Introduction
Deletions, duplications, or other genomic
rearrangements, may result in dosage imbalance of gene(s), which
leads to the loss or gain of genetic material (1). The mechanisms underlying copy number
variant (CNV) formation are based on recombination and replication.
Compared with single-nucleotide polymorphisms (SNPs), the de
novo locus-specific mutation appearance rate for CNVs is
significantly higher (2). CNVs may
cause Mendelian or sporadic traits, or be associated with complex
diseases, and the molecular mechanisms include gene dosage,
disruption, fusion and position effects, among others (3). CNVs are highly significant in human
disease and population diversity (4). Other common complicated
neuropsychiatric disorders, such as autism and schizophrenia, are
also affected by CNVs (5). Various
genome analysis platforms may perform CNV analysis. The golden
standards for CNV detection are array comparative genomic
hybridization (aCGH) and SNP genotyping platforms (6). However, SNP array or aCGH are not
sufficient for detecting smaller CNVs. Multiplex ligation-dependent
probe amplification (MLPA) may make up for the shortcomings of
these technologies (7).
MLPA is an accurate and reliable technique for
identifying CNVs, including large and small deletions, as well as
single-nucleotide aberrations, with several advantages over other
detection methods (8–12). Compared with conventional assays,
including Southern blotting, fluorescence in situ
hybridization and Sanger sequencing, MLPA is a good alternative to
array-based techniques and has high accuracy, specificity and
efficiency. In addition, MLPA is cost-effective and has less
technical complexity relative to array comparative genomic
hybridization, which often requires further validation using other
methods, such as quantitative polymerase chain reaction (qPCR)
(13–22). More importantly, MLPA may overcome
the limitations of CMA and SNP array to some extent. For example,
to our knowledge, the CNVs of the CYP21A2 gene cannot be analyzed
correctly by CMA, SNP array or NGS due to the presence of its
pseudogene (23,24). However, MLPA easily solves this
problem, using the P050-C1 CAH kit (MRC-Holland, Amsterdam, The
Netherlands). At least 300 commercial probe sets are currently
available for detecting relatively common genetic disorders, such
as Duchenne muscular dystrophy and spinal muscular atrophy, as well
as rare genetic conditions, such as antithrombin deficiency and
Birt-Hogg-Dubé syndrome (7).
MLPA mainly involves the separation of amplification
products by size, limiting the maximum number of target sequences
that can be screened in parallel to ~50 (12). However, it does not meet the
requirements for detecting genetic disorders that are caused by
diverse DNA variations. Although MLPA has a higher throughput
compared with qPCR, it is currently not suitable for the
large-scale screening of target regions, although efforts are
currently focused on improving throughput (11,12).
CNV-plex is the most representative of the existing methods, but is
limited by the number of fluorescent groups and fragment length,
with 384 base pairs (bp) as the maximum fragment size that can be
detected in one reaction (21–22,25–29).
The use of additional fluorescent groups also introduces technical
complications into the detection of CNVs, increasing the complexity
of data analysis.
The rapid development of next-generation sequencing
(NGS), which performs sequencing via synthetic processes, provides
a sensitive and accurate tool for detecting known or unknown
genomic variations, including CNVs (25–40).
However, the statistical approaches to CNV identification are
limited, although there are several auto-calculation software types
for CNV detection that utilize data generated from whole-exome or
whole-genome sequencing (32–42).
In the present study, a novel and robust method of
MLPA-based NGS (MLPA-NGS) was introduced, which utilized MLPA
products to construct a library that may be transferred into an NGS
procedure to detect CNVs with improved accuracy and high
throughput. MLPA PCR products with indexed adapters were tested on
an NGS platform, and the resulting data were analyzed by using a
series of analytical software, including FastQC, Burrows-Wheeler
Alignment (BWA) tool and Genome Analysis Toolkit (GATK). The reads
from each probe reflected genetic variations in the target regions,
and fragment differentiation was based on the specific base
composition of the sequences, rather than fragment length, which
was determined by capillary electrophoresis. As such, the probe set
may be designed to be within the same range of lengths, thereby
allowing consistent detection efficiencies among reads.
Furthermore, this approach ensures efficiency in amplification and
purification of PCR products. This method also detects a
significantly higher number of fragments compared with earlier
methods, circumventing the 50 fragment detection limit per run. The
novel approach of the current study also circumvented configuration
of the stuffer sequence for different lengths in the probe. The
synthesized probes did not involve the complexities associated with
preparing long probes. Furthermore, the addition of indices to the
adapters for distinguishing between different samples allowed the
assay to achieve high throughput detection of both sites and
samples, while also ensuring quantitative detection of copy number
with high accuracy. In summary, MLPA-NGS technology not only
possessed all the advantages of MLPA, such as detecting the CNVs of
CYP21A2 gene, but also overcame its limitations.
Materials and methods
Samples
A total of 12 peripheral blood samples were
collected from 12 unrelated subjects (age, 6–12 years) in the
Children's Hospital of Shanghai affiliated to Shanghai Jiao Tong
University (Shanghai, China) between June 2015 and May 2017, and
included four 22q11.2 deletion syndrome (22q11DS; OMIM no. 611867)
samples, five Duchenne muscular dystrophy (DMD; OMIM no. 310200)
samples and three healthy controls. The four 22q11DS samples were
collected from two female and two male patients. All DMD samples
were collected from male patients. The three healthy controls were
collected from two female and one male patient. In the present
experiments, a male 22q11DS sample (termed PC sample) and a female
negative control (termed NC sample) sample are described in detail.
All samples were obtained with written informed consent and the
study was approved by the Research Ethics Committee of the
Children's Hospital of Shanghai. At each collection, a peripheral
blood sample containing 3 ml was collected in BD Vacutainer PPT
K2EDTA tubes (BD Diagnostics, Milan, Italy), genomic DNA was
isolated from fresh blood samples using a DNeasy Blood and Tissue
Extraction kit (Qiagen GmbH, Hilden, Germany), according to the
manufacturer's protocol, and quantified using NanoDrop 1000
spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE,
USA) and stored at −80°C until used.
