Introduction
Coronary artery disease is currently the leading
cause of mortality and morbidity worldwide and acute myocardial
infarction is a major cause of death in numerous countries
(1,2). The most effective strategy for
treating cardiac injury and limiting infarct size is early
restoration of coronary blood flow to the ischemic myocardium.
While essential, this approach is often associated with functional
and structural damage during reperfusion. Although the complex
mechanisms of myocardial ischemia/reperfusion injury (MIRI) are not
well understood, cardiac dysfunction and cardiomyocyte necrosis
during ischemia/reperfusion (I/R) are strongly associated with
intracellular Ca2+ overload and oxidative stress
(3). Additionally, mitochondrial
dysfunction has been proposed to be important to the etiology of
MIRI, since the mitochondria act as the source and target of the
injury process. Indeed, the mitochondria sustain progressive damage
during prolonged cardiac ischemia, and in the process become a
major source of excess reactive oxygen species (ROS) generation
(4,5). ROS have also been demonstrated to
interfere with the Na+-Ca2+ exchanger and to
inhibit Na+,K+-ATPase activity. Impairment of
Na+,K+-ATPase activity results in
Na+ overload, with consequent activation of the
Na+-Ca2+ exchanger that may cause
intracellular Ca2+ overload and accumulation in
mitochondria (6–9). This ultimately results in the
formation of mitochondrial permeability transition pores (mPTPs)
and breakdown of the mitochondrial membrane potential (ΔΨm)
(10). Thus, against this vicious
cycle of perpetual damage by ROS and mitochondrial Ca2+
overload, I/R may preserve the structural and functional integrity
of the mitochondria and other cell structures to reduce cardiac
injury.
The lazaroids are potent scavengers of oxygen free
radicals. The lazaroid derivative U83836E combines the amino
functionalities of the 21-aminosteroids with the antioxidant ring
portion of vitamin E. The antioxidant potency of U83836E is 100
times that of vitamin E (11).
Accumulating evidence has demonstrated that U83836E has a variety
of pharmacological properties, including anti-neurodegenerative
actions (12–14) and the ability to improve
blood-brain barrier function (15). Preliminary studies have
demonstrated that U83836E (5 mg/kg) exerts therapeutic effects
against hemorrhagic shock and limb I/R in rats (16). The protein kinase C (PKC) signaling
pathways are understood to be involved in MIRI protection (17). In hippocampal neurons and glial
cells, PKC is a key enzyme in the signal transduction cascade that
leads to superoxide radical generation (18). The potential protective effects of
U83836E against I/R injury and preservation of functional integrity
of mitochondria remain unclear, in addition to whether this is
mediated by PKC activation. The present study was designed to
confirm the protective effects of U83836E against MIRI in rat
models and to identify the possible mechanisms underlying these
effects.
Materials and methods
Experimental animals
The present study was approved by the Animal Care
and Use Committees of Changzhi Medical College (Changzhi, China). A
total of 120 male Sprague-Dawley (SD) rats (clean grade) of 280–300
g body weight and 8–10 weeks old were obtained from the Shanxi
Medical University Experimental Animal Center (Taiyuan, China). The
animal protocol was designed to minimize pain or discomfort to the
animals. The animals were acclimatized to laboratory conditions
(23°C, 12-h light/dark cyle, 50% humidity, ad libitum access
to food and water) for 2 weeks prior to experimentation.
Intragastric gavage administration was carried out with conscious
animals, using straight gavage needles appropriate for the animal
size (16 gauge, 10 cm length, 1.25 mm ball diameter). All rats were
sacrificed by barbiturate overdose (intravenous injection; 150
mg/kg pentobarbital sodium) for tissue collection.
Establishment of the myocardial I/R
injury model
Lead II electrocardiogram was performed for
continual monitoring using a BL-420F multi-channels physiologic
signal analysis system (Chengdu Taimeng Science and Technology Co.,
Ltd., Chengdu, China). Tracheotomy was performed and an intubating
cannula was connected to a small animal respirator (Taimeng
Technology Co., Ltd.). A polyethylene tube (PE 50; BD Biosciences,
Franklin Lakes, NJ, USA) filled with heparinized saline was
inserted into the left ventricular cavity via the right carotid
artery. The left ventricular systolic pressure (LVSP), left
ventricular end diastolic pressure (LVEDP), and the maximal rate of
increase and decline in left ventricular pressure (± dp/dtmax) were
monitored by the BL-420F signal analysis system. A left thoracotomy
was performed from the third to fourth intercostal space and the
pericardium was opened. The left anterior descending artery (LAD)
was identified and ligated ~2 mm from its origin by a 5/0 curved
suture needle with thread. Ischemia was confirmed by an ST
elevation on the ECG and color change to the myocardial tissue of
the ischemic area. Following 30-min ischemia, the ligature was
loosened and the ischemic myocardium was reperfused for 2 h. At the
end of the protocol, the hearts of the rats were excised while the
animals were anesthetized.
Experimental design and drug
treatment
Male adult SD rats were randomly divided into the
following four groups: i) Sham operation group, the LAD coronary
artery was crossed by thread without ligation; ii) I/R group, the
rats were subjected to LAD occlusion for 30 min followed by
reperfusion for 2 h; iii) U83836E + I/R group, 5 mg/kg U83836E
(PubChem CID 107656; 98%; Enzo Life Sciences, Inc., Farmingdale,
NY, USA) dissolved in physiological saline was administered by tail
vein injection 1 h prior to ischemia; and iv) CHE + U83836E + I/R
group, 1 mg/kg chelerythrine PKC inhibitor (CHE; PubChem CID 72311;
99%; LC Laboratories, Woburn, MA, USA) dissolved in physiological
saline was administered by tail vein injection 15 min prior to
U83836E administration. The sham and I/R groups received an
equivalent volume of physiological saline via tail vein
injection.
Measurement of the myocardial infarct
size
The infarct size was determined by staining with 1%
TTC (99%; Sigma-Aldrich, St. Louis, MO, USA). Following
reperfusion, the heart was rapidly excised from the thorax and
washed with physiological saline at 4°C, then frozen for 10 min at
−20°C. The hearts were subsequently transected into five pieces and
the slices were incubated in 1% TTC in phosphate buffer (pH 7.4) at
37°C for 10 min, followed by post-fixation in 10% formaldehyde
(Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) for 15–20 h
to enhance the contrast between the stained and unstained TTC
tissues. The viable tissue was stained red by TTC, whereas the
infarct area remained pale. The area of infarct size (IS) and area
of the left ventricle (LV) in each slice were imaged with a scanner
(V30; Seiko Epson Corporation, Suwa, Japan) and analyzed by
Image-Pro Plus software, version 7.0 (Media Cybernetics, Inc.,
Rockville, MD, USA). The extent of ischemia in the myocardium was
calculated as the ratio of IS/LV.
