with a reduced
risk of prostate cancer (4).
Genistein has been demonstrated to inhibit growth of tumor cell
lines derived from various malignancies, including breast cancer,
prostate cancer, head and neck squamous cell carcinoma, melanoma
and leukemia (5–11). Genistein is considered to affect
diverse cell functions, for example, it has been demonstrated to
trigger cell cycle arrest and apoptotic cell death through
inactivation of NF-κB and activation of caspase-3 in prostate
cancer cells, as well as to have potent anti-angiogenic activity,
inhibiting tumor cell proliferation (12–14).
Previous studies have also suggested that genistein suppresses
tumor cell growth through the inhibition of tyrosine protein
kinases (15), topoisomerases I
and II (16,17) and the expression of mRNAs of cell
cycle-related genes (18) in
different cell types. By contrast, under other circumstances,
apoptotic cell death was inhibited in the presence of genistein.
Several previous studies revealed that genistein was able to
prevent apoptotic cell death via its antioxidant properties
(19,20). Genistein inhibited UV
irradiation-induced oxidative stresses and neuronal damage
resulting from production of reactive oxygen species (ROS). It also
inhibited methylglyoxal-induced apoptotic cell death in a human
mononuclear cell model, and inhibited methylglyoxal-induced DNA
damage and ROS production in vitro. Animal experiments
further confirmed the protective effect of genistein on
methylglyoxal-induced cell injury (21).
Intracellular ROS, including superoxide, hydrogen
peroxide and hydroxyl radicals, are generated following exposure to
ionizing radiation, selected chemotherapeutic agents (Taxol and
etoposide), hyperthermia, inhibition of antioxidant enzymes
(including thioredoxins, catalase, superoxide dismutases and
glutathione-linked peroxidase) and depletion of cellular
reductants, including nicotinamide adenine dinucleotide phosphate
(NADPH), reducing equivalents and glutathione (GSH) (22–25).
Therefore, ROS are involved in numerous biological and
pathophysiological situations, including aging and inflammation.
ROS have high chemical reactivity and, thus, damage lipids,
proteins, as well as mitochondrial and nuclear DNA, which can lead
to cell cycle arrest (26,27). Furthermore, ROS generation can
induce apoptotic cell death through depletion of intracellular
reduced GSH and protein thiols, and loss of mitochondrial membrane
potential (25,27). The present study used human
promyeloid leukemia HL-60 cells to examine the intracellular signal
mechanisms involved in genistein-induced cell growth arrest and
cell death.
Materials and methods
Cell culture and growth
Genistein and N-acetylcysteine were purchased
from Sigma (St. Louis, MO, USA). A stock solution of genistein was
prepared in dimethyl sulfoxide. Stock solution of N-acetylcysteine
was prepared in phosphate-buffered saline (PBS). Working solutions
were prepared by dilution of stock solutions in culture medium.
HL-60 human promyeloid leukemia cells (4×104/ml) were
grown as suspension cultures in RPMI-1640 medium (Gibco, Scotland,
UK) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan,
UT, USA) and 100 U/ml penicillin/streptomycin (Sigma) in a
humidified atmosphere containing 5% CO2 at 37°C for 24
h. Normal human lymphocytes were isolated from peripheral blood of
healthy human males. Blood was added to Ficoll-Paque (Amersham
Pharmacia Biotech, Uppsala, Sweden) and centrifuged at 400 × g for
20 min. The lymphocyte layer was collected using a micropipette and
diluted with serum-free RPMI-1640[0]. The diluted cell suspension
was centrifuged at 70 × g for 10 min. Lymphocytes
(4×104/ml) were cultured in RPMI-1640 supplemented with
10% FBS, 100 U/ml penicillin/streptomycin and 5 μg/ml
phytohemagglutinin (Sigma) in a humidified atmosphere containing 5%
CO2 at 37°C for 24 h. Cell growth and viability were
determined using a trypan blue (Sigma) exclusion test.
γ-irradiation
Cells (2×105/ml) pretreated with 20 μM
genistein for 6 h and untreated cells were irradiated with a single
dose of 2 Gy (dose rate, 0.2 Gy/min) or 5 Gy (dose rate, 0.5
Gy/min) and then cultured in a humidified atmosphere containing 5%
CO2 at 37°C.
Measurement of intracellular ROS
level
HL-60 cells (4×104 cells/4 ml) were
cultured in T25 flasks and treated with 20 μM genistein for 0, 12,
24 and 48 h. Harvested cells (5×105) were treated with
10 μM chloromethyl-2′,7′-dichlorofluorescein diacetate (DCFH-DA)
for 30 min in the dark and then washed with PBS. The intracellular
ROS level was measured using the FACScan (Beckman-Coulter
Instruments Inc., Brea, CA, USA) and visualized using a
fluorescence microscope (Leica, Heidelberg, Germany).
Measurement of intracellular GSH
level
In order to determine the total intracellular levels
of reduced (GSH) and oxidized (GSSG) forms of GCH, a GSH assay kit
(Cayman Chemical, Ann Arbor, MI, USA) was used. HL-60 cells
(1×107) were used for each experiment. Concentrations of
GSH and GSSG were calculated from the typical standard curves. The
detectable range was 0.2–6.0 nmol/ml.
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
TRIzol® reagent (Invitrogen Life
Technologies, Grand Island, NY, USA) was used to isolate total RNA
from 5×106 HL-60 cells according to the manufacturer’s
instructions. Total RNA (1 mg) was added to a 20-μl reaction
mixture containing Maxime RT PreMix (iNtRON Biotechnology,
Seongnam, Korea) and 10 pmole primers. Primers used were
cICDH, forward 5′-TTGGATCCAAAATGTCCAAAAAA-3′ and reverse
5′-ATGAATTCAAGTAGTCAGAACGT-3′; β-actin, forward 5′-CA TCCTCACCCT
GAAGTACCC-3′ and reverse 5′-AGCCTGGATAGCAACGTACATG-3′. RT-PCR was
performed in a thermal cycler (Apollo, San Diego, CA, USA) under
conditions of 45°C for 30 min, followed by 94°C for 5 min, and 25
cycles of 94°C for 1 min, 52°C for 1 min and 72°C for 1 min. PCR
products were separated on a 1.5% agarose gel and visualized with
ethidium bromide staining.
