1. Introduction
Alzheimer’s disease (AD) is a multi-factorial and
heterogeneous neurodegenerative disorder. It is irreversible,
insidious and characterized by progressive worsening of symptoms,
including a global cognitive decline in memory, orientation,
judgment and reasoning. The cellular etiology of AD is associated
with the loss of neurons and synapses in cortical and limbic
structures, including the hippocampus and amygdala. Currently, AD
has a incidence of 13% in individuals >65 years of age (1); however, its prevalence is expected to
quadruple by 2050. Without a significant therapeutic breakthrough,
1/85 individuals worldwide are likely be living with this disease
(2). Furthermore, the demographics
of patients undergoing surgery under the influence of anesthesia
annually indicates that ~66 million individuals are >65 years
old (1). As life expectancy
continues to increase, the number of patients with AD requiring
surgery and administration of anesthesia may also steadily rise.
Studies have demonstrated that inhaled anesthetics, including
isoflurane, sevoflurane and desflurane, have an impact on the
neuropathogenesis of AD and possibly accelerate the clinical
progression of this neurodegenerative disorder (3,4).
Other previous studies have demonstrated that the administration of
general anesthesia may be a risk factor for the development of AD
(5–9). In addition, the expression levels of
tau protein and certain cytokines in the cerebrospinal fluid (CSF)
following anesthesia and surgery are consistent with those
identified in patients with AD (10). However, certain studies have
arrived at different conclusions and have suggested that anesthesia
and surgery may not contribute to the development of AD (11,12).
Thus, it has been difficult to clinically prove the association
between anesthesia and AD. Evidence from several previous studies
on the impact of isoflurane, sevoflurane and desflurane on the
neurotoxicity and neuropathogenesis of AD may assist in
facilitating and guiding future clinical studies. In the present
review, studies assessing the effects of commonly used inhaled
anesthetics on the processing of amyloid precursor protein (APP),
metabolism of β-amyloid protein (Aβ), tau pathology, synaptic
plasticity and cognitive deficits are discussed. Furthermore, the
molecular mechanisms underlying the anesthetic ally-induced
development of an AD-like neuropathology are reviewed. Finally, the
present review offers a perspective on future studies, which are
required to shed further light on this field of study.
2. Neuropathogenesis of AD
Neuropathogenic hallmarks of AD
The two most important histological features of AD
are the formation of extracellular amyloid plaques, composed
predominantly of Aβ, and intraneuronal neurofibrillary tangles
(NFT), composed of aberrantly hyperphosphorylated tau proteins
assembled into paired helical filaments (PHF) (13). Other characteristics of AD are
dystrophic neurites, extensive neuronal loss and gliosis. Amyloid
plaques are predominantly composed of Aβ40 and Aβ42, which are
generated through the amyloidogenic processing of APP, requiring
the activity of the enzymes β- and γ-secretase (14). Tau is a microtubule-associated
protein, which is normally enriched in the axons. However, during
AD and other tauopathies, it becomes hyperphosphorylated, and is
subsequently relocalized to and aggregates in the somatodendritic
compartment of the affected neurons. Hyperphosphorylated tau
proteins assemble into PHF structures, which in turn induce the
formation of NFTs (13,15,16).
The final outcome of this pathological process, as shown in
Fig. 1, is neuronal cell death and
degeneration, as observed in AD (15).
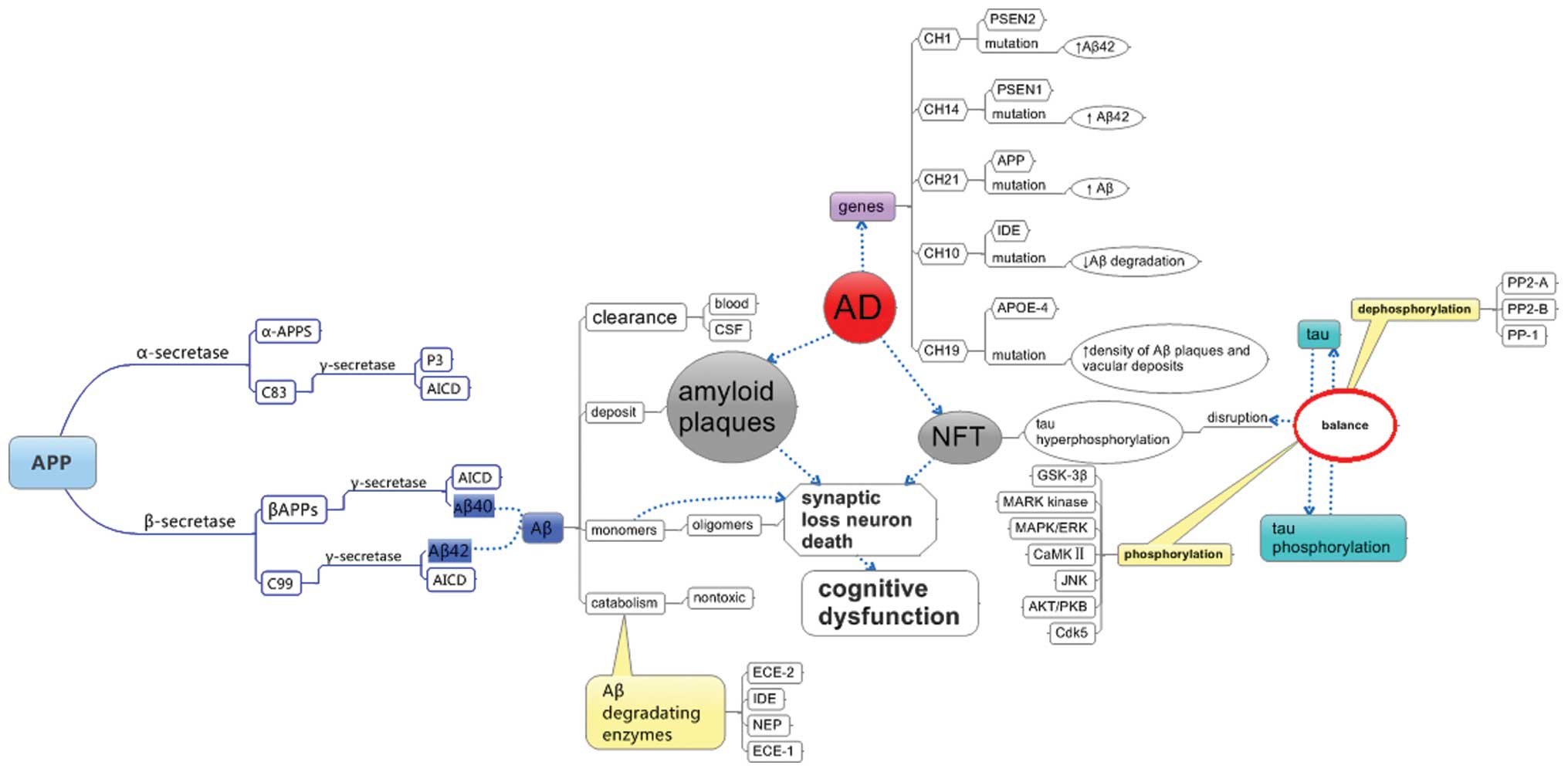 | Figure 1AD-associated genes, hallmarks of AD
and its processing. AD, Alzheimer’s disease; APP, amyloid precursor
protein; APPS, large soluble ectodomain of APP; AICD, β-amyloid
precursor protein intracellular domain; Aβ; β-amyloid; CSF,
cerebrospinal fluid; ECE, endothelin-converting enzyme; IDE,
insulin degrading enzyme; NEP, neprilysin; NFT, neurofibrillary
tangles; PSEN, presenilin; APOE, apolipoprotein E; GSK, glycogen
synthase kinase; MARK, microtubule affinity regulating kinase; ERK,
extracellular regulated kinase; MAPK, mitogen-activated protein
kinase; CaMKII, calcium/calmodulin-depnedent kinase; JNK, c-Jun
N-terminal kinases; PKB, protein kinase B; Cdk, cyclin-dependent
kinase; PP, protein phosphatase. |
APP processing and Aβ metabolism
Aβ is the major component of the amyloid plaques
observed in the brains of patients with AD. The imbalance between
the generation and clearance of Aβ leads to its accumulation, one
of the fundamental molecular features of AD contributing to its
neuropathology.