MLPA-based NGS protocol
An MLPA-based NGS protocol was developed. MLPA was
performed using the MLPA One-Tube MDP-v005 mix (MRC-Holland),
according to the manufacturer's protocol. Briefly, 100 ng isolated
DNA (aforementioned) was denatured at 98°C for 5 min, 3 µl of the
probe mix was added and heated at 95°C for 1 min, and subsequently
hybridized overnight at 60°C. The samples were then treated with
ligase-65 at 54°C for 15 min. The reactions were stopped by
incubating at 98°C for 5 min. PCR amplification was performed with
the specific SALSA FAM PCR primers (a 10 µl mix of 7.5 µl dH2O, 2
µl SALSA PCR primer mix and 0.5 µl SALSA polymerase) in the SALSA
MLPA PCR kit (MRC-Holland). Amplification conditions were: Initial
denaturation at 98°C for 5 min; followed by 35 cycles of 30 sec at
95°C; 30 sec at 60°C; 60 sec at 72°C; and final extension of 20 min
at 72°C; hold at 15°C. As the labeled PCR product may interfere
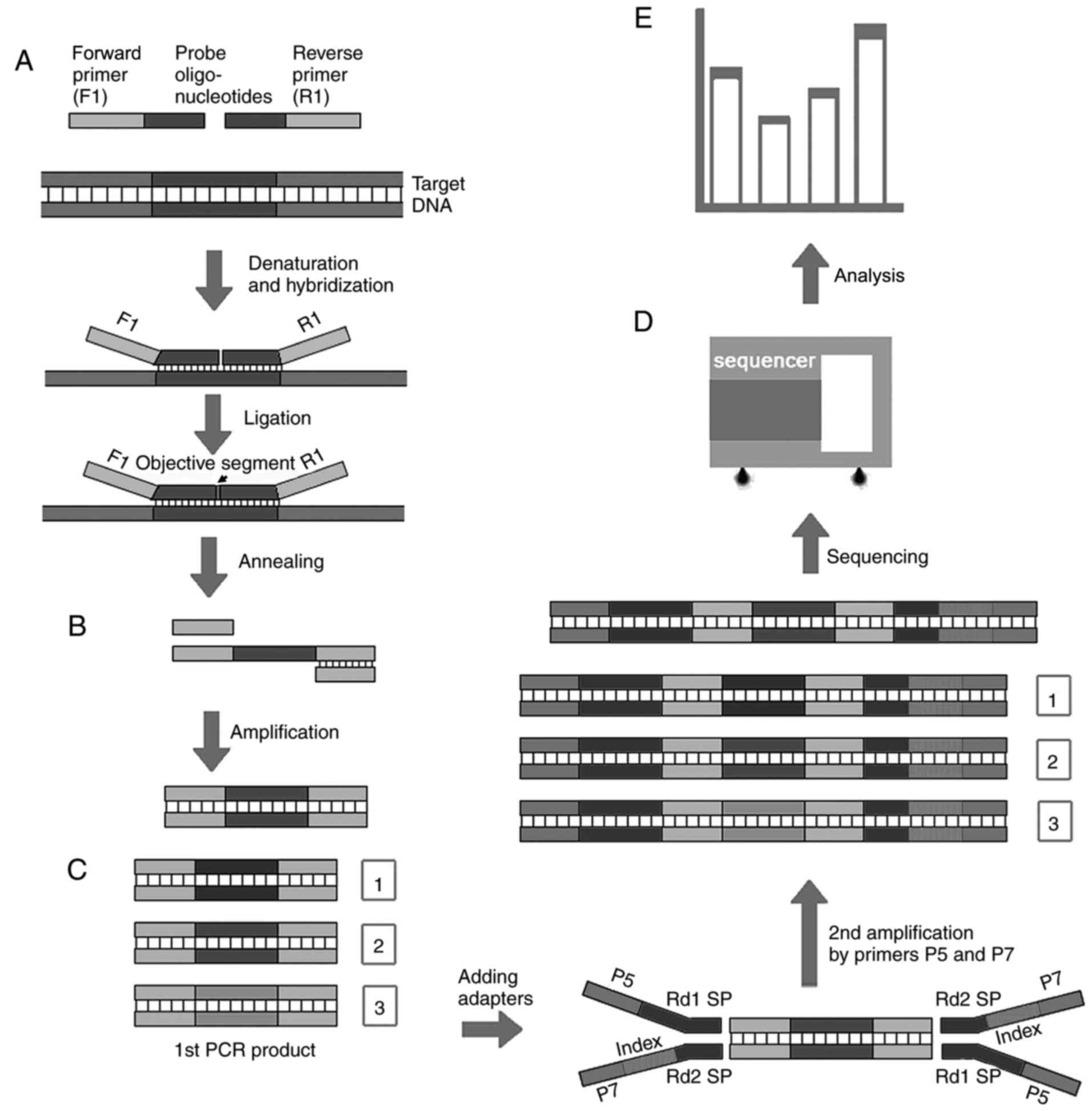
through ligation with NGS adapters (Fig. 1), the PCR products were then
re-amplified by using universal primers without any label (forward,
5′-GGGTTCCCTAAGGGTTGGA-3′ and reverse,
5′-GCGCCAGCAAGATCCAATCTAGA-3′; amplification conditions were the
same as above). PCR fragments were extracted and purified from a 2%
agarose gel (stained with ethidium bromide and visualized under UV
light) using a QIAquick Gel Extraction kit (Qiagen, Inc.),
according to the manufacturer's protocol, and ligated to adapters.
Subsequently, secondary amplification was performed, to obtain the
final NGS templates with labeled MLPA products, as well as the PCR
templates and primers without 6-FAM, to save the MLPA reagents.
Based on the sequencing results, the amplification method did not
interfere with the detection of CNVs. However, it may be advisable
to amplify the MLPA ligation products with non-labeled primers for
one-step PCR, in case of the amplification bias. A subsequent round
of amplification was performed for enrichment using Library
Preparation Kit (Kapa Biosystems; Roche Diagnostics, Basel,
Switzerland), according to the manufacturer's protocol, prior to
testing on the Illumina HiSeq 2500 Analyzer (Illumina, Inc., San
Diego, CA, USA) NGS platform. During data analysis, a large number
of non-human sequences were detected within each MLPA fragment,
including primer sequences used for amplification, as well as the
stuffer sequence derived from the T7 phages that were used to
adjust fragment size. The 61–86 bp long human sequence was aligned
using BWA version 0.6.2 (43) and
the human reference genome sequence (GRCh37/hg19), whereas GATK
version 1.6 (Broad Institute, Cambridge, MA, USA) was used to
calculate the number of reads in the target area, which was set to
a score of five.
MLPA analysis
DNA was isolated using a QIAamp DNA blood mini kit
(Qiagen GmbH) according to the manufacturer's instructions. MLPA
analysis of 22q11DS was performed using 52 pairs of probes in the
SALSA MLPA probemix P064-C1 Mental Retardation-1 (MRC Holland). The
regions targeted by P064-C1 probemix included 1p36, 15q11, 4p16,
16p13, 5p15, 17p13, 5q35, 17p11, 7p21, 20p12, 7q11, 22q11, 8q24,
22q13 and 11p13. The 52 MLPA probes resulted in amplification
products between 130 and 483 nucleotides (nt) in length. MLPA was
then performed according to the manufacturer's instructions.
Briefly, 100 ng DNA was denatured at 98°C for 5 min, then 3 µl of
the probe mix was added, heated at 95°C for 1 min, and then
hybridized overnight at 60°C. The samples were then treated with
ligase-65 at 54°C for 15 min. The reactions were stopped by
incubating at 98°C for 5 min. Finally, PCR amplification was
performed, following the protocol described in the aforementioned
MLPA-based NGS protocol. The amplification products were run on an
ABI PRISM 3500 Dx Genetic Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The raw data from the Genetic Analyzer
were analyzed with Coffalyser.Net (version 14; MRC-Holland).
Briefly, the channel content of the probes was filled with
P064-MR-1-C1-0912 (C1); following fragment analysis and comparative
analysis with default settings, normalization for MLPA fragment
data files was performed. Furthermore, the area encompassing the
CNVs in the PC and NC samples relative to that of the reference was
calculated using Coffalyser.Net. All quality measures and
parameters were within a satisfactory range. All samples were
normalized against multiple runs of the reference sample
(inter-sample normalization), and all probes were adjusted to the
reference probes within each sample (intra-sample
normalization).
MLPA analysis of DMD was performed using P034-B2
DMD-1 & P035-B1 DMD-2 kits, which were also purchased from
MRC-Holland. The analysis was performed using the same procedures
as described above.
Library construction and
sequencing
As MLPA PCR products are labeled with
5-carboxyfluorescein (FAM), which blocks ligation with adapters,
the PCR products were once again amplified using the following
primers: MLPA, forward 5′-GGGTTCCCTAAGGGTTGGA-3′, reverse
5′-GCGCCAGCAAGATCCAATCTAGA-3′. The reaction was performed in system
containing 2 µl MLPA PCR product, 5 µl 10X HS Taq buffer (Takara
Biotechnology Co., Ltd. (Dalian, China), 4 µl dNTPs (2.5 mM), 1 µl
MLPA forward primer (20 pM), 1 µl MLPA reverse primer (20 pM), 2 U
HSTaq (Takara Biotechnology Co., Ltd.), which was made up to a 50
µl with the appropriate volume of ddH2O. PCR was
performed using the same temperature profile as that of the
MLPA-amplified reaction described above.
The PCR products were purified using Agencourt
Ampure XP-PCR purification beads (cat. no. A63880; Beckman Coulter,
Inc., Brea, CA, USA) with a Dynal magnetic bead stand (cat. no.
123-21D; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Briefly, 90 µl Agencourt beads were mixed
with 50 µl PCR product, incubated at room temperature for 15 min,
placed on a magnetic stand, washed twice with 80% (v/v) ethanol,
separated from the ethanol and air-dried for 5 min. The beads were
resuspended and incubated in 22 µl ddH2O. Next, 20 µl
eluate (plus 4 µl of the loading dye) was electrophoresed on a 2%
agarose gel with a 100 bp DNA ladder for 2 h at 120V. DNA fragments
that were within the size range of 40–550 bp were eluted with 25 µl
ddH2O by gel extraction using a QIAquick Gel Extraction
kit (Qiagen, Inc., Valencia, CA, USA), according to the
manufacturer's protocol. The products in the eluates were then
subjected to end repair and A-tailing by a Kapa Library Preparation
kit (Kapa Biosystems), according to the manufacturer's protocol.