Histopathological examination
The hearts were fixed in 10% neutral-buffered
formalin and then dehydrated, decalcified and embedded in paraffin
(Sinopharm Chemical Reagent Co., Ltd.). Cross-sections (5 µm
thick) of the fixed myocardial tissues were cut using a microtome
(Leica RM2235; Leica Microsystems GmbH, Wetzlar, Germany). The
samples were stained with hematoxylin and eosin (ZSGB-BIO Co.,
Ltd., Beijing, China), and observed under a light microscope
(Olympus BX51; Olympus Corporation, Tokyo, Japan) to analyze the
myocardium architecture.
Measurement of creatinine kinase (CK)
activity in plasma
Following 2 h reperfusion, blood samples were
collected from the left ventricle and centrifuged at 3,000 × g for
10 min to isolate the plasma. Myocardial cellular damage was
evaluated by measuring plasma CK activity using assay kits (cat.
no. A032; Nanjing Jiancheng Bioengineering Institute, Nanjing,
China), according to the manufacturer's protocol.
Evaluation of superoxide dismutase (SOD),
glutathione peroxidase (GSH-Px) and malondialdehyde (MDA) in
myocardial tissue
Following 2-h reperfusion, the myocardial samples
were rinsed and homogenized in deionized water (1:10, wt/vol) prior
to centrifugation at 3,000 × g for 5 min. The activities of SOD and
GSH-Px and the concentration of MDA were quantified using detection
kits (cat. nos. A001-1, A005 and A003-1, respectively; Nanjing
Jiancheng Bioengineering Institute) according to the manufacturer's
protocol.
Isolation of mitochondria
Mitochondrial fractions were isolated by
differential centrifugation, as previously described (19). All experiments were performed in
cold conditions. Briefly, the LV tissue (150 mg) was first
suspended in isolation buffer A (250 mM sucrose; 10 mM Tris-HCl; 1
mM EDTA, pH 7.4; Sinopharm Chemical Reagent Co., Ltd.), finely
minced with scissors and then homogenized in deionized water (1:10,
wt/vol). The homogenates were centrifuged at 1,000 × g for 10 min,
and the supernatant was collected and centrifuged at 1,500 × g for
10 min. The supernatant from this centrifugation was subsequently
centrifuged at 10,000 × g for 15 min to yield the cytosolic
fraction. The precipitate, corresponding to the mitochondrial
fraction, was resuspended in isolation buffer B (250 mM sucrose; 10
mM Tris-HCl, pH 7.4). Pellets were maintained on ice prior to
Ca2+ and ΔΨm experiments. The concentration of
mitochondrial proteins was measured using bicinchoninic acid assay
(Beyotime Institute of Biotechnology, Haimen, China).
Measurement of mitochondrial
Ca2+ concentration (m[Ca2+])
The suspensions were adjusted to a concentration of
0.5 mg/ml with buffer B containing the Ca2+ fluoroprobe
Fura2/AM (10 µM) and incubated at 37°C for 30 min, and then
washed to remove the non-bound components. The Fura2/AM
Ca2+ signals were determined with a F4600 fluorescent
spectrophotometer (Hitachi, Ltd., Tokyo, Japan), using an
excitation wavelength of 340 nm and an emission wavelength of 380
nm, as previously described (20).
The Ca2+ levels were calculated using the following
formula: Kd × (R-Rmin)/(Rmax-R);
where Kd=314 nmol/l, R is the resting state,
Rmin is the fluorescence intensity ratio of the 340 nm
and the 380 nm wavelengths to join the ethylene glycol tetraacetic
acid, and Rmax is the fluorescence intensity ratio of
the 340 nm and the 380 nm wavelengths following Triton X-100
(Sinopharm Chemical Reagent Co., Ltd.) addition.
Measurement of ΔΨm
JC-1 dye (Beyotime Institute of Biotechnology) was
used to determine the change in ΔΨm (21). The isolated mitochondria (0.5
mg/ml) were incubated in JC-1 at 37°C for 30 min. The fluorescence
intensity was measured using the F4600 spectrofluorometer with 514
nm excitation and 529 nm emission wavelengths (Hitachi, Ltd.,
Tokyo, Japan). The fluorescence intensity ratio of JC-1 polymers
and monomers reflects the ΔΨm.
Western blot analysis of PKC
Cytosolic proteins and membrane proteins from the
left myocardial samples were extracted using the Membrane and
Cytosol Protein Extraction kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Equal
protein fractions were separated by electrophoresis on 10% SDS-PAGE
gels. Following transfer to polyvinylidene fluoride (PVDF)
membranes, the proteins were probed with a rabbit monoclonal
primary antibody against PKCε (1:1,000; ab124806; Abcam, Cambridge,
UK) overnight at 4°C. The PVDF membranes were washed 3 times with
Tris-buffered saline containing 0.1% Tween-20, and incubated with a
goat anti-rabbit horseradish peroxidase-conjugated secondary
antibody (1:2,000; A0208; Beyotime Institute of Biotechnology) for
2 h at room temperature. β-Actin was detected used a rabbit
polyclonal antibody (1:5,000; ab119716; Abcam) as a loading
control. The proteins were detected with the enhanced
chemiluminescence system (Beyotime Institute of Biotechnology) on
an ImageQuant LAS 4000 (GE Healthcare Life Sciences, Chalfont, UK).
The band intensities were quantified using Image-Pro Plus
software.
Statistical analysis
All quantification data are expressed as the mean ±
standard error. For multiple-group comparisons, one-way analysis of
variance was performed, followed by the least significant
difference t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of U83836E on hemodynamics in I/R
rats
The LVSP, LVEDP and ±dp/dtmax myocardial functional
parameters were measured prior to I/R, following 30 min of
ischemia, and at 5, 30, 60 and 120 min of reperfusion (Fig. 1). These parameters were similar
among the four groups prior to LAD ligation (Fig. 1). Conversely, the rats in the I/R
group exhibited a significant increase in LVEDP, and a significant
decrease in LVSP and ±dp/dtmax, at 30 min of ischemia and at 5, 30,
60 and 120 min of reperfusion (all P<0.01; Fig. 1), as compared with the sham group.
Compared with the I/R group, U83836E treatment significantly
reduced the LVEDP at 30 min of ischemia (P<0.05), and at all
time points of reperfusion (P<0.05). In addition, the LVSP and
±dp/dtmax were significantly increased in the U83836E-treated group
following 30 min of ischemia and at all time points of reperfusion
(P<0.05; Fig. 1). These results
suggest that pretreatment with U83836E may increase cardiac
function following I/R injury. Notably, in the CHE+U83836E+I/R
group, the effects of U83836E on the LVSP, LVDEP and ±dp/dtmax were
partly attenuated by CHE treatment (Fig. 1).
