Introduction
Gallic acid (GA) is found in a variety of fruits and
foods, and is well absorbed in human body (1). GA has been reported to have diverse
biological and pharmacological behaviors, such as antibacterial and
antiviral effects (2,3). However, the most notable role of GA
is associated with its anticancer activity, which has been reported
in lung cancer (4,5), leukemia (6) and prostate cancer (7), as well as breast, gastric, colon,
cervical and esophageal cancers (8,9).
GA-induced apoptosis has been associated with oxidative stress
derived from reactive oxygen species (ROS) (4–6,10).
The central components of ROS are hydrogen peroxide
(H2O2), hydroxyl radical (OH) and superoxide
anion (O2•−), which are generated as
by-products of mitochondrial respiration or certain oxidases
(11). O2•−
is metabolized to H2O2 by superoxide
dismutases, and H2O2 is further detoxified to
O2 and H2O by catalase or glutathione (GSH)
peroxidases (11). Oxidative
stress occurs due to the overproduction of ROS and/or the decreased
decomposition of them. GA-induced cell death has also been
correlated with mitochondrial dysfunction and increased
intracellular Ca2+ level (4,5,12);
however, GA treatment did not lead to cytotoxicity in normal rat
fibroblast and endothelial cells (13). In addition, GA exhibited potential
antiapoptotic effects in normal human lymphocytes (14) and protected rat insulinoma RINm5F
β-cells from glucolipotoxicity through its antiapoptotic mechanism
(15). Controversially, GA was
suggested to have pro-oxidative in addition to antioxidative
properties, depending on the levels of iron or
H2O2 (16,17).
Therefore, additional studies are required to reassess the cellular
properties of GA under diverse conditions.
Vascular endothelial cells (ECs) are involved in the
regulation of inflammation, blood pressure and angiogenesis
(18). Vascular ECs experience a
broad range of oxidative stress, which may ultimately lead to
endothelial dysfunction through the induction of apoptosis
(19). Angiogenesis is a crucial
step in the transition of a dormant tumor into a malignant state;
the proliferation of ECs is an early step during sprouting
angiogenesis. Despite important roles of vascular ECs in tumor
biogenesis and development, the effects of GA on ECs are relatively
poorly understood. In the vasculature, ROS serve physiological and
pathophysiological roles through the regulation of numerous
cellular processes, including cell proliferation, death and
survival (19). Therefore, further
research is required to explore the cellular effects of GA on ECs
in relation to the levels of ROS and GSH expression. Calf pulmonary
arterial ECs (CPAECs) are an established cell model for both
cellular and molecular endothelial cell research (20,21).
The present study investigated the effects of GA
exposure on CPAEC growth and death in relation to ROS and GSH
levels, and examined whether the antioxidant N-acetyl cysteine
(NAC) and the GSH synthesis inhibitor L-buthionine sulfoximine
(BSO) were able to affect GA-induced CPAEC death.
Materials and methods
Cell culture
CPAECs were obtained from Korean Cell Line Bank
(Seoul, Korea) and were cultured in RPMI-1640 medium (GE Healthcare
Life Sciences, Little Chalfont, UK) supplemented with 10% fetal
bovine serum (FBS; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
and 1% penicillin-streptomycin (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). CPAECs were routinely grown in 100 mm
plastic tissue culture dishes (Nalge Nunc International, Penfield,
NY, USA) in humidified incubator containing 5% CO2 at
37°C and harvested with a solution of trypsin-EDTA (Gibco; Thermo
Fisher Scientific, Inc.) while in the logarithmic phase of
growth.
Reagents
GA (Sigma-Aldrich; Merck KGaA) was dissolved in 100%
ethanol to 200 mM, and used at the indicated concentrations. The
pan-caspase inhibitor
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK;
R&D Systems, Inc., Minneapolis, MN, USA) was dissolved in DMSO
(Sigma-Aldrich; Merck KGaA) to 10 mM. NAC and BSO were obtained
from Sigma-Aldrich (Merck KGaA). NAC 200 mM as a stock solution was
dissolved in the buffer of 20 mM
4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid [HEPES; (pH
7.0)], and BSO 100 mM as a stock solution was dissolved in
distilled water. Based on a previous study (22), cells were pre-incubated at 37°C for
1 h with Z-VAD-FMK (15 µM), NAC (2 mM) or BSO (10 µM), followed by
treatment with the GA (25 µM) at 37°C for 24 h before the assays
were performed.
Cell proliferation assay
Viable and dead cell numbers and cell proliferation
were determined by trypan blue staining and MTT dye absorbance by
living cells, respectively, as previously described (23,24).
Briefly, 2×105 cells/well in 24-well plates (Nalge Nunc
International) were seeded for cell counting and 5×103
cells/well in 96-well microtiter plates (Nalge Nunc International)
were seeded for MTT assays. The cells which were cultured in
RPMI-1640 medium (GE Healthcare Life Sciences) supplemented with
10% fetal bovine serum (FBS; Sigma-Aldrich; Merck KGaA) and 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.)
were exposed to the indicated amounts of GA (between 0 and 50 µM)
at 37°C for 24 h and/or 1 h pre-incubation with Z-VAD-FMK (15 µM),
NAC (2 mM) or BSO (10 µM) at 37°C. The plates were incubated for 4
h at 37°C. Medium in plates was removed by pipetting, and 200 µl
DMSO was added to each well to solubilize the formazan crystals.
The optical density was measured at 570 nm using a microplate
reader (Synergy™ 2; BioTek Instruments Inc., Winooski, VT,
USA).
Sub-G1 analysis
Cells at the sub-G1 phase were determined by
staining with propidium iodide [PI; Sigma-Aldrich; Merck KGaA;
excitation (Ex)/emission (Em)=488/617 nm], as previously described
(25). Briefly, 1×106
cells cultured in RPMI-1640 medium (GE Healthcare Life Sciences)
supplemented with 10% FBS (Sigma-Aldrich; Merck KGaA) and 1%
penicillin-streptomycin (Gibco; Thermo Fisher Scientific, Inc.) in
60-mm culture dish (Nalge Nunc International) were exposed to the
indicated amounts of GA and/or Z-VAD-FMK, NAC or BSO at 37°C for 24
h. Cells were then washed in PBS and fixed in 70% ethanol at 4°C
for 1 h. Cells were again washed with PBS and then incubated with
PI (10 µg) with simultaneous treatment with RNase at 37°C for 30
min. Cellular DNA content was measured with a FACStar Flow
Cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed
using lysis II and Cellfit software (version 2.0; BD
Biosciences).
