Introduction
Human oral cancer is a frequently occurring subclass
of head and neck tumours. Surgery, radiation and chemotherapy are
the predominant clinical therapeutic strategies for oral cancer.
However, despite advances in curative multimodal treatments, the
overall survival rate in patients with oral cancer remains
unsatisfactory (1,2). Therefore, it is essential to
determine the most effective therapeutic agents to control this
disease and enhance patient quality of life.
Apoptosis, or programmed cell death, is regulated in
a complex manner by a multitude of factors (3). B-cell lymphoma/leukaemia-2 (Bcl-2)
family proteins are important regulators of cell death, and the
dysregulation of Bcl-2 family members is key in certain human
diseases, including cancer. Myeloid cell leukaemia-1 (Mcl-1) is a
pro-survival member of the Bcl-2 protein family that suppresses
apoptosis by inhibiting the activity of pro-apoptotic proteins
(4,5). Moreover, Mcl-1 expression is highly
amplified in a variety of human cancers and is frequently
associated with chemotherapeutic resistance and recurrence,
suggesting that overexpression of Mcl-1 may contribute to apoptotic
evasion and malignant tumour growth. Therefore, targeting Mcl-1
expression in these cancers, using genetic and pharmacological
approaches, represents a potential means of developing novel
efficacious cancer therapies (6).
Metformin (1,1-dimethylbiguanide hydrochloride) is a
biguanide traditional oral hypoglycaemic agent that is widely used
in the clinic for the treatment of type 2 diabetes mellitus. It
reduces plasma glucose levels by increasing fatty acid oxidation
and glucose utilisation, decreasing hepatic gluconeogenesis
(7). Recently, metformin has
received considerable attention for its antitumour efficacy against
numerous types of malignancies. For example, retrospective studies
in patients with type 2 diabetes showed reduced cancer incidence in
patients who had been treated with metformin for several years
(8–11). Furthermore, other studies have
demonstrated that metformin can inhibit tumour growth in
vitro and increase tumour sensitivity to chemotherapeutic drugs
(12–15). All of these studies strongly
support the clinical development of metformin as a potentially
useful therapeutic agent for cancer.
In the present study, experiments were conducted to
examine the effects of metformin on KB human oral cancer cells and
determine whether metformin induces apoptosis via Bcl-2 family
proteins. Moreover, the present study aimed to further elucidate
the mechanism underlying Mcl-1 regulation.
Materials and methods
Chemicals and reagents
Metformin was purchased from Sigma-Aldrich (St.
Louis, MO, USA). Metformin solution was prepared by dissolving
metformin in phosphate-buffered saline (PBS). Minimum essential
medium (MEM), foetal bovine serum (FBS) and PBS were purchased from
Gibco Life Technologies (Grand Island, NY, USA). An Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis
detection kit was purchased from Keygen Biotech (Nanjing, China).
Anti-Mcl-1 (rabbit anti-human; 1:100), anti-Bax (rabbit anti-human;
1:1,000), anti-Bim (rabbit anti-human; 1:1,000), and anti-caspase-3
(rabbit anti-human; 1:1,000) monoclonal antibodies were obtained
from Abcam (Cambridge, UK). Mouse anti-β-actin antibody (1:2,000)
was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA,
USA). Cells were transfected with miR-26a mimics, negative control
or miR-26a inhibitor purchased from Guangzhou RiboBio (Guangzhou,
China).
Cell lines and cell culture
The KB human oral cancer cell line was obtained from
the Shanghai Cell Bank at the Chinese Academy of Sciences
(Shanghai, China). Cells were cultured in MEM with 10% FBS, and
supplemented with 100 U/ml penicillin and 100 mg/ml streptomycin in
a humidified incubator at 37°C containing 5% CO2. Cells
were passaged at 80% confluence using 0.25% trypsin (Gibco Life
Technologies).
Cell proliferation assay
The effect of metformin on KB cell viability was
evaluated using the 3-(4,5-dimethylthia-zolyl-2)-2-5
diphenyltetrazolium bromide (MTT) assay. KB cells in the
exponential growth phase were plated in 96-well plates at
8×103/well in a final volume of 100 µl. KB cells
were exposed to different concentrations of metformin for 24, 48
and 72 h in a humidified incubator at 37°C with 5% CO2.
For the MTT assay, cells were then incubated with MTT (5 mg/ml in
PBS) for 4 h. Subsequently, the MTT solution was removed and
replaced with 150 µl dimethyl sulphoxide/well, and the
absorbance was measured at 490 nm using an automated microplate
reader (Synergy™ HT; Bio-Tek Instruments, Inc., Winooski, VT, USA).