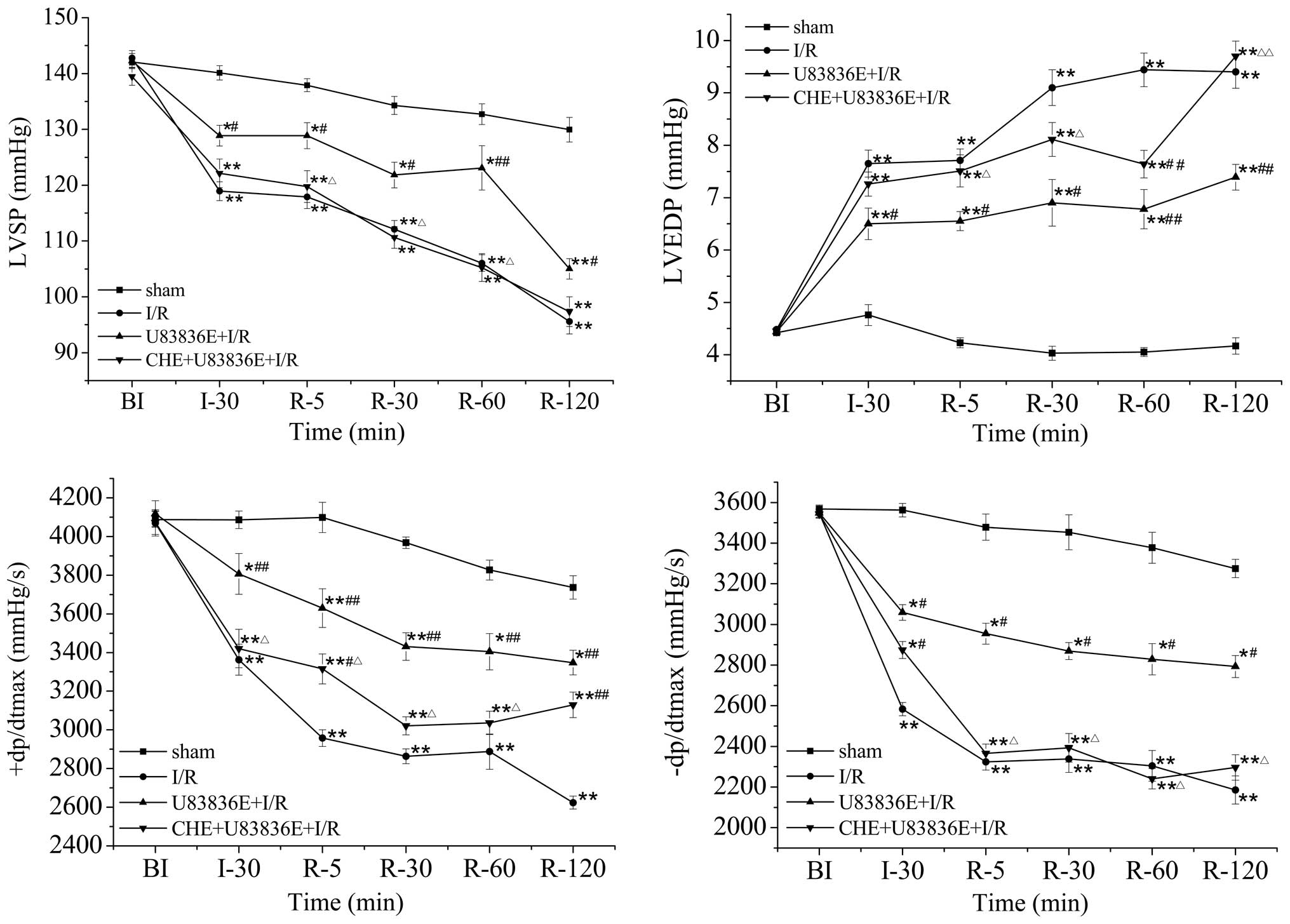 | Figure 1Effects of U83836E on the
hemodynamics in rat hearts subjected to 30 min ischemia followed by
2 h reperfusion. Values are presented as the mean ± standard error,
n=10. *P<0.05, **P<0.01 vs. the sham
group; #P<0.05, ##P<0.01 vs. the I/R
group; ΔP<0.05, ΔΔP<0.01 vs. the
U83836E + I/R group. BI, before ischemia; I, ischemia; R,
reperfusion; I/R, ischemia/reperfusion; LVSP, left ventricular
systolic pressure; LVEDP, left ventricular end diastolic pressure;
+dp/dtmax, maximal rate of increase in left ventricular pressure;
-dp/dtmax, maximal rate of increase and decline in left ventricular
pressure; CHE, chelerythrine. |
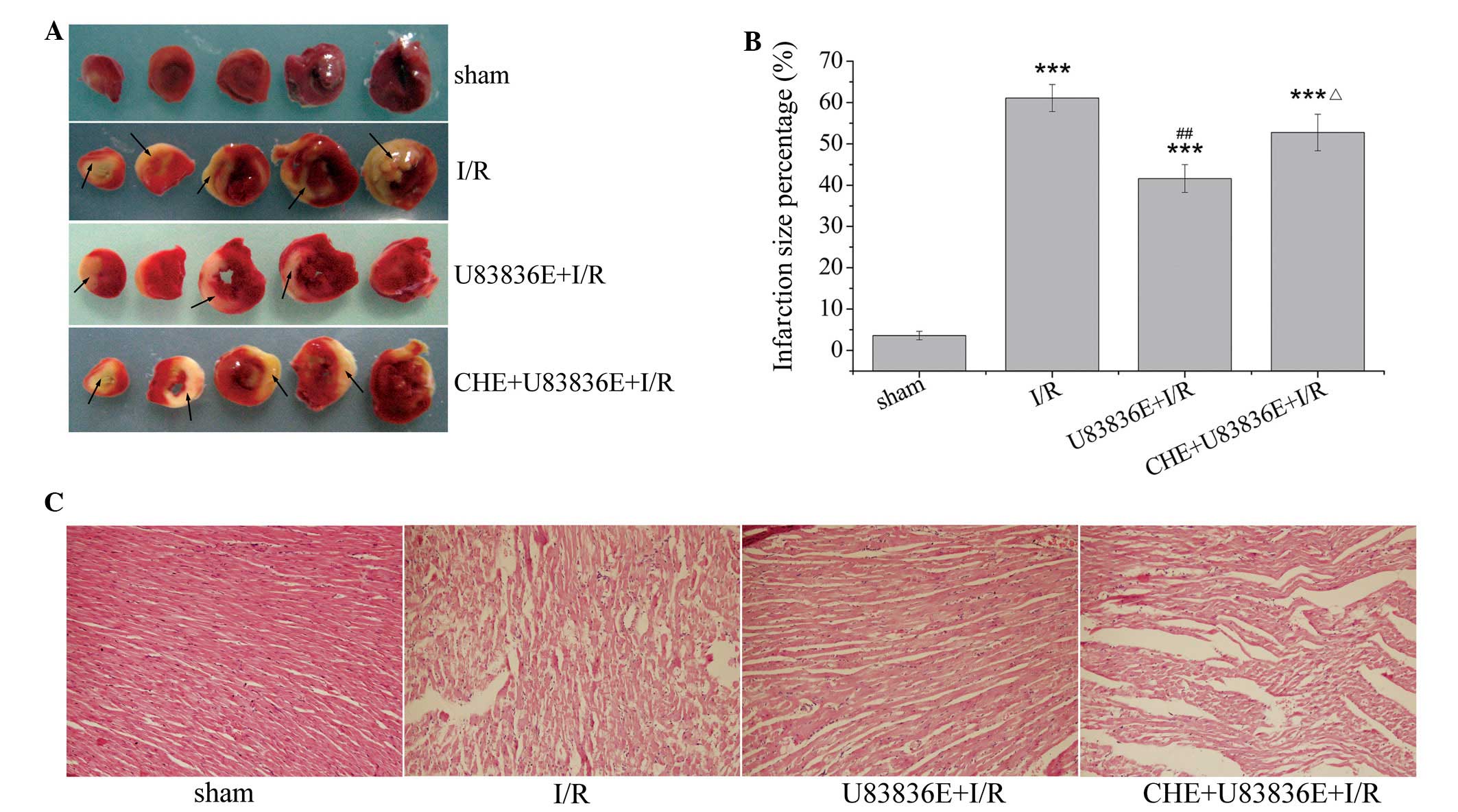
U83836E decreases myocardial infarct size
after I/R injury
As presented in Fig. 2A
and B, I/R induced severe myocardial injury. The infarction
size in the I/R group was 61.08±3.28% (P<0.001 vs. the sham
group). U83836E treatment significantly reduced the infarction size
percentage to 41.60±3.37% compared with the I/R group (P=0.002).
