Introduction
Age-related macular degeneration (AMD) is a retinal
degenerative disease, which causes progressive loss of central
vision in the elderly (1).
Clinically, AMD is comprised of two forms: Dry and wet. Dry AMD is
characterized by drusen formation and geographical atrophy. Wet AMD
is characterized by choroidal neovascularization (CNV) and the
subsequent development of hemorrhage, exudation, scarring or
retinal detachment. The retinal pigment epithelium (RPE) is the
primary target of AMD (2,3). During AMD progression, the RPE is
damaged, accompanied by a disruption of the choroidal blood-eye
barrier and degeneration of photoreceptors. In addition, RPE cells
proliferate and secrete various proangiogenic factors, including
vascular endothelial growth factor (VEGF), which serves an
important role in AMD-associated CNV (4).
Although the precise underlying mechanisms of AMD
are not fully understood, numerous lines of evidence have indicated
that endoplasmic reticulum (ER) stress contributes to the etiology
of RPE cell damage and neovascularization formation (5–7).
Several types of cellular stress, including hypoxia (8), infection (9), nutrient deprivation (10), oxidative stress (11) and dysfunctional calcium homeostasis
(12), may induce accumulation of
unfolded proteins in the ER lumen. This process activates protein
kinase R (PKR)-like endoplasmic reticulum kinase (PERK),
inositol-requiring kinase 1 and activating transcription factor
(ATF) 6, and initiates unfolded protein response (UPR) signaling
pathways (13,14). As one of three primary UPR
effectors, PERK directly phosphorylates eukaryotic initiation
factor 2α (eIF2α), which consequently inhibits initiation of
general translation and reduces ER burden (15). However, eIF2α phosphorylation leads
to selective translation of numerous mRNAs, including ATF4. ATF4
induces expression of the proapoptotic transcription factor
CCAAT/enhancer-binding protein homologous protein (CHOP), which
mediates PERK-induced apoptosis (16). Furthermore, ATF4 serves an
important role in VEGF expression under hypoxia or chemical stress
(17,18). Activation of the PERK/eIF2α/ATF4
signaling pathway has been reported in numerous retinal
degenerative diseases including AMD (16), glaucomatous retinopathy (19) and diabetic retinopathy (20). Disruption of PERK activity has been
demonstrated to reduce hydroquinone-induced apoptosis and
hypoxia-induced VEGF expression in human RPE cells in vitro
(5,21). These findings suggested that
inhibition of the PERK/eIF2α/ATF4 signaling pathway may be a novel
therapeutic strategy for the treatment of AMD.
Screening for PERK inhibitors has led to the
identification of a family of highly selective and potent
molecules, including GSK2606414 (molecular formula,
C24H20F3N5O).
GSK2606414 is an adenosine triphosphate-competitive inhibitor of
PERK with a half maximal inhibitory concentration (IC50)
of 0.4 nM (22). GSK2606414 and
its analogues have previously been demonstrated to inhibit ER
stress, delay tumor growth and prevent neurodegeneration (23–25).
To the best of our knowledge, the effects of GSK2606414 on cell
proliferation and PERK signaling in cultured RPE cells under ER
stress have yet to be reported.
The present study investigated the effects of
GSK2606414 on PERK signaling and VEGF expression levels in RPE
cells under ER stress, and examined the potential underlying
mechanisms of GSK2606414 in RPE proliferation. The results of the
present study provided valuable information for drug development or
potential novel strategies for the treatment of AMD.
Materials and methods
Cell culture
ARPE-19 human RPE cells were purchased from the
American Type Culture Collection (Manassas, VA, USA). Cells were
cultured in a 1:1 mix of Dulbecco's modified Eagle's medium and
F-12 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.). Cells were cultured at 37°C in a humidified
atmosphere containing 5% CO2.
Reagents
GSK2606414 (EMD Millipore, Billerica, MA, USA) and
thapsigargin (TG; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck
KGaA). The concentration of DMSO was <0.1% for all experiments
to avoid cytotoxicity.
Cell Counting kit-8 (CCK8) cell
viability assay
Cell viability was assessed using a CCK8 assay
(Dojindo Molecular Technologies, Inc., Kumamoto, Japan). ARPE-19
cells were seeded into a 96-well plate at a density of
2×103 cells/well and incubated with 0.01, 0.05, 0.1,
0.5, 1, 5, 10 or 50 µM GSK2606414 for 24, 48 or 72 h at 37°C.
Subsequently, the medium was replaced with 100 µl of fresh medium
and CCK8 reagent (10 µl) was added to each well, and cells were
incubated for 2 h at 37°C. Absorbance (optical density) was
measured at a wavelength of 450 nm.
Cell morphology assay
ARPE-19 cells (2×105 cells/well) were
plated on 6-well plates and incubated at 37°C for 24 h. Cells were
subsequently treated with or without 5 µM GSK2606414 for 24 h at
37°C. Cell morphology was observed by phase-contrast microscopy
using an Axiovert 200 inverted microscope (Carl Zeiss AG,
Oberkochen, Germany).