Aβ is produced by the serial proteolysis of APP by
enzymes, including α-secretase, aspartyl protease β-site
APP-cleaving enzyme (BACE), β-secretase and α-secretase, as
reviewed previously (17) and
shown in Fig. 1. α-secretase
cleaves APP at a site close to the transmembrane domain and in the
middle of the Aβ region, to release a large soluble ectodomain
(α-APPs) into the lumen/extracellular space, while retaining a
C-terminal fragment of 83 amino acids (APP-C83) in the membrane.
APP-C83 is further cleaved by α-secretase into p3, an
amino-terminal truncated form of Aβ. By contrast, β-secretase
cleaves APP to generate a 99-residue membrane-associated C-terminal
fragment (APP-C99). This is subsequently cleaved by α-secretase to
release a 4-kDa Aβ protein and the β-amyloid precursor protein
intracellular domain. This cleavage by α-secretase is an unusual
form of proteolysis as the protein is cleaved within the
transmembrane domain (at residue +40 or +42) (18–20).
APP also undergoes caspase-mediated cleavage to generate a 90-kDa
N-terminal APP-caspase fragment. The cleavage of APP into the toxic
Aβ peptides results from the activity of β-secretase. Therefore,
increased proteolysis by α-secretase may alter the balance and lead
to decreased production of Aβ.
Aβ is cleared from the extracellular space and moved
into the blood and CSF, and it can also be degraded into less
neurotoxic metabolites (21). The
enzymes involved in Aβ turnover include, insulin degrading enzyme
(IDE), neprilysin (NEP) (21–25),
endothelin-converting enzyme (ECE)-1, ECE-2 and possibly plasmin,
as previously described (21,23).
NEP, known to be important in Aβ catabolism, is a 90–110 kDa plasma
membrane glycoprotein of the neutral zinc metalloendopeptidase
family (26,27) and has a higher tendency to cleave
Aβ40 than Aβ42 (28). The activity
of NEP is regulated by factors that affect the onset of AD,
including aging, estrogen levels, exercise and environmental
enrichment. IDE, a zinc metalloendopeptidase, is another protease
important in regulating the levels of Aβ in the brain. Certain risk
factors associated with AD, including diabetes mellitus,
hyperinsulinemia and apolipoprotein E (APOE)-ε4 allele, may
facilitate disease onset, at least partly, by affecting the
activity of IDE (29–33). ECE-1 and ECE-2 contribute to the
regulation of steady-state Aβ levels in the brain (23).
Aβ oligomers are formed through the aggregation of
the less toxic Aβ monomers. They lead to synaptic dysfunction and
neuronal damage (34,35), and have been extensively reviewed
(35). Previous studies have
demonstrated a robust correlation between the levels of soluble Aβ
oligomers and the extent of synaptic loss, and the severity of
cognitive impairment (36–41). This suggests that inhibition of Aβ
oligomerization may be a promising approach for the prevention and
treatment of AD (34,35).
Tau pathogenesis
In the normal brain, localization of tau is
restricted to the axonal compartment of neurons (42). Tau proteins are members of a family
of microtubule-associated proteins, which are important in the
assembly of microtubules, contribute to axonal integrity in mature
neurons (43,44), and perform functions at the
dendritic and nuclear level in neurons (45,46).
Hyperphosphorylated and abnormally phosphorylated forms of tau are
the major constituents of the intraneuronal PHFs observed in AD.
They are also detected in similar filaments observed in other
neurodegenerative disorders, termed tauopathies (43,47,48).
Spatiotemporal progression of tau aggregates from the entorhinal
cortex and hippocampus to isocortical areas (49) has been correlated with cognitive
deficits (50), and accumulation
of hyperphosphorylated tau is associated with memory impairment in
several animal models (8,51–53).
These findings support the pivotal role of tau pathology in
AD-associated memory loss.
Homeostasis of tau phosphorylation is maintained
through a balance in the activity of enzymes, which catalyze the
phosphorylation and the dephosphorylation of tau, as shown in
Fig. 1. Tau phosphorylation is
mediated by kinases, including glycogen synthase kinase-3β
(GSK-3β), MARK kinase, mitogen-activated protein
kinase/extracellular signal-regulated kinase (MAPK/ERK),
calcium/calmodulin-dependent protein kinase II, c-Jun N-terminal
kinase, protein kinase B (AKT/PKB) and cyclin-dependent kinase 5
(Cdk5), which includes the catalytic Cdk5 and the p35, p25 and p39
regulatory proteins (54,55). By contrast, dephosphorylation is
mediated by protein phosphatase (PP)-2A, PP-2B and PP-1. PP-2A is
the most important phosphatase accounting for >70% of tau
dephosphorylation in the brain and its activity is downregulated in
AD (15). Disruption in the
homeostasis of tau phosphorylation results from a dysregulation of
tau-associated kinases and phosphatases, eventually leading to the
formation of neurofibrillary tangles and causing the neuronal cell
death exhibited in AD (15). At
the functional level, hyperphosphorylation of tau impairs its
microtubule-binding properties, resulting in its detachment. This
causes destabilization of microtubules, disrupts axonal transport
and eventually leads to the relocalization of tau to the
somatodendritic compartment where NFTs have been identified in AD
and other tauopathies (56–58).
The appearance of tau aggregates is correlated with a loss of
microtubules and the breakdown of normal axonal transport (59). Tau pathology also correlates with
the onset and progression of dementia in AD, and memory loss and
mild cognitive impairment during aging (60).
Aβ, tau and AD
Small and Duff (61) suggested two hypotheses, which may
explain Aβ and tau causality, termed the ‘dual pathway model’ and
‘serial model’ (62,63). According to the dual pathway model,
an insult may induce an increase in the production of Aβ and the
phosphorylation of tau simultaneously, which then independently
leads to synaptic loss and dementia (61). A series of genetic and experimental
findings form the basis of the ‘amyloid hypothesis’, which suggests
a serial model of causality. According to this hypothesis,
increased production and deposition of Aβ is important in
triggering neuronal dysfunction and death in AD (62). Therefore, an increase in Aβ is the
prime pathogenic driver, which in turn leads to the
hyperphosphorylation of tau and other histological and clinical
symptoms of AD, including synaptic loss and dementia. A previous
study demonstrated that soluble Aβ dimers isolated from the cortex
of patients with AD, directly induces the phosphorylation of tau
and neuritic degeneration (64).
This study demonstrated that subnanomolar concentrations of
cortical Aβ dimers from patients with AD, the most abundant form of
soluble oligomers detectable in the human brain, first induced
hyperphosphorylation of tau at AD-relevant epitopes in hippocampal
neurons, subsequently disrupting the microtubule cytoskeleton and
causing degeneration of neuritis (64). Purified, synthetic dimers exerted
identical effects to the natural AD dimers (64). It was also revealed that knocking
down the expression of endogenous tau fully prevented neuritic
changes and by contrast, overexpression of human tau accelerated
these changes (64).
Co-administration of antibodies recognizing the Aβ N-terminus
prevented cytoskeletal disruption (64). Based on these findings, it was
suggested that natural Aβ dimers isolated from the brain of
patients with AD are sufficient to induce AD-type
hyperphosphorylation of tau, followed by neuritic dystrophy.
However, passive immunotherapy mitigates this effect (64). Other previous studies have
demonstrated that Aβ oligomers are also capable of inducing the
phosphorylation of tau (65).
Previous studies in mice have provided evidence to support a model
of the pathogenesis of AD, in which soluble Aβ oligomers trigger
synaptic dysfunction; however, the formation of abnormal tau
aggregates is required to induce neuronal death severe enough to
result in cognitive decline and dementia. This has been reviewed
previously (34). At the
physiological level, it is known that while accumulation of
intraneuronal Aβ causes deficits in long-term synaptic plasticity,
synaptic dysfunction and long-term potentiation deficits manifest
in an age-associated manner prior to the appearance of plaques and
tangles (66).