This result indicates that pretreatment with U83836E may reduce the
size of myocardial infarction in I/R rats. Pretreatment with CHE
abrogated the U83836E-induced decrease in infarct size, and the
infarction size was elevated to 52.74±4.43% (P=0.062 vs. I/R group;
P=0.03 vs. U83836E + I/R group).
Pathological changes in the
myocardium
As demonstrated in Fig.
2C, histopathological examination of the myocardium of the sham
group revealed tightly aligned cardiac muscle, clear transverse
tubules, no evidence of blood vessel enlargement or inflammatory
cell infiltration into the interstitial region. However, in the I/R
group, edema, confluent coagulation necrosis, neutrophil
infiltration and mild inflammation were observed. Rats treated with
U83836E demonstrated marked structural improvement, particularly
with regard to the degree of myonecrosis, inflammatory cell
infiltration and edema compared with the I/R group. In the presence
of CHE, U83836E partially failed to preserve the myocardial
cellular integrity. There was myofibril loss, the appearance of
cracks and an enlarged interstitial region, with minor infiltration
of inflammatory cells.
Effects of U83836E on CK activity in
plasma
CK is a serum biomarker that reflects myocardial
damage. As demonstrated in Fig.
3A, there was a significant increase in CK activity in the I/R
group compared with the sham group (P=0.002). CK activity was
reduced from 29.06±3.14 U/ml in the I/R group to 15.097±1.88 U/ml
following treatment with U83836E (P<0.01). However, CK activity
was elevated to 25.28±2.16 U/ml by pretreatment with CHE (P=0.235
vs. I/R group; P=0.012 vs. U83836E + I/R group).
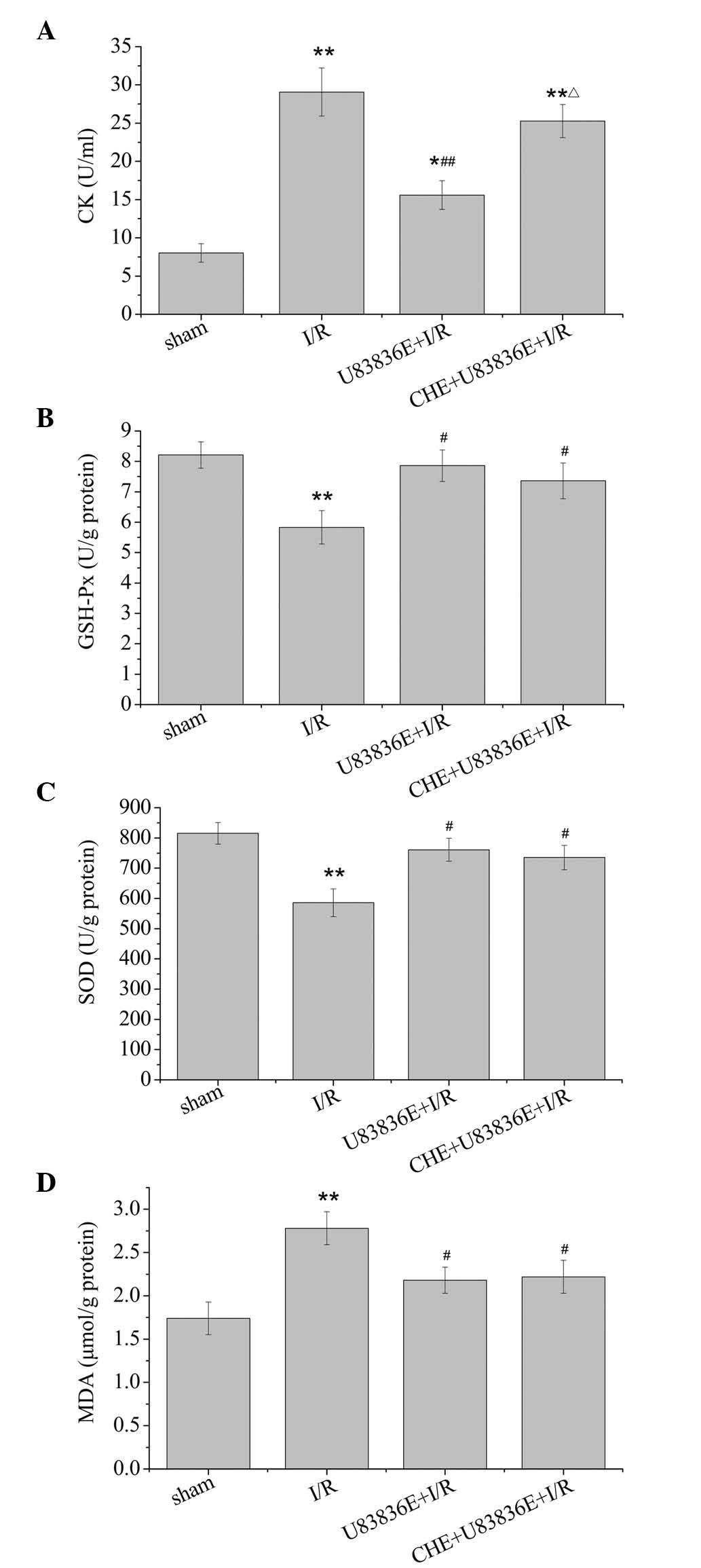 | Figure 3Effects of U83836E on antioxidant and
oxidant production in rat hearts subjected to 30 min of ischemia
followed by 2 h reperfusion. Effect of U83836E (5 mg/kg) on (A) the
CK concentration in the plasma, (B) GSH-Px activity levels in
myocardial tissue, (C) SOD activity levels in myocardial tissue and
(D) MDA levels in myocardial tissue. Values are presented as the
mean ± standard error, n=10. *P<0.05,
**P<0.01 vs. sham group; #P<0.05,
##P<0.01 vs. I/R group; ΔP<0.05 vs.