Quantitative detection of apoptosis by
flow cytometry
Cell apoptosis was measured using the Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) Apoptosis
Detection kit (eBioscience, Inc., San Diego, CA, USA) according to
the manufacturer's protocol. ARPE-19 cells were seeded into a 25
cm2 plate at a density of 3×106 cells/well
for 24 h, and were subsequently treated with various concentrations
of GSK2606414 (0.5–5 µM). Cells treated with 200 µM
H2O2 served as a positive control. After 24 h
treatment, cells were washed once with phosphate-buffered saline
(PBS) and resuspended in 1X binding buffer at a concentration of
1×106 cells/ml. Annexin V-FITC (5 µl) was added to 100
µl cell suspensions, and the cells were incubated at room
temperature for 15 min in the dark. Cells were subsequently washed
in 1X binding buffer and resuspended in 200 µl binding buffer,
followed by the addition of 5 µl PI. Annexin V- and PI-stained
cells were analyzed using a Cytomics FC500 flow cytometer (Beckman
Coulter, Inc., Brea, CA, USA) and CXP Analysis Software version 2.2
(Beckman Coulter, Inc.).
Western blotting
ARPE-19 cells were treated with various
concentrations of GSK2606414 (0.5–1 µM) for 1 h, followed by
stimulation with TG (1 µM) for 2 h at 37°C. For the extraction of
total cellular protein, cells were washed twice in cold PBS and
homogenized in radioimmunoprecipitation assay lysis buffer (Cell
Signaling Technology, Inc., Danvers, MA, USA) containing PhosSTOP™
and protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA).
Lysates were centrifuged at 13,000 × g for 15 min at 4°C.
Protein concentrations were measured using a Bicinchoninic Acid
protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). Samples
(40 µg) were separated by 10% (w/v) SDS-PAGE and subsequently
transferred onto polyvinylidene difluoride membranes (Merck KGaA).
Membranes were blocked in 5% bovine serum albumin (Sigma-Aldrich;
Merck KGaA) for 2 h at room temperature and incubated overnight at
4°C with the following primary antibodies: Anti-eIF2α (1:1,000;
Cell Signaling Technology, Inc.; catalog no. 5324),
anti-phosphorylated (p)-eIF2α (1:1,000; Cell Signaling Technology,
Inc.; catalog no. 3398), and anti-β-actin (1:10,000; Sigma-Aldrich;
Merck KGaA; catalog no. A2228). Membranes were incubated with
horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary
antibody (1:5,000; Abcam, Cambridge, UK; catalog no. ab6721) or
(HRP)-conjugated goat anti-mouse secondary antibody (1:5,000;
Abcam; catalog no. ab6789) for 1 h at room temperature, and the
immunoreactive bands were visualized by an Enhanced
Chemiluminescence detection system (Pierce; Thermo Fisher
Scientific, Inc.). Band densitometry was quantified using Quantity
One image analysis software version 4.62 (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (qPCR)
ARPE-19 cells were treated with various
concentrations of GSK2606414 (0.5–5 µM) for 1 h, followed by TG
stimulation (1 µM) for 24 h at 37°C. Total RNA was isolated using a
PureLink RNA Mini kit (Invitrogen; Thermo Fisher Scientific, Inc.)
and quantified by ultraviolet spectrometry at a wavelength of 260
nm. RNA was reverse transcribed into cDNA in a total reaction
volume of 10 µl using PrimeScript RT Master Mix Perfect Real Time
kit (Takara Bio, Inc., Otsu, Japan). The reaction system contained
200 ng/µl total RNA (2 µl), 5X PrimeScript Buffer (2 µl) and
RNase-free distilled water (6 µl); the reactions were incubated at
37°C for 15 min, followed by 85°C for 5 sec. qPCR reactions were
conducted in a 20 µl reaction volume containing SYBR Green
Real-Time PCR Master Mix (10 µl; Roche Diagnostics), diluted cDNA
(2 µl), 10 µM forward primer (1 µl), 10 µM reverse primer (1 µl)
and 6 µl distilled water. The primers used were as follows: ATF4,
forward 5′-CCCTTCACCTTCTTACAACCTC-3′, reverse
3′-GTCTGGCTTCCTATCTCCTTCA-5′; CHOP, forward
5′-ATGAACGGCTCAAGCAGGAA-3′, reverse 3′-TGTGGGATTGAGGGTCACATC-5′;
VEGF, forward 5′-TCACAGGTACAGGGATGAGGACAC-3′, reverse
3′-TCCTGGGCAACTCAGAAGCA-5′; and β-actin, forward
5′-ACAATGTGGCCGAGGACTTT-3′, reverse 3′-TGTGTGGACTTGGGAGAGGA-5′.
Samples were analyzed in triplicate in a LightCycler 480 Instrument
(Roche Diagnostics GmbH, Mannheim, Germany). The reaction
conditions were as follows: Initial denaturation step at 95°C for 5
min, followed by 50 cycles at 95°C for 10 sec, 60°C for 10 sec,
72°C for 10 sec and a final elongation step at 72°C for 10 min. The
2−ΔΔCq method was applied to estimate relative
transcription levels (26), and
the results were normalized to β-actin. ATF4, CHOP and VEGF mRNA
expression levels are expressed relative to the control group.
Statistical analysis
Data were analyzed with SPSS 19.0 (IBM SPSS, Armonk,
NY, USA). Data are expressed as the mean ± standard deviation.