Experiments were repeated in triplicate, and three parallel samples
were measured each time.
Annexin V-FITC/PI apoptosis assay
Apoptosis was assessed using double staining with
Annexin V-FITC/PI. Briefly, KB cells were treated with metformin at
different doses in a 12-well plate for 24 h. Cells were harvested,
rinsed twice with PBS and re-suspended in 300 µl ice-cold
binding buffer. They were then stained with Annexin V-FITC/PI for
15 min in the dark. Stained cells were analysed using a flow
cytometer (Accuri C6; BD Biosciences, Franklin Lakes, NJ, USA).
Western blot analysis
KB cells grown in 6-well plates at a density of
3×105 cells/well were treated with metformin for various
incubation periods. Cells were collected by centrifugation and
lysed in radioimmunoprecipitation assay buffer (Beyotime Institute
of Biotechnology, Beijing, China) for 30 min on ice. Cell lysates
were centrifuged at 12,000 × g for 30 min at 4°C. Total protein (50
µg) was separated by 15% sodium dodecyl
sulphate-polyacrylamide gel (Beyotime Institute of Biotechnology)
electrophoresis (70 V, 30 min; 120 V, 90 min) and
electrophoretically transferred to polyvinylidene fluoride
membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked
with 5% non-fat dry milk for 4 h at room temperature and
subsequently incubated with the appropriate antibody for 4 h at
room temperature or overnight at 4°C. After washing three times
with Tris-PBS, the membranes were incubated with the corresponding
secondary antibody. β-actin was used as a control for protein
loading.
Plasmid transfection
The pCMV-HA-Mcl-1 plasmid and control plasmid were
obtained from GenePharma (Shanghai, China). KB cells were
transfected using Lipofectamine 2000 reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer's
instructions. After cultivation for 48 h, total cell lysates were
prepared for western blot analysis or an MTT assay.
Reverse transcription-quantitative
polymerase chain reaction for miRNAs
Reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) was performed for miRNA detection. Total RNA was
extracted from KB cells treated with or without metformin using
TRIzol reagent (Invitrogen Life Technologies) and an miRNeasy Mini
kit (Qiagen, Shanghai, China) according to the manufacturer's
instructions. For the detection of miR-26a, mature hsa-miR-26a and
U6 primer from All-in-One™ miRNA qPCR Primer (GeneCopoeia,
Guangzhou, China) was used. RT-qPCR was performed with the
All-in-One™ miRNA qRT-PCR Detection kit (AOMD-Q050, GeneCopoeia) in
an ABI StepOne™ Real-Time PCR System (Applied Biosystems Life
Technologies, Foster City, CA, USA) at 95°C for 10 min, followed by
40 cycles of 95°C for 10 sec, 60°C for 30 sec and 72°C for 15 sec.
PCR data were analysed using the 2−ΔΔCt method and were
normalised against RNU6B expression in each sample. Paired
Student's t-test was performed to appraise the difference in the
level of miRNA expression.
Transfection of miRNA precursor
miR-26a
KB cells were seeded in 6-well plates at a density
of 3×105 cells/well. Cells at 60–80% confluency were
transfected with microRNA (miR)-26a mimics, negative control,
miR-26a inhibitor, or inhibitor control at a final concentration of
20 µM using Lipofectamine 2000 reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer
instructions. Cells were harvested 48 h later for western blot
analysis and PI viability assays.
Plate clone formation assay
Cells were seeded at a density of 1×104
cells/well in 6-well plates with growth medium 48 h after
transfection and incubated in MEM containing 10% FBS at 37°C with
5% CO2 for 7 days. The colonies were washed with PBS,
fixed with 10% formaldehyde for 10 min on ice, and stained with
1.0% crystal violet for 30 min.
Propidium iodide staining
Prior to metformin treatment, cells were plated in
12-well plates and subsequently transfected with miR-26a mimics,
negative control, miR-26a inhibitor, or inhibitor control for 48 h.
Cells were then subjected to PI staining and evaluated using flow
cytometry (Accuri C6).
Statistical analysis
All experiments were repeated at least three times
and the values were expressed as the mean ± standard deviation.
Statistical significance was determined by Student's t-test using
SPSS software, version 13.0 (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Metformin inhibits the proliferation and
induces apoptosis of KB cells
To evaluate the growth inhibitory effect of
metformin on human KB cells in vitro, cells were treated
with different concentrations of metformin for 24, 48 and 72 h. The
rate of cell proliferation was inversely related to the time of
exposure to metformin and the metformin concentration (Fig. 1A).