3. AD-associated genes
The genes encoding APP, presenilin (PSEN)-1 and
PSEN-2, have been demonstrated to harbor autosomal dominant
mutations associated with AD, while the presence of the APOE-4
allele is considered a risk factor for the development of AD
(Table I) (67). APP and PSEN mutations lead to
increased production of Aβ42 peptides, while inheritance of ApoE4
alleles cause an increase in the steady-state levels of Aβ in the
brain, as reviewed previously (14). PSEN-1 mutations, therefore, render
neurons vulnerable to isoflurane toxicity (68). The 3 × Tg-AD mouse model exhibits
mutations in three AD-associated genes, human β-amyloid precursor
protein (APPSwe), PSEN-1 (PS-1M146V) and tau (tauP301L), and
develops Aβ plaques, NFTs and exhibits cognitive impairment
(66,69). Liang et al (68) revealed that neurons exhibiting one
presenilin-1 mutation were susceptible to isoflurane-induced
cytotoxicity and increased cytosolic calcium levels. Certain
studies have suggested that the transgenic mouse models of AD may
be more susceptible to developing neurotoxicity compared with the
wild-type mice following the administration of isoflurane (70) and sevoflurane (71). These findings suggest that patients
exhibiting AD-associated gene mutations may be at an increased risk
of developing anesthesia-induced neurotoxicity. Further evidence
supporting the genetic component of AD etiology comes from
investigations, which correlated genomic variations in close
proximity to the IDE gene with disease severity, plaque and NFT
density (72), and the plasma
levels of Aβ42 in patients with AD (73).
 | Table ICurrently known common Alzheimer’s
disease-associated genes. |
Table I
Currently known common Alzheimer’s
disease-associated genes.
| Chromosome | Protein | Function | Gene defect | Phenotype |
|---|
| 21q21.2 | APP | Aβ generation | APP mutation | Inc. production of
all Aβ peptides or Aβ40 peptide |
| 14q24.3 | PSEN1 | Aβ generation | PSEN1 mutation | Inc. production of
Aβ42 peptide |
| 1q31-q42 | PSEN2 | Aβ generation | PSEN2 mutation | Inc. production of
Aβ42 peptide |
| 10q.23.33 | IDE | Aβ degradation | IDE mutation | Dec. degradation of
Aβ peptides |
| 19q13.2 | APOE | Aβ
clearance/export, Aβ oligomerization | APOE-4
polymorphism | Dec. density of Aβ
plaques and vascular deposits |
4. Effects of inhaled anesthetics on Aβ
Isoflurane
An in vitro study demonstrated that
isoflurane promotes the oligomerization of Aβ and increases its
toxicity (5). A combination of
inhaled anesthetics and hypoxia may activate caspases and induce
apoptosis, increasing the overall level of amyloid proteins
(3,74). Xie et al (3) reported that exposure to 2% isoflurane
for 6 h induces apoptosis, alters the processing of APP and leads
to an increased production of Aβ peptides in H4 human neuroglioma
cells stably transfected to express human wild-type full-length APP
(H4-APP cells). Isoflurane also increases the rate of Aβ
oligomerization and pheochromocytoma cytotoxicity in vitro
(5,75) by exhibiting a preference for
binding small oligomeric species (5). Repetitive exposure to 2% isoflurane
(twice weekly for 3 months) increased the quantity of Aβ aggregates
in APP mice compared with the wild-type (70). A clinically relevant form of
isoflurane anesthetic (1.4% isoflurane for 2 h) was revealed to
induce the activation of caspases with modest increases in the
levels of BACE and Aβ in the mouse brain between 6 and 24 h
following administration (76) In
humans, isoflurane induces an increase in the levels of Aβ40 in the
CSF 24 h following surgery under the influence of the anesthetic
(77). Previous studies have
achieved success in mitigating these effects. For example, the
caspase inhibitor, Z-VAD, has been demonstrated to attenuate
isoflurane-induced caspase activation, APP processing, Aβ
accumulation and apoptosis in H4-APP cells (74). Inhibitors of Aβ aggregation, iAβ5
and clioquinol, selectively attenuate the isoflurane-induced
activation of caspase-3 (74,76).
However, in naïve H4 cells (not overexpressing APP), isoflurane
induces the activation of caspase-3 in the absence of any
detectable alterations in the generation of Aβ, although the latter
may potentiate the activation of caspases (74). These findings suggest that the
caspases activated by isoflurane may in turn increase the activity
of BACE, alter APP processing and increase the levels of Aβ to
trigger further apoptosis (74,76).
The result is a vicious cycle of anesthetic-induced apoptosis,
generation and aggregation of Aβ leading to additional rounds of
apoptosis, and eventually, debilitating levels of
neurodegeneration. This conclusion is also supported by previous
findings where a reduction in the levels of BACE and Aβ were
demonstrated to attenuate the isoflurane-induced activation of
caspase (78). Finally, treatment
of H4-APP cells with a combination (however, not independent
exposure) of 70% nitrous oxide and 1% isoflurane for 6 h induced
the activation of caspase-3 and apoptosis, and increased the levels
of BACE and Aβ peptides (79).
Notably, certain previous studies have failed to
determine an association between exposure to anesthetic during 1–5
years preceding disease onset and the risk of developing AD
(11). In behavioral assays, the
performance of 85Dbo/J transgenic AD mice (APPswe, PSEN1dE9) and
wild-type mice in the Morris Water Maze (MWM) test, improved
significantly 48 h following 5-day exposure to isoflurane (80). The transgenic AD mice made
significantly fewer discrimination errors in the Y maze following
isoflurane administration and no differences were observed compared
with the wild-type, until 5 months following exposure (80). During this period, the quantity of
Aβ plaques and oligomers in the hippocampus were reduced
significantly in the transgenic AD mice (80). These findings suggest that repeated
isoflurane exposure during the pre-symptomatic phase improved
spatial memory in the APP/PS1 transgenic and wild-type mice shortly
following exposure, prevented an age-associated decline in learning
and memory, and attenuated the formation of Aβ plaques and
oligomers in the hippocampus (80).
Sevoflurane
Sevoflurane induces identical cellular and
histological effects to isoflurane. Exposure to 4.1% sevoflurane
for 6 h induces apoptosis, alters APP processing and increases the
production of Aβ in H4-APP human neuroglioma cells. This effect is
attenuated by treatment with Z-VAD and the γ-secretase inhibitor,
L-685,458, and is potentiated by Aβ (81). Previous in vivo studies
exposed naïve mice to 2.5% sevoflurane for 2 h and observed
increased levels of activated caspases, BACE and Aβ aggregates in
the brain at 6, 12, and 24 h following anesthesia (76). Also, a combination of 2.1–3%
sevoflurane and 60% oxygen for either 2 or 6 h successfully induced
caspase activation and apoptosis, altered APP processing, and
increased the levels of Aβ in the brains of 6-day-old mice
(71). Therefore, sevoflurane
appears to act through a vicious cycle similar to isoflurane, by
triggering a cascade of caspase activation, increasing BACE
activity, aberrant APP processing and increasing the generation and
aggregation of Aβ, leading to further apoptosis (81).
By contrast, certain studies have reported that
sevoflurane may either have no deleterious effect or, in certain
cases, a neuroprotective function. For instance, a 4 h exposure to
one minimum alveolar concentration of sevoflurane revealed no
impairment in learning or memory, in young, adult and aged rats,
according to the MWM test (82).
Exposure 2.1% sevoflurane 4- and 16-times and exposure to 3%
sevoflurane 16-times selectively rescued the Δelectroretinograms in
(ΔERG) AD-transgenic flies, in which the ΔERG, climbing ability and
survival rate were lowered; however, no affect was observed in
control flies (83). These
findings led to the conclusion that sevoflurane exerts no
neurotoxic effects on AD-transgenic flies, however, may confer
selective neuroprotection on their retinal function (83). Contradictory results, including
these, warrant further studies to elucidate the effects of
sevoflurane on AD-associated neurotoxicity and subsequent
neuropathology (83).
Desflurane
Desflurane is another commonly used inhaled
anesthetic, which in contrast to isoflurane and sevoflurane, when
supplied to H4-APP cells at a clinically relevant concentration
(12%) for 6 h, revealed no induction in the activation of
caspase-3, aberrant APP processing, or Aβ synthesis (84,85).
