Introduction
Angiotensin II (Ang II) is a major effector peptide
of the renin-angiotensin system (RAS), which controls blood
pressure and the blood volume of the cardiovascular system
(1,2). Previous studies demonstrated that Ang
II is involved in key events in the regulation of various types of
inflammatory mediator (3,4). The effect of Ang II is mediated by
two G protein-coupled receptors, Ang II type 1 receptor (AT1R) and
Ang II type 2 receptor (AT2R), which competitively bind to Ang II.
AT1R stimulates protein phosphorylation, while AT2R stimulates
protein dephosphorylation, which counterbalances the effects of
protein kinases, particularly mitogen-activated protein kinases.
The expression of AT1R and AT2R vary in states of injury, ischemia
and inflammation and differ among tissues and organs (5,6). A
switch in binding to AT1R or AT2R affects the physiological and
pathological functions of Ang II (7).
Evidence suggests that RAS is critical in acute lung
injury (ALI) and acute respiratory distress syndrome (ARDS)
(5,8,9). In
the development of ARDS, the RAS is activated, leading to a
significant increase in levels of Ang II in the bronchoalveolar
lavage fluid and aggravating the lung inflammatory response
(9). In addition,
lipopolysaccharides (LPS) are considered to be important initiation
factors for ARDS. LPSs increase pulmonary endothelial permeability,
which results in pulmonary edema and ALI (10,11).
In addition, a previous study revealed that LPS upregulated the
expression of AT1R in rat pulmonary microvascular endothelial cells
(PMECs) (12); however,
verification in human cells remained to be elucidated. To further
examine this area, the regulation of LPS on Ang II receptors in
human PMECs (HPMECs) was investigated in the present study.
Materials and methods
Reagents
LPS from Escherichia coli 0111:B4 and
synthetic Ang II were purchased from Sigma-Aldrich (St. Louis, MO,
USA). 125I-labeled Ang II (125I-Ang II;
200–300 Ci/mmol) was purchased from Beijing FuRui Biotech Company
(Beijing, China). The mouse anti-human AT1R monoclonal antibodies
(cat. no. 214988) and goat anti-mouse immunoglobulin
(Ig)G/horseradish peroxidase (HRP) (cat. no. FI120611) were
purchased from Abcam (Cambridge, MA, USA). The AccessQuick RT-PCR
system (Promega-A1702)was purchased from Promega Corp. (Madison,
WI, USA). HPMECs (cat. no. 3000), endothelial cell medium (ECM) and
endothelial cell growth supplement (ECGS) were obtained from
ScienCell Research Laboratories (Carlsbad, CA, USA). Finally, human
renal mesangial cells (HRMCs) were purchased from the
Cardiovascular Institute of Southeast University (Nanjing,
China).
Immunofluorescence
HPMECs and HRMCs were identified as endothelial
cells by morphology and were characterized by immunofluorescence
with the following antibodies: Mouse anti-human vWF IgG/fluorescein
isothiocyanate (FITC; cat. no. sc-365712), mouse anti-human desmin
IgG/FITC (cat. no. sc-271677; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), mouse anti-human CD31 IgG/FITC (cat. no.
BYK-0468R) and mouse anti-human cytokeratin IgG/FITC (cat. no.
BYK-1712R; Boye Biotech Co., Ltd., Zhenjing, China). The cells of
passage 6 were used for identification. Monolayers of cells were
grown to confluence on gelatin-coated glass coverslips. The
coverlips were then washed twice with phosphate-buffered saline
(PBS; Shenneng Bocai Biotechnology Co., Ltd., Shanghai, China),
fixed in acetone at 4°C and allowed to air dry. Subsequently, the
cells were stained with the antibodies (anti-vWF-FITC,
anti-desmin-FITC, anti-cytokeratin-FITC and anti-CD31-FITC; 1:1,000
dilution), respectively, on ice for 20 min in the dark.
Subsequently, cells were washed three times with PBS. The detected
cells were viewed under a fluorescence microscope (L2001A; Boshida
Optical Instrument Co., Ltd, Shenzhen, China).
Cell culture and intervention
The culture of HPMECs was performed according to a
modified version of a previously described method (13). Briefly, the frozen HPMECs were
thawed, and inoculated at a density of 5,000 cells/cm2
into a T-25 flask (Sigma-Aldrich) containing 5 ml ECM with 1% ECGS,
penicillin (60 µg/ml), streptomycin (100 µg/ml) and
5% fetal bovine serum (all from ScienCell Research Laboratories) at
37°C. The HPMECs were subsequently placed in an incubator under a
5% CO2 humidified atmosphere. Passages 6–7 were used for
experimentation. LPS at varying concentrations (0, 50, 100 or 200
ng/ml) was added and the cells were incubated for 4, 8, 12 or 16
h.