Unpaired Student's t-test was used to compare differences between
two groups. One-way analysis of variance was used to compare
differences between three or more groups, followed by Bonferroni's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
GSK2606414 suppresses proliferation of
human RPE cells
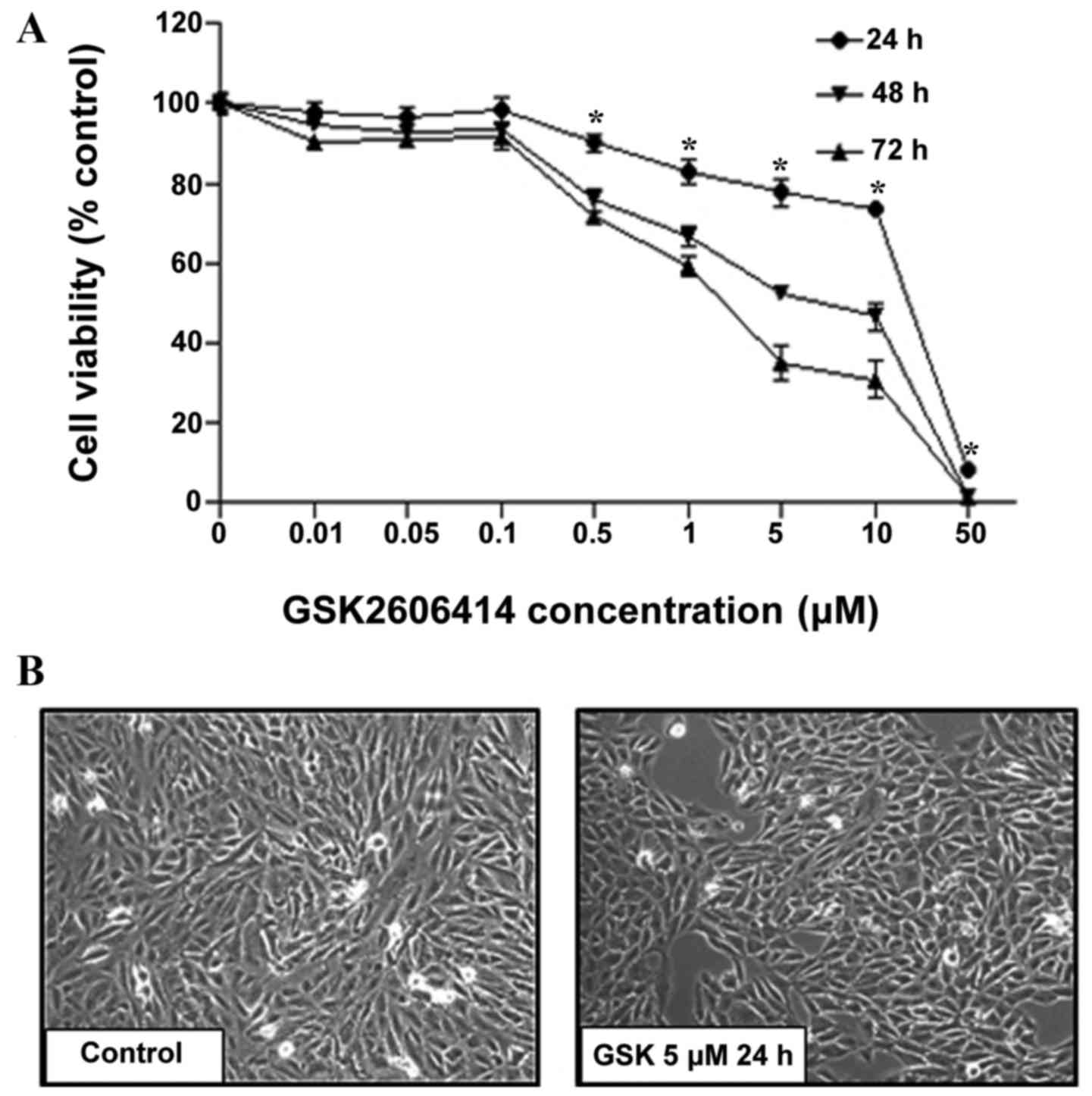
To assess the effects of GSK2606414 on RPE cell
proliferation, ARPE-19 cells were treated with 0.01–50 µM
GSK2606414 for 24, 48 or 72 h. Cell proliferation was quantified by
CCK8 assay. Cell viability was inhibited by GSK2606414 in a dose-
and time-dependent manner (Fig.
1A). At increased concentrations (0.5–50 µM), GSK2606414
significantly inhibited cell proliferation compared with the
control group (P<0.05). The IC50 of GSK2606414 was
1.7 µM in ARPE-19 cells treated with GSK2606414 for 72 h. The
results were confirmed by morphological observation (Fig. 1B), which revealed that GSK2606414
(5 µM) did not cause changes in ARPE-19 cell morphology compared
with control group, but it did significantly reduced cell growth
in vivo.