Isoflurane, however not desflurane, triggers an increase in the
levels of Aβ40 in human CSF 24 h following surgery. In addition,
desflurane, however not isoflurane, was associated with a decrease
in the levels of Aβ42 at 2 h following surgery under anesthesia
(77). Notably, although
desflurane alone cannot increase neuronal cell death, it can
increase the vulnerability of primary cultured neurons to
intracellular and extracellular Aβ1–42 (86). A 6-h exposure to 12% desflurane
under marginally hypoxic conditions (18% O2) was
demonstrated to induce the activation of caspase -3, alter the APP
processing, increase the production of Aβ and increase the activity
of BACE in H4-APP cells. This effect was partially rescued by the
broad caspase inhibitor, benzyloxycarbonyl-VAD, and attenuated by
Clioquinol and L-685,458 (84).
The mechanism of action of inhaled anesthetics
leading to AD-like neuropathology remains to be elucidated. The
data from in vitro human models of AD discussed above, if
confirmed in vivo, may have profound implications in the
field of anesthesiology in elderly patients, particularly those
already diagnosed with AD (84).
Upstream mechanisms
The molecular mechanism underlying the progression
from anesthesia-exposure to neuronal cell death has been
extensively investigated for isoflurane (3,5,9,74–76,79,85,87–91),
sevoflurane (81,82,92)
and desflurane plus hypoxia (84).
These previous studies have demonstrated that inhaled anesthetics
induce caspase activation and cellular apoptosis, increase BACE
levels, affect APP processing, increase the synthesis and
accumulation of Aβ and eventually impair learning and memory.
At the cellular level, signals transmitted by
extracellular anesthetics may manifest into different types of
responses. Firstly, the expression levels of cytokines, including
tumor necrosis factor-α (TNFα), may increase in response to inhaled
anesthetics, leading to the inflammation of neurons. It has been
previously reported that 2.1–3% sevoflurane in combination with 60%
oxygen induces neuroinflammation in the brain of transgenic AD mice
by increasing the expression levels of TNFα (71).
Secondly, isoflurane elevates cytosolic calcium
levels and since calcium is an important secondary messenger,
alterations in its homeostasis is sensed as a danger signal by the
cell, leading to the activation of apoptotic pathways, followed by
neuronal cell death. Previous studies have provided evidence in
favor of this mechanism (92,93).
A previous study demonstrated that the inositol trisphosphate
receptor (IP3R) antagonist, 2-APB, attenuated the
sevoflurane-induced activation of caspase-3 and the accumulation of
Aβ in naïve neonatal mice, suggesting that sevoflurane may act
through IP3R to affect calcium homeostasis (71). Other previous studies have revealed
that isoflurane induces the activation of caspase-3 and the
accumulation of Aβ by increasing cytosolic calcium levels, which
are regulated by memantine, a partial antagonist of the
N-methyl-D-aspartate receptor (NMDAR) (94). A previous study demonstrated that 4
μM memantine inhibits the increase in cytosolic calcium
levels, attenuates the activation of caspase-3 and apoptosis, and
improves cell viability in response to isoflurane exposure
(94). In naïve mice, 20 mg/kg
memantine administered intraperitoneally, reduced the
isoflurane-induced activation of caspase-3 in the brain (94).
The third possible mechanism of action is through
the synthesis of reactive oxygen species (ROS) and the induction of
mitochondrial damage. A previous study demonstrated that isoflurane
increases the generation of ROS, subsequently causing mitochondrial
damage by opening the mitochondrial permeability transition pore,
reducing the mitochondrial membrane potential and decreasing
adenosine triphosphate levels. This sequence of events leads to the
activation of the apoptotic pathways, eventually causing learning
and memory impairment (95). The
results also demonstrated that cyclosporine A, an inhibitor of the
mitochondrial permeability transition pore, inhibits
isoflurane-induced opening of the pore in vitro and rescues
isoflurane-induced deficiencies in learning and memory in mice
(95). By contrast, desflurane
revealed no mitochondrial damage or caspase activation in the mouse
brain and primary neuronal cultures. No impaired learning and
memory was observed in these animals (95). Evidence for the affect of
sevoflurane and isoflurane on mitochondrial membrane permeability
and caspase-3 activation comes from previous studies revealing
their apoptotic effect on T lymphocytes. This mechanism was
identified to be independent of death receptor signaling. By
contrast, desflurane exerted no pro-apoptotic effects (85).
Finally, genetic factors appear to affect the
outcome of exposure to inhaled anesthetics. A previous study
demonstrated that the cytotoxicity of desflurane was caused by the
reduction in miR-214, which normally binds to the 3′ untranslated
region of the pro-apoptotic gene, Bax, and represses the expression
of this protein. Downregulation of miR-214, therefore, leads to
increased expression of Bax and consequently increases neuronal
cytotoxicity by the accumulation of Aβ (86). Other previous studies demonstrated
that a 2 h exposure to sevoflurane causes the accumulation of Aβ in
the brain and exacerbates Alzheimer’s-like pathology by reducing
the levels of low-density lipoprotein receptor-related protein 1,
increasing the expression of receptor for advanced glycation end
products and decreasing the expression levels of IDE and neprilysin
in aged and, to a lesser extent, young rat brain (82). Successful downregulation of BACE,
full-length APP and APP c-terminal fragments by treatment with
small interfering RNAs targeting these transcripts, attenuates the
synthesis and accumulation of Aβ, and isoflurane-induced activation
of caspase-3 (78).
Isoflurane also increases the ratio of Bax/Bcl-2. A
6 h exposure to 2% isoflurane increases the mRNA expression of Bax
and decreases the expression of the anti-apoptotic factor Bcl-2.
This increases the accumulation of ROS, facilitates cytochrome
c release from the mitochondria to the cytosol, induces the
activation of caspase-9 and caspase-3, and leads to apoptotic cell
death (96). This effect can be
attenuated by administration of the intracellular calcium chelator,
BAPTA (96). Isoflurane,
therefore, appears to induce apoptosis by altering the Bax/Bcl-2
ratio and triggering mitochondrial damage through the generation of
ROS (96). This previous study
also confirmed that desflurane does not activate the ROS-mediated
mitochondrial pathway of apoptosis (96).
These findings have partially demonstrated the
upstream mechanisms leading from exposure to isoflurane and
sevoflurane to Aβ generation, caspase activation and apoptosis.
They have also provided evidence to facilitate future studies aimed
at elucidating the mechanism underlying the effect of inhaled
anesthetics on the neuropathogenesis of AD.
5. Effects of inhaled anesthetics on tau
pathology
Isoflurane
Several previous studies have confirmed that
isoflurane can trigger the hyperphosphorylation of tau in
vivo (97,98). Repeated normothermic exposure to
isoflurane significantly increased hippocampal phosphorylation of
tau at the AT180 (pTau213/235) epitope in 3 × Tg-AD mice (97). Dong et al (98) demonstrated that a 2 h exposure to
1.4% isoflurane increased the phosphorylation of tau at serine 262
(Tau-PS262) in AD-transgenic mice, up to 24 h following
administration of anesthesia. An in vitro study revealed
that primary neuronal cultures derived from AD-transgenic mice
exhibited an increase in the expression of Tau-PS262 following a 6
h exposure to 2% isoflurane (98).
At clinically relevant doses, a previous study revealed that
increased levels of phosphorylated-tau was distributed in the
neuropil and cell bodies, increased the levels of insoluble and
aggregated forms of tau, and the detachment of tau from
microtubules (4). The increase in
insoluble tau at 1 week following anesthesia suggested that
anesthetics cause molecular changes in the brain, which trigger
later the development of tauopathy (4). However, as discussed earlier, a
previous study revealed contradictory results, suggesting that
isoflurane was not identified to exhibit a significant affect on
the levels of tau in human CSF (77).
Sevoflurane
Sevoflurane can exhibit identical physiological
effects to isoflurane. At least two independent previous studies
have established a correlation between the exposure to sevoflurane
and persistent postoperative cognitive decline, particularly in
patients >65 years of age (6,7). In
one study, acute exposure to 1.5% sevoflurane was demonstrated to
cause a significant, dose-dependent and reversible
hyperphosphorylation of tau in the hippocampus of 5–6 month-old
C57Bl6/J mice (6). These findings
revealed that repeated exposure to 2.5% sevoflurane under
normothermic conditions leads to persistent hyperphosphorylation of
tau at the Ser396/Ser404 and Thr181 phosphoepitopes. The mice
subsequently developed significant deficiencies in spatial learning
and memory, as assayed using the MWM test (6). Since hyperphosphorylated tau is a
major constituent of neurofibrillary lesions, these previous
studies suggest a possible mechanism by which anesthetics may cause
postoperative cognitive impairment and increase the risk of AD
(7). Nevertheless, a causal
association between the burden of phosphorylated tau and cognitive
decline remains to be elucidated.