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR) assay
Total RNA was extracted from the HPMECs using the RT
system according to the manufacturer's instructions. RT-PCR was
performed with an AccessQuick RT-PCR system (Promega Corp.)
according to the manufacturer's instructions. The primer sequences
were designed and synthesized by (Sangon Biotech, Shanghai, China)
with sequences as follows: Forward, 5′-CCAGCTATAATCCATCGAAA-3′ and
reverse, 5′-TGAATATTTGTGGGGAATC-3′ for AT1R; forward,
5′-CTGCTGTTGTTCTGGCCTTCA-3′ and reverse,
5′-CTCTCTCTTTTCCCTTGGAGTT-3′ for AT2R. GAPDH was
co-amplified as an internal control using the following primer
sequences: Forward, 5′-CTTCATTGACCTAACTACATGG-3′ and reverse,
5′-GAGGGGCCATCCACAGTCTTCTG-3′ for GAPDH. The designed primers were
confirmed using HRMCs as a positive control into which AT1R
and AT2R were transcribed (data not shown). Thermocycling
conditions were as follows: 94°C (2 min) followed by 94°C (30 sec),
57°C (1 min) and 68°C (2 min) for 32 cycles, and 7 min at 72°C. The
PCR products were stored at −20°C prior to quantification by
electrophoresis on agarose gels (Shenneng Bocai Biotechnology Co.,
Ltd.), which were scanned and analyzed using Gel Scan2XL system
(Shimadzu, Kyoto, Japan) for semi-quantitative evaluation. Quantity
and quality of the included RNA were controlled by an additional
PCR from the same reverse-transcription samples using GAPDH as an
internal standard. The PCR products were electrophoresed on agarose
gels.
Western blotting
The cells were harvested, lysed in chilled cell
lysis buffer (radioimmunoprecipitation assay buffer; Shenneng Bocai
Biotechnology Co., Ltd.) at a density of 106 cells/500
µl for 10 min and centrifuged at 15,000 × g at 4°C for 15
min. The precipitate was recovered and stored in aliquots at −80°C.
The protein concentration of samples was estimated using
ultraviolet spectrophotometry (BioSpectrometer; Eppendorf, Hamburg,
Germany). Immunoblotting was performed as previously described
(14). Briefly, the protein
samples were electrophoresed and the gels were transferred onto
polyvinylidene difluoride membranes (Shimadzu, Kyoto, Japan) prior
to blocking for 2 h with 5% bovine serum albumin (BSA) and 0.05%
Tween-20 in PBS (all from Shenneng Bocai Biotechnology Co., Ltd.).
Subsequently, the samples were incubated with mouse anti-human AT1R
or AT2R monoclonal antibodies (1:2,500; 8 µg/ml) in 5% BSA
overnight at 4°C. The blot was washed three times with PBS and then
incubated with HRP-linked goat anti-mouse IgG at 1:2,500 in PBS for
1 h. After the blot was washed three times with PBS, antibodies
were visualized using an enhanced chemiluminescence kit (Cell
Signaling Technology company, Danvers, MA, USA) and signals were
recorded as bands on Kodak X-ray films (Eastman-Kodak, Rochester,
NY, USA). The film was scanned and the band intensity was analyzed
using a densitometer (MSF-300 G Scanner; Microtek, Xinzhu, Taiwan).
β-actin served as an internal reference.
Radioligand binding assay (RLBA)
An RLBA was performed as previously described
(15). Briefly, the cells were
inoculated in 24-well plates at a density of 5×104
cells/well using the medium described above. LPS at varying
concentrations (0, 50, 100 or 200 ng/ml) was added. The cells were
incubated for 4, 8, 12 or 16 h and subsequently kept in chilled PBS
for 10 min. The cells were washed again using 2 ml chilled PBS to
remove endogenous AngII completely. According to protocols of
previous studies (16,17), the cells were incubated with 0.5 ml
binding medium containing various doses of 125I-Ang II
(0.1, 0.2, 0.4, 0.8, 1.6 or 2.0 fmol/well). Nonspecific binding
(NSB) was evaluated in the presence of 5 µg unlabeled Ang II
(10 µg/ml) at which AT1R was entirely saturated. The medium
was removed following incubation at 4°C for 2 h, and the cells were
washed three times with PBS and transferred to a 0.25 mol/l
NaOH-0.05% SDS lysis buffer (Shenneng Bocai Biotechnology Co.,
Ltd.). The radioactivity was measured using a γ-counter system
(GC-911; ZhongJia photoelectricity, Hefei, China). Data on the
total binding (TB) and total ligand (LT) were obtained. Specific
binding (B) and free ligand (F) were calculated as follows: B = TB
− NSB and F = LT − TB. The maximal binding (Bmax) and dissociation
constant (Kd) were determined by Scatchard plot analysis
according to the equation: B / F = −B / Kd + Bmax /
Kd.
