Introduction
Intestinal fibrosis, a serious complication
resulting from inflammatory bowel disease, may result in loss of
intestinal compliance, and thus irreversible intestinal stricture
(1,2). Unfortunately, available medications
have shown disappointing efficacy on prevention and treatment of
intestinal fibrosis; endoscopic or surgical intervention remain the
most practical treatment option for symptomatic intestinal
strictures (2,3). Therefore, it is of great importance
to look for effective means for prevention or treatment of
intestinal fibrosis.
Activated fibroblasts are the main effector cells in
the development of intestinal fibrosis responsible for excessive
extracellular matrix (ECM) production, and have been considered as
a major therapeutic target for antifibrotic therapy (4). Transforming growth factor β1
(TGF-β1), ‘master cytokine’ in fibrosis (5), plays a crucial role in the
pathogenesis of intestinal fibrosis, including fibroblasts
proliferation and differentiation into myofibroblasts, and ECM
production (6,7). Therefore, TGF-β1-stimulated
fibroblasts have been regarded as a good cellular model for
evaluating the efficacy of antifibrotic agents (8,9). It
has been reported that induction of apoptosis and inhibition of
proliferation and profibrogenic effects in fibroblasts have shown
potential in the treatment of intestinal fibrosis (10).
Pirfenidone (PFD), a novel anti-fibrotic drug, has
shown potent antifibrotic efficacy in many organs, such as lung
(11), liver (12), kidney (13), heart (14), and bladder (15). Additionally, it can inhibit cell
proliferation and/or activation of different fibroblasts, including
human Tenon's fibroblasts (16),
rat renal fibroblasts (17), and
rat cardiac fibroblasts (14), as
well as downregulation of a series of cytokines, including TGF-β1
and connective tissue growth factor (CTGF) (18). Recently, this agent has been
approved for clinical use in Europe and Japan due to its safety and
efficacy in the treatment of idiopathic pulmonary fibrosis
(19,20). In the context of the
gastrointestinal tract, PFD has been shown to exert antifibrotic
activities in a newly developed mouse model of intestinal fibrosis
and a murine colitis model (21,22).
In our previous study, we established a rat model of
radiation-induced intestinal fibrosis by an exposure of a single
dose of 20 Gy to the pelvis, and demonstrated that PFD could
attenuate radiation-induced intestinal fibrosis (23).
In the present study, we established ‘fibrotic’
human intestinal fibroblasts (HIFs) through stimulation with TGF-β1
to possibly mimic the in vivo ‘fibrosis’ situation. We
explored the effects of PFD on TGF-β1-induced fibrogenic activities
of HIFs. The results demonstrated that PFD alleviated
TGF-β1-induced HIFs transdifferentiation and collagen production by
inhibiting cell proliferation and inducing apoptosis through
inhibition of Smad and PI3K/AKT pathway.
Materials and methods
Reagents
Normal HIFs were purchased from iCell Bioscience
Inc., (cas no: HUM-CELL-d022; Shanghai, China). PFD was purchased
from Ji'nan Zhuokang Biological Technology Co., Ltd., (cas no:
53179-13-8; Shandong, China). Human recombinant TGF-β1 was
purchased from PeproTech, Inc., (cas no: 100-21; Rock Hill, USA).
Antibodies against α-smooth muscle actin (α-SMA), Collagen I alpha
1 (COLA1), fibronectin, total-Smad2/3 (t-Smad2/3), phospho-Smad2/3
(p-Smad2/3), phosphoinositide 3-kinase (PI3K), total-AKT (t-AKT),
phospho-AKT (p-AKT), Bcl-2, Bad, and GAPDH were purchased from
Bioss Antibodies (Beijing, China).
Cell culture and treatment
PFD was dissolved in dimethyl sulphoxide (DMSO) to
produce 20 µg/µl stock solution; working solution was obtained by
diluting stock solution with Dulbecco's modified minimal essential
medium (DMEM)/F12 solution containing 10% fetal bovine serum (the
final concentration of DMSO was less than 0.1%). No cytotoxic
effect was observed after DMSO treatment. HIFs were grown in
DMEM-F12 supplemented with 10% fetal bovine serum (37°C, 5%
CO2). All experiments were performed after 3–5 cell
passages. Prior to treatment, HIFs were pretreated with PFD (1
mg/ml) for 1 h before subsequent TGF-β1 stimulation (10 ng/ml).
Cytotoxicity and cell proliferation
assays
HIFs were pretreated with DMSO and different
concentrations of PFD (0, 0.01, 0.05, 0.1, 0.5, 1 mg/ml) for 24 h.
Cytotoxicity was assessed with a lactate dehydrogenase (LDH)
Cytotoxicity Assay kit (Beyotime Institute of Biotechnology,
Jiangsu, China) following the manufacturer's instructions.
Cell proliferation assays
HIFs proliferation was examined using Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) assay according to the manufacturer's instructions. Cells in
exponential growth were seeded in 96-well plates, incubated
overnight, then starved by serum deprivation for 24 h, and treated
with concentrations (0.5 mg/ml) of PFD in 10% FCS DMEM/F12 for 24
h. 10 µl CCK-8 solution (5 mg/ml) was added to each well and
incubated at 37°C for 2 h. Then the number of cells was assessed by
measuring optical absorbance at 450 nm using a Multiskan Go
microplate reader (Thermo Fisher Scientific, Inc., Waltham, MA,
USA).
Colony formation assays
HIFs were seeded in 6-well plates at (500 cells per
plate) for 24 h before the addition of drugs, cultured for 14 days,
fixed with 4% paraformaldehyde for 15 min. Cells were stained with
1% crystal violet for 10 min. The number of colonies with diameters
> 1.5 mm were counted.