The Ligation Master mix and the indexed adapters were mixed and
incubated at 2°C for 15 min, 35°C for 15 min and 72°C for 20 min,
and then held at 4°C to produce paired-end libraries.
The post-ligation products were purified using
Agencourt Ampure XP-PCR purification beads (cat. no. A63880;
Beckman Coulter, Inc.) with a Dynal magnetic bead stand (cat. no.
123-21D; Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. During library amplification, the
reaction system was performed in a 0.2 ml tube. Similarly, library
amplification purification was performed using Ampure XP beads,
according to the manufacturer's instructions, in which the product
was quantified using a Qubit DNA HS kit (Thermo Fisher Scientific,
Inc.), and the test for fragment quality was conducted using an
Agilent 2100 (Agilent, Inc., Santa Clara, CA, USA). The test
results generated the expected fragments, which were then
sequenced. The library preparations were sequenced on an Illumina
HiSeq 2500 platform, and 150 bp paired-end reads were
generated.
Analysis of CNVs by SNP array
chip
HumanCytoSNP-12 BeadChip (Illumina, Inc.) was used
to detect CNVs in DNA isolated from PC patient peripheral blood.
The sample DNA was amplified, labeled and hybridized as previously
described (44), and the data were
acquired using Illumina's iScan scanning system. The frequency of
the B allele and the log R ratio were analyzed with Illumina Karyo
Studio (version 1.4.3). The log R ratio is the logged ratio of
observed probe intensity to expected intensity; any deviations from
zero in this metric are evidence of copy number alteration. The
frequency of the B allele is the proportion of the hybridized
sample that carries the B allele, as designated by the Infinium
assay.
Data analysis
Quality control metrics for the NGS raw sequencing
data (FASTQ files) were obtained using FastQC, version 0.10.1
(www.bioinformatics.babraham.ac.uk/projects/fastqc).
The sequences were then aligned to the human reference genome
sequence (GRCh37/hg19) using BWA (version 0.6.2). GATK was used to
compute read depths within the target region. For GATK analyses,
default settings were used, except the mapping quality threshold
(Q=5). The same normalization method treatment with the PC relative
peak area was also performed for the NGS reads of the PC sample to
the NC sample. The adjusted PC reads and the NC reads were compared
using Microsoft Excel (Microsoft Corporation, Redmond, WA,
USA).
MLPA and NGS data were compared to examine the
consistency of the two methods. Following a run on the Genetic
Analyzer, relative peak areas of each sample were calculated and
compared to five sex-matched controls using the Coffalyser.Net
software (MRC-Holland). This program classifies a peak as normal
when the ratio to NC is 0.7–1.3, deletes a peak when the ratio is
<0.7, and designates a peak as duplicated when the ratio is
>1.3. Relative peak area data were extracted from the software
and further analyzed using Microsoft Excel.
The PC relative peak areas were calculated using a
method similar to the normalization method. Briefly, the sum of all
52 peak areas from the NC were compared to the sum of all 52 peak
areas of the PC, thereby resulting in a ratio, and each PC peak
area was then multiplied by that ratio, which was then designated
as the normalized PC area. The adjusted PC and the NC peak areas
were subsequently compared.
Other 22q11DS samples and DMD samples also followed
the same analysis procedures. Data were presented as the mean ±
standard deviation of three repeated experiments.
Results
MLPA analysis
MLPA was performed to validate the NGS-MLPA findings
of the DNA samples from patients with the 22q11 deletion syndrome
or with DMD. For the PC sample, deletions in chromosomal region
22q11, which encompasses the CLTCL1, CDC45, GNB1L, DGCR8, ZNF74,
MED15 and SNAP29 genes, are the most frequent cause of
DiGeorge syndrome (45).
The 52 pairs of MLPA probes in the P064-C1 set were
used to distinguish the seven gene-dosage alterations
aforementioned (Fig. 2). In
addition, nine control fragments were used, which generated
amplicons of <120 nt in size. MLPA was performed using DNA from
the NC and PC samples. Analysis using the Coffalyser.Net software
identified seven peaks with gene dosage alterations that were
clearly distinguishable, using the DNA of the NC sample as a
calibrator (Fig. 2).
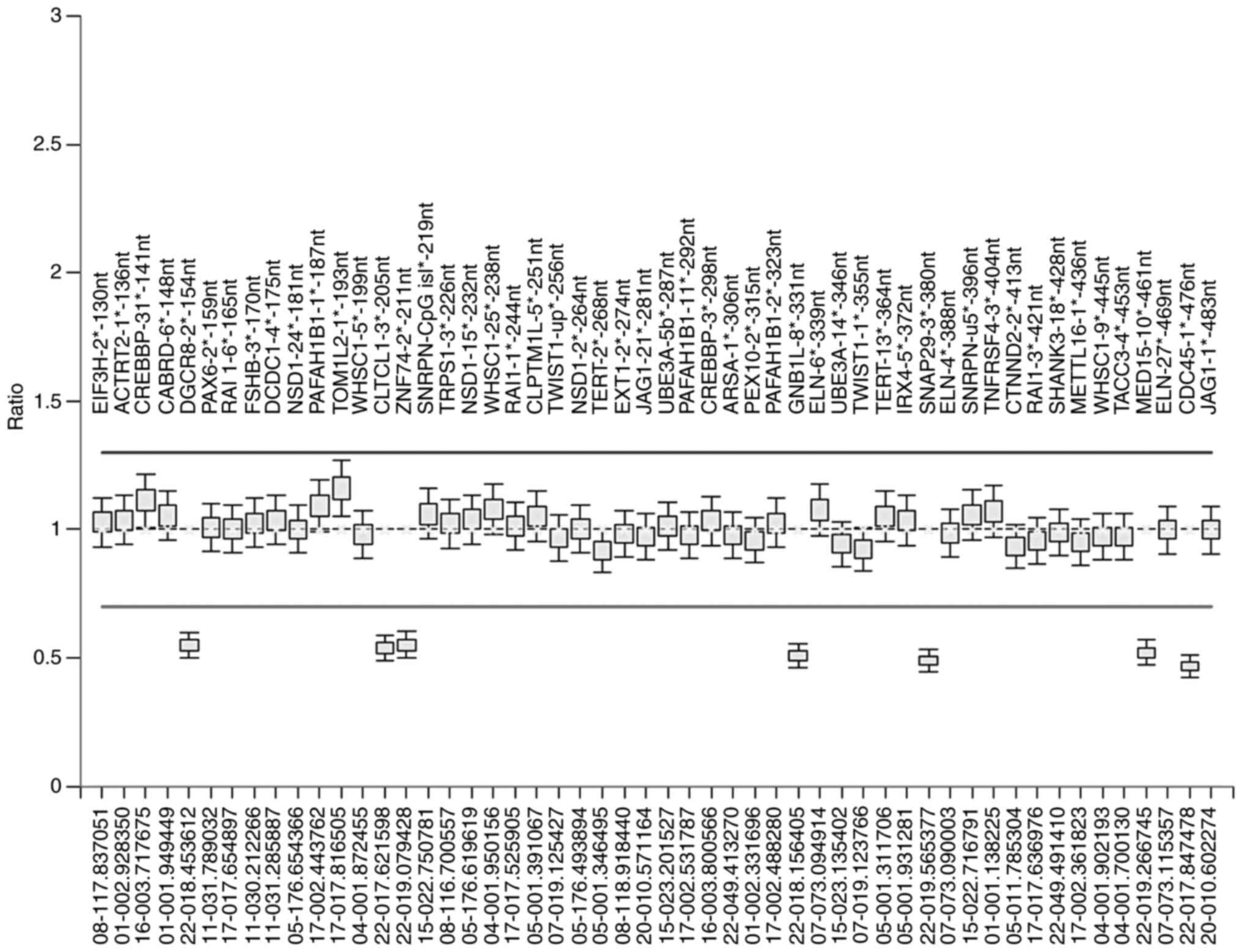 | Figure 2.MLPA analysis. The MLPA data of the
PC sample were calculated using the Coffalyser.Net software with
the NC sample as reference. MLPA represents deletion of the probe
in the 22q11 region, including peaks that correspond to 154, 205,
211, 331, 380, 461 and 476 nt. The error bars represent the
calculated standard deviations for each probe. MLPA, multiplex
ligation-dependent probe amplification; PC, positive control; NC,
negative control; nt, nucleotides. |
For the three other 22q11DS samples, it was
identified that one female patient carried deletions of CLTCL1,
CDC45, GNB1 L and DGCR8 genes, and the other samples
carried the same deletions as the PC sample. Using identical
protocols, it was determined that four DMD samples carried
hemizygous deletions of exons 51, 3–44, 45–48 and 3–11 (PC-Del
sample), and one carried hemizygous duplications of exons 16–44
(PC-Dup sample).