Statistical analysis
Data are expressed as the mean ± standard deviation.
SPSS version 18.0 (SPSS Inc., Chicago, IL, USA) was used for
statistical analyses. Differences between groups were examined
using one-way analysis of variance. Dunnett's test was used for
post hoc analysis and P<0.05 was considered to indicate a
statistically significant difference.
Results
HPMECs and HRMCs characteristics
HPMECs presented with a cobblestone morphology and
were characterized by vWF and CD31 antigen expression as revealed
by immunofluorescence (data not shown). HRMCs exhibited a stellate
or spindle-like morphology, expressed desmin antigen, and did not
express vWF, CD31 or cytokeratin antigen, as indicated by
immunofluorescence.
LPS increases AT1R expression in
HPMECs
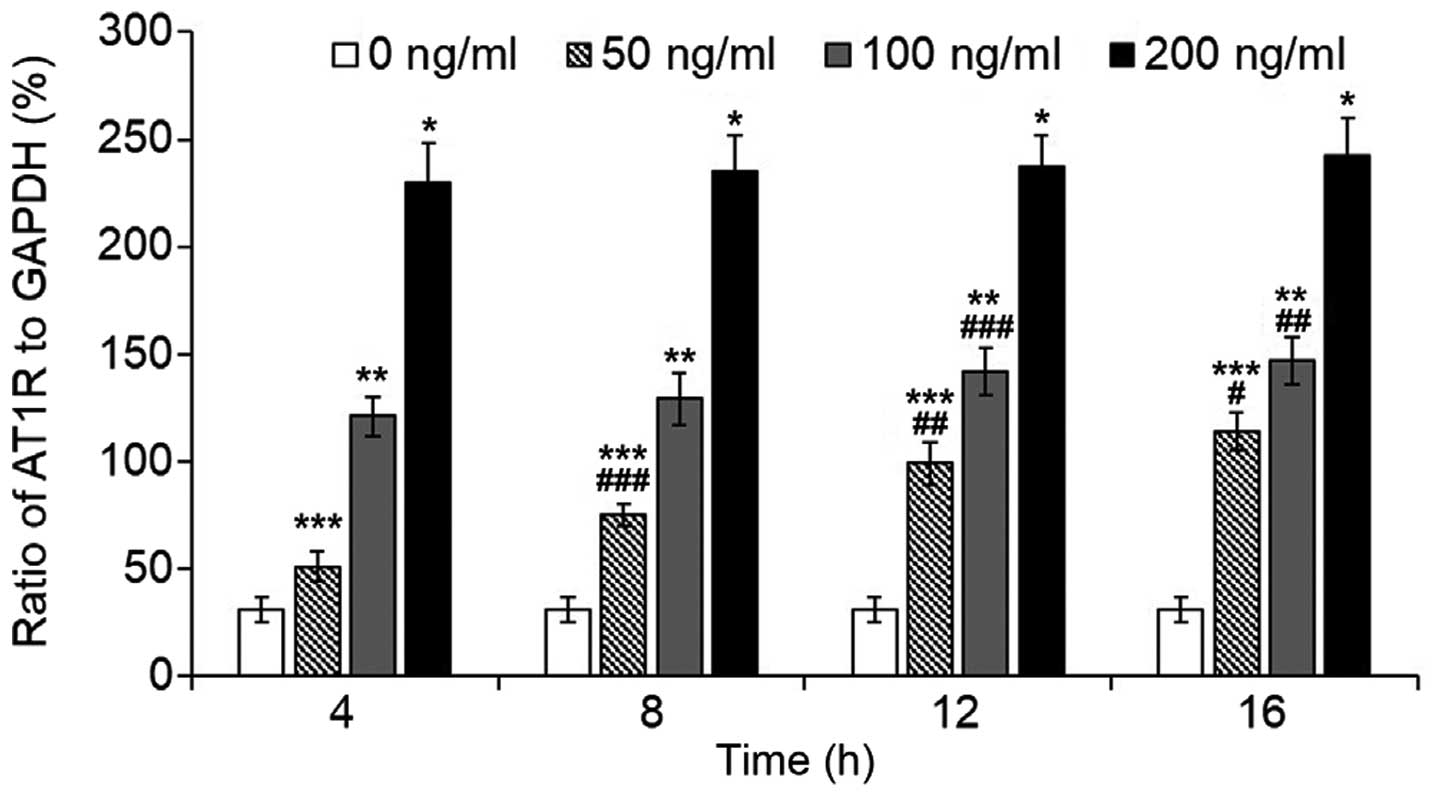
The transcription of AT1R increased
significantly following LPS treatment (Fig. 1). After a 16-h incubation, LPS