Flow cytometry of apoptosis
Cell apoptosis was first detected using Annexin
V-Phycoerythrin (PE) and 7-amino-actinomycin D (7-ADD) Apoptosis
Detection kit I (BD Biosciences, Franklin Lakes, NJ, USA). Briefly,
cells were seeded in 6-well plates (4×104 cells per
well) and incubated for 24 h, and PFD or vehicle was added and
washed twice with PBS. Then, suspension was stained with 5 µl
Annexin V-PE and 7-ADD at room temperature for 15 min in the dark.
The cells were analyzed using a FACSCalibur flow cytometer (BD
Biosciences) within 30 min.
TdT-UTP nick end labeling (TUNEL)
assay
TUNEL staining for apoptotic cells was performed
using One-Step TUNEL Apoptosis Assay kit (Nanjing KeyGen Biotech
Co., Ltd., Nanjing, China) according to the manufacturer's
protocol. Nuclei were stained with DAPI. Fluorescence signals were
detected with a fluorescence microscope (Olympus Corporation,
Tokyo, Japan). Apoptosis index was quantified as the ratio of
(number of TUNEL-positive cells)/(number of total cells) ×100%.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from HIFs using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's instructions. cDNA was generated using M-MLV Reverse
Transcriptase Product (Promega Corporation, Madison, WI, USA).
RT-qPCR was performed in an ABI 7500 Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Primer sequences were
as follows: α-SMA: Forward 5′AGAGTTACGAGTTGCCTGATGG3′, Reverse
5′TTGCGGTGGACAATGGAAGG3′. COL-1A1: Forward
5′TGATGGTGCTACTGGTGCTG3′, Reverse 5′CCTCGCTTTCCTTCCTCTCC3′.
Fibronectin: Forward 5′GTGGTGTGGTCTACTCTGTGG3′, Reverse
5′TCTGGTCGGCATCATAGTTCTG3′, GAPDH: Forward
5′GGAAGGTGAAGGTCGGAGTC3′, Reverse 5′GGCATTGCTGATGATCTTGAGG3′. qPCR
reaction was carried out using SYBR Green reagents (Shanghai
Novland Co., Ltd., Shanghai, China) with the following
thermocycling conditions: Denaturation at 94°C for 2 min, followed
by 94°C for 15 sec, and 62°C for 40 sec for 40 cycles. The relative
expression of each targeted gene was determined using the
2−∆∆Cq comparative method (24) and normalized against that of GAPDH.
Each sample was run as triplicate and repeated in three independent
experiments.
Western blot analysis
Protein extracted from HIFs was quantified by the
BCA assay (Beyotime Institute of Biotechnology). Equal quantities
of protein (40 µg) were separated on 10% sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS/PAGE) and
electrotransferred onto a polyvinylidene fluoride membrane (EMD
Millipore, Billerica, MA, USA). The membrane was then blocked in 5%
BSA+TBST, incubated with primary antibody against α-SMA, COLA1,
fibronectin, Bcl-2, Bad. Smad2/3, p-Smad2/3, PI3K, AKT, and p-AKT,
then washed and incubated in horseradish peroxidase (HRP)
conjugated secondary antibodies. GAPDH acted as the reference
protein for loading control. The signals were detected using an
enhanced chemiluminescence kit (Beyotime Institute of
Biotechnology). Densitometric analysis was conducted using Image J
v.1.43 software (National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
Each experiment was repeated at least three times
and data was expressed as the mean ± standard deviation.
Comparisons between multiple groups were performed using one-way
analysis of variance with the least significant difference post hoc
test to analyze the differences between groups. GraphPad Prism
v.6.01 software (GraphPad Software, Inc., La Jolla, CA, USA) was
used for analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
PFD suppresses cell proliferation in
TGF-β1-induced HIFs
To clarify whether the effects of PFD were mediated
by cytotoxicity, LDH cytotoxicity assay was performed after
administration of DMSO and various concentration of PFD (0, 0.01,
0.05, 0.1, 0.5, 1 mg/ml). No cytotoxic effect of PFD on HIFs was
observed at all tested concentrations (Fig. 1A).