U83836E+I/R group. CK, creatine kinase; GSH-Px, glutathione
peroxidase; SOD, superoxide dismutase; MDA, malondialdehyde; I/R,
ischemia/reperfusion; CHE, chelerythrine. |
Effects of U83836E on GSH-Px activity in
the myocardial tissue
GSH-Px is regarded as an important antioxidant and
free radical scavenger in vivo. As presented in Fig. 3B, the I/R group exhibited a
significant decrease in GSH-Px activity compared with the sham
group (P=0.001). U83836E significantly increased the GSH-Px
activity in myocardial tissue compared with the I/R group
(P=0.018). No significant difference in GSH-Px activity was
observed between the U83836E group and the CHE + U83836E + I/R
group (P=0.481).
Effects of U83836E on SOD activity and
MDA concentration in the myocardial tissue
SOD is an endogenous antioxidant against free
radicals and MDA is important indicator of myocardial oxidative
damage. Data on these parameters are presented in Fig. 3C and D. Compared with the sham
group, the I/R-induced injury resulted in a significant decrease in
SOD activity (P=0.002) and an increase in the MDA concentration
(P=0.003). The administration of U83836E resulted in a significant
decrease in the MDA concentration (P=0.017) and increase in the SOD
enzyme activity (P=0.031) compared with the I/R group. Notably, the
U83836E-induced changes in MDA concentration (P=0.770) and SOD
activity (P=0.819) were not significantly altered by CHE treatment,
compared with the U83836E + I/R group.
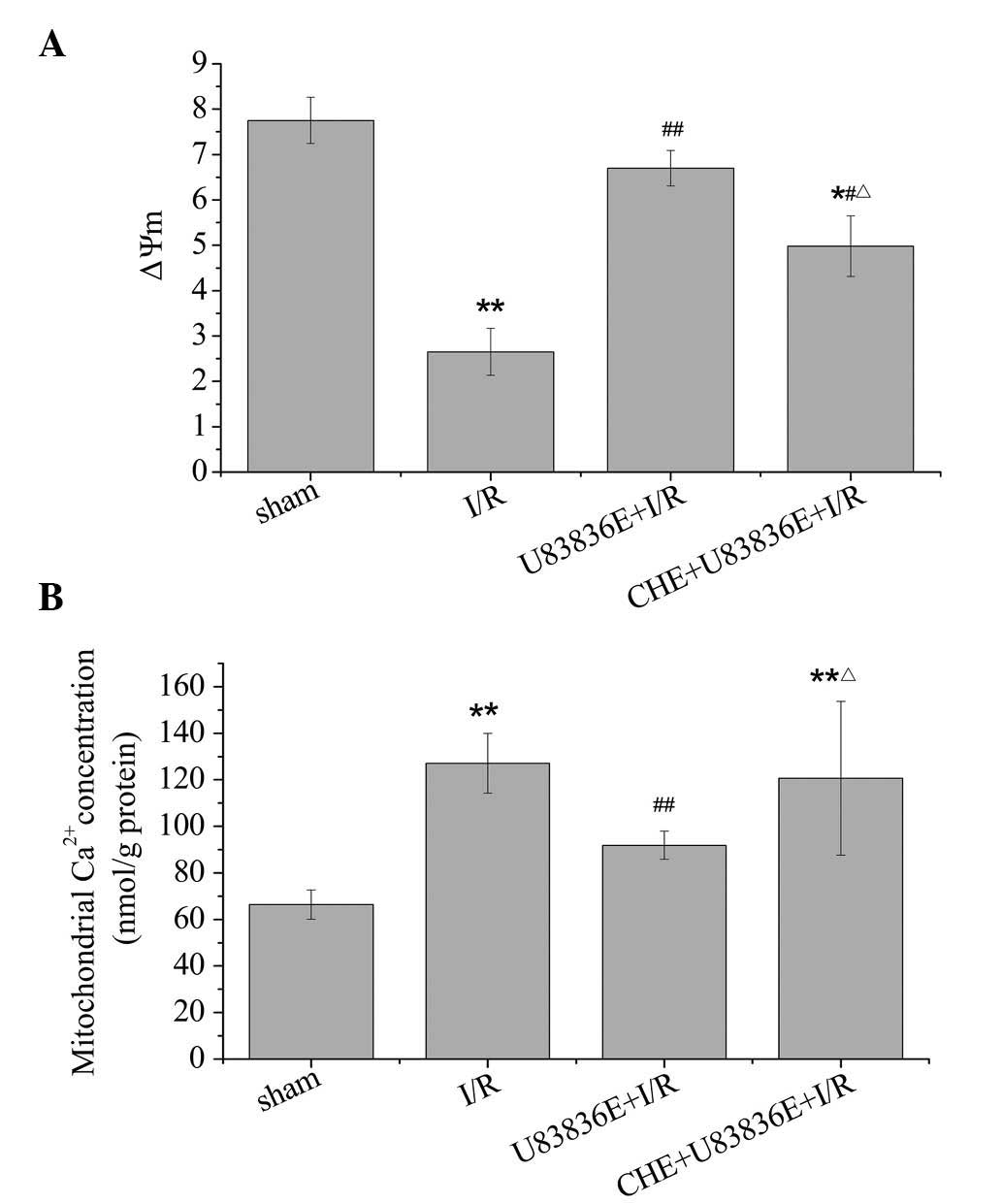
U83836E prevents the loss of ΔΨm
To further determine the effects of U83836E on
mitochondrial function, changes in the ΔΨm were determined. As
demonstrated in Fig. 4A, the
membrane potential in the I/R group was significantly reduced
compared with sham group (2.65±0.52 vs. 7.75±0.51; P=0.003),
indicating a dissipation of ΔΨm in response to I/R injury. Compared
with I/R group, a significant increase of ΔΨm was observed
following treatment with U83836E (P=0.004). Additionally, ΔΨm was
significantly decreased in the CHE + U83836E group compared with
the U83836E + I/R group (P=0.029).