caused an increase of ~8-fold in the 200 ng/ml group, and ~5-fold
increase in the 100 ng/ml group. In the 50 ng/ml group, the
increase of AT1R transcription exhibited a significant
time-dependent effect, whereas in the 100 and 200 ng/ml groups,
AT1R transcription appeared to be stably maintained
following a 4-h incubation with the exception of a marginal
increase after an 8-h incubation in the 100 ng/ml group (16 vs. 4
h, P=0.001; 16 vs. 8 h, P=0.018; 12 vs. 4 h, P=0.011).
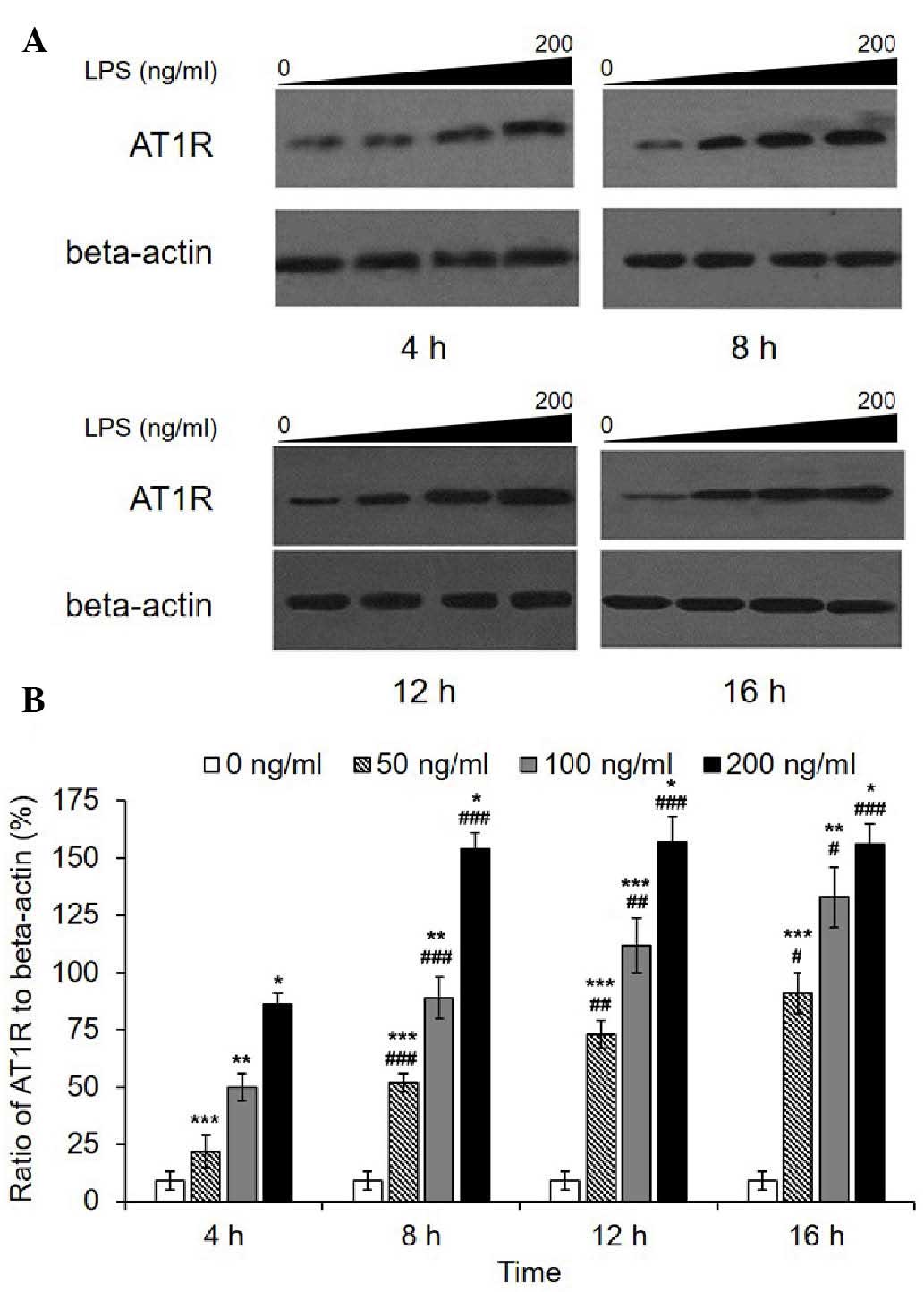
Consistently, western blot analysis revealed a marked increase in
the AT1R protein level following LPS treatment (Fig. 2). A significant dose-dependent
increase was observed in each group for each incubation duration. A
time-dependent increase was observed in the 50 and 100 ng/ml
groups, while in the 200 ng/ml group, AT1R quantities reached a
peak value after an 8-h incubation then remained at the same level.