Annexin V-fluorescein isothiocyanate
(FITC)/PI staining for cell death detection
Cell death was measured by staining cells with
Annexin V-FITC (Ex/Em=488/519 nm; Molecular Probes; Thermo Fisher
Scientific, Inc.) and PI (Ex/Em=488/617 nm; Sigma-Aldrich; Merck
KGaA), as previously described (26). Briefly, 1×106 cells in
60 mm culture dish (Nalge Nunc International) were incubated with
the indicated amounts of GA with or without Z-VAD-FMK, NAC or BSO
at 37°C for 24 h. The prepared cells were washed twice with cold
PBS and then resuspended in 500 µl of binding buffer [10 mM
HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2] at a
concentration of 1×106 cells/ml. Then 5 µl of annexin
V-FITC (BD Biosciences) and 10 µl of 20 µg/ml PI were added to
these cells. Annexin V-FITC/PI staining was analyzed with a FACStar
flow cytometer (BD Biosciences) and CellQuest Pro software (version
5.1; BD Biosciences).
Measurement of mitochondrial membrane
potential (ΔѰm)
ΔѰm was measured using the Rhodamine 123
mitochondrial-specific fluorescent dye (Sigma-Aldrich; Merck KGaA;
Ex/Em=485/535 nm), as previously described (23). Briefly, 1×106 cells in
60 mm culture dish (Nalge Nunc International) were incubated with
the indicated amounts of GA with or without Z-VAD-FMK, NAC or BSO
at 37°C for 24 h. Cells were washed twice with PBS and incubated
with Rhodamine 123 (0.1 µg/ml) at 37°C for 30 min. Rhodamine 123
staining intensity was determined by FACStar flow cytometry (BD
Biosciences) and analyzed using CellQuest Pro software (version
5.1; BD Biosciences). An absence of Rhodamine 123 from cells
indicated the loss of ΔѰm in CPAECs.
Detection of intracellular ROS
levels
Intracellular ROS levels were assessed with the
non-fluorescent probe dye 2′,7′-dichlorodihydrofluorescein
diacetate (H2DCFDA; Ex/Em=495/529 nm; Molecular Probes;
Thermo Fisher Scientific, Inc.); cleavage of the acetate groups
converts H2DCFDA into the highly fluorescent
2′,7′-dichloroflurosceine (DCF). Dihydroethidium (DHE;
Ex/Em=518/605 nm; Molecular Probes; Thermo Fisher Scientific, Inc.)
is a fluorogenic probe that is highly selective for
O2•−. Briefly, 1×106 cells in 60
mm culture dish (Nalge Nunc International) were incubated with the
indicated amounts of GA with or without Z-VAD-FMK, NAC or BSO at
37°C for 24 h. Following incubation, cells were washed with PBS and
incubated with H2DCFDA (20 µM) or DHE (20 µM) at 37°C
for 30 min. DCF and DHE fluorescence was detected with a FACStar
flow cytometer (BD Biosciences). ROS and O2•−
levels were expressed as the mean fluorescence intensity, which was
calculated by CellQuest Pro software (version 5.1; BD
Biosciences).
Detection of the intracellular
glutathione (GSH)
Cellular GSH expression levels were analyzed using
5-chloromethylfluorescein diacetate (CMFDA; Ex/Em=522/595 nm;
Molecular Probes; Thermo Fisher Scientific, Inc.), as previously
described (27). Briefly,
1×106 cells in 60 mm culture dish (Nalge Nunc
International) were incubated with the indicated amounts of GA with
or without Z-VAD-FMK, NAC or BSO at 37°C for 24 h. The cells were
then washed in PBS. They were then incubated with 5 µM CMFDA at
37°C for 30 min. Fluorescence intensity of the cleaved CMF was
determined using a FACStar flow cytometer (BD Biosciences) and
analyzed using CellQuest Pro software (version 5.1; BD
Biosciences).
Statistical analysis
The results represent the mean of at least three
independent experiments ± standard deviation. Student's t-test or
one-way analysis of variance with post hoc analysis using Tukey's
multiple comparison test for parametric data. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of GA on cell growth, cell
death and ΔѰm in CPAECs
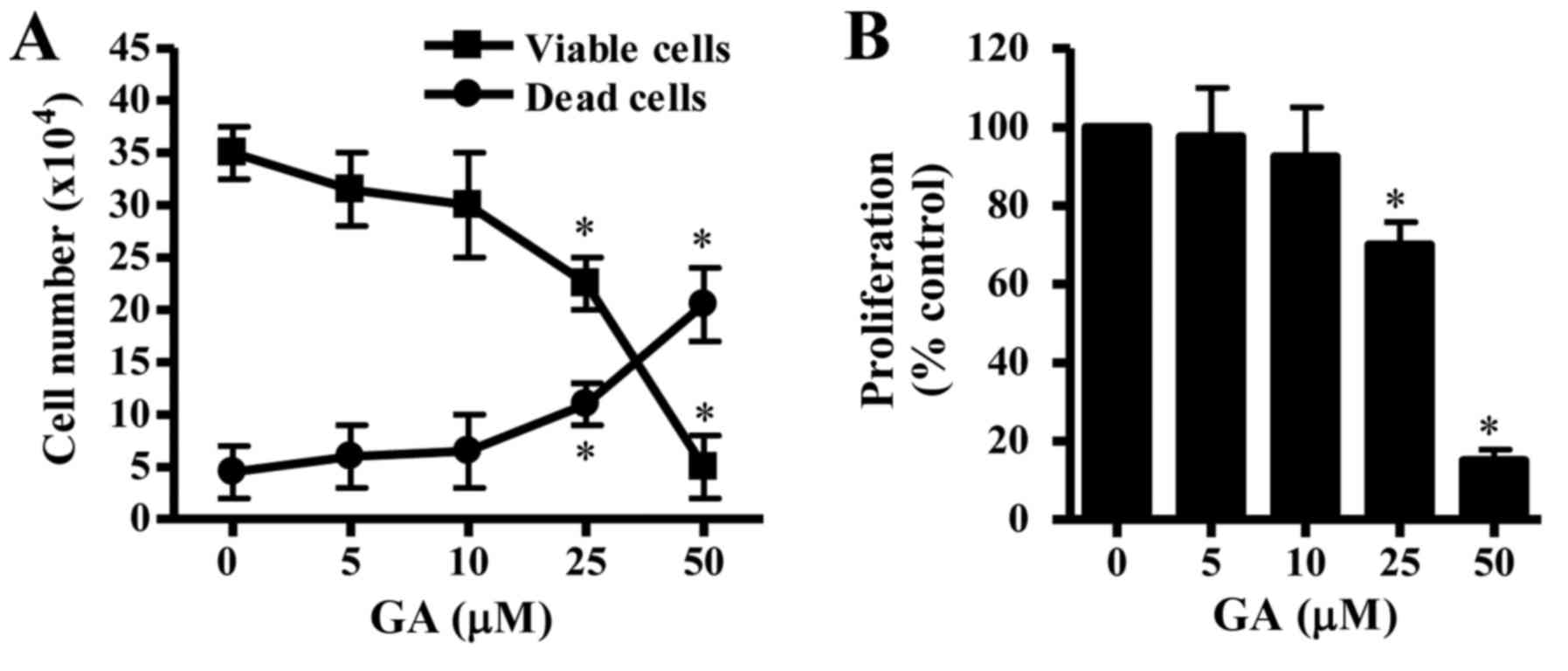
The effects of GA exposure on the growth of CPAECs
were examined by counting the number of trypan blue positive- and
negative-stained cells. Cells treated with various concentrations
of GA for 24 h exhibited a dose-dependent decrease in the
population of viable (trypan blue negative) CPAEC cells (Fig. 1A), whereas the number of dead
(trypan blue positive) cells increased. The ratio of dead cells to
viable cells rose in a dose-dependent manner. Changes in cell
proliferation were assessed by MTT assay. CPAEC cells that were
treated with GA for 24 h exhibited a reduction in proliferation,
and the half-maximal inhibitory concentration (IC50)
value of GA in CPAEC cells was ~30 µM at 24 h (Fig. 1B).