GSK2606414 does not induce apoptosis
in human RPE cells
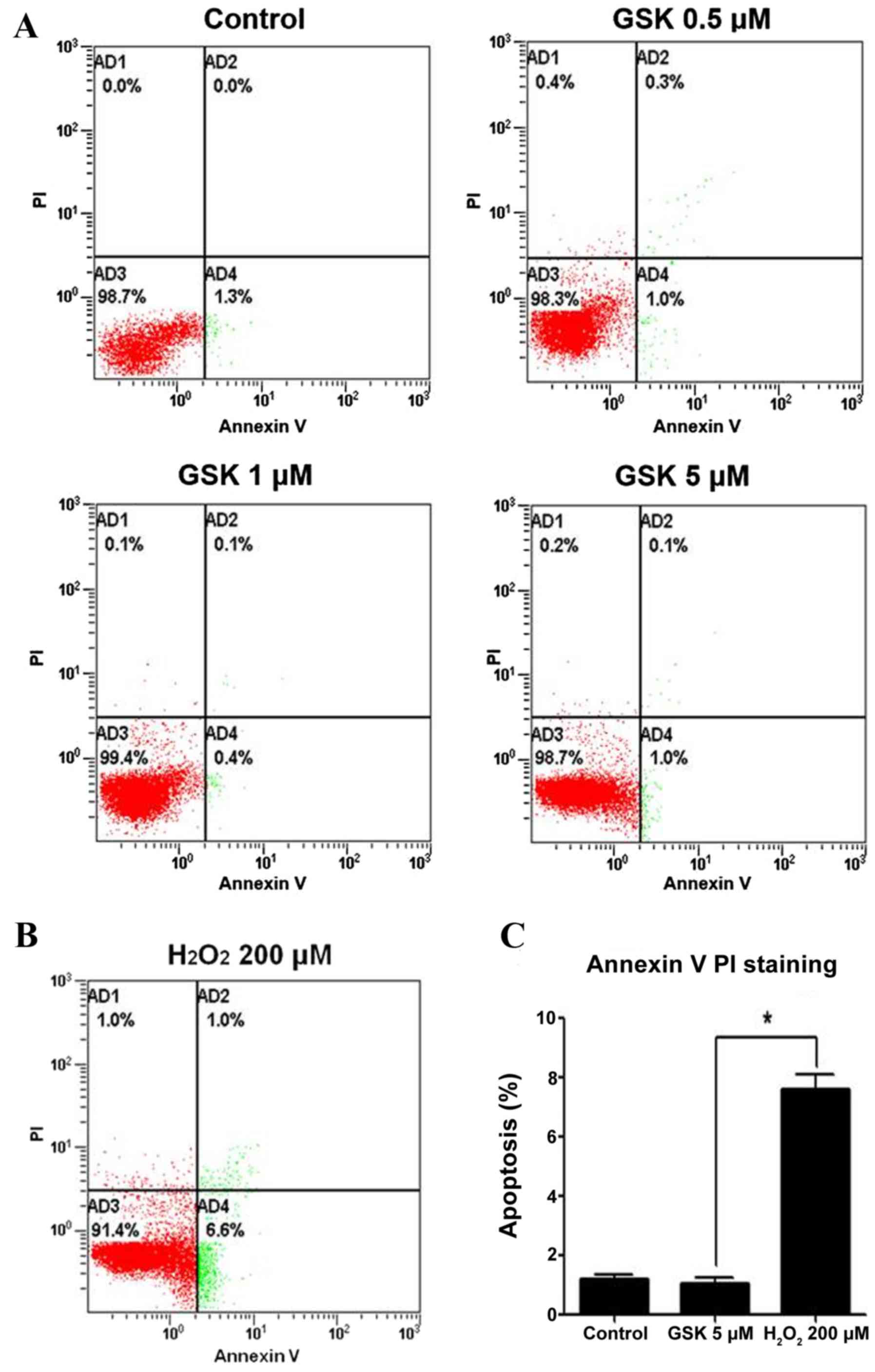
The effects of GSK2606414 on apoptosis were examined
in APRE-19 cells using an Annexin V-FITC/PI Apoptosis Detection
kit. Treatment with 0.5–5 µM GSK2606414 for 24 h did not
significantly induce apoptosis in APRE-19 cells (Fig. 2A). However, apoptosis was
significantly increased in ARPE-19 cells under 200 µM
H2O2-induced oxidative conditions (Fig. 2B) compared with in 5 µM
GSK2606414-treated cells (Fig.
2C).
