Hypoxia-ischemia (H-I) commonly occurs during
myocardial infarction, stroke and perinatal asphyxia. It can lead
to severe injuries such as cerebral palsy (1), H-I brain damage, chronic neurological
and neurodevelopmental disability in children, and even death
(2). In addition, H-I may trigger
massive cellular malfunction and cell death. On the other hand, the
decline of cellular oxygen level during H-I also induces many
compensatory responses, such as neovascularization (3), metabolic regulation and production of
various neurotrophic mediators, which protect neurons from ischemic
death. These processes also form part of an endogenous adaptive
response that aims to defend and help tissues recover from ischemic
injury (4). The rapid restoration
of blood flow in the occluded coronary arteries following H-I is
the most important aspect of the protective mechanism.
Nevertheless, the early opening of an occluded coronary artery may
lead to ischemia/reperfusion (I/R) injury (5,6).
It has been reported that phosphatidylinositol-3
kinase (PI3K)/Akt signaling pathway is involved in H-I. In this
review, we have discussed the potential mechanism of PI3K/Akt
signaling pathway in cellular responses for resisting H-I.
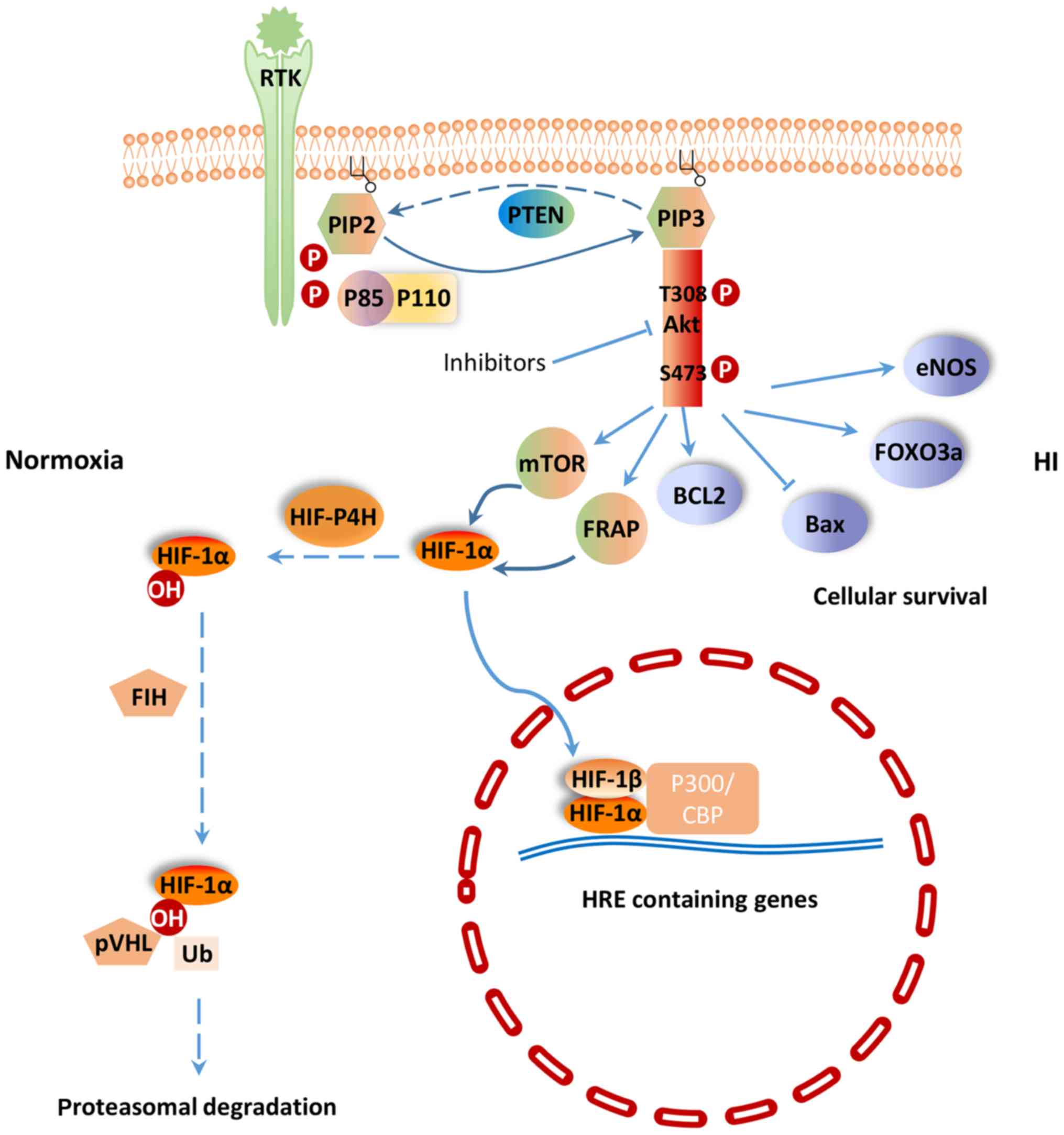
PI3K/Akt signaling pathway regulates a wide range of
cellular activities including cell survival, proliferation,
metabolism, neuroscience, motility and cancer progression (7). PI3K belongs to a lipid kinase family
which is characterized by their ability to phosphorylate inositol
ring 3′-OH group in inositol phospholipids in the plasma membrane
(8). PI3Ks are divided into two
classes: Class-I and II. The function of class-I PI3K is to
phosphorylate PIP-2 to generate the second messenger PIP-3 within
sec (9). PIP-3 can mediate
different cellular functions of PI3K through specific interactions
with pleckstrin homology (PH) domain containing proteins such as
Akt (10). Akt is considered as
the central mediator of the PI3K/Akt signaling pathway, which
ultimately leads to the phosphorylation of some vital downstream
targets (11). Furthermore, some
negative regulators, such as phosphatase and tensin homologue
(PTEN) inhibit PI3K/Akt signaling pathway. PTEN is a lipid
phosphatase that negatively regulates the PI3K/Akt pathway by
hydrolyzing PIP-3 to PIP-2, resulting in a lack of downstream p-Akt
(12) (Fig. 1). PI3K and the downstream effector
Akt belong to a conserved family of signal transduction enzymes,
which are involved in regulating cellular activation, inflammatory
responses and apoptosis (13).