Transcription of AT2R was not detected in the HPMECs with or
without LPS; however, a fault with the primers was excluded, as the
primers had been used effectively in the HRMCs in which AT2R
was transcribed (Data not shown). In addition, western blot
analysis revealed that AT2R was not expressed.
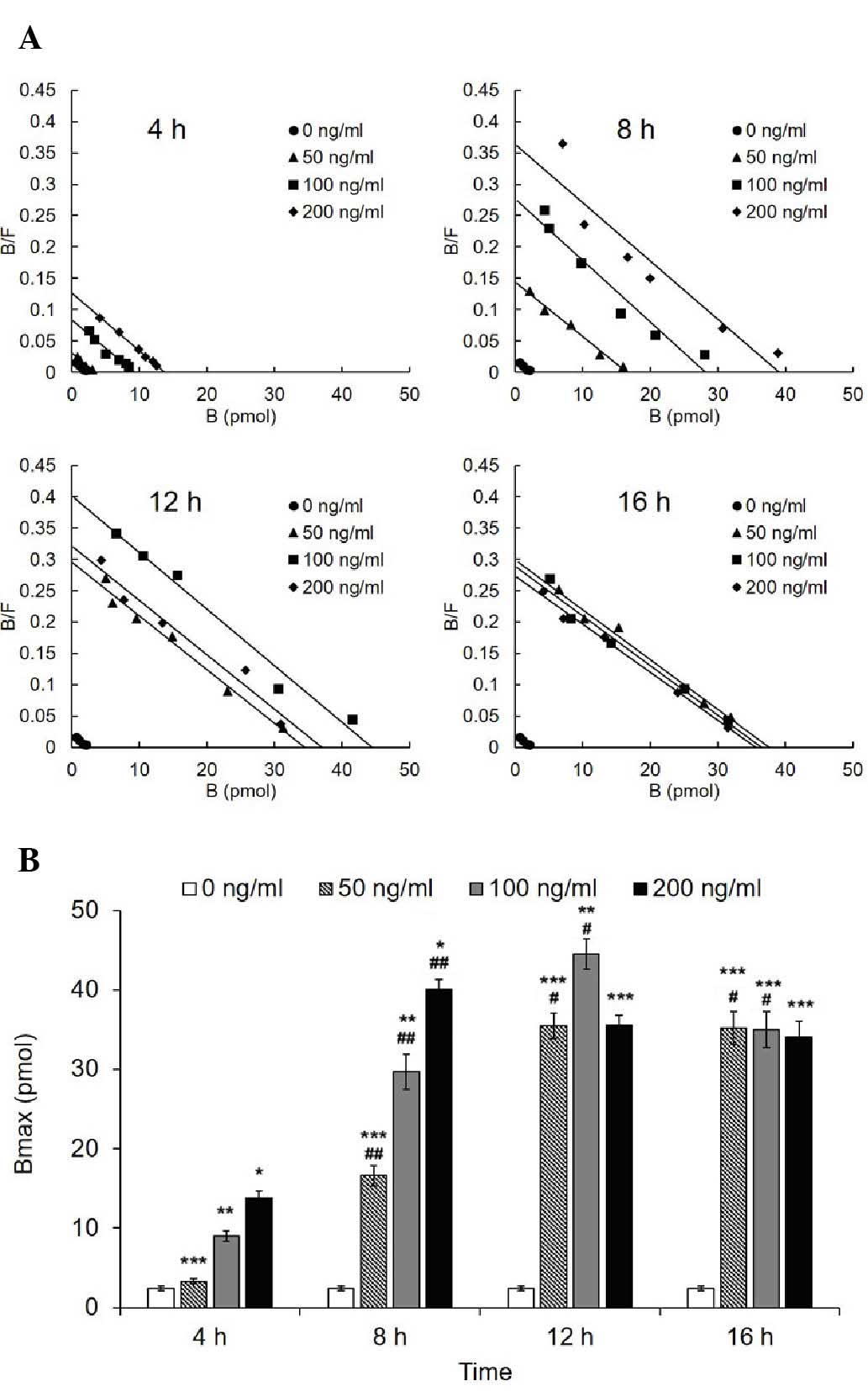
LPS increases Bmax, but not the affinity
of HPMEC-expressed AT1R to Ang II
The Bmax of AT1R of the treated groups (50, 100 and
200 ng/ml) increased significantly compared with the control group
(Fig. 3). Scatchard plotting
revealed a significant linear correlation and regression
(R2=0.9514, b=−0.00092). The Bmax of the 100 and 200
ng/ml groups reached a peak value at 12 and 8 h, respectively, and
the two exhibited a significant decrease thereafter significant
decrease thereafter (12 vs. 16 h in the 100 ng/ml group,
P<0.001; 8 vs. 12 h and 8 vs. 16 h in the 100 ng/ml group,
P<0.001). By contrast, the Bmax of the 50 ng/ml group increased
to the peak value at 12 h in a time-dependent manner, and remained
unchanged at 16 h. In addition, the Bmax did not differ among the
three treated groups at the 16 h time point. Correspondingly, the
Kd representing the affinity exhibited no difference in all
of the groups (Table I).
 | Table IDissociation constant of each group at
varying lipopolysaccharide concentrations (ng/ml). |
Table I
Dissociation constant of each group at
varying lipopolysaccharide concentrations (ng/ml).
| Time (h) | Dissociation constant
(pmol/l)
|
|---|
| 0 ng/ml | 50 ng/ml | 100 ng/ml | 200 ng/ml |
|---|
| 4 | 111±5 | 106±7 | 100±7 | 109±6 |
| 8 | 111±5 | 117±9 | 111±12 | 111±6 |
| 12 | 111±5 | 113±11 | 112±9 | 114±8 |
| 16 | 111±5 | 111±12 | 116±10 | 120±9 |
Discussion
The present study demonstrated that only AT1R was
expressed in HPMECs. LPS increased AT1R expression levels, however,
did not activate AT2R expression. The result is consistent with a
previous study performed using rat pulmonary microvascular