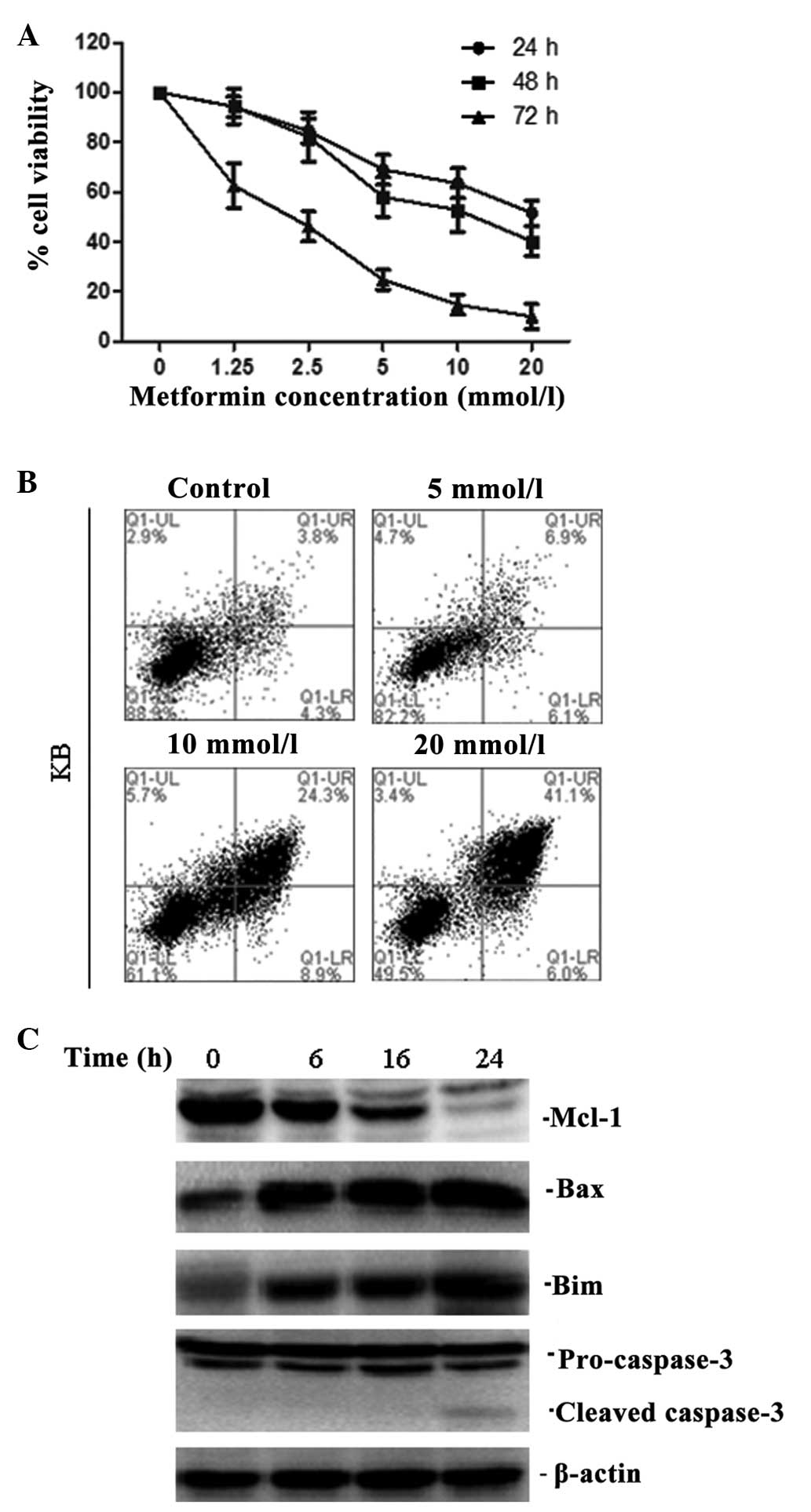 | Figure 1Metformin inhibited the proliferation
and induced apoptosis of KB cells. (A) KB cells were treated with
different concentrations of metformin (0, 1.25, 2.5, 5, 10 and 20
mmol/l) for 24, 48 and 72 h, and the cell viability was analysed
using an 3-(4,5-dimethylthiazolyl-2)-2-5 diphenyltetrazolium
bromide assay. (B) Cells were treated with various concentrations
of metformin (5, 10, 20 mmol/l) and were harvested after incubation
for 24 h before being analysed using Annexin V/PI staining. (C)
Whole cell lysates from KB treated with the combination of
metformin (10 mM) for 0, 6, 16 and 24 h were subjected to western
blot analysis for Mcl-1, Bax, Bim and caspase-3. One representative
blot out of three is shown. |
Flow cytometric analysis revealed that metformin
markedly increased the number of apoptotic KB cells (Fig. 1B). Furthermore, in KB cells,
metformin decreased the expression of Mcl-1 and increased the
expression of Bim and Bax in a time-dependent manner (Fig. 1C). Downstream of apoptosis
signalling pathways, there was a significant activation of
caspase-3 cleavage (Fig. 1C).