MLPA-NGS analysis
All the genes within chromosome 22q11 were detected
as single copies in the PC samples, compared with the same
fragments from the NC samples, in which the read number at each
site was reduced by half. This finding closely matched the MLPA
results (data not shown).
The number of reads of the target sequence that was
calculated using GATK utilized an alignment score, which affected
the final results. For example, the number of NC and PC reads for
the seventh fragment (165 bp; number 16526-L20951, RAI1) were
2,745.19 and 3,780, respectively, at a default score of 20; when
the score was adjusted to 5, those values were converted into
183,901.82 and 190,662.17, respectively. The latter data
demonstrated that on-target rate improved when the alignment score
of ‘5’ was used in this experimental condition, for the sequence of
these added reads is manually verified to be consistent with the
seventh fragment, so we use ‘5’ as the alignment score.
For the PC sample, the PCR products derived from
MLPA were re-amplified, purified, gel extracted, ligated with
indexed adapters and processed with other protocols, and finally
sequenced on an Illumina HiSeq 2500, which generated reads that
were analyzed. The NGS reads contained 52 segments, excluding
fragments <120 bp in size (which refers to the 92 nt benchmark
probe; 64, 70, 76 and 82 bp-long Q-fragments; 88 and 96 bp-long
D-fragments; 100 and 105 bp X & Y fragments, and other quality
control fragments). The PC read fragments were normalized using
similar methods to the relative PC peak area normalization, thereby
resulting in normalized PC reads, which were subsequently compared
with the corresponding original NC reads, for which the ratio was
designated as the reads ratio.
Data from the relative peak areas were extracted
from the Coffalyser.Net software and analyzed in Excel, from which
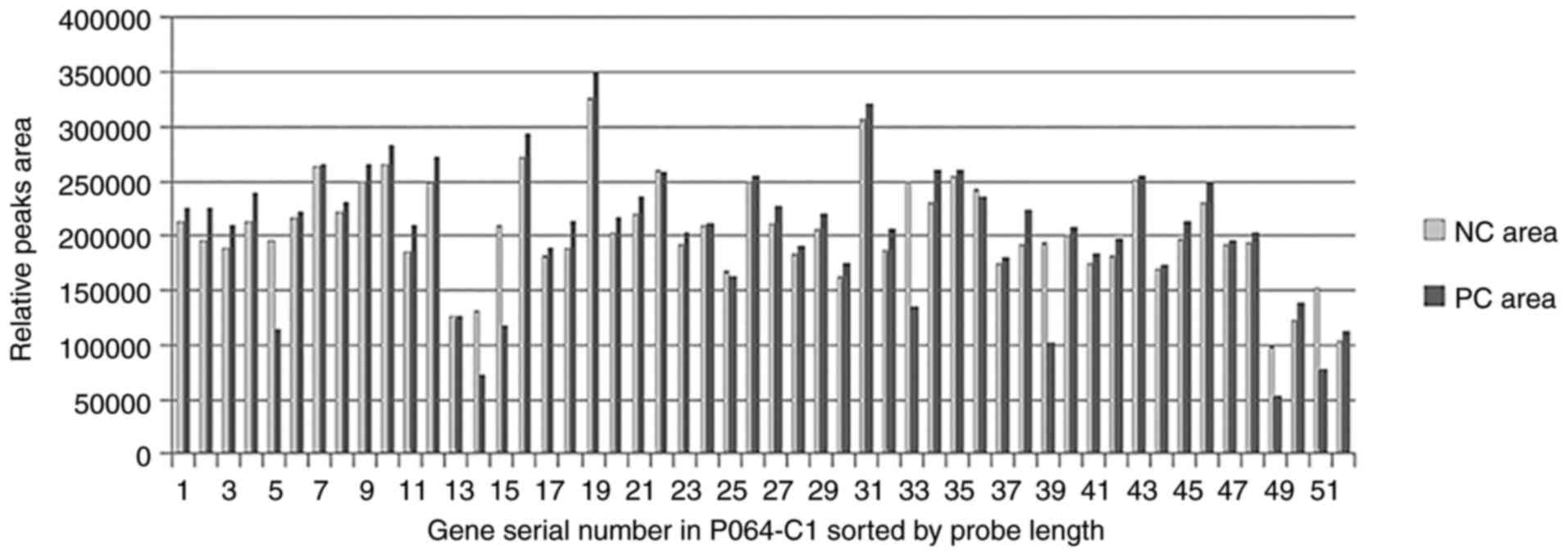
the normalized PC and NC peak areas were compared (Fig. 3). The mean peak area of the
normalized PC ± standard deviation (SD) was 205,609.19±46,075.30
(range, 54,129.87–351,745.90), whereas SD/mean =0.22; the mean peak
area of NC ± SD was 205,609.19±61,965.87 (range, 98,580–326,506),
whereas SD/mean=0.30. The area of the peaks, such as numbers 5, 14,
15, 33, 39, 49 and 51 of the PC sample, were notably smaller
compared with the corresponding peak area of the NC sample,
approximately half of which indicated a gene-dosage mutation, which
was consistent with the results generated by the Coffalyser.Net
software.
For the other 22q11DS samples and DMD sample,
following the same protocols, the read number at each site was
analyzed and the results were consistent with the MLPA results.
Comparison of MLPA area ratio and NGS
reads ratio
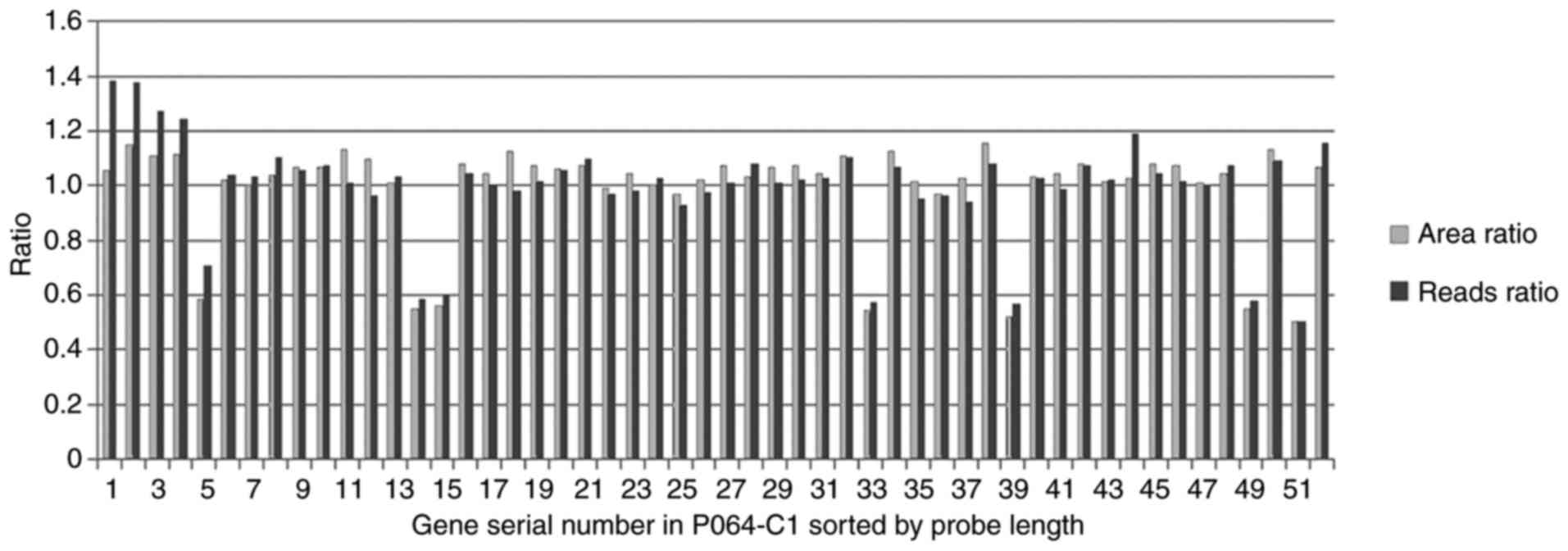
For the PC and NC samples, the area ratio and reads
ratio of each peak was compared (Fig.