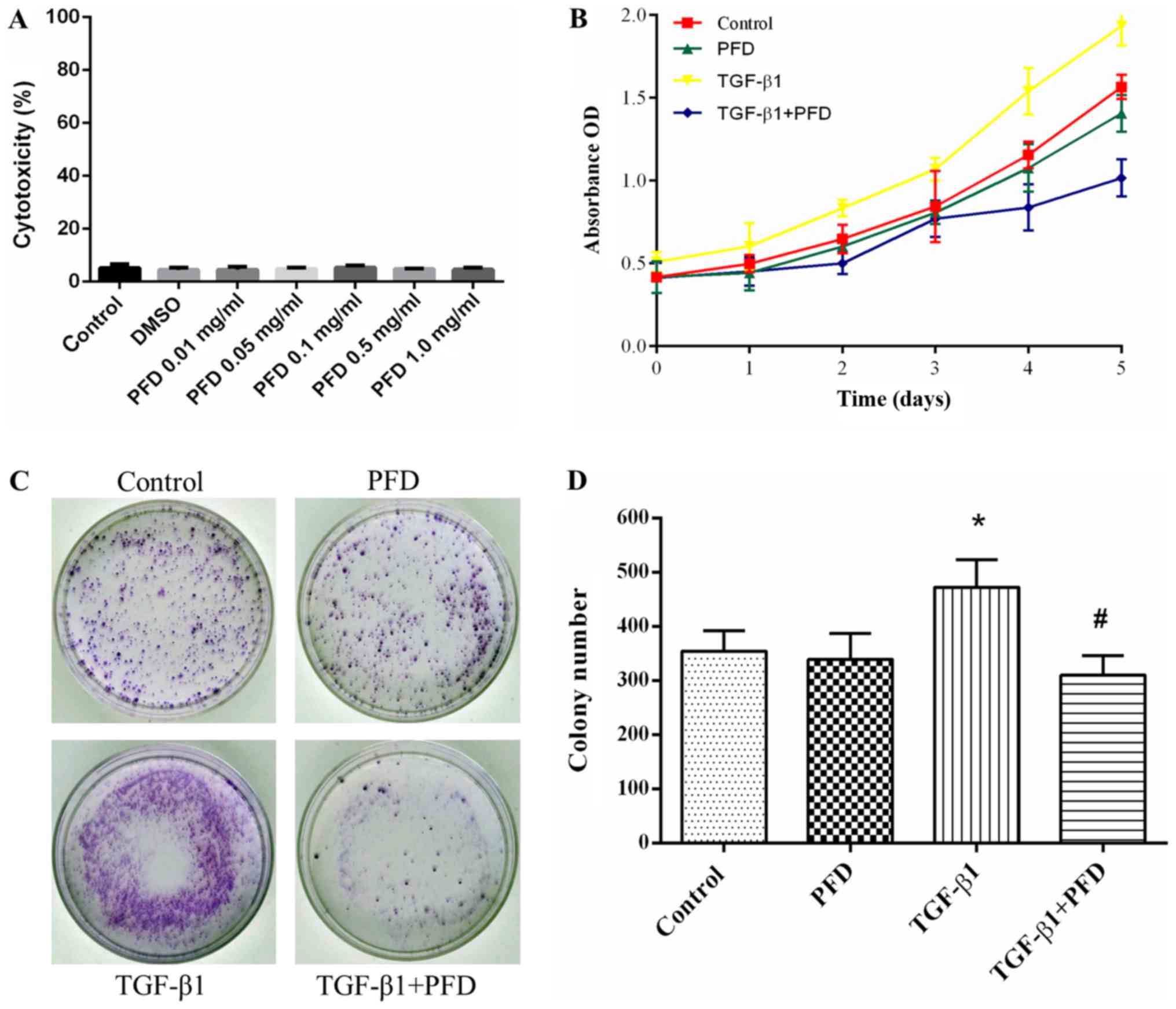 | Figure 1.PFD inhibits cell proliferation in
HIFs following TGF-β1 stimulation. (A) HIFs were pretreated with
DMSO and different concentrations of PFD (0, 0.01, 0.05, 0.1, 0.5
and 1 mg/ml) for 24 h. Percentage of cytotoxicity was evaluated by
a lactate dehydrogenase cytotoxicity assay. (B) HIFs were
pretreated with PFD for 1 h prior to stimulation with TGF-β1 (10
ng/ml) for 1, 2, 3, 4 and 5 days. Cell Counting Kit-8 assays
revealed a robust proliferative response to TGF-β1 when compared
with normal cells, and PFD pretreatment abrogated TGF-β1-induced
proliferation in HIFs, restraining cell numbers to basal levels. (C
and D) HIFs were pretreated with PFD for 1 h prior to stimulation
with TGF-β1 (10 ng/ml) for 14 days. Colony formation assays
demonstrated the mean colony number was significantly increased
following TGF-β1 stimulation in the colony formation assay, and PFD
administration markedly reduced colony growth. Data are presented
as the mean ± standard deviation of triplicate experiments.
*P<0.05 vs. control group; #P<0.05 vs. TGF-β1
group. PFD, pirfenidone; HIFs, human intestinal fibroblasts; TGF,
transforming growth factor; PFD, pirfenidone; OD, optical
density. |
To determine the effect of PFD on cell proliferation
of HIFs, we pretreated HIFs with PFD for 1 h before stimulation
with and without TGF-β1 (10 ng/ml) for 1, 2, 3, 4, and 5 days.
CCK-8 assay demonstrated that HIFs showed a robust proliferative
response to TGF-β1 compared to control group, and PFD pretreatment
abrogated TGF-β1-induced proliferation in HIFs, restraining cell
numbers to basal levels (Fig. 1B).
At 14 days, the colony formation assay demonstrated that the mean
colony number was significantly increased after TGF-β1 stimulation,
and PFD administration remarkably reduced colony growth (Fig. 1C and D). Together, these results
indicated that PFD suppressed cell proliferation in TGF-β1-induced
HIFs.