Propidium iodide staining for analysis of
apoptotic cell death and cell cycle status
HL-60 cells (2×105) were suspended in 2
ml ice-cold 50% ethanol and maintained at 4°C for 40 min. Fixed
cells were harvested by centrifugation, at 1,000 × g for 10 min,
and resuspended in 800 μl PBS. Subsequently, 100 μl RNase (1 mg/ml)
and 100 μl propidium iodide (400 μg/ml) were added to the cell
suspension, and cells were incubated at 37°C for 30 min. This
allowed for the discrimination of live cells from apoptotic and
necrotic cells. Analysis of apoptotic cell death was performed
using a FACScan (Beckman-Coulter Instruments Inc.) equipped with a
single 488-nm argon laser (Beckman-Coulter Instruments Inc.). The
percentages of apoptotic cells and the cell cycle distribution were
calculated using MultiCycle for Windows software (Phoenix Flow
Systems, San Diego, CA, USA).
Western blot analysis
The protein extract sample was separated in a 12.5%
denaturing polyacrylamide gel, followed by transfer onto
nitrocellulose membranes (GE Healthcare Bio-Sciences, Pittsburgh,
PA, USA). Membranes were incubated with monoclonal anti-human
p21wap1/cip1, polyclonal anti-human Bcl-2-associated X
protein (Bax), polyclonal anti-human β-actin (Cell Signaling
Technology, Inc., Danvers, MA, USA) and polyclonal anti-human
B-cell lymphoma 2 (Bcl-2; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) at room temperature for 2 h, and then with secondary
antibodies (anti-mouse or anti-rabbit immunoglobulin G horseradish
peroxidase-conjugated; Cell Signalling Technology, Inc.) at room
temperature for 1 h. Membranes were washed four times with
Tris-buffered saline with Tween 20 and protein bands were
visualized using an ECL detection kit (Amersham Pharmacia Biotech).
Protein concentrations were determined by the Lowry method.
Morphological analysis
Human blood lymphocytes and HL-60 cells were treated
with genistein and γ-radiation, followed by culture for 48 h.
Cytochalasin-B (4 μg/ml, Sigma) was added 20 h after γ-irradiation.
Cells were harvested and resuspended in hypotonic 0.075 M KCl for 3
min. Cells were centrifuged again and Carnoy’s fixative (American
MasterTech, Lodi, CA, USA) was gently added. Cells were then
mounted on clean slides and air-dried. The slides were stained with
Giemsa (Sigma) solution and observed under a light microscope
(Leica).
Results
Effect of genistein on HL-60 cell
proliferation and intracellular ROS generation
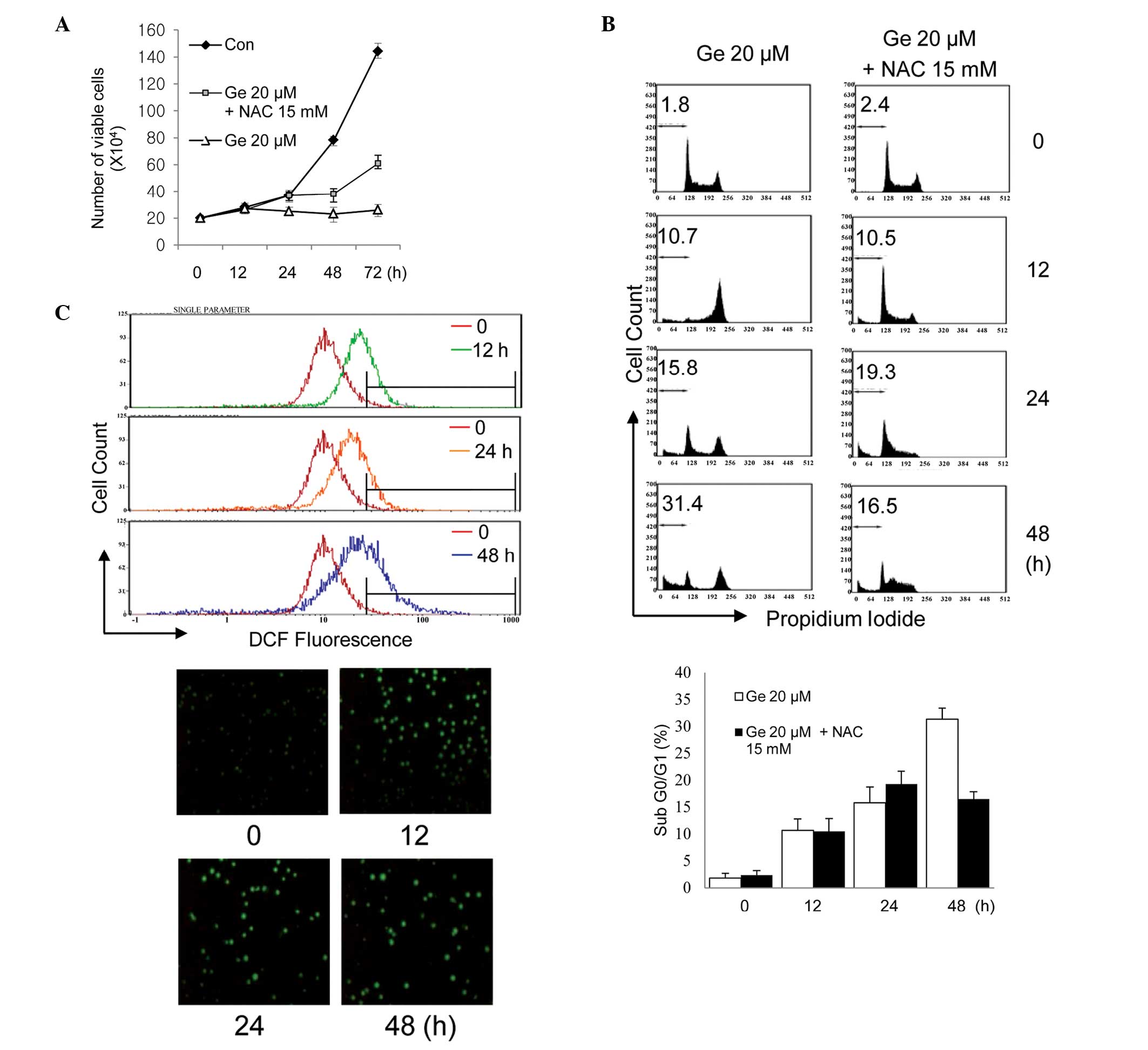
Fig. 1A shows the