In addition, GA treatment enhanced the number of
sub-G1 cells, which indicates cell death, in a dose-dependent
manner (Fig. 2A). At a dose of 25
µM GA, the number of sub-G1 cells increased ~13% compared with the
untreated control CPAECs (Fig.
2A). Whether GA was able to induce apoptosis in CPAECs was
examined further. As shown in Fig.
2B, the number of Annexin V-FITC-stained cells increased in
CPAECs as the concentration of GA increased. At a dose of 25 µM GA,
the number of Annexin V-FITC-stained cells increased by ~16%
compared with the untreated control CPAECs (Fig. 2B). Since apoptosis is closely
related to a collapse of ΔѰm, the effects of GA on
ΔѰm were assessed using rhodamine 123 dye. Treatment
with GA triggered the loss of ΔѰm [that is, (−)
rhodamine 123 cells] in CPAECs in a dose-dependent manner (Fig. 2C). CPAEC cells treated with 25 µM
GA exhibited ~10% decrease in ΔѰm compared with
untreated control cells, whereas 50 µM GA treatment strongly
increased the loss by ~70% (Fig.
2C).
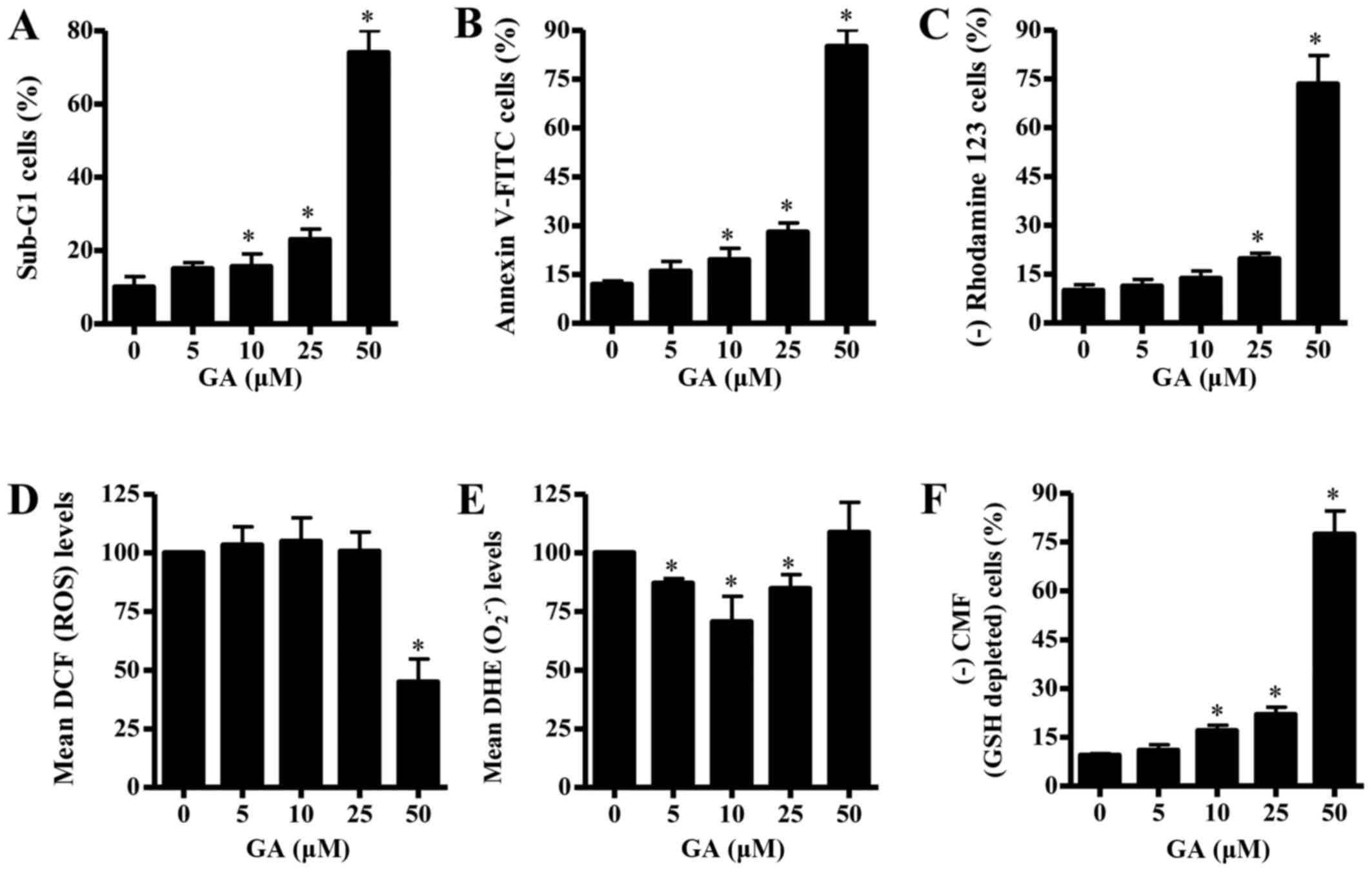 | Figure 2.Effects of GA on cell death,
ΔѰm, ROS and GSH levels in CPAECs. Exponentially growing
CPAEC cells were treated with the indicated concentrations of GA
for 24 h, and various aspects were examined by FACStar flow
cytometry, including: (A) The percentage of cells at sub-G1 phase;
(B) the percentage of cells staining positive for Annexin V-FITC;
(C) the percentage of cells with reduced ΔѰm, as
measured by (−) rhodamine 123 staining; (D) the mean ROS levels, as
measured by DCF fluorescence intensity; (E) the mean
O2•− levels, as measured by DHE fluorescence
intensity; and (F) the percentage of GSH-depleted cells, as
measured by (−) CMF fluorescence. *P<0.05 compared with
untreated (0 µM GA) control group. ΔѰm, mitochondrial
membrane potential; CMF, 5-chloromethylfluorescein; CPAEC, calf
pulmonary arterial endothelial cell; DCF,
2′,7′-dichlorofluorescein; DHE, dihydroethidium; FITC, fluorescein
isothiocyanate; GA, gallic acid; GSH, glutathione;
O2•−, superoxide anion; ROS, reactive oxygen
species. |
Effects of GA on intracellular ROS and
GSH levels in CPAECs
To assess the levels of intracellular ROS in
GA-treated CPAECs at 24 h, H2DCFDA and DHE fluorescent
dyes were used. ROS levels, as measured by mean DCF fluorescence
intensity, were not significantly altered in CPAECs treated with 5,
10 or 25 µM GA, whereas the ROS level was significantly decreased
by 50 µM GA treatment, compared with untreated control cells
(Fig. 2D). The level of red
fluorescence derived from DHE, which indicates the level of
O2•− in the cell, decreased significantly in
CPAECs treated with 5, 10 or 25 µM GA (Fig. 2E); however, the level was not
significantly altered when cells were treated with 50 µM GA.