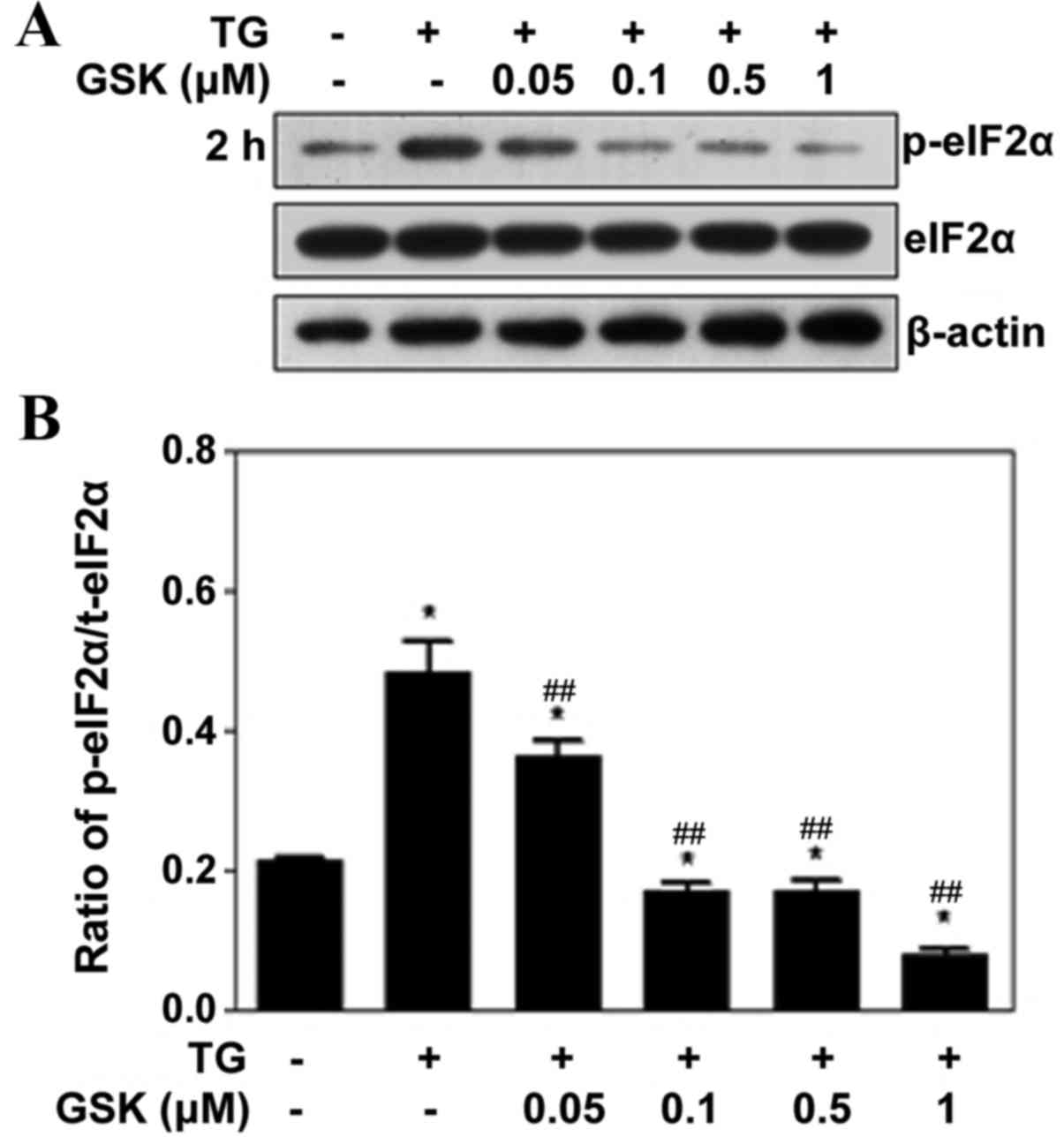
GSK2606414 inhibits eIF2α
phosphorylation in ARPE-19 cells
PERK serves an important role in the UPR by
phosphorylating the translation initiation factor, eIF2α. To
determine the effects of GSK2606414 on eIF2α phosphorylation,
ARPE-19 cells were pretreated with 0.05–1 µM GSK2606414 for 1 h
followed by treatment with the ER stress inducer, TG, for 2 h.
Protein expression levels of p-eIF2α were reduced compared with
eIF2α (Fig. 3A). GSK2606414
treatment dose-dependently inhibited TG-induced eIF2α
phosphorylation (Fig. 3B).
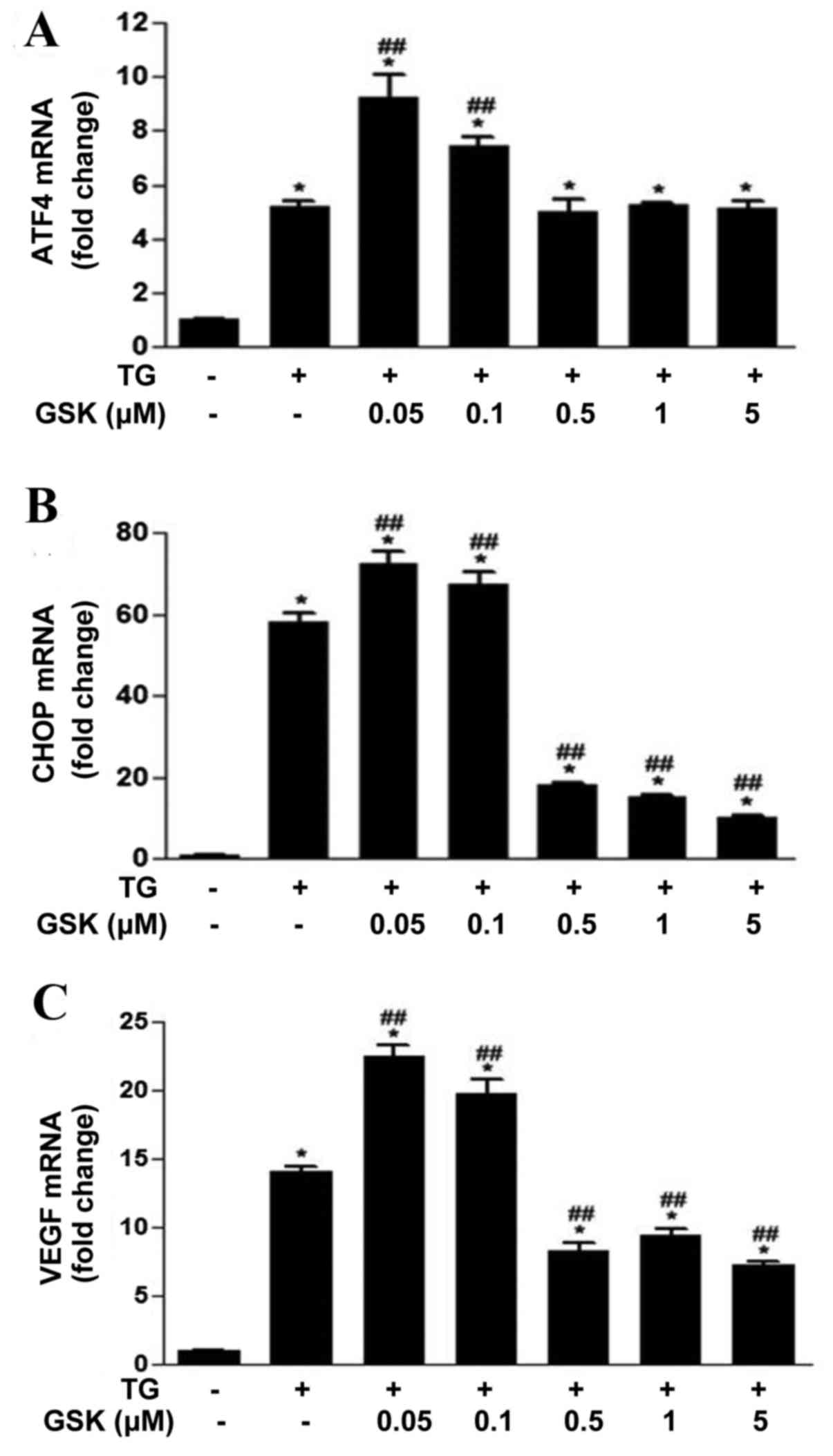
GSK2606414 inhibits ER stress-induced
ATF4, CHOP and VEGF expression in ARPE-19 cells
ATF4 and CHOP are primary transcription factors
involved in the PERK signaling pathway, which serve important roles
in regulating cellular fate and angiogenesis under stress
conditions (15,27). The effects of GSK2606414 on ER
stress-induced ATF4, CHOP and VEGF mRNA expression levels were
examined in ARPE-19 cells. ATF4 (Fig.
4A), CHOP (Fig. 4B) and VEGF
(Fig. 4C) mRNA expression levels
were significantly increased in ARPE-19 cells following TG-induced
ER stress. ATF4, CHOP and VEGF mRNA expression levels were
increased in cells treated with low concentrations of GSK2606414
(0.05–0.1 µM) compared with cells treated with TG alone. CHOP and
VEGF mRNA expression levels were significantly reduced in cells
treated with increased concentrations of GSK2606414 (0.5–5 µM)
compared with cells treated with TG alone.
Discussion
The present study investigated the effects of
GSK2606414 on cell proliferation, apoptosis, and ATF4, CHOP and
VEGF expression levels in human RPE cells under TG-induced ER
stress. GSK2606414 was originally synthesized by GlaxoSmithKline as
a potent and selective inhibitor of PERK, with IC50
values in the low nanomolar range (22,28).