time-dependent response of HL-60 cells to exposure to 20 μM
genistein in the presence or absence of 15 mm N-acetylcysteine, a
sulfur-containing antioxidant compound. Genistein-treated cells
demonstrated significant retardation of cell growth and, based on
time-lapse images, apoptotic cell death was significantly increased
(Fig. 1B). A strictly regulated
cellular level of ROS is essential for the proliferation of tumor
cell growth, therefore an imbalance of ROS affects cell growth
arrest and cell death (28). The
level of cellular ROS in HL-60 cells was examined to determine
whether genistein causes a change in cellular ROS level and is
associated with cell growth inhibition in these cells. The
oxidant-sensitive probe DCFH-DA was used, which permits detection
of various oxygen-derived free radicals by flow cytometry and
fluorescence microscopy (Leica). As shown in Fig. 1C, intracellular ROS were
significantly elevated in HL-60 cells following treatment with 20
μM genistein for 12, 24 and 48 h, compared with the control HL-60
cells. However, cells partly recovered from genistein-induced cell
growth inhibition and death when exposed to genistein in the
presence of N-acetylcysteine, a cell-permeable ROS scavenger. This
finding indicates that genistein led to upregulation of cellular
ROS in HL-60 cells and that this affected cell growth.
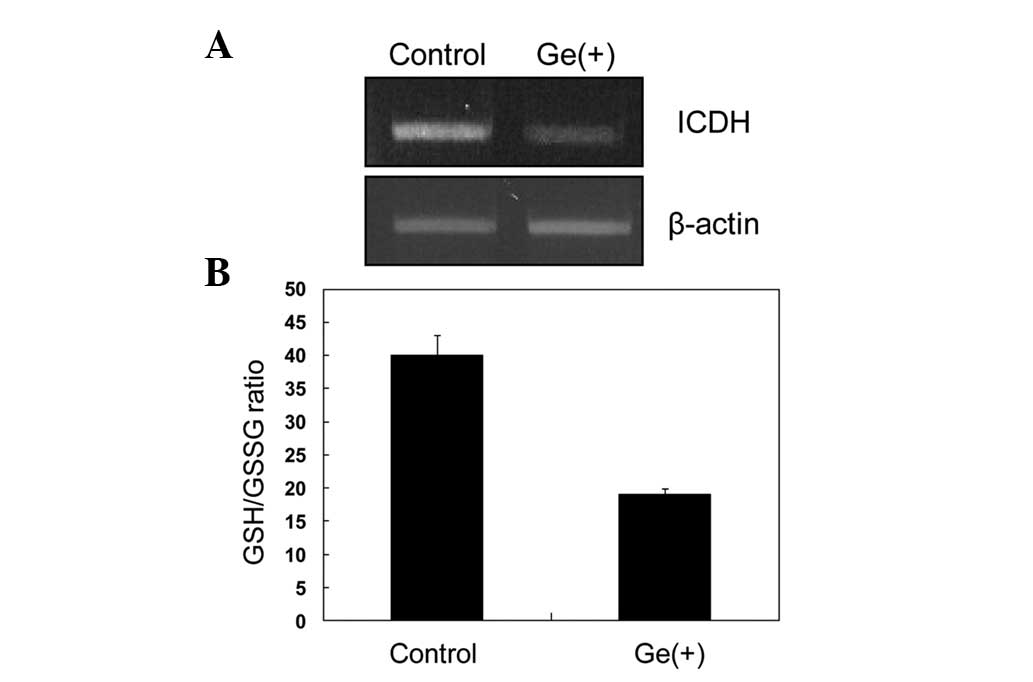
Expression of cICDH
The present study investigated how genistein induced
elevation of the intracellular ROS level. In cells, the cellular
redox potential (GSH/GSSG ratio) is an important factor in the
homeostatic regulation of intracellular ROS, which, in turn, is
important in cell signaling for proliferation. In addition, redox
potential is also a critical factor in the control of cell growth
in various cancer cell lines. Reducing equivalents (NADPH)
generated by cICDH or glucose-6-phosphate dehydrogenase are
indispensable for the regeneration of oxidized GSH, kithioredoxin
and other molecules of this type. Therefore, to ascertain the role
of genistein in the generation of ROS, intracellular redox
potential, as well as cICDH involved in the regulation of
cellular redox status was examined. Genistein treatment decreased
the transcriptional levels of cICDH and, thus, significantly
decreased the GSH/GSSG ratio (Fig. 2A
and B). The level of cICDH gene expression in the
genistein-treated HL-60 cells was only 20% that of the control
cells and, consequently, resulted in a decrement by half in the
GSH/GSSG ratio.