4). The results of the two methods were in agreement, as peaks
5, 14, 15, 33, 39, 49 and 51 had similar area and reads ratios,
both of which were ~0.5-fold lower compared with the other ratios.
However, the ratio of the normalized PC reads and the original NC
reads was slightly higher compared with the ratio of the
corresponding MLPA peak area for peaks 1, 2, 3, and 4, thereby
suggesting an error in the analysis.
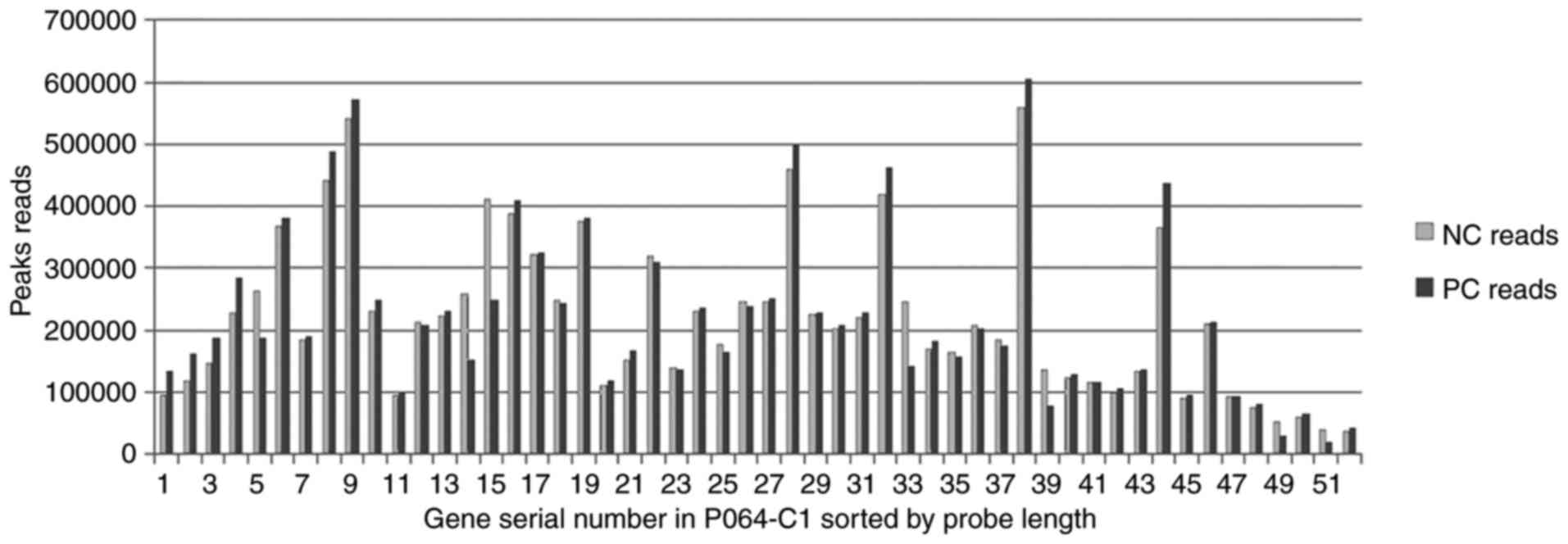
The normalized PC reads and the NC reads were
compared (Fig. 5). The mean peak
reads of the normalized NC ± SD was 22,0881.73±127,415.63 (range,
36,223.14–559,197.7), whereas SD/mean=0.58; the mean peak reads of
PC ± SD was 220,881.73±136,905.55 (range, 19,374.58–604,290.46),
whereas SD/mean =0.62. The SD/mean ratio of the NGS reads was
higher compared with the SD/mean ratio of the MLPA peak area,
thereby illustrating that variations in the NGS data were wider
than those of the MLPA data from peak to peak. However, the reads
of certain peaks in the PC sample were markedly lower than the
corresponding NC peak areas, including peaks 5, 14, 15, 33, 39, 49
and 51, approximately half of which also indicated a gene-dosage
mutation, which coincided with the MLPA peak area.
As for the other three 22q11DS patients and five DMD
patients, the ratio of the normalized PC reads was consistent with
the ratio of the corresponding MLPA peak area for each peak (data
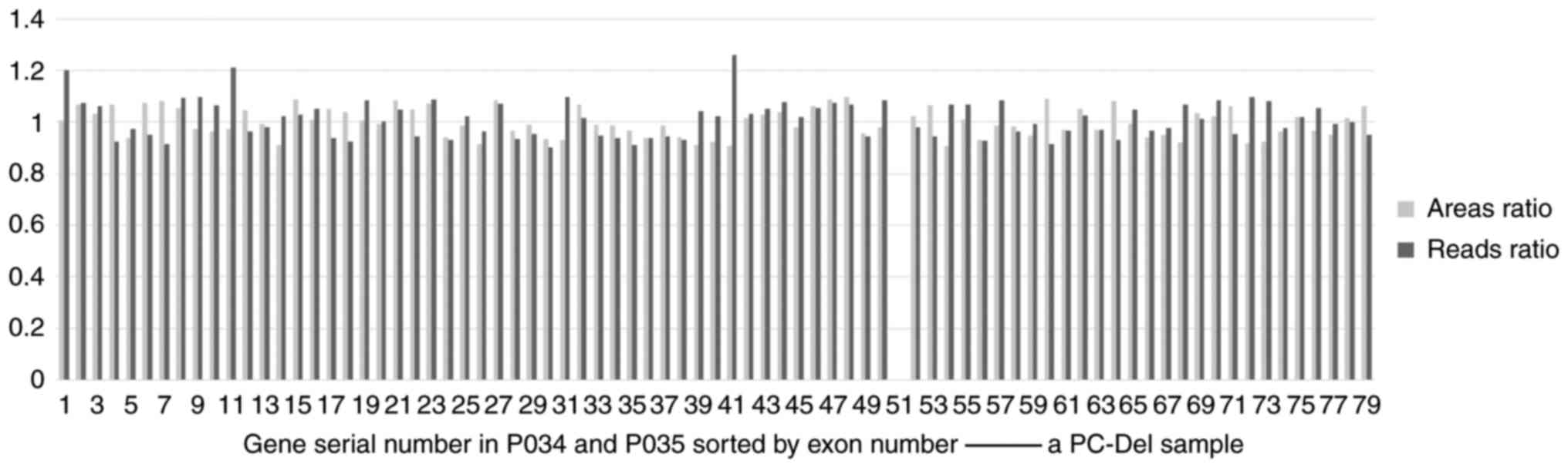
not shown). The hemizygous deletions of exons were not detected by
both the MLPA-NGS and MLPA (Fig.
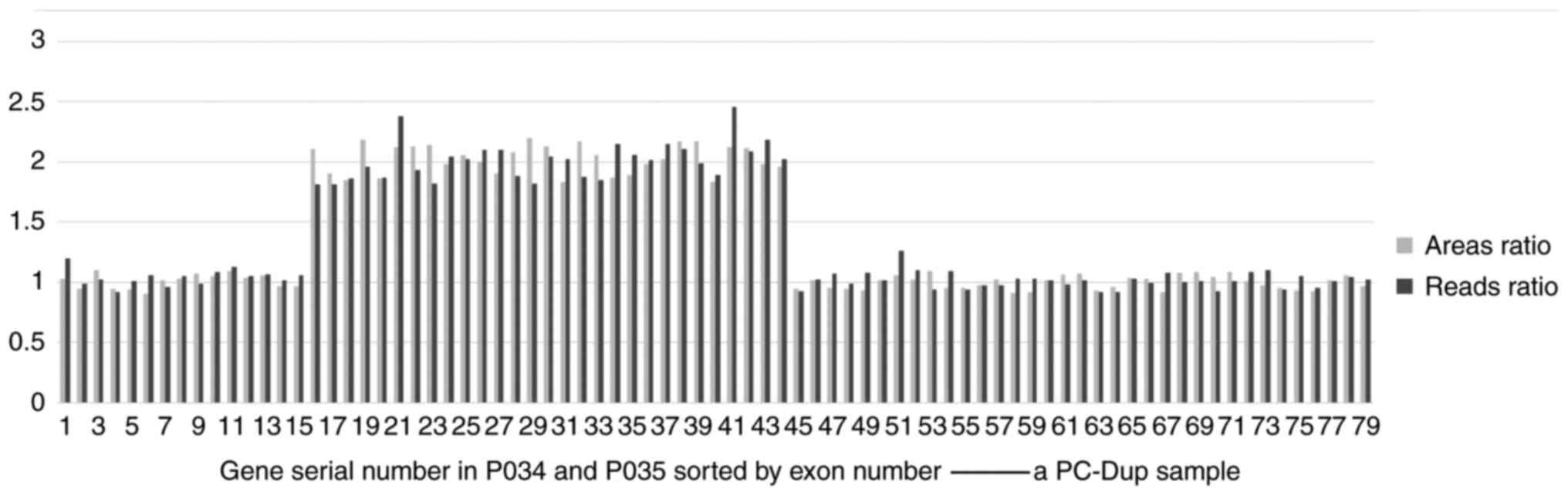
6; this case carries hemizygous deletions of exons 51), while
the hemizygous duplications of exons were detected by both methods
(Fig. 7). As such, it was
concluded that the results generated by the two technologies were
in good agreement.