endothelial cells (12). Notably,
LPS increased the Bmax of AT1R to Ang II. In addition, the effects
of LPS concentration and incubation time on the expression and
affinity of AT1R to Ang II were compared.
In the development of ARDS, the endothelial cells
act as a barrier to pre-inflammatory mediators and are critical in
regulating the exudation of proteins and fluid, vascular tone,
hemostasis and inflammatory cells. Ang II activation in HPMECs is
important in the pathophysiology of ARDS. Ang II mediates
inflammation via AT1R and previous evidence indicates that AT1R is
upregulated under certain types of pathological stimulation, i.e.
by inflammatory factors. Previous studies have identified that
tumor necrosis factor-α, interleukin-1, and interferon-γ upregulate
AT1R expression levels in cardiac fibroblasts and vascular smooth
muscle cells (8,18,19).
The increase of AT1R expression is associated with inflammation and
other pathological effects. Furthermore, the change in AT1R
expression is important in the inflammatory response in the lungs,
which may involve the onset and progression of ARDS. In addition,
it has been reported that AT1R blockers delay the onset of ARDS
(20).
The results of the present study demonstrate that
LPS enhances the expression of AT1R in HPMECs with an increased
stimulating concentration of LPS and a longer time duration.
Notably, it was observed that AT1R expression increased in a
time-dependent manner in the low concentration groups, whereas in
the 200 ng/ml group, the expression appeared to reach a peak value
within a short incubation time. It was therefore hypothesized that
increasing the LPS concentration increased the rate of AT1R
expression. The increase in expression level in the high
concentration groups was particularly rapid, therefore the process
was not captured. The level of AT1R expression reached different
peak values in the three groups within the examined time course.