Finally, changes in the levels of GSH expression in
CPAECs were analyzed using a CMFDA fluorescence dye. GA treatment
led to a dose-dependent increase in the number of GSH-depleted
cells [(−) CMF; Fig. 2F]. CPAECs
treated with 25 µM GA exhibited ~11% increase in the number of
GSH-depleted cells compared with untreated control cells.
Effects of Z-VAD-FMK, NAC and BSO on
cell growth, cell death and ΔѰm in GA-treated
CPAECs
In these experiments, 25 µM GA was used as a
suitable dose to differentiate the levels of proliferation and
apoptosis in the presence or absence of Z-VAD-FMK (15 µM), NAC (2
mM) or BSO (10 µM) since the IC50 value of GA in CPAEC
cells was ~30 µM. Treatment with Z-VAD-FMK, NAC or BSO alone did
not significantly affect the growth of CPAECs at 24 h (Fig. 3A). GA-treated CPAEC cells were
unaffected by co-treatment with either Z-VAD-FMK or BSO, whereas
the GA-induced growth inhibition was strongly enhance by NAC
exposure (Fig. 3A).
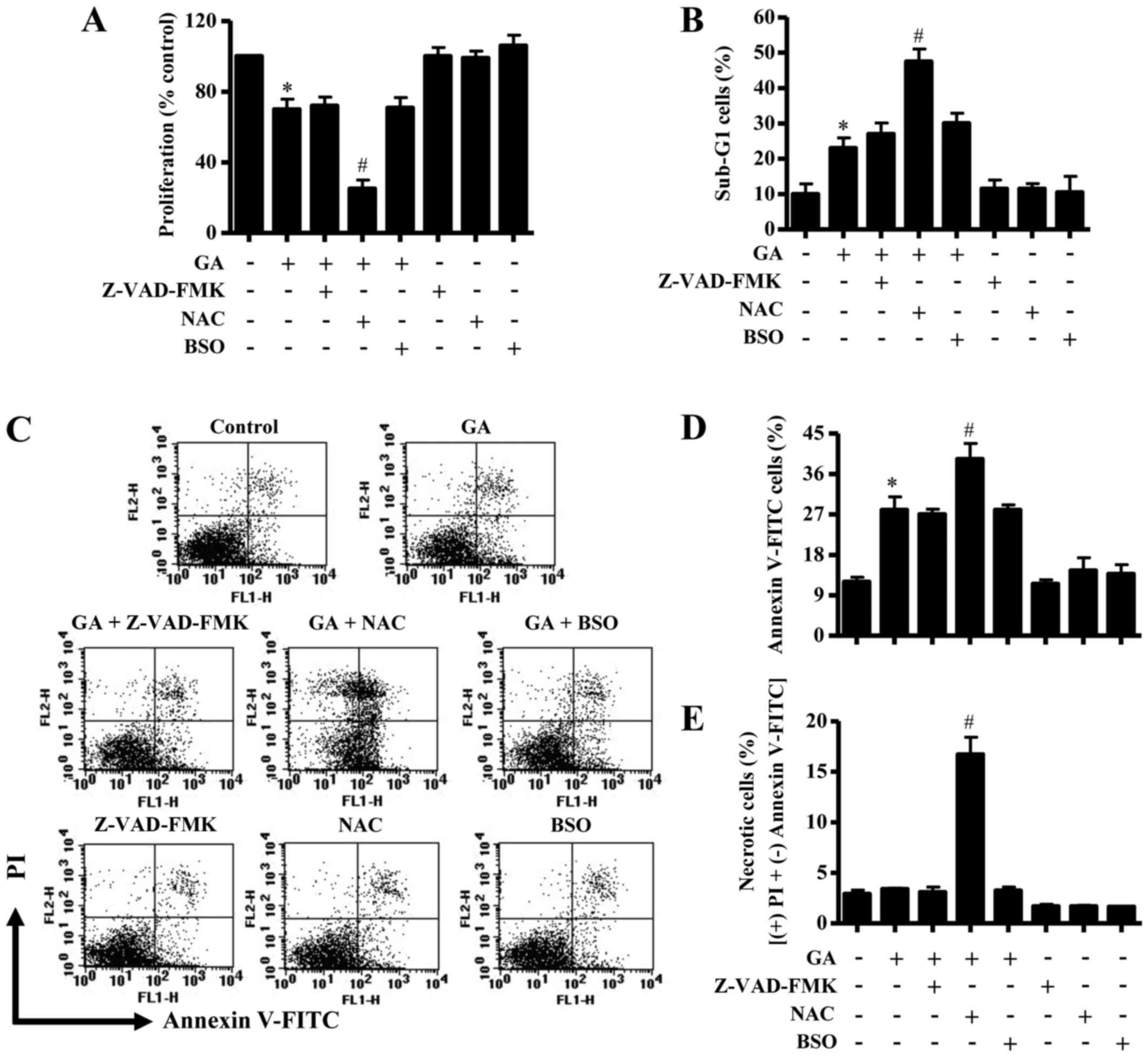 | Figure 3.Effects of Z-VAD-FMK, NAC or BSO on
cell growth and death in GA-treated CPAECs. Exponentially growing
cells were pretreated with Z-VAD-FMK (15 µM), NAC (2 mM) or BSO (10
µM), followed by GA (25 µM) for 24 h. (A) Changes to cell
proliferation were assessed by MTT assay. (B) The percentage of
cells at sub-G1 phase. (C) Representative FACStar flow cytometry
plots for Annexin V-FITC/PI staining in CPAEC cells. (D) Percentage
of Annexin V-FITC-positive staining cells from (C). (E) Percentage
of necrotic cells from (C), necrotic cells are PI-positive and