It has been shown that H-I-induced injuries could be
treated by certain agents that act on the PI3K/Akt signaling
pathway. In cerebral ischemia rats, p-Akt 473 and p-Akt 308 protein
expression was significantly increased after treatment with
silibinin, a compound of flavonolignan with anti-apoptotic,
anti-inflammatory and anti-oxidative functions (14). Phosphorylated Akt promotes the
phosphorylation of downstream molecules, including Bcl-2 apoptosis
related family members, Forkhead box O3 (FoxO3a) transcription
factor, mammalian target of rapamycin (mTOR) and glycogen synthase
kinase-3, in order to protect cells from apoptosis. Bcl-2, an
inhibitor of neuronal apoptosis, is significantly upregulated,
while Bax, which can promote neuronal apoptosis, is significantly
downregulated in cerebral ischemia rats treated with silibinin
(15). Li et al (16) found that the PI3K/Akt/FoxO3a
pathway is involved in neuronal apoptosis in the developing rat
brain. Activated Akt phosphorylates FoxO3a, and leads to the
cytoplasmic localization of FoxO3a and inhibition of apoptosis
(17) (Fig. 1). In addition, sodium tanshinoneIIA
sulfonate and bromelain protect the rat heart from I/R injury via
the activation of PI3K/Akt/FoxO3a pathway (18). In the cytoplasm, mTOR, a
phosphoinositide kinase-related kinase family member, serves as a
Ser/Thr protein kinase (19).
Previous study found that the regulatory mechanism of mTOR activity
is related to the PI3K/Akt signaling pathway (14). Zhong et al (20) were among the first to show that
activation of the epidermal growth factor receptor
(EGFR)/PI3K/AKT/mTOR pathway could positively regulate
hypoxia-induced factor-1α (HIF-1α) at the protein level. Fibroblast
growth factor-2 is a signaling molecular in the PI3K/Akt signaling
pathway. Activation of PI3K/Akt pathway by fibroblast growth
factor-2 prevents reactive oxygen species (ROS)-induced apoptosis
and protects heart from I/R injury by decreasing infarct size and
improving left ventricular function (21).
A key regulator of the response to HI is HIF-1.
HIF-1 is a heterodimeric transcription factor composed of an oxygen
sensitive subunit HIF-1α and an aryl hydrocarbon nuclear
translocator HIF-1β. Under normoxic condition, HIF-1α is
hydroxylated at prolines 402 and 564 by HIF prolyl-4-hydroxylase,
leading to its ubiquitination and proteasomal degradation through
the ubiquitin-proteasome (26S) pathway, which can continuously
provoke proteasomal degradation. Its destruction is caused by the
ubiquitin E3 ligase complex, in which the von Hippel-Lindau tumor
suppressor protein (pVHL) is able to bind to the oxygen-dependent
destruction domain on the subunit, resulting in a short half-life
of the protein under normoxic conditions. In contrast, when HIF
prolyl-4-hydroxylase is less active, HIF-1α is more stable. This
stabilization allows HIF-1α to translocate to the nucleus and
dimerize with its partner HIF-1β (Fig.
1). The HIF-1 dimer subsequently binds to the hypoxia response
element site on DNA, initiating the expression of more than 100
genes that participate in hypoxic adaptation (22,23).
HIF-1α is involved in pathologic conditions such as hypoxia or
ischemia. HIF-1α has also been shown to regulate the expression of
vascular endothelial growth factor (VEGF), erythropoietin (EPO) and
glycolytic enzymes (24) (Fig. 2).
Previous studies have shown that HIF-1α is subjected
to regulation by the PI3K/Akt/mTOR (20,25)
and PI3K/Akt/FRAP (26) signaling
pathways. The p-Akt and HIF-1α protein levels were shown to
increase in response to hypoxia in human mesenchymal stem cells.
Moreover, p-Akt expression peaked earlier than that of HIF-1α.
Interestingly, the PI3K inhibitor LY294002 (27) and Dual PI3K/mTOR inhibitor
NVP-BEZ235 (28) could suppress
the activation of p-Akt and the expression of HIF-1α and VEGF
resulted from H-I. The Akt inhibitor, wortmannin, could also
inhibit the expression of HIF-1α at the protein, but not the mRNA
level (7). mTOR is a
hypoxia/nutrient sensor and a target of Akt during cell cycle
regulation, glycogen metabolism and protein synthesis upon
phosphorylation of its two main targets, eukaryotic initiation
factor 4E-binding protein-1 and ribosomal protein S6 kinase
(29). Moreover, mTOR is an
upstream mediator of HIF-1α activation (30). Based on these previous findings,
the PI3K/Akt signaling pathway could potentially regulates HIF-1α
via mTOR, which could alter HIF-1α post-transcriptional protein
level, but not at the transcriptional mRNA level.
It has been shown that the pVHL mutant fails to
degrade HIF-1α, which implies that pVHL plays an important role in
controlling the stability of HIF-1α (31). In another word, the stabilization
of HIF-1α could be attributed to failure in pVHL-mediated
ubiquitination and proteasomal degradation. The proteasomal
degradation process is often controlled by phosphorylation
(32). Therefore, it is speculated
that HIF-1α activity is under the control of protein kinase
phosphorylation, potentially through the universal phosphorylation
signal transduction pathway of the PI3K/Akt (33).
Under H-I condition, PI3K/HIF pathway plays
important roles in cardio-protection and neuro-protection. The
expression of HIF-1α has been shown to increase significantly in
various ischemic organs and tissues, including myocardium, nervous
system and retina (34). The
protection of HIF-1 has also been widely reported in various H-I
models. For example, HIF-1 has been demonstrated to participate in
neuroprotection during permanent focal ischemia in vivo
(22). Various iron chelators,
such as deferoxamine mesylate and mimosine, protect neurons from
apoptosis through activating HIF-1 in vitro or in
vivo (35,36). These results reveal that induction
of HIF-1 by ischemia itself or via pharmacological channels can
protect against H-I. Furthermore, HIF-1 can regulate the expression
of various genes, including EPO, VEGF, inducible nitric oxide
synthase, hemeoxygenase, and cardiotropin as well as those involved
in glucose metabolism, mitochondrial function, cell apoptosis, and
resistance to oxidative stress that protect or restore cell
functions and facilitate cellular adaptation to H-I (37,38).