Thus, it was proposed that peak value may be associated with LPS
concentration. In addition, HPMECs did not express AT2R and
treatment with LPS failed to activate AT2R. Therefore, the
physiological role of AT2R remains to be clarified. It is
hypothesized that the LPS-regulation of AT2R may be tissue-specific
and AT2R may not be involved in ARDS.
The mechanism that mediates the LPS-induced increase
of AT1R expression in HPMECs remains to be elucidated. Studies have
indicated that nuclear factor (NF)-κB is required for
cytokine-induced upregulation of AT1R transcription in rat cardiac
fibroblasts (21). Preliminary
bioinformatic analysis of the 5′-flanking region of AT1R
revealed two putative NF-κB binding sites at positions −365 and
−2540 (22). LPS, a strong
pre-inflammatory factor, potently activate NF-κB through a
toll-like receptor (23). It was
hypothesized that NF-κB may also be involved in the LPS-induced
upregulation of AT1R transcription in HPMECs, which requires
examination in future studies.
AT1R functions upon binding to Ang II. Therefore,
the affinity and binding capacity of the highly-expressed AT1R was
assessed. It was confirmed by RLBA that LPS-induced AT1R binding
escalated with treatment durations between 4 and 16 h in the 50
ng/ml group. The increased Bmax may have been due to the
upregulation of AT1R expression. Increased binding of AT1R to Ang
II may be involved in the development of ARDS. However, an
explanation for the decrease in binding after 12 h and after 8 h in
the 100 and 200 ng/ml groups, respectively, remains to be
elucidated. The complex mechanism of Ang II binding-induced AT1R
internalization may be a possible explanation; AT1R has long been
considered as an internalizing receptor (24). Internalization of Ang II has been
reported in numerous cells of vascular, adrenal and renal origin,
which express AT1R. The majority of the morphological data indicate
that endocytosis of AT1R occurs via coated endocytic vesicles,
similar to the internalization mechanism of the majority of other G
protein-coupled receptors (25).
Thus, internalization may also be associated with the LPS
concentration, as the Bmax of the 100 and 200 ng/ml groups dropped
more markedly than that of the 50 ng/ml group.
In addition, alteration of the affinity of Ang II
binding to AT1R may alter AT1R pre-inflammatory function. In the
RLBA experiment, the sole function of the cells is the specific
binding with their associated ligand. The critical factor affecting
the affinity is the phosphorylation of the receptor's intracellular
domain. The present RLBA experiment demonstrated that LPS failed to
alter the affinity of Ang II binding to AT1R.
In conclusion, LPS enhanced AT1R expression,
although did not activate AT2R expression in HPMECs. In addition,
LPS increased the binding of AT1R to Ang II, however did not alter
the affinity of AT1R to Ang II. The present results indicate that
AT1R may be involved in LPS-induced ARDS.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 30640012).
References
|
1
|
Li JS, Touyz RM and Schiffrin EL: Effects
of AT1 and AT2 angiotensin receptor antagonists in angiotensin
II-infused rats. Hypertension. 31:487–492. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Griendling KK and Ushio-Fukai M: Reactive
oxygen species as mediators of angiotensin II signaling. Regul
Pept. 91:21–27. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang H, Schmeisser A, Garlichs CD, Plötze
K, Damme U, Mügge A and Daniel WG: Angiotensin II-induced
superoxide anion generation in human vascular endothelial cells:
Role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc Res.
44:215–222. 1999. View Article : Google Scholar
|
|
4
|
Ruiz-Ortega M, Lorenzo O, Ruperez M, König
S, Wittig B and Egido J: Angiotensin II activates nuclear
transcription factor kappaB through AT(1) and AT(2) in vascular
smooth muscle cells: Molecular mechanisms. Circ Res. 86:1266–1272.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marshall RP, Gohlke P, Chambers RC, Howell
DC, Bottoms SE, Unger T, McAnulty RJ and Laurent GJ: Angiotensin II
and the fibroproliferative response to acute lung injury. Am J
Physiol Lung Cell Mol Physiol. 286:L156–L164. 2004. View Article : Google Scholar
|
|
6
|
Marshall RP, McAnulty RJ and Laurent GJ:
Angiotensin II is mitogenic for human lung fibroblasts via
activation of the type 1 receptor. Am J Respir Crit Care Med.