These results confirmed the prediction that metformin induces
apoptosis, and that this is predominantly through the
mitochondria-mediated internal pathway.
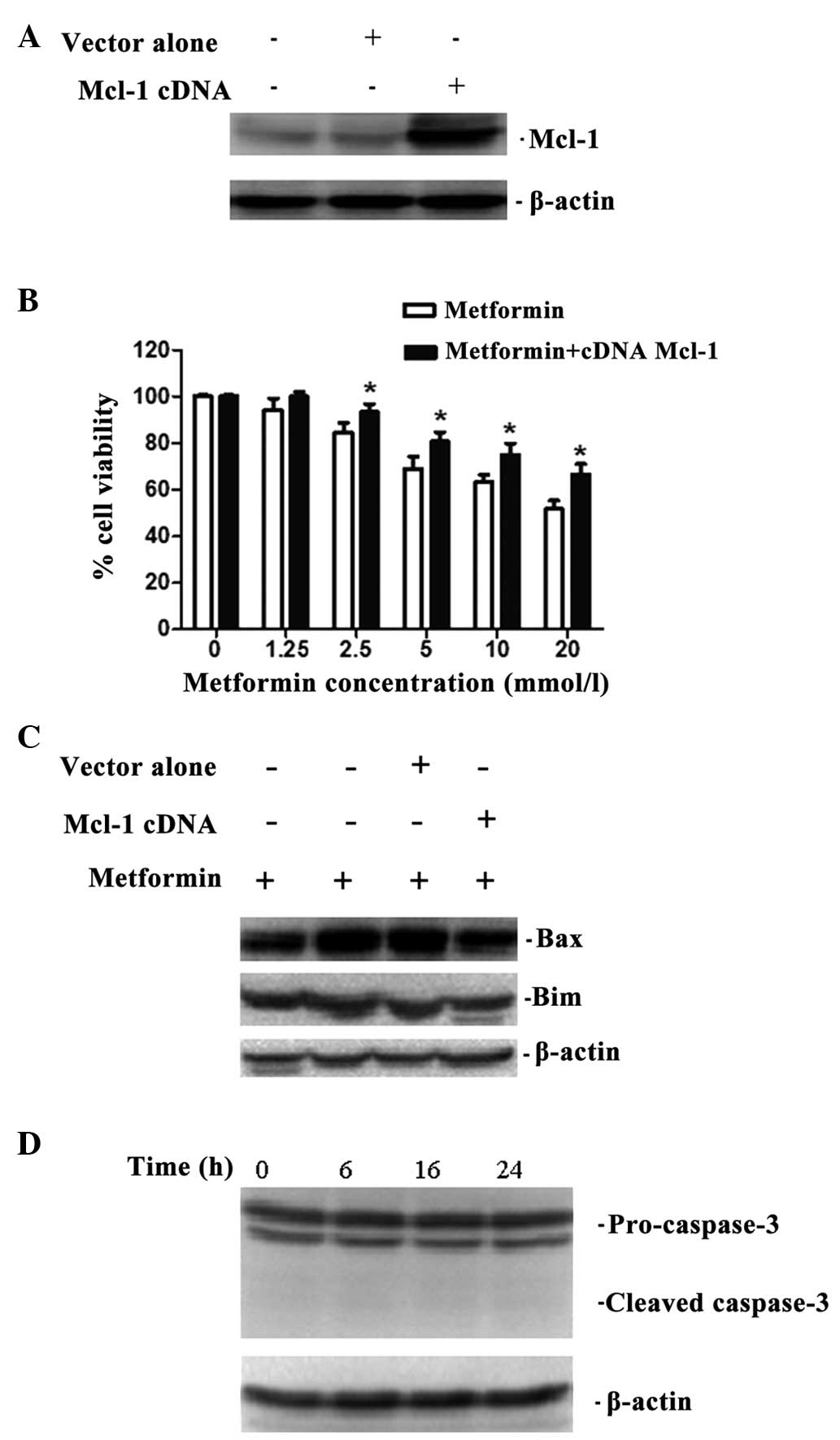
Upregulation of Mcl-1 protects KB cells
from apoptosis
To evaluate the association between Mcl-1 and
metformin-induced apoptosis, an Mcl-1 expression plasmid was
transfected into KB cells (Fig.