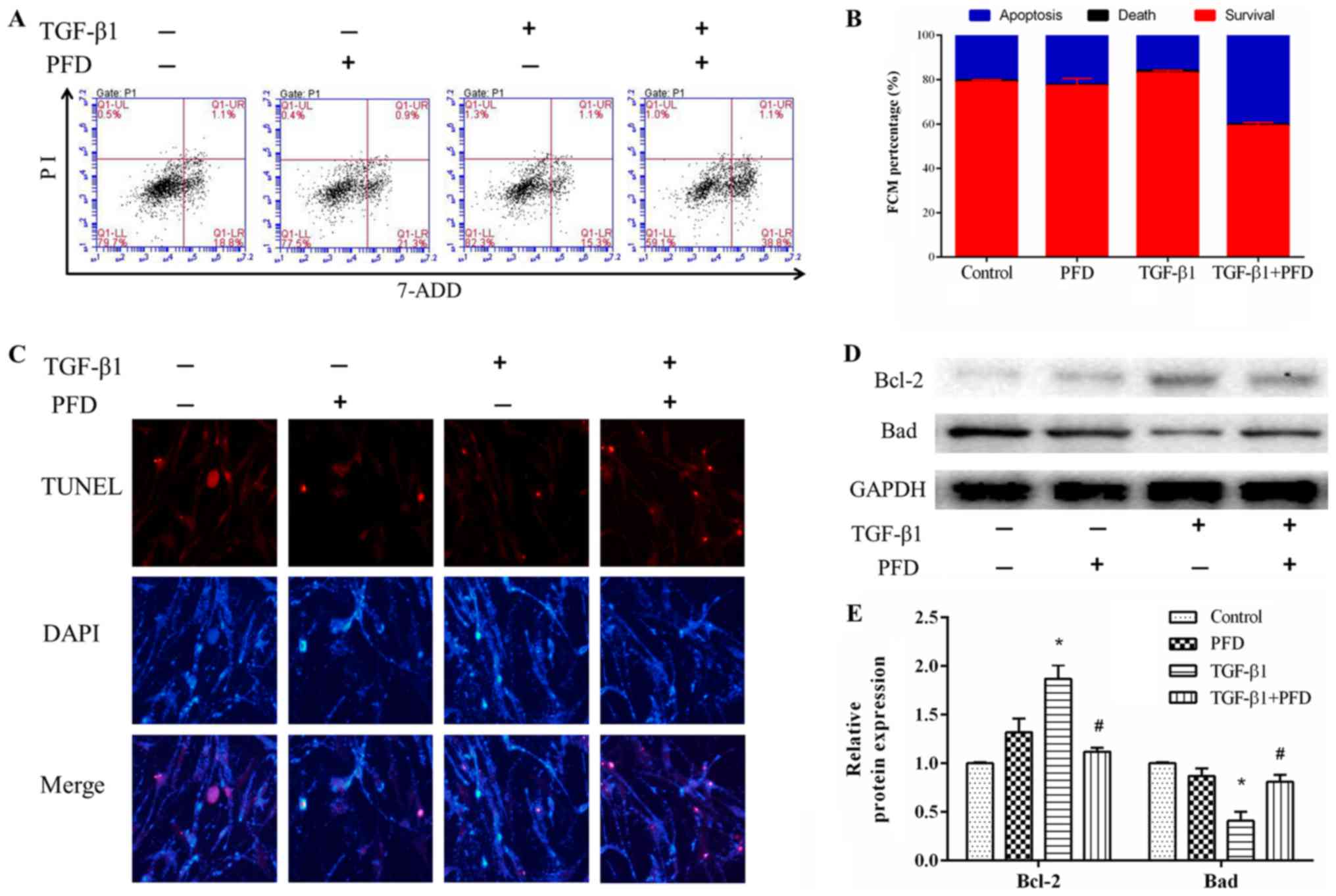
PFD promotes cell apoptosis in
TGF-β1-induced HIFs
The effect of PFD on cell apoptosis was investigated
by using flow cytometry and TUNEL assay. HIFs were pretreated with
PFD for 1 h before stimulation with and without TGF-β1 for 48 h.
Flow cytometry assay indicated that TGF-β1 treatment induced
reduction in cell apoptosis, and pretreatment with PFD reduced
TGF-β1-induced reduction in apoptosis (Fig. 2A and B). TUNEL staining showed that
TGF-β1 decreased the number of apoptotic cells after stimulation
for 48 h, and PFD treatment remarkably increased the number of
TUNEL-positive apoptotic cells (Fig.
2C). Collectively, these data suggested that PFD significantly
inhibited the anti-apoptotic effects of TGF-β1 on HIFs. To further
explore the effect of PFD on apoptosis of HIFs, we determined the
protein expression of Bcl-2 and Bad. These results demonstrated
that TGF-β1 significantly decreased the expression of pro-apoptotic
protein Bad and increased the expression of anti-apoptotic protein
Bcl-2, and this effect was reversed by PFD treatment (Fig. 2D and E).
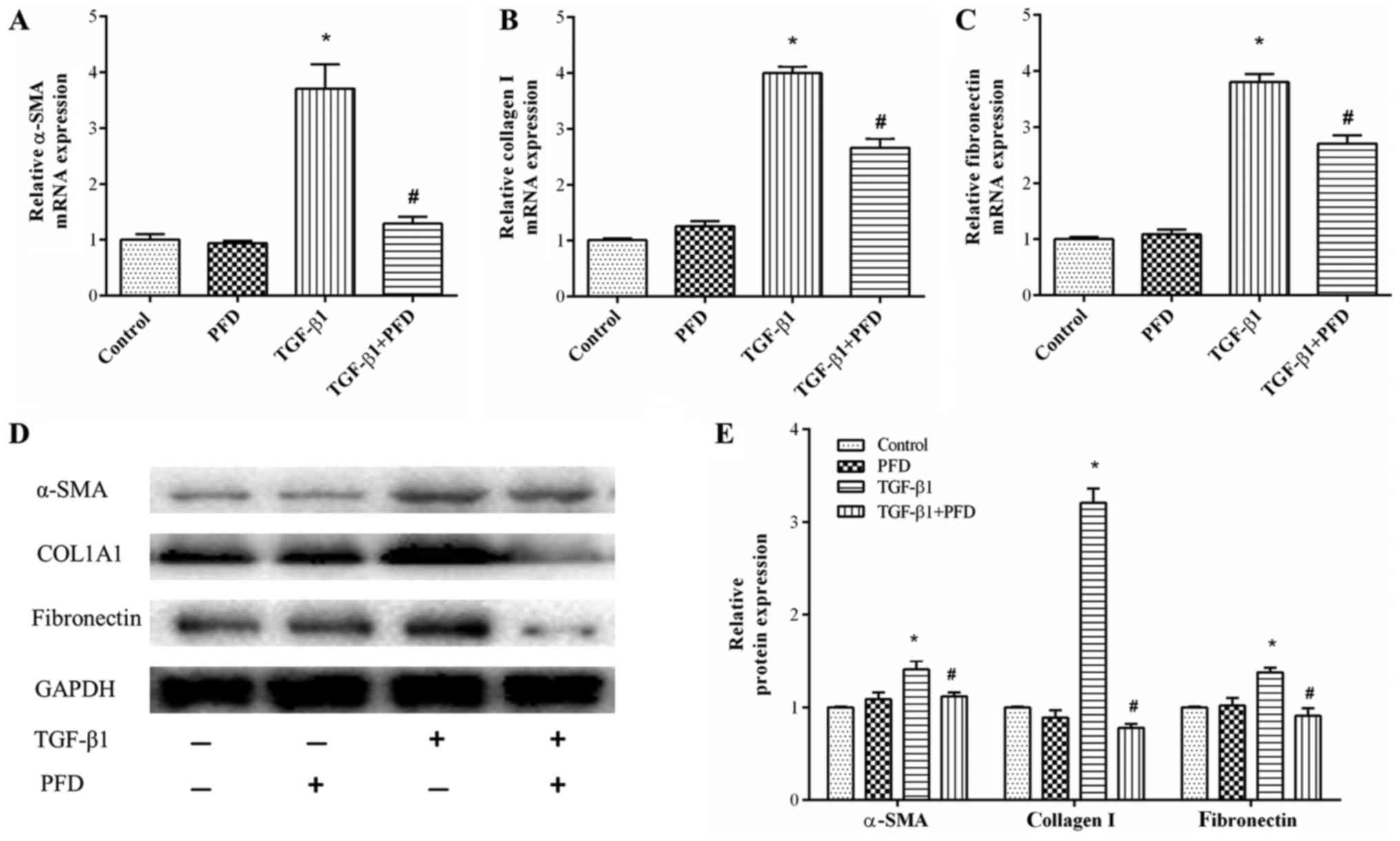
PFD reduces fibroblast differentiation
and collagen synthesis in TGF-β1-induced HIFs
As myofibroblast differentiation is a central event
of the pathogenesis of intestinal fibrosis, we determined the mRNA
and protein expression of α-SMA, a key marker of myofibroblast
differentiation. The results demonstrated that TGF-β1 stimulation
significantly elevated α-SMA mRNA and protein expression and PFD
treatment resulted in a significant inhibition of TGF-β1-induced
expression of α-SMA. However, PFD had no such effects on normal
intestinal fibroblasts, as shown in Fig. 3A, D and E.
Collagen accumulation is another characteristic
feature of myofibroblasts, which are commonly observed in
intestinal fibrosis. To investigate effects of PFD on collagen
synthesis, we examined the protein and mRNA expression of collagen
I and fibronectin. HIFs were pretreated with PFD for 1 h before
TGF-β1 stimulation for 48 h. As shown in Fig. 3B-E, the mRNA expression and protein
production of collagen I and fibronectin were enhanced by TGF-β1,
and PFD pretreatment substantially abrogated this TGF-β1-mediated