A number of mechanisms have been proposed for the
protective effect of HIF-1. HIF-1 has been found to protect cells
from hypoxic injury by promoting nutrient and O2
transport via inducing the expression of downstream proteins such
as VEGF and EPO, which promote angiogenesis and erythropoiesis.
This induction is partly PI3K/Akt inhibition-dependent, suggesting
a close relationship between PI3K/Akt, HIF-1α and the VEGF cascade
in HI (7). EPO promotes the
production and release of red blood cell into blood, thereby,
enhancing the oxygen transport. Meanwhile, the increase in
hemoglobin level also affects oxygen transport capacity, and
ultimately reduces tissue damage. On the other hand, HIF-1 may
prevent apoptotic cell death through inhibiting the release of
cytochrome C, PARP cleavage and caspase activation. In addition,
HIF-1 may maintain cell survival by suppressing p53 activation.
Increased glucose transport and glycolytic flow consequential of
HIF-1 activation by H-I has also been implicated in tissue
viability and cell survival (39).
One important function of glucose metabolism is to
sustain a reducing environment in cells by generating reducing
equivalents through oxidative phosphorylation, glycolysis, and the
pentose phosphate pathway (40).
The switch from aerobic to anaerobic glucose metabolism by
upregulating glucose transporters (GLUTs) and glycolysis-related
enzymes, such as phosphofructokinase 1, fructose-bisphosphate
aldolase, phosphoglycerate kinase 1, pyruvate dehydrogenase kinase
1, and lactate dehydrogenase, is one of the key mechanisms to
maintain cellular energy production and cell survival during
ischemia (39). The expression of
these proteins is mainly controlled by HIFs. HIF-1 activation leads
to increased oxygen and nutrient delivery via enhancing
angiogenesis and erythropoiesis (41,42)
and improving oxygen utilization in metabolism (43). Activated HIF-1 is either directly
or indirectly associated with the upregulation of GLUTs (44) and glycolytic enzymes in glycolysis
and lactate production (45,46)
(Fig. 2). This effect ultimately
leads to the upregulation of aerobic glycolysis in tumor cells,
while dampening the oxidative phosphorylation pathway (47). GLUT1 is upregulated by H-RAS, at
least in part, via PI3K/HIF (48).
Some stimuli, such as insulin, insulin-like growth factor 1,
epidermal growth factor and angiotensin II, are able to increase
HIF-1α level in cells (49). Other
key enzymes involved in metabolism are also upregulated to further
ensure cellular survival (50).
Other studies have further demonstrated that HIF-1 activation could
be attributed to cellular mutations under non-hypoxic conditions.
This phenomenon is resulted from inactivation of various tumor
suppression genes, along with activation of numerous oncogenes,
which then lead to mutations in several growth factor pathways,
such as the loss of pVHL. HIF-1α inactivation is caused by a
physical interaction with pVHL, which elicits the 26S proteasome
response. Studies have also reported that the β domain of pVHL
interacts directly with the HIF-1α subunits. Therefore, any
mutation that affects the β domain of pVHL may prevent its
interaction between HIF-1α and thereby lead to the constitutive
activation of HIF-1 (51).
The glycolysis process is an important metabolic
pathway in mammals. Similar to GLUT, Hexokinase (HK) acts as a
rate-limiting enzyme and is the first glycolytic enzyme which
facilitates the irreversible phosphorylation of glucose to
glucose-6-phosphate in cells, thereby committing the glucose
molecule to the glycolytic cycle. HIF-1 activation can upregulate
the expression level of HK1 and HK2 (52). In addition, HIF-1 has been shown to
effectively upregulate the expression of many other glycolytic
enzymes, leading to enhanced glycolysis. The glycolytic flux
triggered by HIF-1α is also related to the kinetic patterns of the
expressed isoforms of the key glycolytic enzymes, which can further
promote glycolytic energetic capability. Moreover, HIF-1 induces
the transcription of pyruvate dehydrogenase kinase 1, which
effectively inhibits pyruvate dehydrogenase activity, thereby
downregulating acetyl-CoA production and suppressing the TCA cycle
(53). The level of Akt directly
correlates with the rate of glucose uptake into the cell through
the GLUT1 transporter (54). In
addition, Akt can further influence glycolysis via HK2. Akt also
activates FOXO3a to inhibit apoptosis and increases mitochondrial
biogenesis to support a cellular survival (55). PKM2, an isoform of pyruvate kinase,
harbors a hormone response element within its first intron,
indicating that its transcriptional activity is regulated by HIF-1.
PKM2 is also found to interact with HIF-1α in the nucleus and is
believed to act as a transcriptional co-activator. It has been
shown that activated tyrosine kinase inhibits pyruvate kinase,
which further prevents pyruvate from entering into mitochondria and
participating in the TCA cycle (56). Other studies on the glycolytic
cycle have shown that increased pyruvate and lactate result in an
increased expression of the monocarboxylate transporter (MCT) and
lactate dehydrogenase (57).
Although the mechanism is still unknown, reducing the expression of
lactate dehydrogenase may lead to a decrease in production of
lactate. On the other hand, MCT provides rapid transportation of
monocarboxylate compounds, such as pyruvate and lactate, across
plasma membrane, providing essential support for energy metabolism.
Furthermore, activation of these transporters is closely related to
HIF-1α. Previous studies have revealed that the inhibition of MCT1
can suppress lactate-induced HIF-1 activation. Whereas, the
expression of MCT4 is mainly regulated by HIF-1α. Taken together,
metabolism via the aerobic glycolytic pathway appears to be favored
over the oxidative phosphorylation pathway in the presence of
activated HIF. TCA cycle intermediates oxygen molecules and
α-ketoglutarate is responsible for facilitating the degradation of
HIF-1α (58).