2A). Then, overexpression of Mcl-1 significantly increased cell
viability in response to metformin treatment and decreased the
expression of Bim and Bax compared with their respective controls
(Fig. 2B and C). In addition, the
cleaved products of caspase-3 were scarcely activated in the
Mcl-1-overexpressing KB cells (Fig.
2D). These findings suggest that Mcl-1 may be important in
metformin-induced apoptosis as an anti-apoptotic protein.
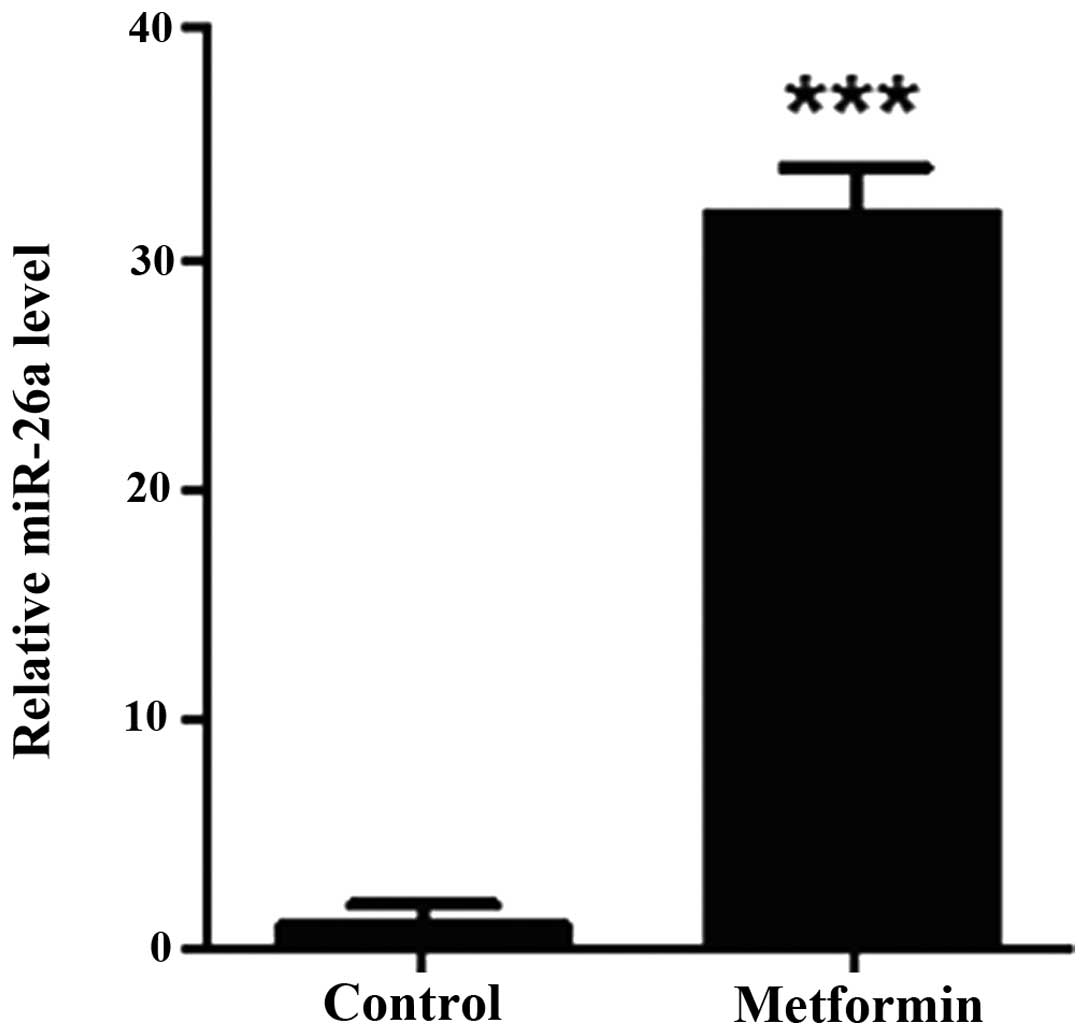
Metformin increases the expression of
miR-26a
Expression of miR-26a was analysed by RT-qPCR and
normalised against an endogenous control (RNU6B). Total RNA was
extracted from KB cells treated with or without 10 mmol/l
metformin. The results indicated that miR-26a was significantly
upregulated in metformin-treated cells compared with non-treated
cells (Fig. 3).