U83836E prevents mitochondrial
Ca2+ overload
Mitochondrial Ca2+ accumulation is
responsible for the cell abnormalities associated with I/R injury.
As demonstrated in Fig. 4B,
compared with the sham group, I/R caused the m[Ca2+] to
increase (P=0.008). The administration of U83836E resulted in a
significant decrease in the m[Ca2+] compared with the
I/R group (P=0.009). The CHE + U83836E + I/R group was observed to
be significantly different from the U83836E + I/R group (P=0.037),
but not the I/R group (P=0.633). Altogether, the findings of the
present study suggest that pretreatment with U83836E may alleviate
the loss of ΔΨm and the m[Ca2+] overload in I/R rats.
However, the effect of U83836E was diminished by CHE.
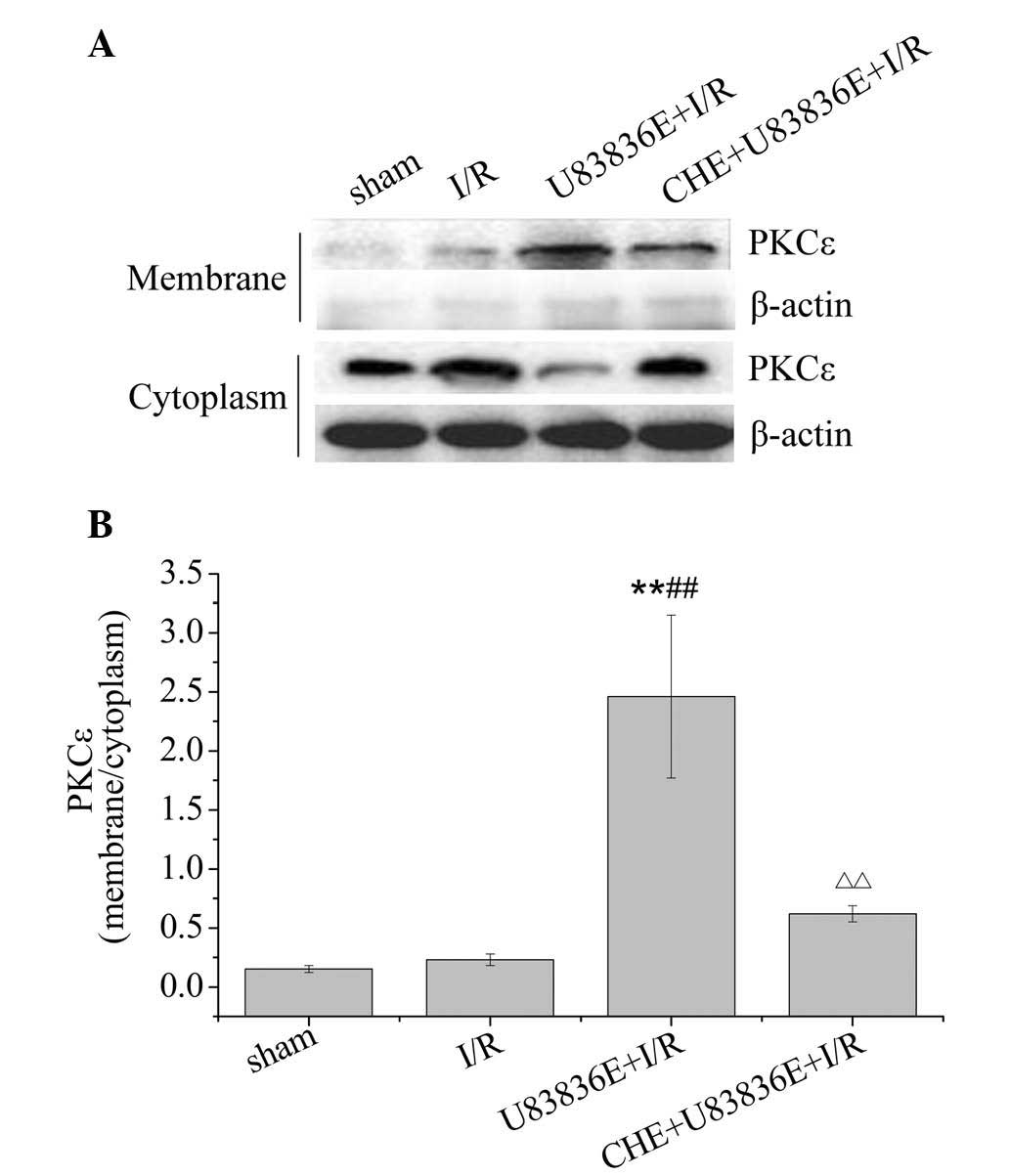
U83836E promotes the translocation of
PKCε from the cytoplasm to the membrane
PKCε is the most abundant PKC isozyme present in the
adult rat cardiac myocytes, and is essential in developing
cardioprotection (22,23). Translocation from the cytoplasm to
the membrane is crucial for PKC activation. To further explore the
relationship between U83836E and PKCε, the cytosolic and membrane
proteins were extracted from the left myocardial samples. As
demonstrated in Fig. 5, the PKCε
protein levels were significantly increased in the U83836E + I/R
group compared with the sham and the I/R groups (P=0.001). CHE
significantly decreased the translocation of PKCε from the
cytoplasm to the membrane compared with the U83836E + I/R group
(P=0.003).
Discussion
Myocardial damage is quickly initiated following
ischemia and is enhanced during the reperfusion period. I/R
disturbs the balance between the formation and elimination of ROS,
resulting in ROS accumulation. The endogenous defense system
against ROS, primarily involving the antioxidant enzymes SOD and
GSH-Px, is critical to provide a line of defense against oxidative
damage under a variety of stress conditions, including I/R injury
(24,25). The present study demonstrated that
U83836E (5 mg/kg) significantly increases SOD and GSH-Px activity,
and suppresses the formation of the lipid peroxidation product,
MDA. These data demonstrate that treatment with U83836E enhances
the activity of antioxidant enzymes in myocardial I/R-challenged
rats. Myocardial enzyme leakage is indicative of myocardial cell
membrane injury. The mass production of oxygen-derived free
radicals can result in myocardial cell membrane injury and
myocardial enzyme leakage from the myocardial cells. In the current
study, it was observed that U83836E treatment significantly
inhibited the CK increase during I/R. In addition, treatment with
U83836E significantly preserved left ventricular function, as
reflected by a significant increase in the indices of contraction
(+dp/dtmax), relaxation (-dp/dtmax), LVSP and a decrease in LVEDP
in the I/R-insulted rat heart. Furthermore, U83836E decreased the
histological damage to the myocardium and reduced the area of
myocardial infarct of the left ventricle. All these data clearly
demonstrate that U83836E exerts protective effects against
myocardial I/R injury in rats.