Annexin V-FITC-negative. *P<0.05 compared with untreated control
group; #P<0.05 compared with GA-treated control
group. BSO, L-buthionine sulfoximine; CPAEC, calf pulmonary
arterial endothelial cell; FITC, fluorescein isothiocyanate; GA,
gallic acid; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NAC,
N-acetyl cysteine; PI, propidium iodine; Z-VAD-FMK,
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone. |
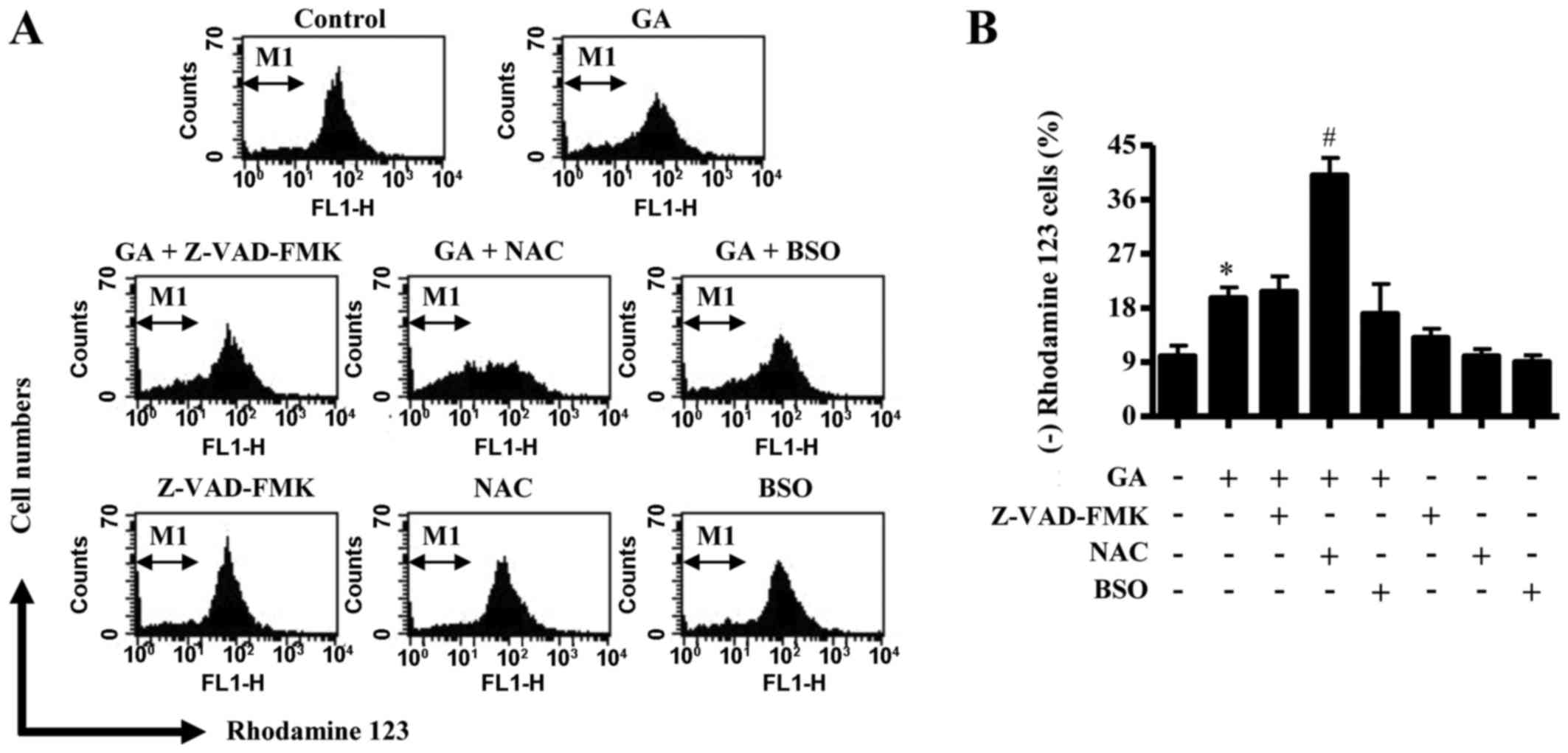
In relation to cell death and ΔѰm, the
number of sub-G1 cells in GA-treated CPAECs that were also treated
with Z-VAD-FMK and BSO did not significantly change, whereas
co-treatment with NAC resulted in an increase in the number of
CPAEC cells at sub-G1 (Fig. 3B).
Similarly, neither Z-VAD-FMK nor BSO treatment affected the number
of Annexin V-FITC-stained cells in GA-treated CPAECs, whereas GA
and NAC co-treatment significantly increased the number in these
apoptotic cells (Fig. 3C and D).
Treatment with 25 µM GA did not significantly trigger necrotic cell
death (that is, cells staining positive for PI and negative for
Annexin V-FITC) in CPAECs (Fig. 3C and
E). However, NAC co-treatment strongly induced necrotic cell
death in CPAECs treated with 25 µM GA, whereas treatment with
Z-VAD-FMK or BSO did not (Fig. 3C and
E). Similarly, neither Z-VAD-FMK nor BSO co-treatment was able
to affect the level of ΔѰm loss [as measured by (−)
rhodamine 123 expression] in GA-treated CPAECs, but co-treatment
with NAC significantly augmented the loss of ΔѰm in
these cells (Fig. 4A and B).