GSK2606414 and its analogue GSK2656157 have previously been
demonstrated to inhibit tumor growth in a murine xenograft model
(21,22). RPE cell proliferation is involved
in late AMD and proliferative retinopathy (4,29–31).
In the present study, GSK2606414 was revealed to inhibit RPE cell
proliferation in a dose- and time-dependent manner. The inhibitory
effects of GSK2606414 on cell proliferation were more potent at
concentrations >0.5 µM. GSK2606414 did not induce RPE cell
apoptosis. Furthermore, GSK2606414 was demonstrated to inhibit
eIF2α phosphorylation in ARPE-19 cells, suggesting that RPE cell
proliferation may be associated with PERK-eIF2α
phosphorylation.
eIF2α is believed to serve a fundamental role in
integrating stress response and cell survival. eIF2α may be
phosphorylated by four kinases including PERK, PKR, general control
non-derepressible 2 and heme-regulated inhibitor in response to
various cellular stressors, including misfolded proteins, oxidative
stress, viral infection and nutrient deprivation (32). PERK is primarily activated by
accumulation of misfolded proteins under ER stress conditions.
eIF2α phosphorylation by PERK may block protein synthesis (32,33).
Axten et al (22) reported
that 0.03 µM GSK2606414 adequately inhibited PERK activity in A549
lung adenocarcinoma cells. The present study demonstrated that the
ER stress inducer TG increased eIF2α phosphorylation in RPE cells,
and eIF2α phosphorylation was inhibited by GSK2606414 at
concentrations >0.1 µM. These results suggested that the novel
PERK inhibitor may inhibit protein synthesis by inhibiting eIF2α
phosphorylation.
eIF2α phosphorylation has been reported to increase
translation of basic leucine zipper transcription factors, such as
ATF4 (32). The present study
examined the effects of GSK2606414 on the expression levels of
numerous ATF4-associated genes in ARPE-19 cells under ER stress.