Oxidative stress and Ca2+ overload are
the two major factors demonstrated to be associated with the
pathology of I/R injury. The importance of the mitochondria as
targets and mediators of I/R is increasingly recognized. ROS are
initially generated at the onset of reperfusion via oxidation of
hypoxanthine by xanthine oxidase, and from the mitochondrial
electron transport chain reactions. More ROS can be generated
through a nicotinamide adenine dinucleotide phosphate (NADPH)
reaction catalyzed by NADPH oxidase during extended reperfusion
(26). Additionally, intracellular
and mitochondrial Ca2+ overload can be caused by various
mechanisms, including membrane lipid peroxidation, the opening of
voltage-sensitive Ca2+ channels, the
Na+/Ca2+ exchanger, the release of
Ca2+ from intracellular endoplasmic and sarcoplasmic
reticulum stores, and the attenuation of Ca2+ uptake by
the sarcoplasmic reticulum Ca2+-ATPase. Ca2+
alters the inner mitochondrial membrane lipid organization by
interacting with the anionic head of cardiolipin, an abundant
component of this membrane. These alterations in membrane
organization may affect the respiratory chain function and favor
monoelectric oxygen reduction to form a superoxide anion at an
intermediate step of the respiratory chain. ROS generation promotes
Ca2+ release from the nearby sarcoplasmic reticulum,
leading to m[Ca2+] accumulation that further increases
the rate of ROS production (27).
These events may be the basis of the findings of the current study
regarding the Ca2+-induced ROS production in I/R injury
and may explain the reduction after treatment with U83836E. Under
physiological conditions, the inner membrane of the mitochondria is
impenetrable to almost all metabolites and ions, and the mPTP
exists in a closed conformation. During the stages of reperfusion,
Ca2+ overload and excessive ROS production can trigger
mPTP opening (28). This opening
leads to the dissipation of ΔΨm, the uncoupling of the respiratory
chain, the inhibition of ATP production, and mitochondrial swelling
and rupture (29). Loss of ΔΨm can
initiate a positive feedback loop by which additional mPTPs open,
resulting in further dissipation of ΔΨm and facilitating persistent
mPTP opening (30). Thus, ΔΨm is
an important parameter in the regulation of mitochondrial function
and is used as an indicator of the mPTP status. The current study
demonstrated that ΔΨm depolarization is induced by I/R injury and
can be significantly alleviated by pretreatment with U83836E. These
results suggest that U83836E exerts a protective effect on the
mitochondria, inhibiting mPTP opening and maintaining mitochondrial
structural integrity during I/R. Overall, U83836E clearly
contributes to mitochondrial protection via prevention of
m[Ca2+] overload and the loss of ΔΨm following
reperfusion.
The PKC enzymes are central to numerous signal
transduction processes, including cardioprotection (31). Activation of PKC was reported to
participate in the protective role of ischemia preconditioning, and
induced cardioprotection in several animal models (17,32).
In addition, PKC protects the mitochondrial ATP-sensitive potassium
channel against Ca2+ overload injury in the rat
myocardium (33–35). PKCε is an abundant PKC isozyme
present in adult rat cardiac myocytes and is essential in
cardioprotection development (22,23).
In the present study, it was observed that the cardioprotective
effect of U83836E was reversed by pretreatment with the CHE PKC
inhibitor. The current study additionally observed that U83836E
promotes translocation of PKCε from the cytoplasm to the membrane.
However, a notable observation in the present study was that
treatment with CHE following U83836E administration did not change
the activities of SOD and GSH-Px, or the MDA concentration. CHE
also did not alter the U83836E-induced antioxidative process. Thus,
it was hypothesized that U83836E exhibits its antioxidative effect
by changing SOD and GSH-Px activities and MDA concentration in a
PKC signaling-independent manner. It is assumed that the ROS may
act upstream of PKC.
It has been previously demonstrated that ROS can
directly activate PKC through sulfhydryl oxidation (36,37).
PKC is considered to be a mediator rather than a trigger of this
process (38). Therefore, the PKC
signaling pathway may not participate in this antioxidative
process.
In conclusion, the present investigation suggests
that the cardioprotective effect of U83836E is mediated by two
different pathways, including the free radical-eliminating
mechanisms and the activation of PKC-dependent signaling. On the
basis of these observations, therapeutic use of U83836E may have
clinical potential for the treatment of cardiac reperfusion
injury.
Acknowledgments
The current study was supported by the Natural
Science Foundation of Shanxi Province (no. 2009011055-3), a Key
Displine Construction Special Fund Projects grant from the Shanxi
Province Colleges and Universities (no. 20131007), and a Science
and Technology Innovation Team Project from Changzhi Medical
College (no. CX201409).
References
|
1
|
Heidenreich PA, Trogdon JG, Khavjou OA,
Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston
SC, Khera A, et al American Heart Association Advocacy Coordinating
Committee; Stroke Council; Council on Cardiovascular Radiology and
Intervention; Council on Clinical Cardiology; Council on
Epidemiology and Prevention; Council on Arteriosclerosis;
Thrombosis and Vascular Biology; Council on Cardiopulmonary;
Critical Care; Perioperative and Resuscitation; Council on
Cardiovascular Nursing; Council on the Kidney in Cardiovascular
Disease; Council on Cardiovascular Surgery and Anesthesia, and
Interdisciplinary Council on Quality of Care and Outcomes Research:
Forecasting the future of cardiovascular disease in the United
States: A policy statement from the American Heart Association.
Circulation. 23:933–944. 2011. View Article : Google Scholar
|
|
2
|
Nabel EG and Braunwald E: A tale of
coronary artery disease and myocardial infarction. N Engl J Med.
366:54–63. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kloner RA, Przyklenk K and Whittaker P:
Deleterious effects of oxygen radicals in ischemia/reperfusion.
Resolved and unresolved issues. Circulation. 80:1115–1127. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chambers JW, Pachori A, Howard S, Iqbal S
and LoGrasso PV: Inhibition of JNK mitochondrial localization and
signaling is protective against ischemia/reperfusion injury in
rats. J Biol Chem. 288:4000–4011. 2013. View Article : Google Scholar :
|
|
5
|
Becker LB: New concepts in reactive oxygen
species and cardiovascular reperfusion physiology. Cardiovasc Res.
61:461–470. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Renlund DG, Gerstenblith G, Lakatta EG,
Jacobus WE, Kallman CH and Weisfeldt ML: Perfusate sodium during
ischemia modifies post-ischemic functional and metabolic recovery
in the rabbit heart. J Mol Cell Cardiol. 16:795–801. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reeves JP, Bailey CA and Hale CC: Redox
modification of sodium-calcium exchange activity in cardiac
sarcolemmal vesicles. J Biol Chem. 1986.261:4948–4955. PubMed/NCBI
|
|
8
|
Kim MS and Akera T: O2 free radicals:
Cause of ischemia-reper-fusion injury to cardiac
Na+-K+-ATPase. Am J Physiol. 252:H252–H257.