Angiogenesis is a key step in oxygen and nutrient
transport. Therapeutic angiogenesis is an attractive approach for
curing or alleviating ischemic cardiovascular disease (59). Angiogenesis plays an important role
in the repair of tissues subjected to ischemic insult.
Neovascularization is expected to reduce ventricular dysfunction
and remodeling after myocardial infarction (MI) (60). The PI3K/Akt signaling pathway is
crucial to inducing vascularization of heart and inhibiting
cardiomyocyte apoptosis after MI (61,62).
PI3K has several different isoforms (p110α, p110β, and p110δ), but
only p110α is selectively required for angiogenesis (63). Interestingly, the protein kinase,
Akt, has also been implicated as a mediator of cardio-protection
(64). The activation of Ras and
EGFR, a transmembrane receptor tyrosine kinase (RTK) that belongs
to the HER family of receptors, upregulates HIF-1α via the PI3K/Akt
signaling pathway (65).
EGFR/PI3K/AKT/mTOR pathway increases VEGF and endothelial cell NO
synthase (eNOS) expression by upregulating HIF-1α. VEGF, an
endothelial-specific mitogen and survival factor, is one of the
most potent angiogenic factors, and plays key roles in both
angiogenesis and vasculogenesis. Hypoxia can increase eNOS
phosphorylation by activating the PI3K/AKT pathway (66). HIF-1α can also directly influence
the expression of eNOS, which can be activated by phosphorylation
of the serine 1177 residue, thereby, triggering migration and
angiogenesis (67) (Fig. 2). Accumulating evidence has shown
that HIF-1α acts as a potential therapeutic proangiogenic molecule
in experimental models (68,69).
Furthermore, EGFR amplification and PTEN mutation exert an additive
effect on increasing VEGF promoter activity in human glioblastoma
cells. A recent study that explored the role of PTEN in
hepatocellular carcinoma also found similar inhibition of
angiogenesis (70). Elevated
levels of VEGF can increase vascular permeability, leading to
vessel leakage, sluggish blood flow, and elevated interstitial
pressure. One of the potent stimuli for increased VEGF production
is hypoxia (71). Binding of both
STAT3 and HIF-1α to the VEGF promoter has been demonstrated to be
essential for maximum transcription of VEGF mRNA under hypoxia
(72). Therefore, therapies that
affect HIF-1α expression could potentially induce neoangiogenesis
in ischemic heart.
The reintroduction of oxygen after H-I is
inevitable, nevertheless, reperfusion is associated with
exacerbation of I/R tissue injury caused by inflammatory responses
and ROS production. Therefore, the alleviation of I/R injury is a
popular strategy for treating diseases associated with H-I. Factors
such as high mobility group box 1 (HMGB1) may exert its protective
effect by upregulating the protein expression of HIF-1α in the
ischemic myocardium via enhancing Akt phosphorylation through the
PI3K/Akt signaling pathway. Treatment with LY294002 inhibits
HMGB1-induced expression of HIF-1α and eliminates the
cardioprotective effects exerted by intravenous HMGB1 in an I/R rat
model. ROS can directly damage the cell membrane and cause cell
death during I/R. Furthermore, ROS-mediated apoptosis and necrosis
can be a determinant of infarct size. HMGB1 reduces the myocardial
content of MDA and increases the activity of SOD induced by I/R,
whereas LY294002 eliminates these effects (34). Guo et al (73) demonstrated that inhibiting HIF-1α
expression by HIF-1α-specific small interfering RNA transfection
increases ROS generation and promotes cell death.
Cardiomyocyte-specific HIF-1α gene deletion leads to reduced
contractility and vascularization, along with altering the
expression of multiple genes in normoxic heart. I/R significantly
increases the myocardial expression of HIF-1α, while HMGB1 also
markedly upregulates the expression of HIF-1α. Furthermore,
consistent with the increased expression of HIF-1α, the myocardial
injury induced by I/R was inhibited by HMGB1. It was also found
that intravenous HMGB1 increases SOD activity in the I/R
myocardium, which suggests that these changes may be occurring
downstream of its effects on HIF-1α overexpression. Thus,
intravenous HMGB1 may exert its cardioprotective effects through
increasing the expression of HIF-1α (34). In addition, increasing HIF-1α
expression by drugs such as desferrioxamine, can induce a more
reducing environment and decrease cell death. These results suggest
that maintenance of cellular redox status via HIF-1 can protect
cells from H-I mediated injuries (74).
Although HIF-1 exerts protective effects, it may
also contribute to cellular and tissue damage. It has been reported
that HIF-1 may mediate apoptosis in embryonic stem cells under
hypoxic conditions (75).
Similarly, it has been observed that HIF-1 signaling elicits
delayed death via p53 in ischemic primary cortical neurons in
vivo (76) and in vitro
(77). Chen et al (78) has shown that inhibition of HIF-1
decreases the expression of VEGF and BCL2 interacting protein 3
(BNIP3) and thereby offering protection against delayed cell death.
BNIP3 reduces increased levels of ROS via HIF-1-inducible
mitochondrial autophagy (79),
meanwhile causing mitochondrial dysfunction, opening of the
mitochondrial permeability transition pores, membrane
depolarization and cell death. Two h of ischemia has been shown to
result in damage of brain cortex and blood-brain barrier in the
non-infarcted ventromedial striatum and preoptic area. BNIP3 is
induced in the brain under H-I condition as a master regulator in
hypoxia. Suppression of HIF-1α and VEGF has been shown to reduce
acute hyperglycemia-induced HT in the ischemic brain (80). Moreover, the various protective
effects through PI3K/AKT and HIF-1 pathways may become reverse in
cancer hypoxic microenvironment. Multiple members of the lysyl
oxidase family induced in an HIF-1-dependent manner are involved in
Metastatic niche formation (81,82).
It was shown that HIF-1 is involved in almost every key step of the
breast cancer metastatic process including epithelial-mesenchymal
transition, invasion, intravasation, extravasation, and metastatic
niche formation (83).
Despite the relatively high incidence of ischemic
cerebrovascular and cardiovascular disease, limited therapies are
currently available for its prevention and treatment (92,93).