Notably, the inhibitory effect of GSK2606414 on gene expression did
not correspond to its inhibitory effect on eIF2α phosphorylation.
GSK2606414 at 0.05 and 0.1 µM inhibited eIF2α phosphorylation;
however, GSK2606414 at these doses upregulated ATF4, CHOP and VEGF
mRNA expression levels. At increased concentrations (>0.5 µM),
GSK2606414 consistently inhibited eIF2α phosphorylation and CHOP
and VEGF mRNA expression levels. These results suggested that a
concentration >0.5 µM may be required for GSK2606414 to inhibit
ATF4 signaling. CHOP, which is a proapoptotic transcription factor,
is a key downstream target of ATF4 (13,34).
Downregulation of CHOP has been reported to serve a neuroprotective
role against stress-induced injury (13,35).
The present study demonstrated that increased concentrations of
GSK2606414 inhibited CHOP expression without significantly
inhibiting ATF4 expression in RPE cells. This result may explain
why GSK2606414 did not induce apoptosis. Therefore, GSK2606414 may
be useful in preventing ER stress-induced apoptosis in RPE cells.
In addition, VEGF secretion by RPE cells is thought to be a key
factor responsible for ocular neovascularization. Subretinal
injection of adeno-associated virus-VEGF was reported to induce
subretinal neovascularization and RPE cell proliferation in rats
(36). Conversely, knockdown of
VEGF by various pharmacologic agents effectively inhibited
neovascularization in animal models of CNV (37,38).
PERK/eIF2α/ATF4 signaling has previously been reported to regulate
VEGF expression (6,17). The present study demonstrated that
VEGF mRNA expression levels were decreased in RPE cells treated
with GSK2606414. This result may explain, at least in part, why
GSK2606414 inhibited RPE cell proliferation without inducing
apoptosis. These findings additionally indicated that GSK2606414
may serve as a relatively safe antiproliferative and antiangiogenic
drug for the treatment of AMD and numerous retinal proliferative
diseases. The in vivo effects of GSK2606414 on the treatment
of AMD require further examination in animal models in future
studies.
In conclusion, the results of the present study
demonstrated that GSK2606414 inhibits eIF2α phosphorylation, and
downregulates the expression levels of CHOP and VEGF in RPE cells,
which serves an important role in AMD. These findings suggested
that GSK2606414 may function as a potential neuroprotective and
antiangiogenic drug for the treatment of AMD.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81441025)
and the Guangdong Science and Technology Plan Project (grant no.
2012B031800380).
References
|
1
|
Bressler NM: Age-related macular
degeneration is the leading cause of blindness. JAMA.
291:1900–1901. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ramkumar HL, Zhang J and Chan CC: Retinal
ultrastructure of murine models of dry age-related macular
degeneration (AMD). Prog Retin Eye Res. 29:169–190. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ambati J and Fowler BJ: Mechanisms of
age-related macular degeneration. Neuron. 75:26–39. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jager RD, Mieler WF and Miller JW:
Age-related macular degeneration. N Engl J Med. 358:2606–2617.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen C, Cano M, Wang JJ, Li J, Huang C, Yu
Q, Herbert TP, Handa JT and Zhang SX: Role of unfolded protein
response dysregulation in oxidative injury of retinal pigment
epithelial cells. Antioxid Redox Signal. 20:2091–2106. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salminen A, Kauppinen A, Hyttinen JM,
Toropainen E and Kaarniranta K: Endoplasmic reticulum stress in
age-related macular degeneration: Trigger for neovascularization.
Mol Med. 16:535–542. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Libby RT and Gould DB: Endoplasmic
reticulum stress as a primary pathogenic mechanism leading to
age-related macular degeneration. Adv Exp Med Biol. 664:403–409.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bi M, Naczki C, Koritzinsky M, Fels D,
Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al: ER
stress-regulated translation increases tolerance to extreme hypoxia
and promotes tumor growth. EMBO J. 24:3470–3481. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Häcker G: ER-stress and apoptosis:
Molecular mechanisms and potential relevance in infection. Microbes
Infect. 16:805–810. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mei Y, Thompson MD, Cohen RA and Tong X:
Endoplasmic reticulum stress and related pathological processes. J
Pharmacol Biomed Anal. 1:10001072013.PubMed/NCBI
|
|
11
|
Dandekar A, Mendez R and Zhang K: Cross
talk between ER stress, oxidative stress, and inflammation in
health and disease. Methods Mol Biol. 1292:205–214. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rizzuto R, Pinton P, Carrington W, Fay FS,
Fogarty KE, Lifshitz LM, Tuft RA and Pozzan T: Close contacts with
the endoplasmic reticulum as determinants of mitochondrial Ca2+
responses. Science. 280:1763–1766. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hiramatsu N, Chiang WC, Kurt TD, Sigurdson
CJ and Lin JH: Multiple mechanisms of unfolded protein
response-induced cell death. Am J Pathol. 185:1800–1808. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu H, Ng BS and Thibault G: Endoplasmic
reticulum stress response in yeast and humans. Biosci Rep. 34:pii:
e00118. 2014. View Article : Google Scholar
|
|
15
|
Liu Z, Lv Y, Zhao N, Guan G and Wang J:
Protein kinase R-like ER kinase and its role in endoplasmic