Pro-oxidant activity of genistein results
in G2/M phase arrest and apoptosis
Genistein was suggested to induce cell cycle arrest
in the G2/M phase, which leads to inhibition of cell growth
(29). To investigate whether ROS
are involved in genistein-induced G2/M phase transition and cell
death in the HL-60 cell line, cell cycle progression was analyzed.
HL-60 cells were treated for 48 h with 20 μM genistein. Following
12 h of genistein treatment, cell cycle progression into the G2/M
phase was most prominent. In total, 63% of HL-60 cells treated with
genistein were in the G2/M phase, with a concomitant decrease in
cells in the G0/G1 phase from 32 to 1%. An increase in the
sub-G0/G1 peak (hypodiploid apoptotic cells) was also noted. Cell
death exponentially increased 48 h after genistein treatment. By
contrast, addition of N-acetylcysteine inhibited or delayed
genistein-induced G2/M phase progression and prevented apoptotic
cell death. N-acetylcysteine also significantly induced S
phase arrest, enabling repair of genistein-induced damage (Table I). These data indicated that
genistein-induced G2/M phase arrest is caused by elevated
intracellular ROS. Based on these findings, the levels of
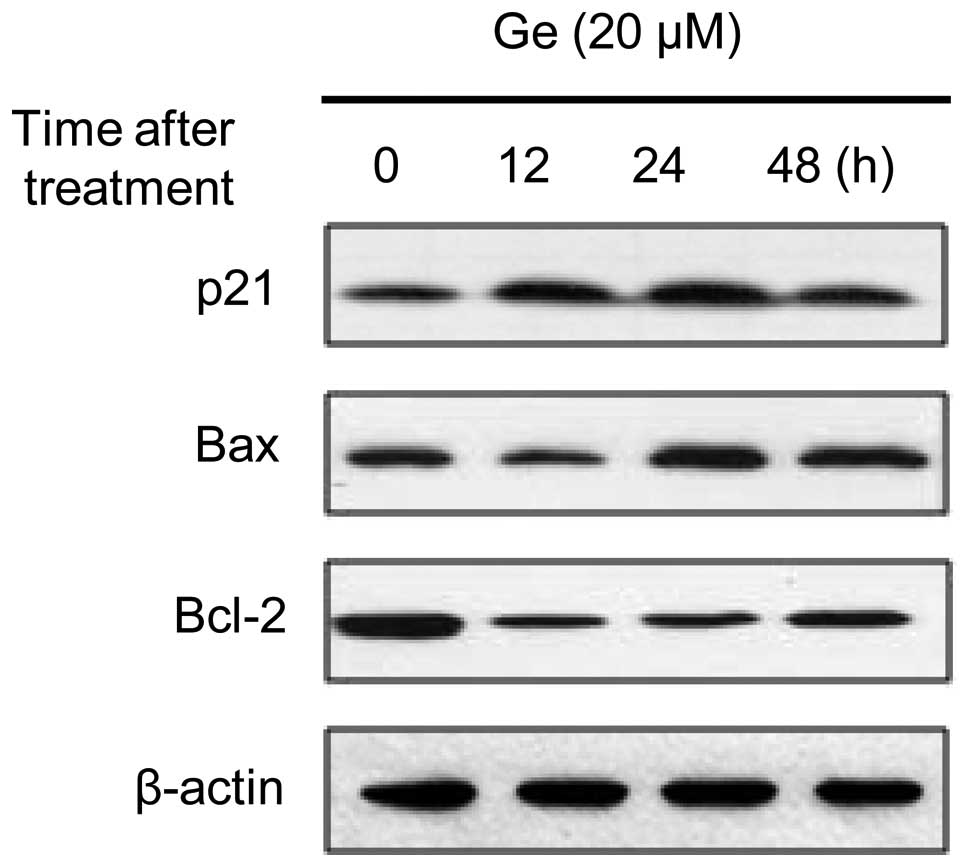
expression of p21WAF1/Cip1 and cyclin B1, two molecules
involved in cell cycle progression, were evaluated by western blot
analysis. As shown in Fig. 3,
genistein increased the level of p21WAF1/Cip1
after 12, 24 and 48 h of treatment, resulting in a 2–3-fold
increase in expression. The effect of genistein on the Bcl-2 family
of proteins, which are associated with apoptotic cell death, in
HL-60 cells was also examined. Upregulation of the proapoptotic
protein Bax in genistein-treated cells and downregulation of the
antiapoptotic protein Bcl-2 was observed.
 | Table ICell cycle distribution of HL-60
cells following treatment with genistein and
N-acetylcysteine. |
Table I
Cell cycle distribution of HL-60
cells following treatment with genistein and
N-acetylcysteine.