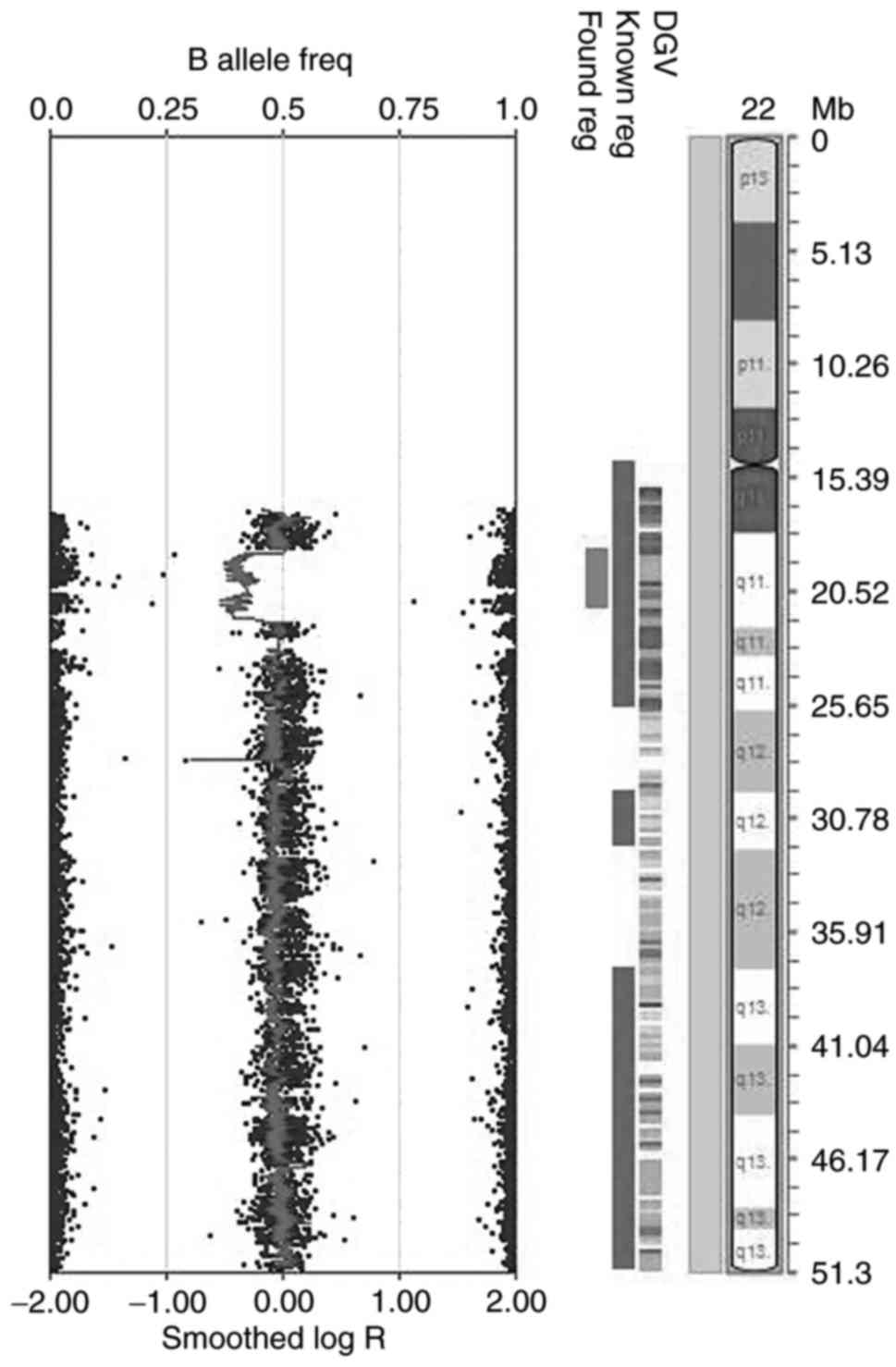
SNP array results
For the PC Sample, the CNVs in the DNA extracted
from peripheral blood were detected by using HumanCytoSNP-12
BeadChip. A 3 Mb deletion within the 22q11 region was observed
(Fig. 8), which coincided with the
observed absence in the MLPA results.
Discussion
In the present study, an MLPA product was sequenced
using NGS. Following read analysis, large differences were observed
among fragments relative to the MLPA results, as indicated by
higher standard deviations and mean values from read ratios
following NGS, compared with those indicated by the peak area
ratios using MLPA capillary electrophoresis. The relatively large
standard deviation value reduced confidence in the analysis of the
initial copy number of the template. Provided that the number of
reads for each fragment is proportional to the amount of the
initial template, a fragment containing CNVs may be deduced based
on the relative read values of each amplified fragment when no
reference sample is present (28).
Unfortunately, the read standard deviation for different segments
was too excessive to allow this rigorous form of analysis.
Several aspects that contributed to these
differences were considered. Initially, amplification bias may have
occurred during the first PCR amplification in MLPA and the second
amplification with labeled MLPA products as the templates and
non-labeled primers to get non-labeled MLPA products. Subsequent
PCR steps were performed to ligate adapters to the fragments,
further increasing heterogeneity in the fragment number. In
addition, the gel extraction step after PCR was performed to remove
fragments of the wrong size, in order to improve target segment
detection. Taq polymerase was used for the aforementioned
steps, which may have caused greater bias. There are many
Taq enzymes used in the amplification of high-throughput
sequencing, which have high requirements for fidelity and bias, but
this enzyme is not included (44).
Gel electrophoresis revealed that fragments over a certain size
range were distributed upstream and downstream of the peak rather
than within the peak, and were the brightest sites following
ethidium bromide staining. Furthermore, fragments that were closer
to the peak were also more abundant. The majority of the fragments
were evidently not within the size range (88–480 bp) and were thus
discarded during gel extraction, based on the boundaries of 40–550
bp. Thus, the reads for these fragments were relatively low.
Furthermore, it was almost impossible to maintain fragment sizes
during alignment, particularly for different samples used in gel
extraction. The reads of a few peaks at one edge exhibited greater
deviation when the position of the gel piece was slightly offset.
Overall, the ratio of the normalized PC reads to the original NC
reads was >1.2 for several smaller segments, such as 130, 136,
141 and 148 bp, which may have been caused by inconsistent cutting
sites. Thus, it was deduced that gel extraction should be excluded
from this approach. Fragment purification, which was performed
using magnetic beads, caused fragment retention during library
construction, which resulted in low yields for very small fragments
and subsequently relatively few reads for small fragments.
Additionally, there was a large number of base-pair alterations in
the probes during the alignment, as not all reads could be aligned
with the real target area during sequence alignment against the
human reference genome sequence (GRCh37/hg19), using BWA. Thus, the
number of reads of the target sequence that was calculated using
GATK utilized a revised alignment score, thereby affecting the
final results. Lastly, errors in sequencing may have resulted in
further errors, based on the length of the 150 bp pair-end reads
generated in both directions. In regards to the small fragments,
such as the 130 bp fragment, the homologous sequences could be
sequenced twice, thereby doubling the resulting number of reads.