collagen I and fibronectin production.
PFD inhibits TGF-β1-simulated
phosphorylation of TGF-β1/Smad2/3 and PI3K/AKT signaling pathways
in HIFs
As TGF-β1 is the most crucial mediator in the
pathogenesis of intestinal fibrosis, we determined the protein
expression in TGF-β1 signaling pathway. In the canonical signaling
pathway, TGF-β1 activates down-streaming Smad2/3 pathways to exert
its pro-fibrotic effects. To explore the effect of PFD on Smad2/3,
we detected the protein expression of p-Smad2/3. HIFs were
pretreated with PFD for 1 h before TGF-β1 stimulation for 48 h. As
shown in Fig. 4A, the expression
of p-Smad2/3 protein was significantly increased after TGF-β1
stimulation (P<0.05), and PFD significantly reduced
TGF-β1-induced phosphorylation of Smad2/3 (Fig. 4A and D).
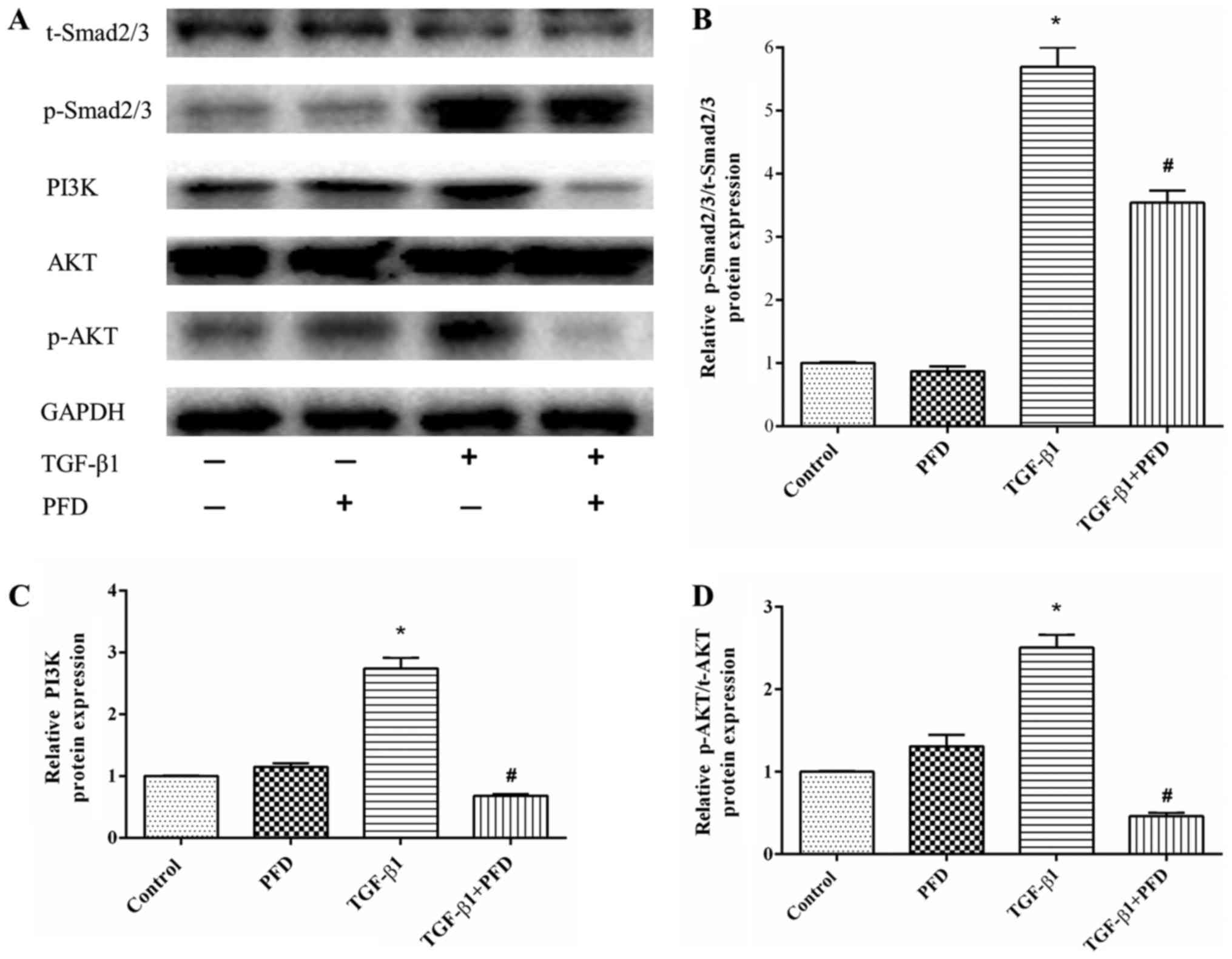 | Figure 4.PFD exerts antifibrotic effects by
inhibiting Smad and PI3K/AKT signaling activation in HIFs incubated
with TGF-β1. (A) Western blotting analysis of Smad2/3, p-Smad2/3,
PI3K, AKT and p-AKT protein expression. HIFs were pretreated with
PFD for 1 h prior to TGF-β1 stimulation for 48 h. (B-D)
Quantitative analysis of (B) p-Smad2/3 to Smad2/3 ratio, (C) PI3K
and (D) p-AKT to t-AKT ratio protein expression. Data are presented
as the mean ± standard deviation of triplicate experiments.
*P<0.05 vs. control group; #P<0.05 vs. TGF-β1
group. PFD, pirfenidone; HIFs, human intestinal fibroblasts; TGF,
transforming growth factor; Smad, mothers against decapentaplegic
homolog; p-, phosphorylated; AKT, protein kinase B; PI3K,
phosphoinositide 3-kinase; t-, total. |
In addition, TGF-β1 can also activate the
non-canonical PI3K/AKT pathway to modulate its downstream
pro-fibrotic signaling and related protein expression (25,26).
As shown in Fig. 4A and B, TGF-β1
significantly increased the protein expression of PI3K and p-AKT,
while pretreatment with PFD markedly reduced PI3K and p-AKT
expression. These results suggested that PFD inhibited PI3K/AKT
activation in response to TGF-β1 in HIFs, which might help to
alleviate intestinal fibrosis.
Discussion
Intestinal fibrosis is still considered as a
progressive and irreversible process, and currently, no effective