Effects of Z-VAD-FMK, NAC or BSO on
ROS, O2•- and GSH levels in GA-treated CPAECs
ROS and GSH levels in GA-treated CPAECs were
examined to assess any changes in their expression levels due to
treatment with Z-VAD-FMK, NAC or BSO at 24 h. Neither Z-VAD-FMK nor
NAC treatment resulted in a change in ROS levels (as determined by
DCF fluorescence intensity) in GA-treated CPAECs (Fig. 5A); although co-treatment with BSO
appeared to slightly increase the ROS level, this increase was not
significant. Cells treated with NAC alone appeared to exhibit a
mild reduction in the basal level of ROS, but this was not
significant (Fig. 5A). In relation
to O2•− levels (as measured by mean DHE
fluorescence intensity), none of the co-treatments with Z-VAD-FMK,
NAC or BSO significantly affected the GA-induced reduction in
O2•− levels in CPAEC cells (Fig. 5B). Cells treated BSO alone
exhibited a significant increase in O2•−
level compared with untreated control CPAECs (Fig. 5B). Although co-treatment with
Z-VAD-FMK or BSO did not significantly affect the levels of GSH
depletion (as measured by absence of CMF fluorescence) in
GA-treated CPAECs, cells co-treated with NAC significantly
increased the number of GSH-depleted cells (Fig. 5C and D). Notably, cells treated
with BSO alone exhibited an increased number of GSH-depleted cells
compared with untreated control CPAECs (Fig. 5C and D).
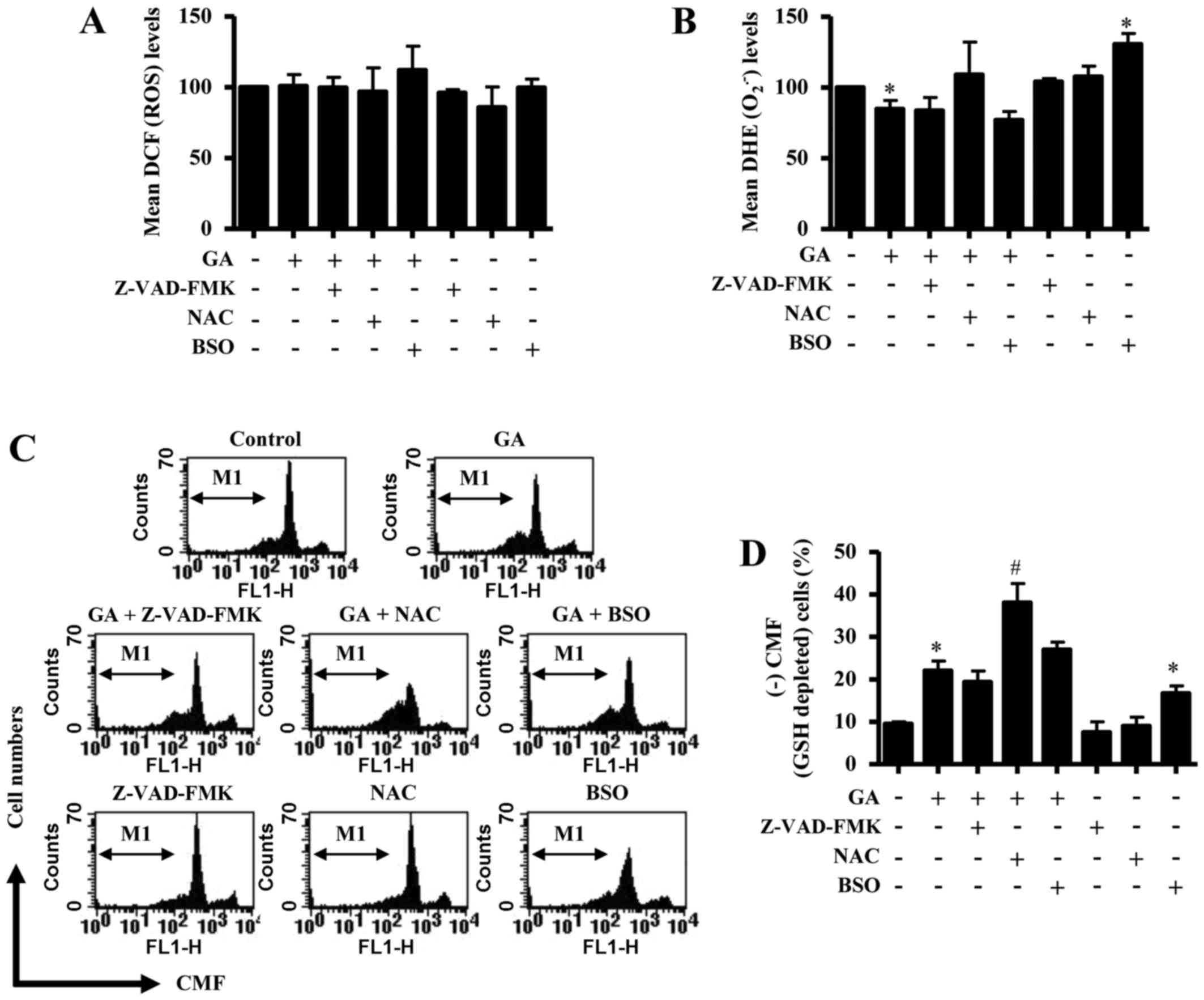 | Figure 5.Effects of Z-VAD-FMK, NAC or BSO on
ROS and GSH levels in GA-treated CPAECs. Exponentially growing
cells were pretreated with Z-VAD-FMK (15 µM), NAC (2 mM) or BSO (10
µM), followed by GA (25 µM) for 24 h; ROS and GSH levels were
measured using a FACStar flow cytometer. (A) Mean ROS levels, as
measured by DCF fluorescence intensity. (B) Mean
O2•− levels, as measured by DHE fluorescence
intensity. (C) Representative flow cytometry plots for (+) CMF
staining in CPAECs. (D) Percentage of GSH-depleted cells, as
measured by (−) CMF fluorescence from the M1 region in C.
*P<0.05 compared with untreated (0 µM GA) control group;
#P<0.05 compared with GA-treated control group. BSO,
L-buthionine sulfoximine; CMF, 5-chloromethylfluorescein; CPAEC,
calf pulmonary arterial endothelial cell; DCF,
2′,7′-dichlorofluorescein; DHE, dihydroethidium; GA, gallic acid;
GSH, glutathione; NAC, N-acetyl cysteine;
O2•−, superoxide anion; ROS, reactive oxygen
species; Z-VAD-FMK,
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone. |
Discussion
A number of previous studies have reported that GA
exposure inhibited the growth of HeLa cervical cancer cells and
Calu-6 and A549 lung cancer cells, and the IC50 was
between 30 and 150 µM in these cell lines (4,5,9). In
the present study, GA treatment decreased the growth of CPAECs in a
dose-dependent manner, with the IC50 value of ~30 µM at
24 h. Conversely, GA treatment was previously demonstrated to have
no relative cytotoxicity in normal fibroblasts and ECs (13,28,29).
CPAECs appeared less resistant to GA treatment compared with other
normal cells, and instead they were similar to cancer cells in
their susceptibility to GA exposure. These differences may be due
to the differing activities and antioxidant systems in each cell
line.