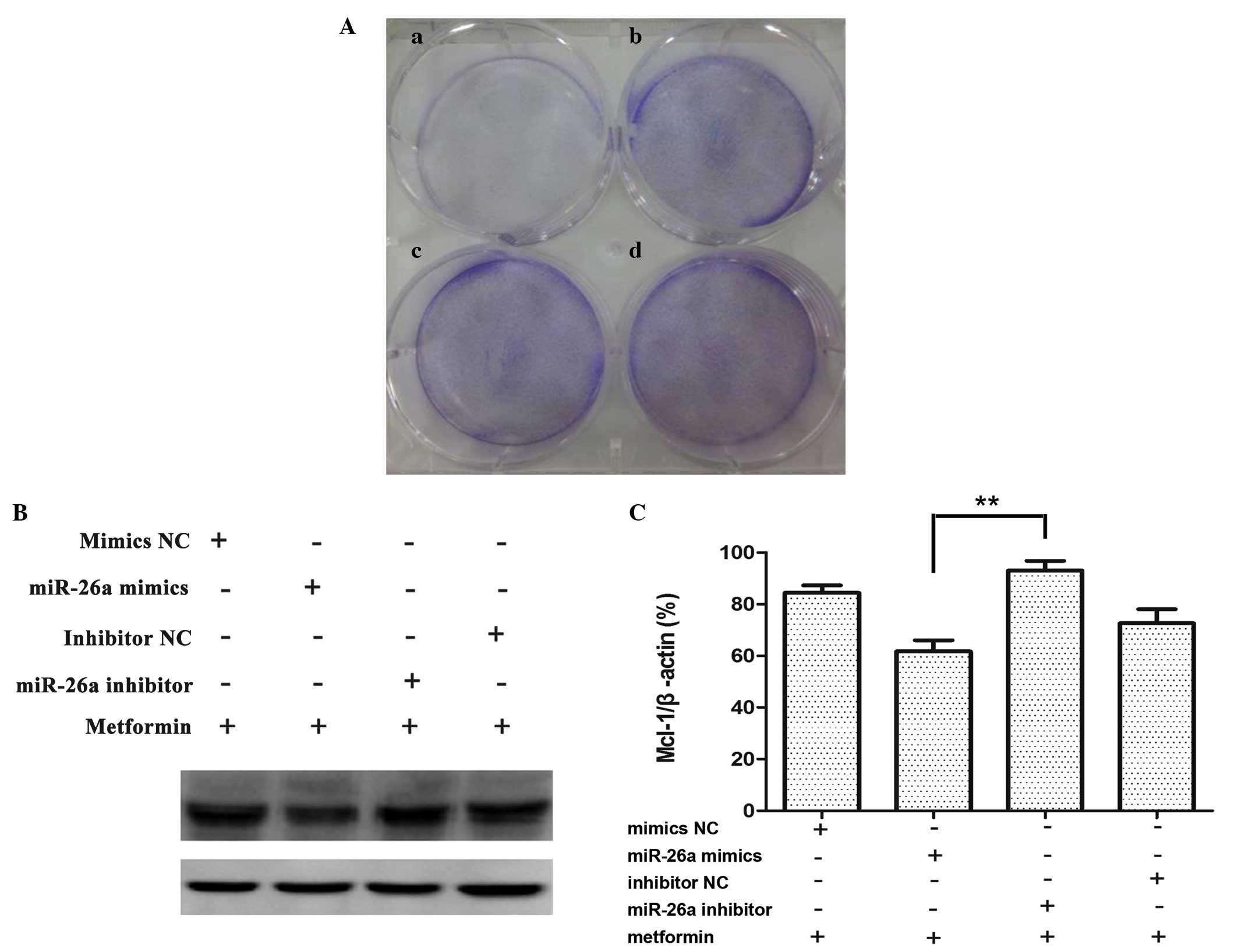
Overexpression of miR-26a reduces cell
viability and regulated Mcl-1 expression in KB cells
In order to confirm the biological function of
miR-26a, KB cells were transfected with miR-26a mimics or
corresponding negative controls for 7 days, miR-26a inhibited KB
cell growth compared with miR-Ctrl (Fig. 4A). As shown in Fig. 4B, the level of Mcl-1 decreased in
cells transfected with miR-26a mimics compared with cells
transfected with the corresponding negative control. By contrast,
Mcl-1 was increased in KB cells transfected with anti-miR-26a
inhibitor compared with cells transfected with the corresponding
negative control. These experiments confirm that the expression of
Mcl-1 can be regulated by miR-26a.