161:1999–2004. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bechara RI, Pelaez A, Palacio A, Joshi PC,
Hart CM, Brown LA, Raynor R and Guidot DM: Angiotensin II mediates
glutathione depletion, transforming growth factor-beta1 expression
and epithelial barrier dysfunction in the alcoholic rat lung. Am J
Physiol Lung Cell Mol Physiol. 289:L363–L370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan
B, Yang P, Sarao R, Wada T, Leong-Poi H, et al:
Angiotensin-converting enzyme 2 protects from severe acute lung
failure. Nature. 436:112–116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Idell S, Kueppers F, Lippmann M, Rosen H,
Niederman M and Fein A: Angiotensin converting enzyme in
bronchoalveolar lavage in ARDS. Chest. 91:52–56. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brasier AR, Recinos A III and Eledrisi MS:
Vascular inflammation and the renin-angiotensin system.
Arterioscler Thromb Vasc Biol. 22:1257–1266. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Masaki H, Kurihara T, Yamaki A, Inomata N,
Nozawa Y, Mori Y, Murasawa S, Kizima K, Maruyama K, Horiuchi M, et
al: Cardiac-specific overexpression of angiotensin II AT2 receptor
causes attenuated response to AT1 receptor-mediated pressor and
chronotropic effects. J Clin Invest. 101:527–535. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H and Sun GY: Expression and
regulation of AT1 receptor in rat lung microvascular endothelial
cell. J Surg Res. 134:190–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garcia JG, Liu F, Verin AD, Birukova A,
Dechert MA, Gerthoffer WT, Bamberg JR and English D: Sphingosine
1-phosphate promotes endothelial cell barrier integrity by
Edg-dependent cytoskeletal rearrangement. J Clin Invest.
108:689–701. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Birukova AA, Wu T, Tian Y, Meliton A,
Sarich N, Tian X, Leff A and Birukov KG: Iloprost improves
endothelial barrier function in lipopolysaccharide-induced lung
injury. Eur Respir J. 41:165–176. 2013. View Article : Google Scholar :
|
|
15
|
Li H, Qiu H, Xu D, Liu L, Wang L, Dig H,
Yang Y and Zhou S: Establishment of radioligand binding assay for
angiotensin II type 1 receptor of human pulmonary microvascular
endothelial cell after lipopolysaccharide stimulation. Int J
Respir. 27:1761–1764. 2007.
|
|
16
|
Dincer HE, Gangopadhyay N, Wang R and Uhal
BD: Norepinephrine induces alveolar epithelial apoptosis mediated
by alpha-, beta- and angiotensin receptor activation. Am J Physiol
Lung Cell Mol Physiol. 281:L624–L630. 2001.PubMed/NCBI
|
|
17
|
Li X, Shu R, Filippatos G and Uhal BD:
Apoptosis in lung injury and remodeling. J Appl Physiol (1985).
97:1535–1542. 2004. View Article : Google Scholar
|
|
18
|
Chan LY, Leung JC, Tang SC, Choy CB and
Lai KN: Tubular expression of angiotensin II receptors and their
regulation in IgA nephropathy. J Am Soc Nephrol. 16:2306–2317.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li JY, Avallet O, Berthelon MC, Langlois D
and Saez JM: Transcriptional and translational regulation of
angiotensin II type 2 receptor by angiotensin II and growth
factors. Endocrinology. 140:4988–4994. 1999.PubMed/NCBI
|
|
20
|
Raiden S, Nahmod K, Nahmod V, Semeniuk G,
Pereira Y, Alvarez C, Giordano M and Geffner JR: Nonpeptide
antagonists of AT1 receptor for angiotensin II delay the onset of
acute respiratory distress syndrome. J Pharmacol Exp Ther.
303:45–51. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sasamura H, Nakazato Y, Hayashida T,
Kitamura Y, Hayashi M and Saruta T: Regulation of vascular type 1
angiotensin receptors by cytokines. Hypertension. 30:35–41. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li D, Yang B, Philips MI and Mehta JL:
Proapoptotic effects of ANG II in human coronary artery endothelial
cells: Role of AT1 receptor and PKC activation. Am J Physiol.
276:H786–H792. 1999.PubMed/NCBI
|
|
23
|
Trowbridge IS, Collawn JF and Hopkins CR:
Signal-dependent membrane protein trafficking in the endocytic
pathway. Annu Rev Cell Biol. 9:129–161. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mukherjee S, Ghosh RN and Maxfield FR:
Endocytosis. Physiol Rev. 77:759–803. 1997.PubMed/NCBI
|
|
25
|
Marsh M and McMahon HT: The structural era
of endocytosis. Science. 285:215–220. 1999. View Article : Google Scholar : PubMed/NCBI
|