GA treatment has previously been demonstrated to
induce apoptosis in cancer cells through mitochondrial dysfunction
(4,5,10).
In the present study, GA exposure appeared to induce apoptosis in
CPAECs, as evidenced by the increase in the number of sub-G1 cells
and Annexin V-FITC-positive staining cells, and it triggered the
loss of ΔѰm. The dose-dependent loss of ΔѰm
corresponded to the dose-dependent rise in Annexin V-FITC staining
cells, supporting the hypothesis that GA-induced cell death may be
tightly correlated with the collapse of ΔѰm. In
particular, the tested dose of 15 µM Z-VAD-FMK used in the present
study did not affect cell death or ΔѰm in GA-treated
CPAECs. However, 15 µM Z-VAD-FMK was reported to significantly
reduce cell death and ΔѰm loss in CPAECs treated with
pyrogallol, a derivative of GA (20). Therefore, this result suggested
that the activation of caspases is not closely related to
GA-induced apoptosis in CPAECs. It has also been reported that
Z-VAD-FMK effectively prevented GA-induced apoptosis in lung cancer
cells (30). Therefore, the modes
of caspase activation during GA-induced apoptosis may be dependent
on cell types (e.g., normal cells vs. cancer cells) and the
difference requires further study.
GA exhibits both pro- and antioxidative properties
(16,17). Results from a number of previous
studies suggested that GA-induced apoptosis may be associated with
oxidative stress derived from ROS (4–6,10).
However, treatment with the lower doses of GA (5–25 µM) did not
increase ROS levels in CPAECs in the present study, whereas a dose
of 50 µM GA induced CPAEC death and decreased ROS levels.
Conversely, 5 to 25 µM GA treatment significantly decreased
O2•− levels in CPAECs, whereas exposure to 50
µM GA had no effect. Therefore, in the present study, GA treatment
appeared to exhibit more antioxidative than pro-oxidative
properties in CPAECs. Notably, although NAC exposure did not affect
ROS levels in GA-treated CPAECs, NAC co-treatment did result in
reduced cell proliferation and increased cell death and
ΔѰm loss in these cells; NAC treatment also strongly
induced necrotic cell death in CPAECs treated with GA. A previous
study reported a similar NAC-induced increase in growth inhibition
and apoptotic death in GA-treated lung cancer cells (5). In the present study, neither
Z-VAD-FMK nor BSO treatment significantly influenced ROS levels or
cell death in GA-treated CPAECs. Cells treated with BSO alone
exhibited increased O2•− levels without
triggering cell death. Overall, the results from the present study
suggest that GA-mediated CPAEC death is not related to oxidative
stress. The precise roles of ROS in GA-induced CPAEC death require
further investigation.
In general, apoptotic effects have been reported to
be inversely proportional to GSH content in cells (22,31,32).
The intracellular GSH content may have a decisive effect on
GA-induced cell death (4,5,33).
Similarly, results from the present study demonstrated that GA
treatment dose-dependently decreased the number of GSH-positive
cells in CPAECs. Z-VAD-FMK co-treatment did not affect the
GA-induced depletion of GSH in CPAECs. In addition, NAC treatment,
which showed increases in both necrotic and apoptotic cell deaths
by GA, significantly augmented GSH depletion in these cells.
Although it is recognized that NAC contains a thiol group and is a
GSH precursor, this agent did not seem to be a GSH precursor in
GA-treated CPAECs. However, NAC significantly attenuated both GSH
depletion and cell death in propyl gallate- and MG132-treated
CPAECs (21,34). Thus, NAC may or may not be a GSH
precursor depending on other agents used in co-treatments. In
addition, when treated with BSO alone, there was a significant
increase in the number of GSH-depleted cells; however, co-treatment
with GA did not augment GSH depletion CPAECs. By contrast, previous
studies have reported that BSO co-treatment enhanced GSH depletion
and cell death in GA-treated lung cancer cells, fibroblasts and
normal ECs (5,28,33),
and decreased GSH levels in MG132-treated CPAECs (35). Therefore, these data suggested that
GSH content may serve a vital role in GA-induced cell death, and
that BSO exposure may influence GSH levels dependent on cell type
and co-incubation drugs.
In conclusion, GA exposure induced growth inhibition
and death in CPAECs, which were revealed to be cause by GSH
depletion rather than changes in ROS levels. The present data may
provide useful information to understand the antiproliferative
effects of GA in ECs, particularly CPAECs, in relation to the
cellular changes in ROS and GSH levels.
Acknowledgements
The present study was supported by a grant from the
National Research Foundation of Korea funded by the Korean
government (Ministry of Science ICT and Future Planning; grant no.
2016R1A2B4007773).
Glossary
Abbreviations
Abbreviations:
|
ΔѰm
|
mitochondrial membrane potential
|
|
BSO
|
l-buthionine sulfoximine
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
CPAEC
|
calf pulmonary arterial endothelial
cell
|
|
DHE
|
dihydroethidium
|
|
EC
|
endothelial cell
|
|
FITC
|
fluorescein isothiocyanate
|
|
GA
|
gallic acid
|
|
GSH
|
glutathione
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
NAC
|
N-acetyl cysteine
|
|
PI
|
propidium iodine
|
|
ROS
|
reactive oxygen species
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone.
|
References
|
1
|
Shahrzad S, Aoyagi K, Winter A, Koyama A
and Bitsch I: Pharmacokinetics of gallic acid and its relative
bioavailability from tea in healthy humans. J Nutr. 131:1207–1210.
2001.PubMed/NCBI
|
|
2
|
Kang MS, Oh JS, Kang IC, Hong SJ and Choi
CH: Inhibitory effect of methyl gallate and gallic acid on oral
bacteria. J Microbiol. 46:744–750. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kratz JM, Andrighetti-Fröhner CR, Leal PC,
Leal PC, Nunes RJ, Yunes RA, Trybala E, Bergström T, Barardi CR and
Simões CM: Evaluation of anti-HSV-2 activity of gallic acid and
pentyl gallate. Biol Pharm Bull. 31:903–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
You BR, Kim SZ, Kim SH and Park WH: Gallic
acid-induced lung cancer cell death is accompanied by ROS increase
and glutathione depletion. Mol Cell Biochem. 357:295–303. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
You BR and Park WH: Gallic acid-induced
lung cancer cell death is related to glutathione depletion as well
as reactive oxygen species increase. Toxicol In Vitro.