Discussion
Metformin is widely used for the treatment of
diabetes mellitus, however, recent epidemiological and preclinical
studies have demonstrated that metformin is also a promising
anticancer agent. In this study, exposure of KB oral cancer cells
to metformin led to a time- and concentration-dependent inhibition
of proliferation. Moreover, metformin was found to induce
apoptosis.
Apoptosis is an activated cellular death process
that is induced by physiological or pathological factors to
eliminate redundant and damaged cells. Furthermore, apoptosis is an
important defence against cancer (16). When cells are exposed to various
stress stimuli, Bim relays apoptotic signals to the mitochondria
through the activation of Bax, resulting in an increase in
mitochondrial outer membrane permeabilisation and the consequent
release of cytochrome c into the cytosol. Cytochrome
c then binds to Apaf-1 to ensure the formation of the
apoptosome that leads to activation of caspase-9 and the induction
of the apoptosis-promoting caspase cascade (17). By contrast, anti-apoptotic Bcl-2
proteins, such as Mcl-1 serve to inhibit apoptosis. Generally,
promotion of cell survival by Mcl-1 is hypothesised to be due to
sequestration of Bim, thereby inhibiting Bax/Bim interaction in the
mitochondrial outer membrane. Alternatively, Mcl-1 may directly
bind to Bax and maintain it in an inactive conformation. This
competition leads to the suppression of cytochrome c release
from the mitochondria and a reduction in Apaf-1-dependent
activation of caspase-3. Therefore, Mcl-1 acts in a significant
regulatory role that can modulate the expression of pro-apoptotic
proteins and control cell fate decisions (18). Consistent with this theory, the
results of the present study demonstrate that metformin
downregulated the expression of Mcl-1 and upregulated the
expression of Bim and Bax. Conversely, overexpression of Mcl-1
downregulated Bim and Bax expression, suggesting that Mcl-1 is
involved in KB cell apoptosis, and predominantly through
mitochondria-mediated pathways.
In vitro and in vivo studies have
shown that the expression of various miRNAs was markedly altered by
treatment with metformin (19). By
binding to the 3′-untranslated region (UTR) of target mRNAs, miRNAs
act as endogenous sequence-specific suppressors that regulate gene
expression by eliciting mRNA degradation or inhibition of mRNA
translation (20). Therefore,
miRNAs are important regulators of tumourigenicity, proliferation,
apoptosis, invasion and metastasis. Numerous studies have
highlighted the role of miRNAs in Mcl-1 regulation (21–23).
Analysis of candidate target genes for miR-26a using miRBase
previously revealed perfect complementarity between miR-26a and the
3′ UTR of Mcl-1 over the first 9 nucleotides (21,23).
In the current study, it was demonstrated that miR-26a
significantly downregulated Mcl-1, which led to the apoptosis of KB
cells. Furthermore, miR-26a overexpression suppressed in
vitro cell proliferation. Conversely, downregulation of miR-26a
inhibited apoptosis and promoted proliferation. These results
indicate that miR-26a may be a novel tumour suppressor that is
important in the regulation of tumoural Mcl-1 expression.
In conclusion, the results of this study confirm
that metformin restricted tumour growth and induced apoptosis
predominantly by regulating the expression of Bcl-2 family members
in the KB human oral cancer cell line. Furthermore, it was
demonstrated that metformin increased expression of miR-26a in KB
cells and miR-26a itself mediated inhibition of proliferation and
induction of apoptosis. In combination, these findings suggest that
metformin induces apoptosis in human oral cancer cells by
downregulating Mcl-1 via miR-26a and these results provide in
vitro evidence to support the use of metformin as a novel and
efficient candidate for the treatment of human oral cancer.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81000992 and 81072207); the
Natural Science Foundation of Anhui Province (grant no.
KJ2012A201); and Graduate Scientific Research and Innovation
Projects of Bengbu Medical College of the Anhui Province (grant no.
Byycx1315).
References
|
1
|
Cruz GD, Ostroff JS, Kumar JV and Gajendra
S: Preventing and detecting oral cancer. Oral health care
providers' readiness to provide health behavior counseling and oral
cancer examinations. J Am Dent Assoc. 136:594–601. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rogers SN, Brown JS, Woolgar JA, Lowe D,
Magennis P, Shaw RJ and Vaughan D: Survival following primary
surgery for oral cancer. Oral Oncol. 45:201–211. 2009. View Article : Google Scholar
|
|
3
|
Plati J, Bucur O and Khosravi-Far R:
Apoptotic cell signaling in cancer progression and therapy. Integr
Biol (Camb). 3:279–296. 2011. View Article : Google Scholar
|
|
4
|
Perciavalle RM and Opferman JT: Delving
deeper: MCL-1′s contributions to normal and cancer biology. Trends
Cell Biol. 23:22–29. 2013. View Article : Google Scholar
|
|
5
|
Thomas LW, Lam C and Edwards SW: Mcl-1;
the molecular regulation of protein function. FEBS Lett.