| Time following
treatment (h) | Percentage of cells
in |
|---|
|
|---|
| Sub-G0/G1 | G0/G1 | S | G2/M |
|---|
| Genisteina | | | | |
| 0 | 1.8 | 32.6 | 49.7 | 15.9 |
| 12 | 10.7 | 1.4 | 24.6 | 63.3 |
| 24 | 15.8 | 30.6 | 23.9 | 29.7 |
| 48 | 31.4 | 19.5 | 11.1 | 38.0 |
| Genistein +
N-acetylcysteineb | | | | |
| 0 | 2.4 | 34.9 | 47.0 | 15.7 |
| 12 | 10.5 | 41.2 | 41.0 | 7.3 |
| 24 | 19.3 | 32.1 | 46.2 | 2.4 |
| 48 | 16.5 | 12.6 | 66.8 | 4.1 |
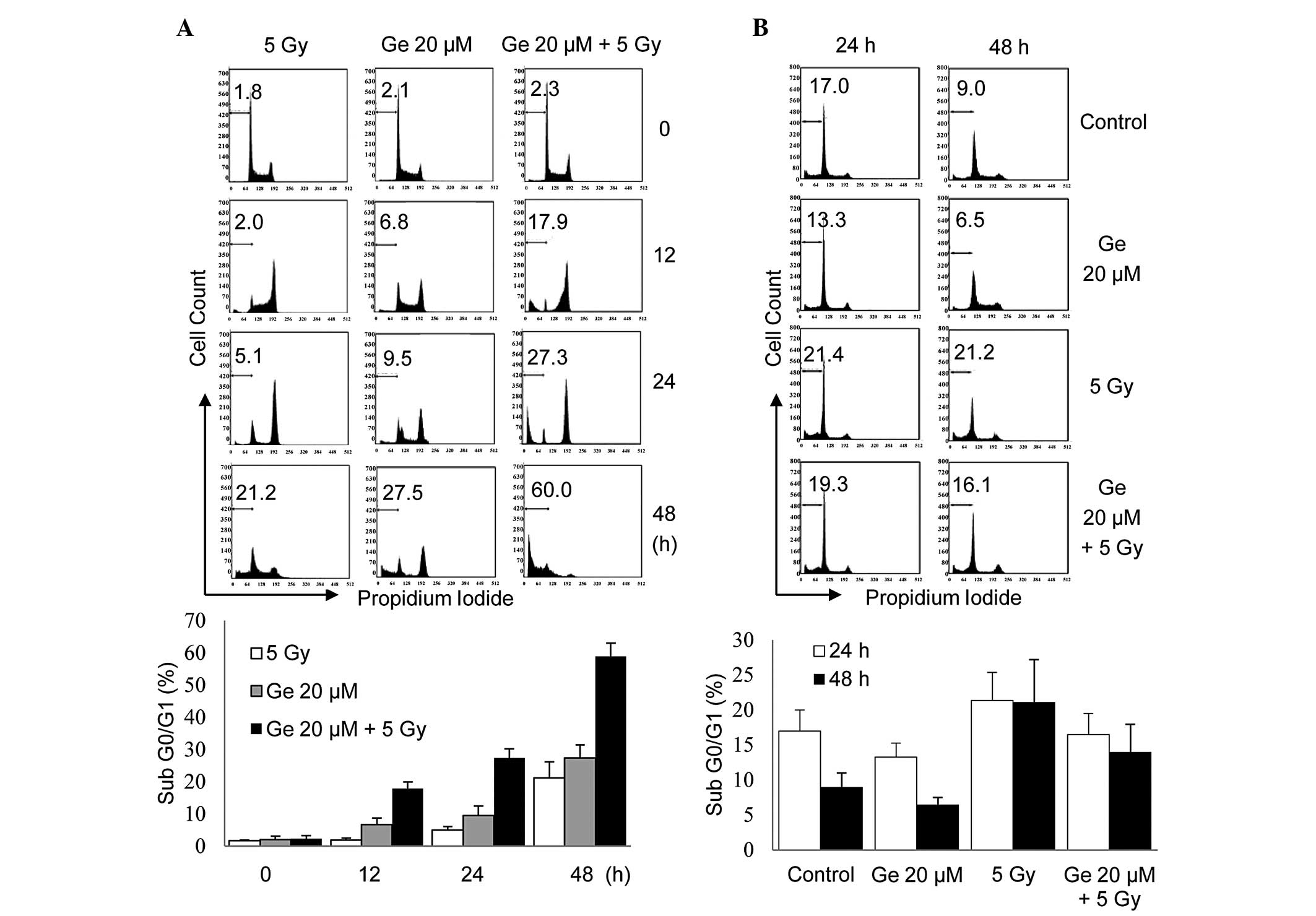
Effects of γ-irradiation on human
promyeloid leukemia HL-60 cells and normal human lymphocytes
As shown in Fig. 4,
the effect of sensitization to γ-radiation in apoptotic cell death
was investigated in genistein-treated HL-60 cells by measuring the
change in hypodiploid content. The effect of genistein on
radiation-induced damage in normal lymphocytes was also
investigated. Following γ-irradiation at a dose of 5 Gy (dose rate,
0.5 Gy/min), HL-60 cells progressed into the G2/M phase and
arrested there. After 48 h, cells either undergo cell death or are
repaired and re-enter the G1 phase; at that time point, ~21% of
cells underwent cell death. Genistein-treated HL-60 cells also
progressed into the G2/M phase and, 48 h after genistein treatment,
cell death was observed in 27% of cells. When administered
together, genistein and γ-radiation synergistically increased cell
death to a higher level than either agent alone (Fig. 4A). Notably, no such synergistic
effect was observed in normal human lymphocytes. Compared with its
radiosensitizing effect on HL-60 leukemia cells, genistein had a
radioprotective effect on normal lymphocytes after 24 and 48 h of
treatment (Fig. 4B).