For larger-sized fragments, homologous sequences may only be
measured from one end, whereas medium-sized fragments are likely to
be partially sequenced repeatedly, thereby increasing deviations in
length differences between the reads following statistical analysis
and finding the actual fragment number.
Overall, there was a high level of diversity of
reads among the different fragments following NGS, which was mainly
due to the broad range in fragment length, whereas there were only
slight differences among the same fragments from different samples.
The number of reads of the same fragment from the two samples was
highly similar when no CNVs were detected within the area. Taken
together, these findings supported the conclusions reached by the
MLPA-NGS method, which demonstrated that, relative to the results
of MLPA and SNP array chip, the PC sample harbored a single copy of
the 22q11 region. These consistent results prove the reliability of
the MLPA-NGS results.
The results of other three 22q11DS samples and five
DMD samples turned out that a secondary amplification with labeled
PCR products as the templates and non-labeled primers would not
interfere with accurate CNV detection, as long as the gels were cut
accurately. It was assumed that the PCR bias was caused by the
standard Taq polymerase, instead of the secondary
amplification. Using a specific Taq polymerase for NGS
library construction, such as High-Fidelity 2X PCR MasterMix
specified by Illumina, Inc., would likely avoid PCR bias, thus
improving the accuracy in CNV detection.
The MLPA-NGS method described herein was a reliable
method that would be suitable for detecting the CNVs of target
genes at a large scale when performing sample detection together
with normal controls. This reliability was based on a certain depth
of sequencing. In general, the depth in this experiment was
>1,000-fold higher compared with that of ordinary whole-exome
sequencing (average depth of 100×), although the absolute extent by
which sequencing depth reduced dependability on the final results
is unclear.
In the MLPA-NGS method, several further studies are
required to improve the assay: First, in the probe designing stage,
the MLPA products obtained should all be roughly of the same
length, considering that there is no need to distinguish different
fragments based on length during capillary electrophoresis. This
ensures amplification and purification efficiency of the different
fragments, thus rendering consistent detection efficiency for all
reads, as well as eliminating the requirement for the number of
fragments to be <50 per run. The addition of indices to this
high-throughput method may also increase the number of samples in
an assay, although this has yet to be verified. To further improve
accuracy, limiting amplification to a one-step PCR method by
redesigning the primers may be useful. By using ligase-65 at 54°C
during the ligation step of MLPA and stopping the reaction at 98°C,
it was possible to remove the stuffer sequences from the ligated
product as the adapters were being simultaneously added. Following
PCR of the ligated product, the 5′-terminus was subsequently used
as an adapter for the NGS, which was made possible by using new
adapters for amplifying ligated products, thereby simplifying the
MLPA-NGS process. This allowed sample fragments to be detected on
the NGS platform following amplification and purification, which
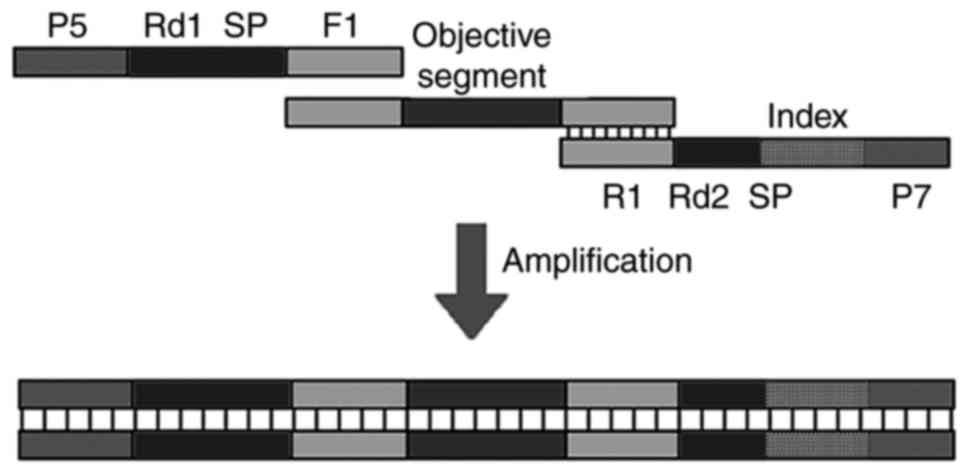
was beneficial in reducing both PCR bias and workload. The forward
primer (comprising P5, Rd1 SP and F1) and the reverse primer
(consisting of R1, Rd2 SP, index or barcode, and P7) contained PCR
sequences that were later used in library construction, sample
differentiation and NGS sequencing (Fig. 9). Second, due to the simplicity of
MLPA-NGS, this method may serve as a powerful tool in classifying
tumors or genetic disorders caused by CNVs. MLPA-NGS may be used
for the analysis of both CNVs and certain types of variations in
genomic DNA derived from peripheral blood, along with various
genetic disorders, such as 22q11 deletion syndrome. In addition,
point mutation-specific MLPA probes may be designed to detect
currently known single nucleotide variants (28). Finally, CNV detection by MLPA-NGS,
as well as other types of DNA variations, can be simultaneously
analyzed on the same NGS platform. Samples marked with different
indices will not interfere with their respective sequencing.
Appropriate correction or algorithm optimization will render it
more adaptable to data analysis, and the supporting software for
this method can be exploited.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Shanghai
Children's Hospital (grant no. 2012M007) and The Shanghai Municipal
Commission of Health and Family Planning (grant no.
2015ZB0203).
Availability of data and materials
The datasets generated and analyzed in the presenr
study are available from the corresponding author upon reasonable
request.
Authors' contributions
YY and HZ designed the experiments. DW, KX, JJ, ZZ,
CX and WX performed the experiments. ZZ and YY analyzed the data.
YY and CX wrote the paper. HZ reviewed and edited the manuscript.
All the authors have read and approved the final version of this
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shaikh TH: Copy number variation
disorders. Curr Genet Med Rep. 5:183–190. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hastings PJ, Lupski JR, Rosenberg SM and
Ira G: Mechanisms of change in gene copy number. Nat Rev Genet.
10:551–564. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang F, Gu W, Hurles ME and Lupski JR:
Copy number variation in human health, disease, and evolution. Annu
Rev Genomics Hum Genet. 10:451–481. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Girirajan S, Campbell CD and Eichler EE:
Human copy number variation and complex genetic disease. Annu Rev
Genet. 45:203–226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iourov IY, Vorsanova SG and Yurov YB:
Molecular cytogenetics and cytogenomics of brain diseases. Curr
Genomics. 9:452–465. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang X, Du R, Li S, Zhang F, Jin L and
Wang H: Evaluation of copy number variation detection for a SNP
array platform. BMC Bioinformatics. 15:502014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stuppia L, Antonucci I, Palka G and Gatta
V: Use of the MLPA assay in the molecular diagnosis of gene copy
number alterations in human genetic diseases. Int J Mol Sci.
13:3245–3276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsuchiya KD, Shaffer LG, Aradhya S,
Gastier-Foster JM, Patel A, Rudd MK, Biggerstaff JS, Sanger WG,
Schwartz S, Tepperberg JH, et al: Variability in interpreting and
reporting copy number changes detected by array-based technology in
clinical laboratories. Genet Med. 11:866–873. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Manning M and Hudgins L: Professional
Practice and Guidelines Committee: Array-based technology and
recommendations for utilization in medical genetics practice for
detection of chromosomal abnormalities. Genet Med. 12:742–745.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schouten JP, McElgunn CJ, Waaijer R,
Zwijnenburg D, Diepvens F and Pals G: Relative quantification of 40
nucleic acid sequences by multiplex ligation-dependent probe
amplification. Nucleic Acids Res. 30:e572002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Banerjee S, Oldridge D, Poptsova M,
Hussain WM, Chakravarty D and Demichelis F: A computational
framework discovers new copy number variants with functional
importance. PLoS One. 6:e175392011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eijk-Van Os PG and Schouten JP: Multiplex
ligation-dependent probe amplification (MLPA®) for the
detection of copy number variation in genomic sequences. Methods
Mol Biol. 688:97–126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deveson IW, Chen WY, Wong T, Hardwick SA,
Andersen SB, Nielsen LK, Mattick JS and Mercer TR: Representing
genetic variation with synthetic DNA standards. Nat Methods.
13:784–791. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang X, Xu Y, Liu D, Geng J, Chen S,
Jiang Z, Fu Q and Sun K: A modified multiplex ligation-dependent
probe amplification method for the detection of 22q11.2 copy number
variations in patients with congenital heart disease. BMC Genomics.