584:2981–2989. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Quinn BA, Dash R, Azab B, Sarkar S, Das
SK, Kumar S, Oyesanya RA, Dasgupta S, Dent P, Grant S, et al:
Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig
drugs. 20:1397–1411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Foretz M and Viollet B: New promises for
metformin: Advances in the understanding of its mechanisms of
action. Med Sci (Paris). 30:82–92. 2014.In French. View Article : Google Scholar
|
|
8
|
Evans JM, Donnelly LA, Emslie Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Noto H, Goto A, Tsujimoto T and Noda M:
Cancer risk in diabetic patients treated with metformin: A
systematic review and meta-analysis. PLoS One. 7:e334112012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: A systematic review and meta-analysis. Cancer
Prev Res (Phila). 3:1451–1461. 2010. View Article : Google Scholar
|
|
11
|
Landman GW, Kleefstra N, van Haterenm KJ,
Groenier KH, Gans RO and Bilo HJ: Metformin associated with lower
cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care.
33:322–326. 2010. View Article : Google Scholar :
|
|
12
|
Wang F, Xu J, Xia F, Liu Z, Zhao S, Liu H
and Jiang Z: Effects of metformin on human oral cancer KB cell
proliferation and apoptosis in vitro. Nan Fang Yi Ke Da Xue Xue
Bao. 34:159–163. 2014.In Chinese. PubMed/NCBI
|
|
13
|
Marini C, Salani B, Massollo M, Amaro A,
Esposito AI, Orengo AM, Capitanio S, Emionite L, Riondato M,
Bottoni G, et al: Direct inhibition of hexokinase activity by
metformin at least partially impairs glucose metabolism and tumor
growth in experimental breast cancer. Cell Cycle. 12:3490–3499.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mohammed A, Janakiram NB, Brewer M,
Ritchie RL, Marya A, Lightfoot S, Steele VE and Rao CV:
Antidiabetic drug metformin prevents progression of pancreatic
cancer by targeting in part cancer stem cells and mTOR signaling.
Transl Oncol. 6:649–659. 2013. View Article : Google Scholar
|
|
15
|
Miyoshi H, Kato K, Iwama H, Maeda E,
Sakamoto T, Fujita K, Toyota Y, Tani J, Nomura T, Mimura S, et al:
Effect of the anti-diabetic drug metformin in hepatocellular
carcinoma in vitro and in vivo. Int J Oncol. 45:322–332.
2014.PubMed/NCBI
|
|
16
|
Melet A, Song K, Bucur O, Jagani Z,
Grassian AR and Khosravi-Far R: Apoptotic pathways in tumor
progression and therapy. Adv Exp Med Biol. 615:47–79. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Del Gaizo Moore V and Letai A: BH3
profiling-measuring integrated function of the mitochondrial
apoptotic pathway to predict cell fate decisions. Cancer Lett.
332:202–205. 2013. View Article : Google Scholar
|
|
18
|
Karnak D and Xu L: Chemosensitizaition of
prostate cancer by modulating Bcl-2 family proteins. Curr Drug
Targets. 11:699–707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li W, Yuan Y, Huang L, Qiao M and Zhang Y:
Metformin alters the expression profiles of microRNAs in human
pancreatic cancer cells. Diabetes Res Clin Pract. 96:187–195. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O'Hara SP, Mott JL, Splinter PL, Gores GJ
and LaRusso NF: MicroRNAs: Key modulators of posttranscriptional
gene expression. Gastroenterology. 136:17–25. 2009. View Article : Google Scholar
|
|
21
|
Yang X, Liang L, Zhang XF, Jia HL, Qin Y,
Zhu XC, Gao XM, Qiao P, Zheng Y, Sheng YY, et al: MicroRNA-26a
suppresses tumor growth and metastasis of human hepatocellular
carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology.
58:158–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang YK, Wang H, Leng Y, Li ZL, Yang YF,
Xiao FJ, Li QF, Chen XQ and Wang LS: Overexpression of microRNA-29b
induces apoptosis of multiple myeloma cells through down regulating
Mcl-1. Biochem Biophys Res Commun. 414:233–239. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao J, Li L, Wu M, Liu M and Xie X, Guo J,
Tang H and Xie X: MiR-26a inhibits proliferation and migration of
breast cancer through repression of MCL-1. PLoS One. 8:e651382013.
View Article : Google Scholar : PubMed/NCBI
|