Although the survival rate for pre-term infants has been increased,
neurological conditions such as cerebral palsy still occur in most
survivors (94). PI3K/HIF pathway
is important for both the mechanistic understanding and therapeutic
intervention of diseases associate with H-I such as stroke,
cardiovascular disease, cerebral ischemia and perinatal asphyxia.
Interestingly, HIF-1 and PI3K/Akt appears to be involved in the
cellular responses to H-I, but with a double-edged sword effect,
which could possibly be dependent on the degree and duration of
H-I. Therefore, therapies for hypoxic injury should be selected
with this caveat in mind, and further study is necessary to find
the optimal hypoxic pattern of different cell types. Understanding
the mechanism of HIF-1 and PI3K/Akt accumulation would undoubtedly
provide important insight into its role in H-I and provide
potential approaches to regulate its expression.
The authors would like to thank Dr Qiang Lin
(Department of Rehabilitation, Fifth Affiliated Hospital of
Guangzhou Medical University, Guangdong, China) for his linguistic
work on this manuscript.
The present study was supported by the Natural
Science Foundation of Guangdong Province (grant no. 2015A030313254)
and the Science and Technology Planning Project of Guangdong
Province (grant no. 2014A020212493).
All data generated or analyzed during the present
study are included in this published article.
ZZ, LY and JY designed the study and drafted the
manuscript. ZZ and ZW performed the data collection and generated
the figures. ZZ and GD conceived the research and were in charge of
overall direction and planning. All authors approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Movsas TZ, Weiner RL, Greenberg MB,
Holtzman DM and Galindo R: Pretreatment with human chorionic
gonadotropin protects the neonatal brain against the effects of
hypoxic-ischemic injury. Front Pediatr. 5:2322017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferriero DM: Neonatal brain injury. N Engl
J Med. 351:1985–1995. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adluri RS, Thirunavukkarasu M, Zhan L,
Akita Y, Samuel SM, Otani H, Ho YS, Maulik G and Maulik N:
Thioredoxin 1 enhances neovascularization and reduces ventricular
remodeling during chronic myocardial infarction: A study using
thioredoxin 1 transgenic mice. J Mol Cell Cardiol. 50:239–247.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Greenberg DA and Jin K: From angiogenesis
to neuropathology. Nature. 438:954–959. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li L, Qu Y, Mao M, Xiong Y and Mu D: The
involvement of phosphoinositid 3-kinase/Akt pathway in the
activation of hypoxia-inducible factor-1alpha in the developing rat
brain after hypoxia-ischemia. Brain Res. 1197:152–158. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fruman DA, Meyers RE and Cantley LC:
Phosphoinositide kinases. Annu Rev Biochem. 67:481–507. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pawson T and Nash P: Protein-protein
interactions define specificity in signal transduction. Genes Dev.
14:1027–1047. 2000.PubMed/NCBI
|
|
11
|
Zhang F, Ding T, Yu L, Zhong Y, Dai H and
Yan M: Dexmedetomidine protects against oxygen-glucose
deprivation-induced injury through the I2 imidazoline
receptor-PI3K/AKT pathway in rat C6 glioma cells. J Pharm
Pharmacol. 64:120–127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ciuffreda L, Falcone I, Incani UC, Del
Curatolo A, Conciatori F, Matteoni S, Vari S, Vaccaro V, Cognetti F
and Milella M: PTEN expression and function in adult cancer stem
cells and prospects for therapeutic targeting. Adv Biol Regul.
56:66–80. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han JQ, Yu KY and He M: Effects of
puerarin on the neurocyte apoptosis and p-Akt (Ser473) expressions
in rats with cerebral ischemia/reperfusion injury. Zhongguo Zhong
Xi Yi Jie He Za Zhi. 32:1069–1072. 2012.(In Chinese). PubMed/NCBI
|
|
15
|
Liu BN, Han BX and Liu F: Neuroprotective
effect of pAkt and HIF-1α on ischemia rats. Asian Pac J Trop Med.
7:221–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li D, Qu Y, Mao M, Zhang X, Li J, Ferriero
D and Mu D: Involvement of the PTEN-AKT-FOXO3a pathway in neuronal
apoptosis in developing rat brain after hypoxia-ischemia. J Cereb
Blood Flow Metab. 29:1903–1913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Wang Y, Ye J, Lu X, Cheng Y, Xiang
L, Chen L, Feng W, Shi H, Yu X, et al: bFGF attenuates endoplasmic
reticulum stress and mitochondrial injury on myocardial
ischaemia/reperfusion via activation of PI3K/Akt/ERK1/2 pathway. J
Cell Mol Med. 19:595–607. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu MH, Li GH, Peng LJ, Qu SL, Zhang Y,
Peng J, Luo XY, Hu HJ, Ren Z, Liu Y, et al: PI3K/Akt/FoxO3a
signaling mediates cardioprotection of FGF-2 against hydrogen
peroxide-induced apoptosis in H9c2 cells. Mol Cell Biochem.
414:57–66. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Correia SC, Cardoso S, Santos RX, Carvalho
C, Santos MS, Perry G, Smith MA and Moreira PI: New insights into
the mechanisms of mitochondrial preconditioning-triggered
neuroprotection. Curr Pharm Des. 17:3381–3389. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhong H, Chiles K, Feldser D, Laughner E,
Hanrahan C, Georgescu MM, Simons JW and Semenza GL: Modulation of
hypoxia-inducible factor 1alpha expression by the epidermal growth
factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human
prostate cancer cells: Implications for tumor angiogenesis and
therapeutics. Cancer Res 60: 1541–1545, 2000. Cancer Res 60:
1541–1545, 2000. 60: 1541–1545, 2000:1541-1545, 2000–1545, 2000.
2000.
|
|
21
|
Wang Z, Zhang H, Xu X, Shi H, Yu X, Wang
X, Yan Y, Fu X, Hu H, Li X and Xiao J: bFGF inhibits ER stress
induced by ischemic oxidative injury via activation of the PI3K/Akt
and ERK1/2 pathways. Toxicol Lett. 212:137–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kunze R, Zhou W, Veltkamp R, Wielockx B,
Breier G and Marti HH: Neuron-specific prolyl-4-hydroxylase domain
2 knockout reduces brain injury after transient cerebral ischemia.