Desflurane
Desflurane has not been indicated to significantly
affect the levels of tau in human CSF between 2 and 24 h following
surgery under spinal anesthesia (77). However, whether desflurane can
directly or indirectly induce the hyperphosphorylation of tau in
vivo remains to be elucidated.
Role of anesthesia-induced
hypothermia
Previous studies have demonstrated that
anesthesia-induced hypothermia directly induces aberrant
hyperphosphorylation of tau (99,100). At a temperature <37°C, an 80%
increase in the phosphorylation of tau is observed with each degree
of decline in temperature, an effect that was rescued in
vivo by preventing hypothermia (100). Planel et al (99) demonstrated that restoration of core
body temperature to normal reversed the increase in the
hyperphosphorylation of tau caused not by anesthesia per se,
but by anesthesia-induced hypothermia. Mechanistically, this type
of phosphorylation was not due to the activation of tau kinase, but
rather a secondary consequence of the direct inhibition of PP-2A
activity by the hypothermic conditions (99,100). Other previous studies have
confirmed these findings in different in vivo models. For
example, Tan et al (8)
demonstrated that exposure of Sprague-Dawley rats to 1.5%
isoflurane without temperature control inhibited the activity of
PP-2A and increased the hyperphosphorylation of tau at the Thr-205
and Ser-396 epitopes in the hippocampus. This was, in turn,
associated with the deficits in spatial learning and memory
observed in hypothermic rats (8).
In humans, body temperature is suggested to be a risk factor for AD
and hypothermia (common in the elderly) and is hypothesized to
increase the pathology of AD (101). However, the impairment in
learning and memory in response to isoflurane with temperature
control is not accompanied by changes in the expression levels of
the total tau protein or phosphorylated tau at Thr231 and Ser396
(102). Further studies are
required to ascertain the direct, causal association between
anesthesia-induced hypothermia and memory impairment.
Upstream mechanisms
Le Freche et al (6) demonstrated that the activation of
AKT, ERK and GSK-3 signaling in response to anesthesia contributes
to its pathological consequences. Specifically, this study revealed
that normothermic sevoflurane exposure can lead to the
dysregulation of the MAPK and AKT/GSK3 pathways (6). As discussed, aberrant
hyperphosphorylation of tau due to anesthesia-induced hypothermia
and hypothermia-induced hyperphosphorylation of tau results from
the direct inhibition of PP-2A activity by the hypothermic
conditions rather than the activation of tau kinase (99,100). A study using the NMDAR
antagonist, memantine, in a mouse model of tauopathy suggested that
NMDAR-mediated signaling may be important in the development of the
pathology of tau following isoflurane-induced hypothermia (103). However, other receptors may also
contribute to this phenomenon. The mechanism by which memantine
antagonizes the deleterious effects of isoflurane leading to tau
pathology remains to be elucidated and warrants further
investigation.
6. Inhaled anesthesia, Aβ, tau
hyperphosphorylation and AD
Dong et al (98) first reported that clinically
relevant exposure to isoflurane increased the expression levels of
phosphorylated tau, most likely due to anesthetic-induced caspase
activation and generation of Aβ aggregates. These findings
demonstrated that the increase in Tau-PS262 levels in brain tissues
and primary neurons derived from AD-transgenic mice [B6.Cg-Tg
(APPswe, PSEN1dE9)85Dbo/J] following isoflurane exposure is
attenuated by Z-VAD and L-685,458 (98). An animal study demonstrated that a
combination of 70% nitrous oxide and 1.2% isoflurane is associated
with a long-term deficit in learning and memory in young and aged
rats (87). Notably, in a previous
study on human subjects, desflurane, in contrast to isoflurane, was
not observed to induce a decline in cognitive function (104). There are also studies that have
suggested that isoflurane may impair learning and memory
independent of the accumulation of Aβ, suggesting an alternative
pathway upstream of neurodegeneration (105). Other previous studies have
revealed that isoflurane, however not desflurane, induces the
activation of caspase and the synthesis of Aβ (76,84,96).
Notably, AD-like pathology has been demonstrated to recover over
time in vivo. In 85Dbo/J AD-transgenic mice (APPswe,
PSEN1dE9), the Aβ plaques and oligomers observed in the hippocampus
decreased significantly during the 5 months following isoflurane
exposure (80). Further studies
are required to elucidate the association between inhaled
anesthetics, Aβ accumulation, tau hyperphosphorylation and the
onset of AD.
7. Future studies
To date, the effects of isoflurane, sevoflurane and
desflurane on Aβ aggregation, tau hyperphosphorylation and
cognitive function have been determined. However, several of these
independent conclusions are inconsistent and often contradictory.
Resolving these discrepancies is required by specifically
addressing the following issues: i) The cause-effect association
between inhaled anesthetics and Aβ accumulation, tau
hyperphosphorylation and neurobehavioral deficits. ii) The effect
of anesthetics on the expression levels and activity of the enzymes
involved in Aβ degradation (IDE and neprilysin), which may aid in
clarifying whether Aβ accumulation is the result of increased
generation and/or decreased degradation of Aβ peptides. iii) The
upstream mechanisms by which inhaled anesthetics induce caspase
activation and cause mitochondrial damage. iv) Comparative analysis
of the effects of commonly used inhaled anesthetics, including
isoflurane, sevoflurane and desflurane, on the neuropathogenesis
and cognitive function of AD in order to identify those with the
least deleterious effects. v) Contrasting mechanism of action of
different anesthetics, for example, isoflurane versus desflurane,
on caspase activation and Aβ accumulation, which may lead to
targeted approaches to prevent or treat anesthesia-induced
neurotoxicity. vi) It is clear that cognitive decline following
major surgery is associated with gliosis, Aβ accumulation and tau
phosphorylation in aged mice (106). Since anesthesia also increases
the accumulation of Aβ, it is essential to determine possible
synergistic effects of anesthetics, including isoflurane, together
with surgical procedures, which may lead to more severe cognitive
dysfunction. vii) Although there is preclinical evidence supporting
the ability of inhaled anesthetics to exacerbate the pathology of
tau in a dose-dependent manner, further studies are required to
investigate the impact of prolonged or repeated anesthesia exposure
in humans, particularly those diagnosed with or at risk of
developing AD. viii) Mechanisms by which tau hyperphosphorylation
and aggregation leads to synaptic dysfunction and neurotoxicity,
and the contribution of anesthetics to this phenomenon. ix) A
direct causal link between increased hyperphosphorylation of tau
and impaired cognition remains to be elucidated.
8. Conclusion
The present review has summarized previous findings
on the effects of the commonly used inhaled anesthetics,
isoflurane, sevoflurane and desflurane, on the accumulation of Aβ
peptides, tau hyperphosphorylation and other AD-like pathologies,
in vitro and in vivo. It is clear that isoflurane and
sevoflurane can induce pro-apoptotic signaling, including caspase
activation, and cause aberrant APP processing, increased synthesis
and accumulation of Aβ, and hyperphosphorylation of tau in cell
lines, primary neurons and in vivo in the brain. Findings on
the effect of desflurane on the generation of Aβ and
hyperphosphorylation of tau are contradictory. Several mechanisms
have been suggested to explain the role of inhaled anesthetics in
inducing caspase activity, Aβ generation, tau hyperphosphorylation
and cognitive impairment. These include disruption of calcium
homeostasis, mitochondrial damage and downregulation of miR-214.
Although previous studies have revealed the deleterious effects of
inhaled anesthetics on patients with AD, further studies are
required to fully elucidate anesthesia-induced neurotoxicity.
Further investigation may assist in the development of future
guidelines for the safe administration of anesthetics to patients
with AD to avoid worsening cognitive dysfunction.
References
|
1
|
Alzheimer’s Association: Alzheimer’s
disease facts and figures. Alzhemier’s Dement. 7:208–244. 2011.