24:1356–1362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inoue M, Sakaguchi N, Isuzugawa K, Tani H
and Ogihara Y: Role of reactive oxygen species in gallic
acid-induced apoptosis. Biol Pharm Bull. 23:1153–1157. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Agarwal C, Tyagi A and Agarwal R: Gallic
acid causes inactivating phosphorylation of cdc25A/cdc25C-cdc2 via
ATM-Chk2 activation, leading to cell cycle arrest, and induces
apoptosis in human prostate carcinoma DU145 cells. Mol Cancer Ther.
5:3294–3302. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Faried A, Kurnia D, Faried LS, Usman N,
Miyazaki T, Kato H and Kuwano H: Anticancer effects of gallic acid
isolated from Indonesian herbal medicine, Phaleria macrocarpa
(Scheff.) Boerl, on human cancer cell lines. Int J Oncol.
30:605–613. 2007.PubMed/NCBI
|
|
9
|
You BR, Moon HJ, Han YH and Park WH:
Gallic acid inhibits the growth of HeLa cervical cancer cells via
apoptosis and/or necrosis. Food Chem Toxicol. 48:1334–1340. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen HM, Wu YC, Chia YC, Chang FR, Hsu HK,
Hsieh YC, Chen CC and Yuan SS: Gallic acid, a major component of
Toona sinensis leaf extracts, contains a ROS-mediated anti-cancer
activity in human prostate cancer cells. Cancer Lett. 286:161–171.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Apak R, Özyürek M, Güçlü K and Çapanoğlu
E: Antioxidant activity/capacity measurement. 3. Reactive oxygen
and nitrogen species (ROS/RNS) scavenging assays, oxidative stress
biomarkers, and chromatographic/chemometric assays. J Agric Food
Chem. 64:1046–1070. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hsieh SC, Wu CH, Wu CC, Yen JH, Liu MC,
Hsueh CM and Hsu SL: Gallic acid selectively induces the necrosis
of activated hepatic stellate cells via a calcium-dependent calpain
I activation pathway. Life Sci. 102:55–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Inoue M, Suzuki R, Sakaguchi N, Li Z,
Takeda T, Ogihara Y, Jiang BY and Chen Y: Selective induction of
cell death in cancer cells by gallic acid. Biol Pharm Bull.
18:1526–1530. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sohi KK, Mittal N, Hundal MK and Khanduja
KL: Gallic acid, an antioxidant, exhibits antiapoptotic potential
in normal human lymphocytes: A Bcl-2 independent mechanism. J Nutr
Sci Vitaminol (Tokyo). 49:221–227. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sameermahmood Z, Raji L, Saravanan T,
Vaidya A, Mohan V and Balasubramanyam M: Gallic acid protects
RINm5F beta-cells from glucolipotoxicity by its antiapoptotic and
insulin-secretagogue actions. Phytother Res. 24 Suppl 1:S83–S94.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Strlic M, Radovic T, Kolar J and Pihlar B:
Anti- and prooxidative properties of gallic acid in fenton-type
systems. J Agric Food Chem. 50:6313–6317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sakagami H and Satoh K: Prooxidant action
of two antioxidants: Ascorbic acid and gallic acid. Anticancer Res.
17:221–224. 1997.PubMed/NCBI
|
|
18
|
Bassenge E: Endothelial function in
different organs. Prog Cardiovasc Dis. 39:209–228. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Irani K: Oxidant signaling in vascular
cell growth, death, and survival: A review of the roles of reactive
oxygen species in smooth muscle and endothelial cell mitogenic and
apoptotic signaling. Circ Res. 87:179–183. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han YH and Park WH: Pyrogallol-induced
calf pulmonary arterial endothelial cell death via
caspase-dependent apoptosis and GSH depletion. Food Chem Toxicol.
48:558–563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han YH, Moon HJ, You BR and Park WH:
Propyl gallate inhibits the growth of calf pulmonary arterial
endothelial cells via glutathione depletion. Toxicol In Vitro.
24:1183–1189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park WH: Pyrogallol induces the death of
human pulmonary fibroblast cells through ROS increase and GSH
depletion. Int J Oncol. 49:785–792. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
You BR, Kim SH and Park WH: Reactive
oxygen species, glutathione, and thioredoxin influence suberoyl
bishydroxamic acid-induced apoptosis in A549 lung cancer cells.
Tumour Biol. 36:3429–3439. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park WH: Anti-apoptotic effect of caspase
inhibitors on H2O2-treated HeLa cells through early suppression of
its oxidative stress. Oncol Rep. 31:2413–2421. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han YH, Kim SZ, Kim SH and Park WH:
Apoptosis in pyrogallol-treated Calu-6 cells is correlated with the
changes of intracellular GSH levels rather than ROS levels. Lung
Cancer. 59:301–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
You BR, Shin HR, Han BR and Park WH: PX-12
induces apoptosis in Calu-6 cells in an oxidative stress-dependent
manner. Tumour Biol. 36:2087–2095. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han YH, Kim SH, Kim SZ and Park WH:
Caspase inhibitor decreases apoptosis in pyrogallol-treated lung
cancer Calu-6 cells via the prevention of GSH depletion. Int J
Oncol. 33:1099–1105. 2008.PubMed/NCBI
|
|
28
|
Park WH and Kim SH: Involvement of
reactive oxygen species and glutathione in gallic acid-induced
human umbilical vein endothelial cell death. Oncol Rep. 28:695–700.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
You BR and Park WH: Gallic acid-induced
human pulmonary fibroblast cell death is accompanied by increases
in ROS level and GSH depletion. Drug Chem Toxicol. 34:38–44. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ohno Y, Fukuda K, Takemura G, Toyota M,
Watanabe M, Yasuda N, Xinbin Q, Maruyama R, Akao S, Gotou K, et al:
Induction of apoptosis by gallic acid in lung cancer cells.
Anticancer Drugs. 10:845–851. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han YH, Kim SZ, Kim SH and Park WH:
Intracellular GSH level is a factor in As4.1 juxtaglomerular cell
death by arsenic trioxide. J Cell Biochem. 104:995–1009. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
You BR and Park WH: Enhancement of gallic
acid-induced human pulmonary fibroblast cell death by N-acetyl
cysteine and L-buthionine sulfoximine. Hum Exp Toxicol. 30:992–999.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
You BR and Park WH: MG132, a proteasome
inhibitor-induced calf pulmonary arterial endothelial cell growth
and death, are changed by MAPK inhibitors. Drug Chem Toxicol.
34:45–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han YH, Kim SZ, Kim SH and Park WH:
Reactive oxygen species and glutathione level changes by a
proteasome inhibitor, MG132, partially affect calf pulmonary
arterial endothelial cell death. Drug Chem Toxicol. 33:403–409.
2010. View Article : Google Scholar : PubMed/NCBI
|