Stroke. 43:2748–2756. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Semenza GL: Hypoxia-inducible factors in
physiology and medicine. Cell. 148:399–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jain T, Nikolopoulou EA, Xu Q and Qu A:
Hypoxia inducible factor as a therapeutic target for
atherosclerosis. Pharmacol Ther. 183:22–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao Y, Peng H, Hong C, Chen Z, Deng X,
Wang A, Yang F, Yang L, Chen C and Qin X: PDGF promotes the warburg
effect in pulmonary arterial smooth muscle cells via activation of
the PI3K/AKT/mTOR/HIF-1α signaling pathway. Cell Physiol Biochem.
42:1603–1613. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Laughner E, Taghavi P, Chiles K, Mahon PC
and Semenza GL: HER2 (neu) signaling increases the rate of
hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel
mechanism for HIF-1-mediated vascular endothelial growth factor
expression. Mol Cell Biol. 21:3995–4004. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang XM, Wang YS, Zhang J, Li Y, Xu JF,
Zhu J, Zhao W, Chu DK and Wiedemann P: Role of PI3K/Akt and MEK/ERK
in mediating hypoxia-induced expression of HIF-1alpha and VEGF in
laser-induced rat choroidal neovascularization. Invest Ophthalmol
Vis Sci. 50:1873–1879. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karar J, Cerniglia GJ, Lindsten T,
Koumenis C and Maity A: Dual PI3K/mTOR inhibitor NVP-BEZ235
suppresses hypoxia-inducible factor (HIF)-1α expression by blocking
protein translation and increases cell death under hypoxia. Cancer
Biol Ther. 13:1102–1111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
van den Beucken T, Koritzinsky M and
Wouters BG: Translational control of gene expression during
hypoxia. Cancer Biol Ther. 5:749–755. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hudson CC, Liu M, Chiang GG, Otterness DM,
Loomis DC, Kaper F, Giaccia AJ and Abraham RT: Regulation of
hypoxia-inducible factor 1alpha expression and function by the
mammalian target of rapamycin. Mol Cell Biol. 22:7004–7014. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ivan M and Kaelin WG Jr: The von
Hippel-Lindau tumor suppressor protein. Curr Opin Genet Dev.
11:27–34. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bai S, Datta J, Jacob ST and Ghoshal K:
Treatment of PC12 cells with nerve growth factor induces
proteasomal degradation of T-cadherin that requires tyrosine
phosphorylation of its cadherin domain. J Biol Chem.
282:27171–27180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dimova EY, Michiels C and Kietzmann T:
Kinases as upstream regulators of the HIF system: their emerging
potential as anti-cancer drug targets. Curr Pharm Des.
15:3867–3877. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yao HC, Zhou M, Zhou YH, Wang LH, Zhang
DY, Han QF, Liu T, Wu L, Tian KL and Zhang M: Intravenous high
mobility group box 1 upregulates the expression of HIF-1α in the
myocardium via a protein kinase B-dependent pathway in rats
following acute myocardial ischemia. Mol Med Rep. 13:1211–1219.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zaman K, Ryu H, Hall D, O'Donovan K, Lin
KI, Miller MP, Marquis JC, Baraban JM, Semenza GL and Ratan RR:
Protection from oxidative stress-induced apoptosis in cortical
neuronal cultures by iron chelators is associated with enhanced DNA
binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased
expression of glycolytic enzymes, p21 (waf1/cip1), and
erythropoietin. J Neurosci 19: 9821–9830, 1999. J Neurosci 19:
9821–9830, 1999. 19: 9821–9830, 1999:9821-9830, 1999–9830, 1999.
1999.
|
|
36
|
Hamrick SE, McQuillen PS, Jiang X, Mu D,
Madan A and Ferriero DM: A role for hypoxia-inducible factor-1alpha
in desferoxamine neuroprotection. Neurosci Lett. 379:96–100. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sharp FR and Bernaudin M: HIF1 and oxygen
sensing in the brain. Nat Rev Neurosci. 5:437–448. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Semenza GL: Angiogenesis in ischemic and
neoplastic disorders. Annu Rev Med. 54:17–28. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Blanco Pampin J, Garcia Rivero SA, Otero
Cepeda XL, Vázquez Boquete A, Forteza Vila J and Hinojal Fonseca R:
Immunohistochemical expression of HIF-1alpha in response to early
myocardial ischemia. J Forensic Sci. 51:120–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shi H: Hypoxia inducible factor 1 as a
therapeutic target in ischemic stroke. Curr Med Chem. 16:4593–4600.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Manalo DJ, Rowan A, Lavoie T, Natarajan L,
Kelly BD, Ye SQ, Garcia JG and Semenza GL: Transcriptional
regulation of vascular endothelial cell responses to hypoxia by
HIF-1. Blood. 105:659–669. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Minet E, Michel G, Remacle J and Michiels
C: Role of HIF-1 as a transcription factor involved in embryonic
development, cancer progression and apoptosis (review). Int J Mol
Med. 5:253–259. 2000.PubMed/NCBI
|
|
43
|
Semenza GL: Regulation of cancer cell
metabolism by hypoxia-inducible factor 1. Semin Cancer Biol.
19:12–16. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Adekola K, Rosen ST and Shanmugam M:
Glucose transporters in cancer metabolism. Curr Opin Oncol.
24:650–654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Semenza GL: Hypoxia-inducible factor 1:
Regulator of mitochondrial metabolism and mediator of ischemic
preconditioning. Biochim Biophys Acta. 1813:1263–1268. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim JW, Tchernyshyov I, Semenza GL and
Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase
kinase: a metabolic switch required for cellular adaptation to
hypoxia. Cell Metab. 3:177–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Simon MC: Coming up for air: HIF-1 and
mitochondrial oxygen consumption. Cell Metab. 3:150–151. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen C, Pore N, Behrooz A, Ismail-Beigi F
and Maity A: Regulation of glut1 mRNA by hypoxia-inducible
factor-1. Interaction between H-ras and hypoxia. J Biol Chem.