View Article : Google Scholar
|
|
2
|
Brookmeyer R, Johnson E, Ziegler-Graham K
and Arrighi HM: Forecasting the global burden of Alzheimer’s
disease. Alzheimers Dement. 3:186–191. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xie Z, Dong Y, Maeda U, Alfille P, Culley
DJ, Crosby G and Tanzi RE: The common inhalation anesthetic
isoflurane induces apoptosis and increases amyloid beta protein
levels. Anesthesiology. 104:988–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Planel E, Bretteville A, Liu L, Virag L,
Du AL, Yu WH, Dickson DW, Whittington RA and Duff KE: Acceleration
and persistence of neurofibrillary pathology in a mouse model of
tauopathy following anesthesia. FASEB J. 23:2595–2604. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eckenhoff RG, Johansson JS, Wei H, Carnini
A, Kang B, Wei W, Pidikiti R, Keller JM and Eckenhoff MF: Inhaled
anesthetic enhancement of amyloid-beta oligomerization and
cytotoxicity. Anesthesiology. 101:703–709. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Le Freche H, Brouillette J,
Fernandez-Gomez FJ, et al: Tau phosphorylation and sevoflurane
anesthesia: An association to postoperative cognitive impairment.
Anesthesiology. 116:779–787. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Run X, Liang Z, Zhang L, Iqbal K,
Grundke-Iqbal I and Gong CX: Anesthesia induces phosphorylation of
tau. J Alzheimers Dis. 16:619–626. 2009.PubMed/NCBI
|
|
8
|
Tan W, Cao X, Wang J, Lv H, Wu B and Ma H:
Tau hyperphosphorylation is associated with memory impairment after
exposure to 1.5% isoflurane without temperature maintenance in
rats. Eur J Anaesthesiol. 27:835–841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xie Z and Tanzi RE: Alzheimer’s disease
and post-operative cognitive dysfunction. Exp Gerontol. 41:346–359.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang JX, Baranov D, Hammond M, Shaw LM,
Eckenhoff MF and Eckenhoff RG: Human Alzheimer and inflammation
biomarkers after anesthesia and surgery. Anesthesiology.
115:727–732. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gasparini M, Vanacore N, Schiaffini C,
Brusa L, Panella M, Talarico G, Bruno G, Meco G and Lenzi GL: A
case-control study on Alzheimer’s disease and exposure to
anesthesia. Neurol Sci. 23:11–14. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Knopman DS, Petersen RC, Cha RH, Edland SD
and Rocca WA: Coronary artery bypass grafting is not a risk factor
for dementia or Alzheimer disease. Neurology. 65:986–990. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung
YC, Zaidi MS and Wisniewski HM: Microtubule-associated protein tau.
A component of Alzheimer paired helical filaments. J Biol Chem.
261:6084–6089. 1986.PubMed/NCBI
|
|
14
|
Selkoe DJ: Alzheimer’s disease: Genes,
proteins, and therapy. Physiol Rev. 81:741–766. 2001.PubMed/NCBI
|
|
15
|
Iqbal K and Grundke-Iqbal I: Alzheimer
neurofibrillary degeneration: Significance, etiopathogenesis,
therapeutics and prevention. J Cell Mol Med. 12:38–55. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Trojanowski JQ and Lee VM: Paired helical
filament tau in Alzheimer’s disease. The kinase connection. Am J
Pathol. 144:449–453. 1994.PubMed/NCBI
|
|
17
|
Xie Z and Xu Z: General anesthetics and
β-amyloid protein. Prog Neuropsychopharmacol Biol Psychiatry.
47:140–1446. 2013. View Article : Google Scholar
|
|
18
|
Gu Y, Misonou H, Sato T, Dohmae N, Takio K
and Ihara Y: Distinct intramembrane cleavage of the beta-amyloid
precursor protein family resembling gamma-secretase-like cleavage
of Notch. J Biol Chem. 276:35235–35238. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sastre M, Steiner H, Fuchs K, Capell A,
Multhaup G, Condron MM, Teplow DB and Haass C: Presenilin-dependent
gamma-secretase processing of beta-amyloid precursor protein at a
site corresponding to the S3 cleavage of Notch. EMBO Rep.
2:835–841. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu C, Kim SH, Ikeuchi T, Xu H, Gasparini
L, Wang R and Sisodia SS: Characterization of a presenilin-mediated
amyloid precursor protein carboxyl-terminal fragment gamma.
Evidence for distinct mechanisms involved in gamma-secretase
processing of the APP and Notch1 transmembrane domains. J Biol
Chem. 276:43756–43760. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miners JS, Baig S, Palmer J, Palmer LE,
Kehoe PG and Love S: Abeta-degrading enzymes in Alzheimer’s
disease. Brain Pathol. 18:240–252. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bates KA, Verdile G, Li QX, Ames D, Hudson
P, Masters CL and Martins RN: Clearance mechanisms of Alzheimer’s
amyloid-beta peptide: Implications for therapeutic design and
diagnostic tests. Mol Psychiatry. 14:469–486. 2009. View Article : Google Scholar
|
|
23
|
Eckman EA and Eckman CB: Abeta-degrading
enzymes: Modulators of Alzheimer’s disease pathogenesis and targets
for therapeutic intervention. Biochem Soc Trans. 33:1101–1105.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Higuchi M, Iwata N and Saido TC:
Understanding molecular mechanisms of proteolysis in Alzheimer’s
disease: Progress toward therapeutic interventions. Biochim Biophys
Acta. 1751:60–67. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang DS, Dickson DW and Malter JS:
beta-Amyloid degradation and Alzheimer’s disease. J Biomed
Biotechnol. 2006:584062006. View Article : Google Scholar
|
|
26
|
Turner AJ and Tanzawa K: Mammalian
membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J.
11:355–364. 1997.PubMed/NCBI
|
|
27
|
Turner AJ, Isaac RE and Coates D: The
neprilysin (NEP) family of zinc metalloendopeptidases: Genomics and
function. Bioessays. 23:261–269. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kanemitsu H, Tomiyama T and Mori H: Human
neprilysin is capable of degrading amyloid beta peptide not only in
the monomeric form but also the pathological oligomeric form.
Neurosci Lett. 350:113–116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ho L, Qin W, Pompl PN, et al: Diet-induced
insulin resistance promotes amyloidosis in a transgenic mouse model
of Alzheimer’s disease. FASEB J. 18:902–904. 2004.PubMed/NCBI
|
|
30
|
Kuusisto J, Koivisto K, Mykkänen L,
Helkala EL, Vanhanen M, Hänninen T, Kervinen K, Kesäniemi YA,
Riekkinen PJ and Laakso M: Association between features of the
insulin resistance syndrome and Alzheimer’s disease independently
of apolipoprotein E4 phenotype: Cross sectional population based
study. BMJ. 315:1045–1049. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Edland SD, Wavrant-De Vriesé F, Compton D,
Smith GE, Ivnik R, Boeve BF, Tangalos EG and Petersen RC: Insulin
degrading enzyme (IDE) genetic variants and risk of Alzheimer’s
disease: Evidence of effect modification by apolipoprotein E
(APOE). Neurosci Lett. 345:21–24. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Raber J, Huang Y and Ashford JW: ApoE
genotype accounts for the vast majority of AD risk and AD
pathology. Neurobiol Aging. 25:641–650. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cook DG, Leverenz JB, McMillan PJ, Kulstad
JJ, Ericksen S, Roth RA, Schellenberg GD, Jin LW, Kovacina KS and
Craft S: Reduced hippocampal insulin-degrading enzyme in late-onset
Alzheimer’s disease is associated with the apolipoprotein
E-epsilon4 allele. Am J Pathol. 162:313–319. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ashe KH and Zahs KR: Probing the biology
of Alzheimer’s disease in mice. Neuron. 66:631–645. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sakono M and Zako T: Amyloid oligomers:
Formation and toxicity of Abeta oligomers. FEBS J. 277:1348–1358.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Caughey B and Lansbury PT: Protofibrils,
pores, fibrils, and neurodegeneration: Separating the responsible
protein aggregates from the innocent bystanders. Annu Rev Neurosci.