16:3642015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gross SJ, Ryan A and Benn P: Noninvasive
prenatal testing for 22q11.2 deletion syndrome: Deeper sequencing
increases the positive predictive value. Am J Obstet Gynecol.
213:254–255. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chung JH, Cai J, Suskin BG, Zhang Z,
Coleman K and Morrow BE: Whole-genome sequencing and integrative
genomic analysis approach on two 22q11.2 deletion syndrome family
trios for genotype to phenotype correlations. Hum Mutat.
36:797–807. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang H, Nettleton D and Ying K: Copy
number variation detection using next generation sequencing read
counts. BMC Bioinformatics. 15:1092014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bunyan DJ, Skinner AC, Ashton EJ,
Sillibourne J, Brown T, Collins AL, Cross NC, Harvey JF and
Robinson DO: Simultaneous MLPA-based multiplex point mutation and
deletion analysis of the dystrophin gene. Mol Biotechnol.
35:135–140. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Naoufal R, Legendre M, Couet D,
Gilbert-Dussardier B, Kitzis A, Bilan F and Harbuz R: Association
of structural and numerical anomalies of chromosome 22 in a patient
with syndromic intellectual disability. Eur J Med Genet.
59:483–487. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiong B, Tan K, Tan YQ, Gong F, Zhang SP,
Lu CF, Luo KL, Lu GX and Lin G: Using SNP array to identify
aneuploidy and segmental imbalance in translocation carriers. Genom
Data. 2:92–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Belfield EJ, Brown C, Gan X, Jiang C,
Baban D, Mithani A, Mott R, Ragoussis J and Harberd NP:
Microarray-based optimization to detect genomic deletion mutations.
Genom Data. 2:53–54. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gilbert DC, McIntyre A, Summersgill B,
Missiaglia E, Goddard NC, Chandler I, Huddart RA and Shipley J:
Minimum regions of genomic imbalance in stage I testicular
embryonal carcinoma and association of 22q loss with relapse. Genes
Chromosomes Cancer. 50:186–195. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chan LF, Campbell DC, Novoselova TV, Clark
AJ and Metherell LA: Whole-exome sequencing in the differential
diagnosis of primary adrenal insufficiency in children. Front
Endocrinol (Lausanne). 6:1132015.PubMed/NCBI
|
|
24
|
Nimkarn S, Gangishetti PK, Yau M and New
MI: 21-hydroxylase-deficient congenital adrenal
hyperplasiaGeneReviews®. Adam MP, Ardinger HH, Pagon RA,
Wallace SE, Bean LJH, Stephens K and Amemiya A: Seattle (WA):
1993
|
|
25
|
Wong A, Martin Lese C, Heretis K, Ruffalo
T, Wilber K, King W and Ledbetter DH: Detection and calibration of
microdeletions and microduplications by array-based comparative
genomic hybridization and its applicability to clinical genetic
testing. Genet Med. 7:264–271. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aten E, White SJ, Kalf ME, Vossen RH,
Thygesen HH, Ruivenkamp CA, Kriek M, Breuning MH and den Dunnen JT:
Methods to detect CNVs in the human genome. Cytogenet Genome Res.
123:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen Y and Wu BL: Designing a simple
multiplex ligation-dependent probe amplification (MLPA) assay for
rapid detection of copy number variants in the genome. J Genet
Genomics. 36:257–265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bremer A, Giacobini M, Nordenskjöld M,
Brøndum-Nielsen K, Mansouri M, Dahl N, Anderlid B and Schoumans J:
Screening for copy number alterations in loci associated with
autism spectrum disorders by two-color multiplex ligation-dependent
probe amplification. Am J Med Genet B Neuropsychiatr Genet.
153B:280–285. 2010.PubMed/NCBI
|
|
29
|
Cai G, Edelmann L, Goldsmith JE, Cohen N,
Nakamine A, Reichert JG, Hoffman EJ, Zurawiecki DM, Silverman JM,
Hollander E, et al: Multiplex ligation-dependent probe
amplification for genetic screening in autism spectrum disorders:
Efficient identification of known microduplications and
identification of a novel microduplication in ASMT. BMC Med
Genomics. 1:502008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Slater H, Bruno D, Ren H, La P, Burgess T,
Hills L, Nouri S, Schouten J and Choo KH: Improved testing for
CMT1A and HNPP using multiplex ligation-dependent probe
amplification (MLPA) with rapid DNA preparations: Comparison with
the interphase FISH method. Hum Mutat. 24:164–171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Herodez Stangler S, Zagradisnik B, Skerget
Erjavec A, Zagorac A and Vokac Kokalj N: Molecular diagnosis of
PMP22 gene duplications and deletions: Comparison of different
methods. J Int Med Res. 37:1626–1631. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Q and Keleş S: CNV-guided multi-read
allocation for ChIP-seq. Bioinformatics. 30:2860–2867. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Duan J, Zhang JG, Deng HW and Wang YP:
Detection of common copy number variation with application to
population clustering from next generation sequencing data. Conf
Proc IEEE Eng Med Biol Soc. 2012:1246–1249. 2012.PubMed/NCBI
|
|
34
|
de Ligt J, Boone PM, Pfundt R, Vissers LE,
de Leeuw N, Shaw C, Brunner HG, Lupski JR, Veltman JA and Hehir-Kwa
JY: Platform comparison of detecting copy number variants with
microarrays and whole-exome sequencing. Genom Data. 2:144–146.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
de Ligt J, Boone PM, Pfundt R, Vissers LE,
Richmond T, Geoghegan J, O'Moore K, de Leeuw N, Shaw C, Brunner HG,
et al: Detection of clinically relevant copy number variants with
whole-exome sequencing. Hum Mutat. 34:1439–1448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tan R, Wang Y, Kleinstein SE, Liu Y, Zhu
X, Guo H, Jiang Q, Allen AS and Zhu M: An evaluation of copy number
variation detection tools from whole-exome sequencing data. Hum
Mutat. 35:899–907. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Samarakoon PS, Sorte HS, Kristiansen BE,
Skodje T, Sheng Y, Tjønnfjord GE, Stadheim B, Stray-Pedersen A,
Rødningen OK and Lyle R: Identification of copy number variants
from exome sequence data. BMC Genomics. 15:6612014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fromer M, Moran JL, Chambert K, Banks E,
Bergen SE, Ruderfer DM, Handsaker RE, McCarroll SA, O'Donovan MC,
Owen MJ, et al: Discovery and statistical genotyping of copy-number
variation from whole-exome sequencing depth. Am J Hum Genet.
91:597–607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu J, Grzeda KR, Stewart C, Grubert F,
Urban AE, Snyder MP and Marth GT: Copy number variation detection
from 1000 Genomes project exon capture sequencing data. BMC
Bioinformatics. 13:3052012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo Y, Sheng Q, Samuels DC, Lehmann B,
Bauer JA, Pietenpol J and Shyr Y: Comparative study of exome copy
number variation estimation tools using array comparative genomic
hybridization as control. Biomed Res Int. 2013:9156362013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sathya B, Dharshini AP and Kumar GR: NGS
meta data analysis for identification of SNP and INDEL patterns in
human airway transcriptome: A preliminary indicator for lung
cancer. Appl Transl Genom. 4:4–9. 2014.PubMed/NCBI
|
|
42
|
Liu B, Madduri RK, Sotomayor B, Chard K,
Lacinski L, Dave UJ, Li J, Liu C and Foster IT: Cloud-based
bioinformatics workflow platform for large-scale next-generation
sequencing analyses. J Biomed Inform. 49:119–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li H and Durbin R: Fast and accurate short
read alignment with burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brandariz-Fontes C, Camacho-Sanchez M,
Vilà C, Vega-Pla JL, Rico C and Leonard JA: Effect of the enzyme
and PCR conditions on the quality of high-throughput DNA sequencing
results. Sci Rep. 5:80562015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen CP, Huang JP, Chen YY, Chern SR, Wu
PS, Su JW, Chen YT, Chen WL and Wang W: Chromosome 22q11.2 deletion
syndrome: Prenatal diagnosis, array comparative genomic
hybridization characterization using uncultured amniocytes and
literature review. Gene. 527:405–409. 2013. View Article : Google Scholar : PubMed/NCBI
|