276:9519–9525. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lu H, Forbes RA and Verma A:
Hypoxia-inducible factor 1 activation by aerobic glycolysis
implicates the Warburg effect in carcinogenesis. J Biol Chem.
277:23111–23115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Semenza GL: HIF-1 mediates the Warburg
effect in clear cell renal carcinoma. J Bioenerg Biomembr.
39:231–234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Maxwell PH, Pugh CW and Ratcliffe PJ:
Activation of the HIF pathway in cancer. Curr Opin Genet Dev.
11:293–299. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Semenza GL: HIF-1: Upstream and downstream
of cancer metabolism. Curr Opin Genet Dev. 20:51–56. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nagy MA: HIF-1 is the commander of
gateways to cancer. J Cancer Sei Ther. 3:35–40. 2011.
|
|
54
|
Courtnay R, Ngo DC, Malik N, Ververis K,
Tortorella SM and Karagiannis TC: Cancer metabolism and the Warburg
effect: The role of HIF-1 and PI3K. Mol Biol Rep. 42:841–851. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dang CV: Links between metabolism and
cancer. Genes Dev. 26:877–890. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Martini M, De Santis MC, Braccini L,
Gulluni F and Hirsch E: PI3K/AKT signaling pathway and cancer: an
updated review. Ann Med. 46:372–383. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Koukourakis MI, Giatromanolaki A, Sivridis
E, Gatter KC and Harris AL; Tumour Angiogenesis Research Group, :
Lactate dehydrogenase 5 expression in operable colorectal cancer:
Strong association with survival and activated vascular endothelial
growth factor pathway-a report of the Tumour Angiogenesis Research
Group. J Clin Oncol. 24:4301–4308. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Solaini G, Baracca A, Lenaz G and Sgarbi
G: Hypoxia and mitochondrial oxidative metabolism. Biochim Biophys
Acta. 1797:1171–1177. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Samuel SM, Akita Y, Paul D,
Thirunavukkarasu M, Zhan L, Sudhakaran PR, Li C and Maulik N:
Coadministration of adenoviral vascular endothelial growth factor
and angiopoietin-1 enhances vascularization and reduces ventricular
remodeling in the infarcted myocardium of type 1 diabetic rats.
Diabetes. 59:51–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bai WW, Xing YF, Wang B, Lu XT, Wang YB,
Sun YY, Liu XQ, Guo T and Zhao YX: Tongxinluo improves cardiac
function and ameliorates ventricular remodeling in mice model of
myocardial infarction through enhancing angiogenesis. Evid Based
Complement Alternat Med 2013. 8132472013.
|
|
61
|
Patten RD, Pourati I, Aronovitz MJ, Baur
J, Celestin F, Chen X, Michael A, Haq S, Nuedling S, Grohe C, et
al: 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in
vitro via activation of phospho-inositide-3 kinase/Akt signaling.
Circ Res. 95:692–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
He Z, Opland DM, Way KJ, Ueki K, Bodyak N,
Kang PM, Izumo S, Kulkarni RN, Wang B, Liao R, et al: Regulation of
vascular endothelial growth factor expression and vascularization
in the myocardium by insulin receptor and PI3K/Akt pathways in
insulin resistance and ischemia. Arterioscler Thromb Vasc Biol.
26:787–793. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Graupera M, Guillermet-Guibert J, Foukas
LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J,
Cutillas PR, et al: Angiogenesis selectively requires the p110alpha
isoform of PI3K to control endothelial cell migration. Nature.
453:662–666. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sumida A, Horiba M, Ishiguro H, Takenaka
H, Ueda N, Ooboshi H, Opthof T, Kadomatsu K and Kodama I: Midkine
gene transfer after myocardial infarction in rats prevents
remodelling and ameliorates cardiac dysfunction. Cardiovasc Res.
86:113–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Dutta PR and Maity A: Cellular responses
to EGFR inhibitors and their relevance to cancer therapy. Cancer
Lett. 254:165–177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen JX and Meyrick B: Hypoxia increases
Hsp90 binding to eNOS via PI3K-Akt in porcine coronary artery
endothelium. Lab Invest. 84:182–190. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hirota K and Semenza GL: Regulation of
angiogenesis by hypoxia-inducible factor 1. Crit Rev Oncol Hematol.