26:267–298. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haass C and Selkoe DJ: Soluble protein
oligomers in neurodegeneration: Lessons from the Alzheimer’s
amyloid beta-peptide. Nat Rev Mol Cell Biol. 8:101–112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
LaFerla FM, Green KN and Oddo S:
Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev
Neurosci. 8:499–509. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Klein WL, Krafft GA and Finch CE:
Targeting small Abeta oligomers: The solution to an Alzheimer’s
disease conundrum? Trends Neurosci. 24:219–224. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chiti F and Dobson CM: Protein misfolding,
functional amyloid, and human disease. Annu Rev Biochem.
75:333–366. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ferreira ST, Vieira MN and De Felice FG:
Soluble protein oligomers as emerging toxins in Alzheimer’s and
other amyloid diseases. IUBMB Life. 59:332–345. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Binder LI, Frankfurter A and Rebhun LI:
The distribution of tau in the mammalian central nervous system. J
Cell Biol. 101:1371–1378. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Buée L, Bussière T, Buée-Scherrer V,
Delacourte A and Hof PR: Tau protein isoforms, phosphorylation and
role in neurodegenerative disorders. Brain Res Brain Res Rev.
33:95–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sergeant N, Bretteville A, Hamdane M, et
al: Biochemistry of Tau in Alzheimer’s disease and related
neurological disorders. Expert Rev Proteomics. 5:207–224. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ittner LM, Ke YD, Delerue F, et al:
Dendritic function of tau mediates amyloid-beta toxicity in
Alzheimer’s disease mouse models. Cell. 142:387–397. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sultan A, Nesslany F, Violet M, et al:
Nuclear tau, a key player in neuronal DNA protection. J Biol Chem.
286:4566–4575. 2011. View Article : Google Scholar :
|
|
47
|
Buée L, Troquier L, Burnouf S, et al: From
tau phosphorylation to tau aggregation: What about neuronal death?
Biochem Soc Trans. 38:967–972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Iqbal K, Liu F, Gong CX and Grundke-Iqbal
I: Tau in Alzheimer disease and related tauopathies. Curr Alzheimer
Res. 7:656–664. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Braak H and Braak E: Staging of
Alzheimer’s disease-related neurofibrillary changes. Neurobiol
Aging. 16:271–284. 1995. View Article : Google Scholar
|
|
50
|
Grober E, Dickson D, Sliwinski MJ, Buschke
H, Katz M, Crystal H and Lipton RB: Memory and mental status
correlates of modified Braak staging. Neurobiol Aging. 20:573–579.
1999. View Article : Google Scholar
|
|
51
|
Van der Jeugd A, Ahmed T, Burnouf S, et
al: Hippocampal tauopathy in tau transgenic mice coincides with
impaired hippocampus-dependent learning and memory, and attenuated
late-phase long-term depression of synaptic transmission. Neurobiol
Learn Mem. 95:296–304. 2011. View Article : Google Scholar
|
|
52
|
Polydoro M, Acker CM, Duff K, Castillo PE
and Davies P: Age-dependent impairment of cognitive and synaptic
function in the htau mouse model of tau pathology. J Neurosci.
29:10741–10749. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kimura T, Yamashita S, Fukuda T, Park JM,
Murayama M, Mizoroki T, Yoshiike Y, Sahara N and Takashima A:
Hyperphosphorylated tau in parahippocampal cortex impairs place
learning in aged mice expressing wild-type human tau. EMBO J.
26:5143–5152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Maccioni RB, Otth C, Concha II and Muñoz
JP: The protein kinase Cdk5. Structural aspects, roles in
neurogenesis and involvement in Alzheimer’s pathology. Eur J
Biochem. 268:1518–1527. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tatebayashi Y, Planel E, Chui DH, et al:
c-jun N-terminal kinase hyperphosphorylates R406W tau at the PHF-1
site during mitosis. FASEB J. 20:762–764. 2006.PubMed/NCBI
|
|
56
|
Feinstein SC and Wilson L: Inability of
tau to properly regulate neuronal microtubule dynamics: A
loss-of-function mechanism by which tau might mediate neuronal cell
death. Biochim Biophys Acta. 1739:268–279. 2005. View Article : Google Scholar
|
|
57
|
Mandelkow EM, Stamer K, Vogel R, Thies E
and Mandelkow E: Clogging of axons by tau, inhibition of axonal
traffic and starvation of synapses. Neurobiol Aging. 24:1079–1085.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Trojanowski JQLV and Lee VM: Paired
helical filament tau in Alzheimer’s disease. The kinase connection
Am J Pathol. 144:449–453. 1994.
|
|
59
|
Terry RD: The pathogenesis of Alzheimer
disease: An alternative to the amyloid hypothesis. J Neuropathol
Exp Neurol. 55:1023–1025. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Guillozet AL, Weintraub S, Mash DC and
Mesulam MM: Neurofibrillary tangles, amyloid, and memory in aging
and mild cognitive impairment. Arch Neurol. 60:729–736. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Small SA and Duff K: Linking Abeta and tau
in late-onset Alzheimer’s disease: A dual pathway hypothesis.
Neuron. 60:534–542. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer’s disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tanzi RE and Bertram L: Twenty years of
the Alzheimer’s disease amyloid hypothesis: A genetic perspective.
Cell. 120:545–555. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jin M, Shepardson N, Yang T, Chen G, Walsh
D and Selkoe DJ: Soluble amyloid beta-protein dimers isolated from
Alzheimer cortex directly induce Tau hyperphosphorylation and
neuritic degeneration. Proc Natl Acad Sci USA. 108:5819–5824. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ma QL, Yang F, Rosario ER, et al:
β-amyloid oligomers induce phosphorylation of tau and inactivation
of insulin receptor substrate via c-Jun N-terminal kinase
signaling: Suppression by omega-3 fatty acids and curcumin. J
Neurosci. 29:9078–9089. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Oddo S, Caccamo A, Shepherd JD, Murphy MP,
Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y and LaFerla
FM: Triple-transgenic model of Alzheimer’s disease with plaques and
tangles: Intracellular Abeta and synaptic dysfunction. Neuron.
39:409–421. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bertram L and Tanzi RE: Thirty years of
Alzheimer’s disease genetics: The implications of systematic
meta-analyses. Nat Rev Neurosci. 9:768–778. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liang G, Wang Q, Li Y, Kang B, Eckenhoff
MF, Eckenhoff RG and Wei H: A presenilin-1 mutation renders neurons
vulnerable to isoflurane toxicity. Anesth Analg. 106:492–500. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Filali M, Lalonde R, Theriault P, Julien
C, Calon F and Planel E: Cognitive and non-cognitive behaviors in
the triple transgenic mouse model of Alzheimer’s disease expressing
mutated APP, PS1, and Mapt (3xTg-AD). Behav Brain Res. 234:334–342.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Perucho J, Rubio I, Casarejos MJ, Gomez A,
Rodriguez-Navarro JA, Solano RM, De Yébenes JG and Mena MA:
Anesthesia with isoflurane increases amyloid pathology in mice
models of Alzheimer’s disease. J Alzheimers Dis. 19:1245–1257.
2010.
|
|
71
|
Lu Y, Wu X, Dong Y, Xu Z, Zhang Y and Xie
Z: Anesthetic sevoflurane causes neurotoxicity differently in
neonatal naïve and Alzheimer disease transgenic mice.
Anesthesiology. 112:1404–1416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Prince JA, Feuk L, Gu HF, Johansson B,
Gatz M, Blennow K and Brookes AJ: Genetic variation in a haplotype
block spanning IDE influences Alzheimer disease. Hum Mutat.
22:363–371. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ertekin-Taner N, Allen M, Fadale D,
Scanlin L, Younkin L, Petersen RC, Graff-Radford N and Younkin SG:
Genetic variants in a haplotype block spanning IDE are
significantly associated with plasma Abeta42 levels and risk for
Alzheimer disease. Hum Mutat. 23:334–342. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xie Z, Dong Y, Maeda U, Moir RD, Xia W,
Culley DJ, Crosby G and Tanzi RE: The inhalation anesthetic
isoflurane induces a vicious cycle of apoptosis and amyloid
beta-protein accumulation. J Neurosci. 27:1247–1254. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Xie Z, Dong Y, Maeda U, Moir R, Inouye SK,
Culley DJ, Crosby G and Tanzi RE: Isoflurane-induced apoptosis: A
potential pathogenic link between delirium and dementia. J Gerontol
A Biol Sci Med Sci. 61:1300–1306. 2006. View Article : Google Scholar
|
|
76
|
Xie Z, Culley DJ, Dong Y, Zhang G, Zhang
B, Moir RD, Frosch MP, Crosby G and Tanzi RE: The common inhalation
anesthetic isoflurane induces caspase activation and increases
amyloid beta-protein level in vivo. Ann Neurol. 64:618–627. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang B, Tian M, Zheng H, Zhen Y, Yue Y,
Li T, Li S, Marcantonio ER and Xie Z: Effects of anesthetic
isoflurane and desflurane on human cerebrospinal fluid Aβ and τ
level. Anesthesiology. 119:52–60. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Dong Y, Xu Z, Zhang Y, McAuliffe S, Wang
H, Shen X, Yue Y and Xie Z: RNA interference-mediated silencing of
BACE and APP attenuates the isoflurane-induced caspase activation.