reticulum stress-decided cell fate. Cell Death Dis. 6:e18222015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jing G, Wang JJ and Zhang SX: ER stress
and apoptosis: A new mechanism for retinal cell death. Exp Diabetes
Res. 2012:5895892012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pollreisz A, Afonyushkin T, Oskolkova OV,
Gruber F, Bochkov VN and Schmidt-Erfurth U: Retinal pigment
epithelium cells produce VEGF in response to oxidized phospholipids
through mechanisms involving ATF4 and protein kinase CK2. Exp Eye
Res. 116:177–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Y, Alam GN, Ning Y, Visioli F, Dong
Z, Nör JE and Polverini PJ: The unfolded protein response induces
the angiogenic switch in human tumor cells through the PERK/ATF4
pathway. Cancer Res. 72:5396–5406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Doh SH, Kim JH, Lee KM, Park HY and Park
CK: Retinal ganglion cell death induced by endoplasmic reticulum
stress in a chronic glaucoma model. Brain Res. 1308:158–166. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miranda S, González-Rodriguez Á,
Garcia-Ramirez M, Revuelta-Cervantes J, Hernández C, Simó R and
Valverde ÁM: Beneficial effects of fenofibrate in retinal pigment
epithelium by the modulation of stress and survival signaling under
diabetic conditions. J Cell Physiol. 227:2352–2362. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Atkins C, Liu Q, Minthorn E, Zhang SY,
Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, et al:
Characterization of a novel PERK kinase inhibitor with antitumor
and antiangiogenic activity. Cancer Res. 73:1993–2002. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Axten JM, Medina JR, Feng Y, Shu A,
Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, et
al: Discovery of
7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p
yrrolo [2,3-d]pyrimidin-4-amine (GSK2606414), a potent and
selective first-in-class inhibitor of protein kinase R (PKR)-like
endoplasmic reticulum kinase (PERK). J Med Chem. 55:7193–7207.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moreno JA, Halliday M, Molloy C, Radford
H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA
and Mallucci GR: Oral treatment targeting the unfolded protein
response prevents neurodegeneration and clinical disease in
prion-infected mice. Sci Transl Med. 5:206ra1382013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Krishnamoorthy J, Rajesh K, Mirzajani F,
Kesoglidou P, Papadakis AI and Koromilas AE: Evidence for eIF2α
phosphorylation-independent effects of GSK2656157, a novel
catalytic inhibitor of PERK with clinical implications. Cell Cycle.
13:801–806. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Axten JM, Romeril SP, Shu A, Ralph J,
Medina JR, Feng Y, Li WH, Grant SW, Heerding DA, Minthorn E, et al:
Discovery of GSK2656157: An optimized PERK inhibitor selected for
preclinical development. Acs Med Chem Lett. 4:964–968. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pereira ER, Liao N, Neale GA and
Hendershot LM: Transcriptional and post-transcriptional regulation
of proangiogenic factors by the unfolded protein response. PLoS
One. 5:pii: e12521. 2010. View Article : Google Scholar
|
|
28
|
Wang H, Blais J, Ron D and Cardozo T:
Structural determinants of PERK inhibitor potency and selectivity.
Chem Biol Drug Des. 76:480–495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pereira ER, Frudd K, Awad W and Hendershot
LM: Endoplasmic reticulum (ER) stress and hypoxia response pathways
interact to potentiate hypoxia-inducible factor 1 (HIF-1)
transcriptional activity on targets like vascular endothelial
growth factor (VEGF). J Biol Chem. 289:3352–3364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liegl R, Koenig S, Siedlecki J, Haritoglou
C, Kampik A and Kernt M: Temsirolimus inhibits proliferation and
migration in retinal pigment epithelial and endothelial cells via
mTOR inhibition and decreases VEGF and PDGF expression. PloS One.
9:e882032014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Campochiaro PA: Ocular neovascularization.
J Mol Med (Berl). 91:311–321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Donnelly N, Gorman AM, Gupta S and Samali
A: The eIF2α kinases: Their structures and functions. Cell Mol Life
Sci. 70:3493–3511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
Embo Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang SX, Sanders E, Fliesler SJ and Wang
JJ: Endoplasmic reticulum stress and the unfolded protein responses
in retinal degeneration. Exp Eye Res. 125:30–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang F, Rendahl KG, Manning WC, Quiroz D,
Coyne M and Miller SS: AAV-mediated expression of vascular
endothelial growth factor induces choroidal neovascularization in
rat. Invest Ophthalmol Vis Sci. 44:781–790. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cao J, Zhao L, Li Y, Liu Y, Xiao W, Song
Y, Luo L, Huang D, Yancopoulos GD, Wiegand SJ and Wen R: A
subretinal matrigel rat choroidal neovascularization (CNV) model
and inhibition of CNV and associated inflammation and fibrosis by
VEGF trap. Invest Ophthalmol Vis Sci. 51:6009–6017. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saishin Y, Saishin Y, Takahashi K, Silva
Lima e R, Hylton D, Rudge JS, Wiegand SJ and Campochiaro PA:
VEGF-TRAP(R1R2) suppresses choroidal neovascularization and
VEGF-induced breakdown of the blood-retinal barrier. J Cell
Physiol. 195:241–248. 2003. View Article : Google Scholar : PubMed/NCBI
|