antifibrotic therapies are available. Intestinal fibroblasts are
the main effector cells of intestinal fibrosis, and considered as a
crucial target in treatment of intestinal fibrosis (5). In the present study, PFD inhibited
cell proliferation and enhanced apoptosis of HIFs; these effects
were accompanied by inhibition of TGF-β1-induced fibrotic
activities, including myofibroblast differentiation (α-SMA) and
collagen production (collagen I and fibronectin). Furthermore, the
results showed that the antifibrotic effects of PFD in vitro
may be mediated by via Smad and PI3K/AKT signaling pathway.
Collectively, the present results suggested that PFD might be a
potential therapeutic agent for intestinal fibrosis.
As the major ECM-producing cells, myofibroblasts
play a pivotal role in the development of intestinal fibrosis, and
myofibroblasts differentiation has been accepted as a central event
in the intestinal fibrosis pathogenesis (4). To possibly mimic the ‘in vivo’
situation, we established a ‘fibrotic’ culture via stimulation with
TGF-β1, the ‘master cytokine’ in fibrosis (5). We demonstrated here that TGF-β1
induced myofibroblast differentiation and increased mRNA and
protein expression of α-SMA, and such effects were significantly
inhibited by PFD pretreatment. This finding was in line with
observations in previous researches focused on effects of PFD on
cardiac, renal, and hepatic fibroblasts (14,17,12).
Activated myofibroblasts were demonstrated to exhibit profibrogenic
effects, such as increased proliferative, migratory and secretory
activities (4). Herein, we
demonstrated that PFD treatment alone did not significantly affect
the mRNA and protein of collagen I and fibronectin; conversely, PFD
inhibited TGF-β1-induced synthesis of collagen I and fibronectin.
Collectively, our data proved that PFD inhibited the
TGF-β1-mediated fibrotic process in vitro, which supported
effects of PFD observed in hepatic stellate cells, leiomyoma cells,
and alveolar epithelial cells (12,27,28).
Excessive fibroblast/myofibroblasts accumulation,
which is secondary to increased proliferation and/or inhibited
apoptosis, gradually leads to excessive ECM synthesis and
deposition and thus progressive distortion of intestinal structure.