59:15–26. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kido M, Du L, Sullivan CC, Li X, Deutsch
R, Jamieson SW and Thistlethwaite PA: Hypoxia-inducible factor
1-alpha reduces infarction and attenuates progression of cardiac
dysfunction after myocardial infarction in the mouse. J Am Coll
Cardiol. 46:2116–2124. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Khan M, Varadharaj S, Ganesan LP, Shobha
JC, Naidu MU, Parinandi NL, Tridandapani S, Kutala VK and Kuppusamy
P: C-phycocyanin protects against ischemia-reperfusion injury of
heart through involvement of p38 MAPK and ERK signaling. Am J
Physiol Heart Circ Physiol. 290:H2136–H2145. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Tian T, Nan KJ, Wang SH, Liang X, Lu CX,
Guo H, Wang WJ and Ruan ZP: PTEN regulates angiogenesis and VEGF
expression through phosphatase-dependent and -independent
mechanisms in HepG2 cells. Carcinogenesis. 31:1211–1219. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Forsythe JA, Jiang BH, Iyer NV, Agani F,
Leung SW, Koos RD and Semenza GL: Activation of vascular
endothelial growth factor gene transcription by hypoxia-inducible
factor 1. Mol Cell Biol. 16:4604–4613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Gray MJ, Zhang J, Ellis LM, Semenza GL,
Evans DB, Watowich SS and Gallick GE: HIF-1alpha, STAT3, CBP/p300
and Ref-1/APE are components of a transcriptional complex that
regulates Src-dependent hypoxia-induced expression of VEGF in
pancreatic and prostate carcinomas. Oncogene. 24:3110–3120. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Guo S, Miyake M, Liu KJ and Shi H:
Specific inhibition of hypoxia inducible factor 1 exaggerates cell
injury induced by in vitro ischemia through deteriorating cellular
redox environment. J Neurochem. 108:1309–1321. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang Z and Si LY: Hypoxia-inducible
factor-1α and vascular endothelial growth factor in the
cardioprotective effects of intermittent hypoxia in rats. Ups J Med
Sci. 118:65–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Carmeliet P, Dor Y, Herbert JM, Fukumura
D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R,
Maxwell P, et al: Role of HIF-1alpha in hypoxia-mediated apoptosis,
cell proliferation and tumour angiogenesis. Nature. 394:485–490.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Halterman MW and Federoff HJ: HIF-1alpha
and p53 promote hypoxia-induced delayed neuronal death in models of
CNS ischemia. Exp Neurol. 159:65–72. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Halterman MW, Miller CC and Federoff HJ:
Hypoxia-inducible factor-1alpha mediates hypoxia-induced delayed
neuronal death that involves p53. J Neurosci. 19:6818–6824. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Chen C, Hu Q, Yan J, Lei J, Qin L, Shi X,
Luan L, Yang L, Wang K, Han J, et al: Multiple effects of 2ME2 and
D609 on the cortical expression of HIF-1alpha and apoptotic genes
in a middle cerebral artery occlusion-induced focal ischemia rat
model. J Neurochem. 102:1831–1841. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang H, Bosch-Marce M, Shimoda LA, Tan
YS, Baek JH, Wesley JB, Gonzalez FJ and Semenza GL: Mitochondrial
autophagy is an HIF-1-dependent adaptive metabolic response to
hypoxia. J Biol Chem. 283:10892–10903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sun Y, Chen X, Zhang X, Shen X, Wang M,
Wang X, Liu WC, Liu CF, Liu J, Liu W and Jin X: β2-adrenergic
receptor-mediated HIF-1α upregulation mediates blood brain barrier
damage in acute cerebral ischemia. Front Mol Neurosci. 10:2572017.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wong CC, Gilkes DM, Zhang H, Chen J, Wei
H, Chaturvedi P, Fraley SI, Wong CM, Khoo US, Ng IO, et al:
Hypoxia-inducible factor 1 is a master regulator of breast cancer
metastatic niche formation. Proc Natl Acad Sci USA. 108:pp.
16369–16374. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wong CC, Zhang H, Gilkes DM, Chen J, Wei
H, Chaturvedi P, Hubbi ME and Semenza GL: Inhibitors of
hypoxia-inducible factor 1 block breast cancer metastatic niche
formation and lung metastasis. J Mol Med (Berl). 90:803–815. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Liu ZJ, Semenza GL and Zhang HF:
Hypoxia-inducible factor 1 and breast cancer metastasis. J Zhejiang
Univ Sci B. 16:32–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Baranova O, Miranda LF, Pichiule P,
Dragatsis I, Johnson RS and Chavez JC: Neuron-specific inactivation
of the hypoxia inducible factor 1 alpha increases brain injury in a
mouse model of transient focal cerebral ischemia. J Neurosci.
27:6320–6332. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Helton R, Cui J, Scheel JR, Ellison JA,
Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S, et al:
Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces
rather than increases hypoxic-ischemic damage. J Neurosci.
25:4099–4107. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Filippi I, Morena E, Aldinucci C, Carraro
F, Sozzani S and Naldini A: Short-term hypoxia enhances the
migratory capability of dendritic cell through HIF-1α and PI3K/Akt
pathway. J Cell Physiol. 229:2067–2076. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hu X, Wei L, Taylor TM, Wei J, Zhou X,
Wang JA and Yu SP: Hypoxic preconditioning enhances bone marrow
mesenchymal stem cell migration via Kv2.1 channel and FAK
activation. Am J Physiol Cell Physiol. 301:C362–C372. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hu X, Yu SP, Fraser JL, Lu Z, Ogle ME,
Wang JA and Wei L: Transplantation of hypoxia-preconditioned
mesenchymal stem cells improves infarcted heart function via
enhanced survival of implanted cells and angiogenesis. J Thorac
Cardiovasc Surg. 135:799–808. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Jian KT, Shi Y, Zhang Y, Mao YM, Liu JS
and Xue FL: Time course effect of hypoxia on bone marrow-derived
endothelial progenitor cells and their effects on left ventricular
function after transplanted into acute myocardial ischemia rat. Eur
Rev Med Pharmacol Sci. 19:1043–1054. 2015.PubMed/NCBI
|
|
90
|
Ginouvès A, Ilc K, Macias N, Pouysségur J
and Berra E: PHDs overactivation during chronic hypoxia
‘desensitizes’ HIFalpha and protects cells from necrosis. Proc Natl
Acad Sci USA. 105:pp. 4745–4750. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Poitz DM, Augstein A, Hesse K, Christoph
M, Ibrahim K, Braun-Dullaeus RC, Strasser RH and Schmeißer A:
Regulation of the HIF-system in human macrophages-differential
regulation of HIF-α subunits under sustained hypoxia. Mol Immunol.
57:226–235. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Higgins RD, Raju T, Edwards AD, Azzopardi
DV, Bose CL, Clark RH, Ferriero DM, Guillet R, Gunn AJ, Hagberg H,
et al: Hypothermia and other treatment options for neonatal
encephalopathy: An executive summary of the Eunice Kennedy Shriver
NICHD workshop. J Pediatr. 159:851–858.e1. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Shankaran S, Pappas A, McDonald SA, Vohr
BR, Hintz SR, Yolton K, Gustafson KE, Leach TM, Green C, Bara R, et
al: Childhood outcomes after hypothermia for neonatal
encephalopathy. N Engl J Med. 366:2085–2092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Huang J, Zhang L, Qu Y, Zhou Y, Zhu J, Li
Y, Zhu T, Zhao F, Tang J and Mu D: Histone acetylation of
oligodendrocytes protects against white matter injury induced by
inflammation and hypoxia-ischemia through activation of BDNF-TrkB
signaling pathway in neonatal rats. Brain Res. 1688:33–46. 2018.
View Article : Google Scholar : PubMed/NCBI
|