Med Gas Res. 1:52011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhen Y, Dong Y, Wu X, Xu Z, Lu Y, Zhang Y,
Norton D, Tian M, Li S and Xie Z: Nitrous oxide plus isoflurane
induces apoptosis and increases β-amyloid protein levels.
Anesthesiology. 111:741–752. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Su D, Zhao Y, Xu H, Wang B, Chen X, Chen J
and Wang X: Isoflurane exposure during mid-adulthood attenuates
age-related spatial memory impairment in APP/PS1 transgenic mice.
PLoS One. 7:e501722012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Dong Y, Zhang G, Zhang B, Moir RD, Xia W,
Marcantonio ER, Culley DJ, Crosby G, Tanzi RE and Xie Z: The common
inha-lational anesthetic sevoflurane induces apoptosis and
increases beta-amyloid protein levels. Arch Neurol. 66:620–631.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Callaway JK, Jones NC, Royse AG and Royse
CF: Sevoflurane anesthesia does not impair acquisition learning or
memory in the Morris water maze in young adult and aged rats.
Anesthesiology. 117:1091–1101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chen CW, Lin WY, Chen KB, Wu YS, Kuo YC,
Liu HP and Li CY: Inhalational anesthetic sevoflurane rescues
retina function in Alzheimer’s disease transgenic Drosophila. Curr
Alzheimer Res. 10:1005–1014. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhang B, Dong Y, Zhang G, et al: The
inhalation anesthetic desflurane induces caspase activation and
increases amyloid beta-protein levels under hypoxic conditions. J
Biol Chem. 283:11866–11875. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Loop T, Dovi-Akue D, Frick M, et al:
Volatile anesthetics induce caspase-dependent,
mitochondria-mediated apoptosis in human T lymphocytes in vitro.
Anesthesiology. 102:1147–1157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yu Y and Zhang Y: Desflurane accelerates
neuronal cytotoxicity of Aβ by downregulating miR-214. Neurosci
Lett. 554:28–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Culley DJ, Baxter MG, Yukhananov R and
Crosby G: Long-term impairment of acquisition of a spatial memory
task following isoflurane-nitrous oxide anesthesia in rats.
Anesthesiology. 100:309–314. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kvolik S, Glavas-Obrovac L, Bares V and
Karner I: Effects of inhaled anesthetics halothane, sevoflurane,
and isoflurane on human cell lines. Life Sci. 77:2369–2383. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wei H, Kang B, Wei W, Liang G, Meng QC, Li
Y and Eckenhoff G: Isoflurane and sevoflurane affect cell survival
and BCL-2/BAX ratio differently. Brain Res. 1037:139–147. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Matsuoka H, Kurosawa S, Horinouchi T, Kato
M and Hashimoto Y: Inhaled anesthetics induce apoptosis in normal
peripheral lymphocytes in vitro. Anesthesiology. 95:1467–1472.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Brambrink AM, Evers AS, Avidan MS, Farber
NB, Smith DJ, et al: Isoflurane-induced neuroapoptosis in the
neonatal rhesus macaque brain. Anesthesiology. 112:834–841. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wei H, Liang G, Yang H, Wang Q, Hawkins B,
Madesh M, Wang S and Eckenhoff RG: The common inhalational
anesthetic isoflurane induces apoptosis via activation of inositol
1,4,5-trisphosphate receptors. Anesthesiology. 108:251–260. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhang G, Dong Y, Zhang B, Ichinose F, Wu
X, Culley DJ, Crosby G, Tanzi RE and Xie Z: Isoflurane-induced
caspase-3 activation is dependent on cytosolic calcium and can be
attenuated by memantine. J Neurosci. 28:4551–4560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhang G, Dong Y, Zhang B, Ichinose F, Wu
X, Culley DJ, Crosby G, Tanzi RE and Xie Z: Isoflurane-induced
caspase-3 activation is dependent on cytosolic calcium and can be
attenuated by memantine. J Neurosci. 28:4551–4560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhang Y, Xu Z, Wang H, Dong Y, Shi HN,
Culley DJ, Crosby G, Marcantonio ER, Tanzi RE and Xie Z:
Anesthetics isoflurane and desflurane differently affect
mitochondrial function, learning, and memory. Ann Neurol.
71:687–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhang Y, Dong Y, Wu X, Lu Y, Xu Z, Knapp
A, Yue Y, Xu T and Xie Z: The mitochondrial pathway of anesthetic
isoflurane-induced apoptosis. J Biol Chem. 285:4025–4037. 2010.
View Article : Google Scholar :
|
|
97
|
Tang JX, Mardini F, Caltagarone BM,
Garrity ST, Li RQ, Bianchi SL, Gomes O, Laferla FM, Eckenhoff RG
and Eckenhoff MF: Anesthesia in presymptomatic Alzheimer’s disease:
A study using the triple-transgenic mouse model. Alzheimers Dement.
7:521–531.e1. 2011. View Article : Google Scholar
|
|
98
|
Dong Y, Wu X, Xu Z, Zhang Y and Xie Z:
Anesthetic isoflurane increases phosphorylated tau levels mediated
by caspase activation and Aβ generation. PLoS One. 7:e393862012.
View Article : Google Scholar
|
|
99
|
Planel E, Richter KE, Nolan CE, et al:
Anesthesia leads to tau hyperphosphorylation through inhibition of
phosphatase activity by hypothermia. J Neurosci. 27:3090–3097.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Planel E, Miyasaka T, Launey T, Chui DH,
Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y and
Takashima A: Alterations in glucose metabolism induce hypothermia
leading to tau hyperphosphorylation through differential inhibition
of kinase and phosphatase activities: Implications for Alzheimer’s
disease. J Neurosci. 24:2401–2411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Holtzman A and Simon EW: Body temperature
as a risk factor for Alzheimer’s disease. Med Hypotheses.
55:440–444. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Liu W, Xu J, Wang H, et al:
Isoflurane-induced spatial memory impairment by a mechanism
independent of amyloid-beta levels and tau protein phosphorylation
changes in aged rats. Neurol Res. 34:3–10. 2012. View Article : Google Scholar
|
|
103
|
Menuet C, Borghgraef P, Voituron N,
Gestreau C, Gielis L, Devijver H, Dutschmann M, Van Leuven F and
Hilaire G: Isoflurane anesthesia precipitates tauopathy and upper
airways dysfunction in pre-symptomatic Tau.P301L mice: Possible
implication for neurodegenerative diseases. Neurobiol Dis.
46:234–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhang B, Tian M, Zhen Y, Yue Y, Sherman J,
Zheng H, Li S, Tanzi RE, Marcantonio ER and Xie Z: The effects of
isoflurane and desflurane on cognitive function in humans. Anesth
Analg. 114:410–415. 2012. View Article : Google Scholar
|
|
105
|
Bianchi SL, Tran T, Liu C, Lin S, Li Y,
Keller JM, Eckenhoff RG and Eckenhoff MF: Brain and behavior
changes in 12-month-old Tg2576 and nontransgenic mice exposed to
anesthetics. Neurobiol Aging. 29:1002–1010. 2008. View Article : Google Scholar
|
|
106
|
Wan Y, Xu J, Meng F, et al: Cognitive
decline following major surgery is associated with gliosis,
β-amyloid accumulation, and τ phosphorylation in old mice. Crit
Care Med. 38:2190–2198. 2010. View Article : Google Scholar : PubMed/NCBI
|