Given that apoptosis is responsible for mediating the reduction in
myofibroblasts number, induction of myofibroblasts apoptosis would
have an antifibrotic effect during the resolution of fibrosis
(29). Suppression of activation
and proliferation and induction of apoptosis could be helpful to
reduce activated fibroblasts and their profibrogenic effects. In
this context, we examined effects of PFD on the regulation of cell
proliferation and apoptosis of HIFs in vitro. PFD inhibited
the proliferative activity of HIFs in response to TGF-β1, as
demonstrated by CCK-8 assay and colony formation assay. We ruled
out that antiproliferative effects were mediated by drug toxicity,
as evidenced by LDH cytotoxicity assay. These results were
consistent with findings reported previously (9,14,16).
Additionally, PFD also induced HIFs cell apoptosis and decreased
the number of TUNEL-positive cells after TGF-β1 stimulation, which
was accompanied by the increase of Bad protein expression, and the
decrease of Bcl-2 protein level. Taken together, these results
suggested that PFD reduced the accumulation of intestinal
fibroblasts/myofibroblasts by reducing proliferation and increasing
apoptosis of HIFs.
TGF-β1 plays a central role in the development of
intestinal fibrosis; it can augment the proliferative, migratory,
and collagen-producing abilities of intestinal fibroblasts by
inducing myofibroblast differentiation (6). TGF-β1 can activate both
Smad-dependent and -independent pathways. Smad2/3s are activated
after phosphorylation, and the activated form of Smad2/3s is
released and translocated into the nucleus, and then induce the
subsequent profibrogenic effects (30). The potent antifibrotic properties
of PFD including TGF-β1 suppression were demonstrated in many
studies (9,14,22,31).
In the present work, we analyzed the key participants in
TGF-β1/Smad signaling pathway to explore the potential mechanisms
responsible for the antifibrotic effects of pirfenidone. PFD
treatment was observed to attenuate TGF-β1-induced Smad2/3
phosphorylation, which was consistent with previous results
(22,31).
In addition to canonical pathway, TGF-β1-mediated
fibrotic process could be mediated through non-canonical signaling
pathways. Previous studies have showed that PI3K/AKT signal pathway
is involved in the TGF-β1 regulating pathway, and TGF-β1 could
induce activation of the PI3K/AKT pathway (25,26).
The PI3K/AKT signaling pathway is a prototypic survival pathway, in
which AKT is a downstream target and plays an important role in
cell growth or survival. When activated, AKT may produce its
anti-apoptotic effects via phosphorylation of both anti-apoptotic
and pro-apoptotic substrates, such as Bcl-2 and Bad (32). Consistent with previous studies,
the up-regulation of PI3K and p-AKT indicated that PI3K/AKT
signaling pathway was activated after TGF-β1 treatment, and these
changes were accompanied by the increase of Bad expression and the
decrease of Bcl-2 expression. In addition, this effect was reversed
by PFD treatment, which means that PFD alleviates TGF-β1-induced
profibrotic process at least in part by inducing apoptosis of HIFs
through PI3K/AKT signaling pathway. Further studies that
investigate the underlying mechanism in more detail are
warranted.
The present study demonstrated that pirfenidone, an
orally potent antifibrotic agent, could alleviate TGF-β1-induced
fibrogenic activities, such as myofibroblast differentiation
(α-SMA) and collagen production (collagen I and fibronectin), as
wells as pro-proliferative and anti-apoptotic effects of TGF-β1 on
HIFs. In addition, the antifibrotic effects of PFD on HIFs may be
mediated by inhibiting phosphorylation of Smad2/3 and AKT
pathways.
Acknowledgements
The authors would like to thank the Fujian Medical
University Stem Cell Research Institute (Fujian, China) for their
technical assistance.
Funding
The present study was supported by National Clinical
Key Specialty Construction Project (General Surgery) of China
(grant no. 2012-649) and Guiding Key Project of Social Development
by the Fujian Provincial Science and Technology Department (grant
no. 2015Y0058).
Availability of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contribution
YS and PC conceived and designed the study. YS and
YZ performed the experiments and were major contributors in writing
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rieder F and Fiocchi C: Intestinal
fibrosis in IBD-a dynamic, multifactorial process. Nat Rev
Gastroenterol Hepatol. 6:228–235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stacey R and Green JT: Radiation-induced
small bowel disease: Latest developments and clinical guidance.
Ther Adv Chronic Dis. 5:15–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hamama S, Delanian S, Monceau V and
Vozenin MC: Therapeutic management of intestinal fibrosis induced
by radiation therapy: From molecular profiling to new intervention
strategies et vice et versa. Fibrogenesis Tissue Repair. 5 Suppl
1:S132012.PubMed/NCBI
|
|
4
|
Kendall RT and Feghali-Bostwick CA:
Fibroblasts in fibrosis: Novel roles and mediators. Front
Pharmacol. 5:1232014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zeisberg M and Kalluri R: Cellular
mechanisms of tissue fibrosis. 1. Common and organ-specific
mechanisms associated with tissue fibrosis. Am J Physiol Cell
Physiol. 304:C216–C225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Speca S, Giusti I, Rieder F and Latella G:
Cellular and molecular mechanisms of intestinal fibrosis. World J
Gastroenterol. 18:3635–3661. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Y, Liu H, Liang Y, Peng P, Ma X and
Zhang X: DKK3 regulates cell proliferation, apoptosis and collagen
synthesis in keloid fibroblasts via TGF-β1/Smad signaling pathway.