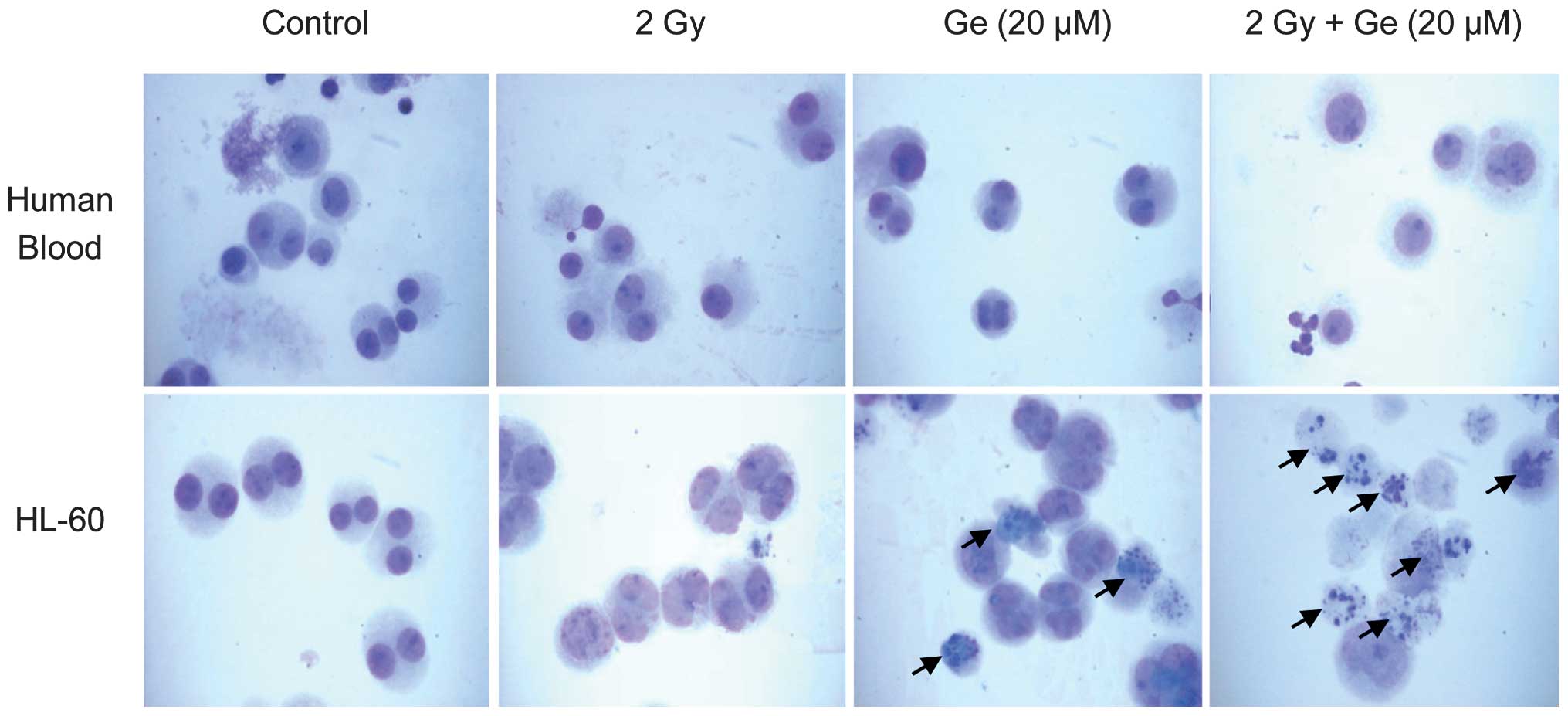
Differences in morphology of human
promyeloid leukemia HL-60 cells and normal human lymphocytes
Finally, it was confirmed that genistein had
different effects on radiation-induced damage in promyeloid
leukemia HL-60 cells and normal human lymphocytes by the detection
of apoptotic bodies. At a dose of 2 Gy, a negligible number of
γ-radiation-induced apoptotic bodies were detected in normal
lymphocytes. However, radiation treatment partially induced
initiation of apoptosis in HL-60 cells. Genistein clearly induced
the formation of apoptotic bodies in certain HL-60 cells. However,
it did not affect apoptotic body formation in normal lymphocytes.
Genistein (20 μM) and γ-radiation synergistically increased
apoptotic body formation in HL-60 cells. Furthermore, this
combination treatment resulted in the formation of apoptotic bodies
in HL-60 cells. However, significant numbers of apoptotic bodies
were not observed in normal lymphocytes under any condition
(Fig. 5).
Discussion
Genistein is known to induce differentiation, cell
cycle arrest, apoptosis and inhibition of tumor cell growth, and
also possesses anti-angiogenesis and antioxidant activities
(9–11). Cell cycle arrest is a
characteristic of eukaryotic cells and the cell cycle progresses
through different phases commonly referred to as checkpoints
(12). Cell cycle checkpoints are
transient delays in G1/S or G2/M transition
that constitute a response to DNA damage by cellular stressors,
including ROS, and allow time for the activation of repair
mechanisms (13). Following repair
of damaged DNA, cells resume cell cycle progression. However, if
the damage is too severe, the cells may undergo apoptosis or
irreversible senescence (14).
Similarly to genistein, ROS cause DNA damage and induce G2/M phase
arrest and apoptosis (22,25). The present study investigated
whether ROS are involved in genistein-induced cell cycle arrest and
cell death in the HL-60 cell line. To date, it has been
hypothesized that genistein eliminates oxygen free radicals
generated by toxic agents and hydrogen peroxide (28–31).
In addition, it is well known that genistein inhibits topoisomerase
II activity, which leads to its cleavage and thus induced G2/M
phase arrest and apoptosis (30–32).
However, additional mechanisms underlying the antioxidant activity
and induction of apoptotic cell death by genistein remain to be
elucidated. The present study concluded that, in human promyeloid
leukemia HL-60 cells, genistein affects the cellular redox
potential level, which is known to be important in the regulation
of cellular physiology, including cell growth and
differentiation.
The result from the present study that
G2/M arrest in response to genistein treatment
sensitizes HL-60 cells to γ-radiation-induced cell death,
corroborates previous studies in DU145 human prostate cancer cells
(33) and cervical cancer cells
(34). Cells in the
G2/M phase have been demonstrated to be more
radiosensitive than cells in other phases of the cell cycle
(35–37). Pretreatment with genistein arrests
cells in G2/M, and thus, may increase their
radiosensitivity, resulting in increased cell death, in addition to
the direct cytotoxic effects of genistein and γ-radiation. The
present study further addresses the role of the cellular redox
potential and reducing equivalents-generating enzyme, cICDH,
in the mechanism by which genistein enhances intracellular ROS and
radiation-induced cell death. Intracellular GSH depletion or low
GSH/GSSG ratio caused excessive intracellular ROS accumulation.
Alternatively, the downregulation of the enzymes involved in GSH
synthesis, and maintenance of the reduced GSH level may also result
in ROS accumulation and, thus, sensitivity to γ-radiation and
anticancer drugs. In the present study, total GSH increased with
genistein treatment (data not shown). Furthermore, the level of the
antioxidant enzyme thioredoxin also increased (data not shown).