1987.PubMed/NCBI
|
|
9
|
Hearse DJ: Stunning: A radical review.
Cardiovasc Drugs Ther. 5:853–876. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tanaka-Esposito C, Chen Q and Lesnefsky
EJ: Blockade of electron transport before ischemia protects
mitochondria and decreases myocardial injury during reperfusion in
aged rat hearts. Transl Res. 160:207–216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McCall JM, Braughler JM and Hall ED: A new
class of compounds for stroke and trauma: Effects of
21-aminosteroid on lipid peroxidation. Acta Anaesthesiol Belg.
38:417–420. 1987.
|
|
12
|
Karlsson J, Love RM, Clarke DJ and Brundin
P: Effects of anaesthetics and lazaroid U-83836E on survival of
transplanted rat dopaminergic neurons. Brain Res. 821:546–550.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sayyed SG, Kumar A and Sharma SS: Effects
of U83836E on nerve functions, hyperalgesia and oxidative stress in
experimental diabetic neuropathy. Life Sci. 79:777–783. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang H, Zheng Z, Gong Y, Zhu B and Xu X:
U83836E inhibits retinal neurodegeneration in early-stage
streptozotocin-induced diabetic rats. Ophthalmic Res. 46:19–24.
2011. View Article : Google Scholar
|
|
15
|
Giese H, Mertsch K and Blasig IE: Effect
of MK-801 and U83836E on a porcine brain capillary endothelial cell
barrier during hypoxia. Neurosci Lett. 191:169–172. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Labruto F, Song X, Valen G and Vaage J:
Lazaroid U-83836E improves tolerance to hemorrhagic shock and limb
ischemia and reperfusion in rats and increases cardiac heat shock
protein 72. Acad Emerg Med. 13:7–12. 2006. View Article : Google Scholar
|
|
17
|
Li H and Lang XE: Protein kinase C
signaling pathway involvement in cardioprotection during isof
lurane pretreatment. Mol Med Rep. 11:2683–2688. 2015.
|
|
18
|
Hongpaisan J, Winters CA and Andrews SB:
Strong calcium entry activates mitochondrial superoxide generation,
upregulating kinase signaling in hippocampal neurons. J Neurosci.
24:10878–10887. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Argaud L, Gateau-Roesch O, Chalabreysse L,
Gomez L, Loufouat J, Thivolet-Béjui F, Robert D and Ovize M:
Preconditioning delays Ca2+-induced mitochondrial
permeability transition. Cardiovasc Res. 61:115–122. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lukács GL and Kapus A: Measurement of the
matrix free Ca2+ concentration in heart mitochondria by
entrapped fura-2 and quin2. Biochem J. 248:609–613. 1987.
View Article : Google Scholar
|
|
21
|
Li D, Liu M, Tao TQ, Song DD, Liu XH and
Shi DZ: Panax quinquefolium saponin attenuates cardiomyocyte
apoptosis and opening of the mitochondrial permeability transition
pore in a rat model of ischemia/reperfusion. Cell Physiol Biochem.
34:1413–1426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Okubo S, Tanabe Y, Fujioka N, Takeda K and
Takekoshi N: Differential activation of protein kinase C between
ischemic and pharmacological preconditioning in the rabbit heart.
Jpn J Physiol. 53:173–180. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Holzerová K, Hlaváčková M, Žurmanová J,
Borchert G, Neckář J, Kolář F, Novák F and Nováková O: Involvement
of PKCepsilon in cardioprotection induced by adaptation to chronic
continuous hypoxia. Physiol Res. 64:191–201. 2015.
|
|
24
|
Jaeschke H and Woolbright BL: Current
strategies to minimize hepatic ischemia reperfusion injury by
targeting reactive oxygen species. Transplant Rev (Orlando).
26:103–114. 2012. View Article : Google Scholar
|
|
25
|
Cheng Z, He W, Zhou X, Lv Q, Xu X, Yang S,
Zhao C and Guo L: Cordycepin protects against cerebral
ischemia/reperfusion injury in vivo and in vitro. Eur J Pharmacol.
664:20–28. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao ZQ: Oxidative stress-elicited
myocardial apoptosis during reperfusion. Curr Opin Pharmacol.
4:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Limón-Pacheco J and Gonsebatt ME: The role
of antioxidants and antioxidant-related enzymes in protective
responses to environmentally induced oxidative stress. Mutat Res.
674:137–147. 2009. View Article : Google Scholar
|
|
28
|
Baines CP: The mitochondrial permeability
transition pore and ischemia-reperfusion injury. Basic Res Cardiol.
104:181–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Javadov S and Karmazyn M: Mitochondrial
permeability transition pore opening as an endpoint to initiate
cell death and as a putative target for cardioprotection. Cell
Physiol Biochem. 20:1–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Halestrap AP, Clarke SJ and Javadov SA:
Mitochondrial permeability transition pore opening during
myocardial reperfusion - a target for cardioprotection. Cardiovasc
Res. 61:372–385. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Budas GR, Church ill EN and Mochly-Rosen
D: Cardioprotective mechanisms of PKC isozyme-selective activators
and inhibitors in the treatment of ischemia-reperfusion injury.
Pharmacol Res. 55:523–536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kawamura S, Yoshida K, Miura T, Mizukami Y
and Matsuzaki M: Ischemic preconditioning translocates PKC-delta
and -epsilon, which mediate functional protection in isolated rat
heart. Am J Physiol. 275:H2266–H2271. 1998.PubMed/NCBI
|
|
33
|
Wang Y and Ashraf M: Role of protein
kinase C in mitochondrial KATP channel-mediated protection against
Ca2+ overload injury in rat myocardium. Circ Res.
84:1156–1165. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Agudo-López A, Miguel BG, Fernández I and
Martínez AM: Role of protein kinase C and mitochondrial
permeability transition pore in the neuroprotective effect of
ceramide in ischemia-induced cell death. FEBS Lett. 585:99–103.
2011. View Article : Google Scholar
|
|
35
|
Wang C, Hu SM, Xie H, Qiao SG, Liu H and
Liu CF: Role of mitochondrial ATP-sensitive potassium
channel-mediated PKC-ε in delayed protection against myocardial
ischemia/reperfusion injury in isolated hearts of
sevoflurane-preconditioned rats. Braz J Med Biol Res. 48:528–536.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang X, Cohen MV and Downey JM: Mechanism
of cardio-protection by early ischemic preconditioning. Cardiovasc
Drugs Ther. 24:225–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Otani H: Reactive oxygen species as
mediators of signal transduction in ischemic preconditioning.
Antioxid Redox Signal. 6:449–469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cohen MV and Downey JM: Adenosine: Trigger
and mediator of cardioprotection. Basic Res Cardiol. 103:203–215.
2008. View Article : Google Scholar
|