Biomed Pharmacother. 91:174–180. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Conte E, Gili E, Fagone E, Fruciano M,
Iemmolo M and Vancheri C: Effect of pirfenidone on proliferation,
TGF-β-induced myofibroblast differentiation and fibrogenic activity
of primary human lung fibroblasts. Eur J Pharm Sci. 58:13–19. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abe Y, Murano M, Murano N, Morita E, Inoue
T, Kawakami K, Ishida K, Kuramoto T, Kakimoto K, Okada T, et al:
Simvastatin attenuates intestinal fibrosis independent of the
anti-inflammatory effect by promoting fibroblast/myofibroblast
apoptosis in the regeneration/healing process from TNBS-induced
colitis. Dig Dis Sci. 57:335–344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe
S, Nakata K, Taguchi Y, Nagai S, Itoh H, Ohi M, et al:
Double-blind, placebo-controlled trial of pirfenidone in patients
with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
171:1040–1047. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Di Sario A, Bendia E, Svegliati Baroni G,
Ridolfi F, Casini A, Ceni E, Saccomanno S, Marzioni M, Trozzi L,
Sterpetti P, et al: Effect of pirfenidone on rat hepatic stellate
cell proliferation and collagen production. J Hepatol. 37:584–591.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miric G, Dallemagne C, Endre Z, Margolin
S, Taylor SM and Brown L: Reversal of cardiac and renal fibrosis by
pirfenidone and spironolactone in streptozotocin-diabetic rat. Br J
Pharmacol. 133:687–694. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi Q, Liu X, Bai Y, Cui C, Li J, Li Y, Hu
S and Wei Y: In vitro effects of pirfenidone on cardiac
fibroblasts: Proliferation, myofibroblast differentiation,
migration and cytokine secretion. PLoS One. 6:e281342011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Duan L, Qi J, Huang T, Gu X, Xu D, Kong X
and Qian XQ: Pirfenidone attenuates bladder fibrosis and mitigates
deterioration of bladder function in a rat model of partial bladder
outlet obstruction. Mol Med Rep. 12:3639–3647. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin X, Yu M, Wu K, Yuan H and Zhong H:
Effects of pirfenidone on proliferation, migration, and collagen
contraction of human Tenon's fibroblasts in vitro. Invest
Ophthalmol Vis Sci. 50:3763–3770. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hewitson TD, Kelynack KJ, Tait MG, Martic
M, Jones CL, Margolin SB and Becker GJ: Pirfenidone reduces in
vitro rat renal fibroblast activation and mitogenesis. J Nephrol.
14:453–460. 2001.PubMed/NCBI
|
|
18
|
Chan AL, Rafii R, Louie S and Albertson
TE: Therapeutic update in idiopathic pulmonary fibrosis. Clin Rev
Allergy Immunol. 44:65–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cottin V: The role of pirfenidone in the
treatment of idiopathic pulmonary fibrosis. Respir Res. 14 Suppl
1:S52013.PubMed/NCBI
|
|
20
|
Takeda Y, Tsujino K, Kijima T and
Kumanogoh A: Efficacy and safety of pirfenidone for idiopathic
pulmonary fibrosis. Patient Prefer Adherence. 8:361–370. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meier R, Lutz C, Cosín-Roger J, Fagagnini
S, Bollmann G, Hünerwadel A, Mamie C, Lang S, Tchouboukov A, Weber
FE, et al: Decreased fibrogenesis after treatment with pirfenidone
in a newly developed mouse model of intestinal fibrosis. Inflamm
Bowel Dis. 22:569–582. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li G, Ren J, Hu Q, Deng Y, Chen G, Guo K,
Li R, Li Y, Wu L, Wang G, et al: Oral pirfenidone protects against
fibrosis by inhibiting fibroblast proliferation and TGF-β signaling
in a murine colitis model. Biochem Pharmacol. 117:57–67. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun YW, Zhang YY, Ke XJ, Wu XJ, Chen ZF
and Chi P: Pirfenidone prevents radiation-induced intestinal
fibrosis in rats by inhibiting fibroblast proliferation and
differentiation and suppressing the TGF-β1/Smad/CTGF signaling
pathway. Eur J Pharmacol. 822:199–206. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ni BB, Li B, Yang YH, Chen JW, Chen K,
Jiang SD and Jiang LS: The effect of transforming growth factor β1
on the crosstalk between autophagy and apoptosis in the annulus
fibrosus cells under serum deprivation. Cytokine. 70:87–96. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu L, Zhang Q, Mo W, Feng J, Li S, Li J,
Liu T, Xu S, Wang W, Lu X, et al: Quercetin prevents hepatic
fibrosis by inhibiting hepatic stellate cell activation and
reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways. Sci
Rep. 7:92892017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takano M, Yamamoto C, Yamaguchi K, Kawami
M and Yumoto R: Analysis of TGF-β1- and drug-induced
epithelial-mesenchymal transition in cultured alveolar epithelial
cell line RLE/Abca3. Drug Metab Pharmacokinet. 30:111–118. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee BS, Margolin SB and Nowak RA:
Pirfenidone: A novel pharmacological agent that inhibits leiomyoma
cell proliferation and collagen production. J Clin Endocrinol
Metab. 83:219–223. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luna J, Masamunt MC, Lawrance IC and Sans
M: Mesenchymal cell proliferation and programmed cell death: Key
players in fibrogenesis and new targets for therapeutic
intervention. Am J Physiol Gastrointest Liver Physiol.
300:G703–G708. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Choi K, Lee K, Ryu SW, Im M, Kook KH and
Choi C: Pirfenidone inhibits transforming growth factor-β1-induced
fibrogenesis by blocking nuclear translocation of Smads in human
retinal pigment epithelial cell line ARPE-19. Mol Vis.
18:1010–1020. 2012.PubMed/NCBI
|
|
32
|
Downward J: PI 3-kinase, Akt and cell
survival. Semin Cell Dev Biol. 15:177–182. 2004. View Article : Google Scholar : PubMed/NCBI
|