Despite elevated levels of these factors, genistein increased
sensitivity to γ-radiation-induced cell death. These findings
suggest that cellular redox potential (GSH/GSSG) may be a critical
factor in this process. Although levels of GSH and thioredoxin
increased, if the oxidized form is converted to the reduced form,
cellular redox potential is not maintained at a steady state.
NADP+-dependent ICDH is necessary for the
maintenance of the cellular redox potential level at a steady state
by production of the reducing equivalents (NADPH) (38). Therefore, the present study
examined the expression of the ICDH gene by RT-PCR and
confirmed that the expression level was significantly lower in
genistein-treated cells compared with the controls.
It has been reported that genistein treatment
combined with radiation enhances radiosensitivity in numerous
cancer cell lines (37,38). In the present study, it was
demonstrated that genistein also has a synergistic effect with
γ-radiation on apoptosis in HL-60 cells. By contrast, genistein has
a protective effect on normal lymphocytes. Cells respond to
DNA-damaging agents by activating cell-cycle checkpoints, and cells
in the G2/M phase of the cell cycle have been
demonstrated to be more radiosensitive than cells in other phases
(33–35). Several types of cancer cells are
hypersensitive to γ-radiation in the G2/M phase, compared with
normal cells, as they are deficient in DNA repair capacity
(39–41). However, in normal human
lymphocytes, neither genistein nor radiation alone promoted a
decrease in the percentage of cells in G0/G1
and a concomitant increase in the percentage of cells in
G2/M. This indicated that DNA damage by genistein or
radiation is not critical in normal lymphocytes and, thus, cell
cycle transition and arrest for repair is not required. This may
explain why genistein did not have a synergistic effect on
radiation-induced cell death. By contrast, genistein had a
radioprotective effect in normal human lymphocytes as G2/M phase
arrest did not occur. In conclusion, the results from the present
study suggest that genistein does not act as an antioxidant, but as
a pro-oxidant, in human promyeloid leukemia HL-60 cells. The
pro-oxidant activity of genistein caused a rapid transition of
HL-60 cells into the G2/M phase and, thus, inhibited cell
proliferation and apoptotic cell death. In addition, the
combination of genistein treatment and γ-irradiation demonstrated a
synergistic effect on cell death in HL-60 cells, whereas genistein
exhibited a radioprotective effect in normal lymphocytes.
Acknowledgements
This study was supported by grants from the Ministry
of Science, ICT and Future Planning (Nuclear Research and
Development Program) of the Republic of Korea and by a creative
program of the Korea Atomic Energy Research Institute.
References
|
1
|
MacGregor JT: Genetic toxicology of
dietary flavonoids. Prog Clin Biol Res. 206:33–43. 1986.PubMed/NCBI
|
|
2
|
Soulinna EM, Buchsbaum RN and Racker E:
The effect of flavonoids on aerobic glycolysis and growth of tumor
cells. Cancer Res. 35:1865–1872. 1975.PubMed/NCBI
|
|
3
|
Adlercreutz CH, Goldin BR, Gorbach SL, et
al: Soybean phytoestrogen intake and cancer risk. J Nutr.
125:757S–770S. 1995.PubMed/NCBI
|
|
4
|
Giovannucci E: Epidemiologic
characteristics of prostate cancer. Cancer. 75:1766–1777. 1995.
View Article : Google Scholar
|
|
5
|
Sarkar FH and Li Y: The role of
isoflavones in cancer chemoprevention. Front Biosci. 9:2714–2724.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pagliacci MC, Smacchia M, Migliorati G, et
al: Growth inhibitory effect of the natural phytoe-strogen
genistein in MCF-7 human breast cancer cells. Eur J Cancer.
30:1675–1682. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kyle E, Neckers L, Takimoto C, Curt G and
Bergan R: Genistein-induced apoptosis of prostate cancer cells is
preceded by a specific decrease in focal adhesion kinase activity.
Mol Pharmacol. 51:193–200. 1997.PubMed/NCBI
|
|
8
|
Spinnozi F, Pagliacci M, Migliorati G, et
al: The natural tyrosine kinase inhibitor genistein produces cell
cycle arrest and apoptosis in Jurkat T-leukemia cells. Leuk Res.
18:431–439. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y, Upadhyay S, Bhuiyan M and Sarkar FH:
Induction of apoptosis in breast cancer cells MDA-MB-231 by
genistein. Oncogene. 18:3166–3172. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Bhuiyan M and Sarkar FH: Induction
of apoptosis and inhibition of c-erbB-2 in MDA-MB-435 cells by
genistein. Int J Oncol. 15:525–533. 1999.PubMed/NCBI
|
|
11
|
Alhasan SA, Pietrasczkiwicz H, Alonso MD,
Ensley J and Sarkar FH: Genistein-induced cell cycle arrest and
apoptosis in a head and neck squamous cell carcinoma cell line.
Nutr Cancer. 34:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kumi-Diaka J, Sanderson NA and Hall A: The
mediating role of caspase-3 protease in the intracellular mechanism
of genistein-induced apoptosis in human prostatic carcinoma cell
lines, DU145 and LNCaP. Biol Cell. 92:595–604. 2000. View Article : Google Scholar
|
|
13
|
Davis N, Kucuk O and Sarkar FH: Genistein
inhibits NF-κB activation in prostate cancer cells. Nutr Cancer.
35:167–174. 1999.
|
|
14
|
Fotsis T, Pepper MS, Aktas E, et al:
Flavonoids, dietary-derived inhibitors of cell proliferation and in
vitro angiogenesis. Cancer Res. 57:2916–2921. 1997.PubMed/NCBI
|
|
15
|
Gradzka I, Buraczewska I, Kuduk-Jaworska
J, Romaniewska A and Szumiel I: Radiosensitizing properties of
novel hydroxydicarboxylatoplatinum (II) complexes with high or low
reactivity with thiols: two modes of action. Chem Biol Interact.
146:165–177. 2003. View Article : Google Scholar
|
|
16
|
Salti GI, Grewal S, Mehta RR, et al:
Genistein induces apoptosis and topoisomerase II-mediated DNA
breakage in colon cancer cells. Eur J Cancer. 36:796–802. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okura A, Arakawa H, Oka H, Yoshinari T and
Monnden Y: Effect of genistein on topoisomerase activity and on the
growth of (Val 12) Ha-ras-transformed NIH 3T3 cells. Biochem
Biophys Res Commun. 157:183–189. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Davis JN, Singh B, Bhuiyan M and Sarkar
FH: Genistein-induced upregulation of p21WAF1,
downregulation of cyclin B, and induction of apoptosis in prostate
cancer cells. Nutr Cancer. 32:123–131. 1998.PubMed/NCBI
|
|
19
|
Chan WH and Yu JS: Inhibition of UV
irradiation-induced oxidative stresses and apoptotic biochemical
changes in human epidermal carcinoma A431 cells by genistein. J
Cell Biochem. 78:73–84. 2000. View Article : Google Scholar
|
|
20
|
Liang HW, Qiu SF, Shen J, et al: Genistein
attenuates oxidative stress and neuronal damage following transient
global cerebral ischemia in rat hippocampus. Neurosci Lett.
438:116–120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu HJ and Chan WH: Genistein protects
methylglyoxal-induced oxidative DNA damage and cell injury in human
mononuclear cells. Toxicol In Vitro. 21:335–342. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Varbiro G, Veres B, Gallyas F Jr and
Sumegi B: Direct effect of Taxol on free radical formation and
mitochondrial permeability transition. Free Radic Biol Med.
31:548–558. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kinnula VL, Paakko P and Soini Y:
Antioxidant enzymes and redox regulating thiol proteins in
malignancies of human lung. FEBS Letters. 569:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nguyen TD, Maquart F and Monboisse J:
Ionizing radiations and collagen metabolism: from oxygen free
radicals to radio-induced late fibrosis. Radia Phys Chem.
72:381–386. 2005. View Article : Google Scholar
|
|
25
|
Meister A and Anderson ME: Glutathione.
Annu Rev Biochem. 52:711–760. 1983. View Article : Google Scholar
|
|
26
|
Singh KK: Mitochondria damage checkpoint,
aging, and cancer. Ann NY Acad Sci. 1067:182–190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Burhans WC and Hwintz NH: The cell cycle
is a redox cycle: linking phase-specific target to cell fate. Free
Radic Biol Med. 47:1282–1293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zielonka J, Gebicki J and Grynkiewicz G:
Radical scavenging properties of genistein. Free Radic Biol Med.
35:958–965. 2003. View Article : Google Scholar
|
|
29
|
Wei H, Bowen R, Cai Q, Barnes S and Wang
Y: Antioxidant and antipromotional effects of the soybean
isoflavone genistein. Proc Soc Exp Biol Med. 208:124–130. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Polkowski K and Mazurek AP: Biological
properties of genistein. A review of in vitro and in vivo data.
Acta Pol Pharm. 57:135–155. 2000.PubMed/NCBI
|
|
31
|
Setchell KD and Cassidy A: Dietary
isoflavones: biological effects and relevance to human health. J
Nutr. 129:758S–767S. 1999.PubMed/NCBI
|
|
32
|
Schmidt F, Knobbe CB, Frank B, Wolburg H
and Weller M: The topoisomerase II inhibitor, genistein, induces
G2/M arrest and apoptosis in human malignant glioma cell lines.
Oncol Rep. 19:1061–1066. 2008.PubMed/NCBI
|
|
33
|
Wang Y, Raffoul JJ, Che M, et al: Prostate
cancer treatment is enhanced by genistein in vitro and in vivo in a
syngeneic orthotopic tumor model. Radiat Res. 166:73–80. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shin JI, Shim JH, Kim KH, et al:
Sensitization of the apoptotic effect of gamma-irradiation in
genistein-pretreated CaSki cervical cancer cells. J Microbiol
Biotechnol. 18:523–531. 2008.PubMed/NCBI
|
|
35
|
Matsukawa Y, Marui N, Sakai T, et al:
Genistein arrests cell cycle progression at G2-M. Cancer Res.
1328–1331. 1993.PubMed/NCBI
|
|
36
|
Cappelletti V, Fioravanti L, Miodini P and
Di Fronzo G: Genistein blocks breast cancer cells in the G(2)M
phase of the cell cycle. J Cell Biochem. 79:594–600. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kumi-Diaka JK, Merchant K, Haces A, et al:
Genistein-selenium combination induces growth arrest in prostate
cancer cells. J Med Food. 13:842–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schafer FQ and Buettner GR: Redox
environment of the cell as viewed through the redox state of the
glutathione disulfide/glutathione couple. Free Radic Biol Med.
30:1191–1212. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Szumiel I, Kapiszewska M, John A, et al:
Caffeine-inhibitable control of the radiation-induced G2 arrest in
L5178Y-S cells deficient in non-homologous end-joining. Radiat
Environ Biophys. 40:137–143. 2001. View Article : Google Scholar
|
|
40
|
Theron T, Binder A, Verheye-Dua F and
Boehm L: The role of G2-block abrogation, DNA double-strand break
repair and apoptosis in the radiosensitization of melanoma and
squamous cell carcinoma cell lines by pentoxyfylline. Int J Radiat
Biol. 76:1197–1208. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Abbott DW, Freeman ML and Holt JT:
Double-strand break repair deficiency and radiation sensitivity in
BRCA2 mutant cancer cells. J Natl Cancer Inst. 90:978–985. 1998.
View Article : Google Scholar : PubMed/NCBI
|