Introduction
Pancreatic ductal adenocarcinoma (PDAC), which
accounts for ~80% of all pancreatic tumors, is an aggressive
malignancy and the fourth leading cause of cancer-associated
mortality worldwide (1,2). With the rising pancreatic cancer
incidence rates, the American Cancer Society estimates that 55,440
new cases will occur in the United States with 44,330
PDAC-associated deaths in 2018 (3). Treatment of pancreatic cancer
consists of surgery, radiotherapy, chemotherapy and palliative
care. Appropriate treatments are selected depending on disease
stage. Compared with other treatments, surgical resection is the
best curative option to significantly prolong patient survival
(4). Unfortunately, numerous
studies have reported that prognosis remains poor following radical
surgical resection, with a median survival of 10–20 months and
5-year survival rates of only 10–25% in patients who receive
pancreaticoduodenectomy (5–11);
the survival rates are even lower for patients who receive
pancreatectomy (12,13). For patients who undergo radical
resection, gemcitabine-based adjuvant chemotherapy is currently the
standard treatment following surgery. If the patients exhibit nodal
involvement or microscopic residual disease following resection,
radiotherapy is recommended (14).
Despite substantial advancements in screening, diagnosis and
treatment, PDAC has an extremely dismal prognosis, with a median
survival of 2–8 months and a 5-year survival rate as low as 8%
(15). Using prognostic biomarkers
to select the most suitable adjuvant therapies for patients may
improve clinical outcomes and reduce the toxicity caused by
ineffective therapies.
As a class of single-stranded small non-coding RNAs
(typically 14–25 nucleotides), microRNAs (miRNAs/miRs) suppress
protein translation at the post-transcriptional level via
translational inhibition or degradation of their target mRNAs, and
subsequently affect various biological processes (16–18).
miRNA dysregulation has been reported in a range of human
malignancies, and identified as an important mechanism that
regulates cancer-associated genes and signaling pathways (19–22).
Recently, the vital roles of miRNAs in PDAC pathogenesis have been
revealed. Certain miRNAs, which are potentially oncogenic, are
upregulated in PDAC tissues compared with normal tissues, including
miR-10b, miR-17 and miR-21, whereas several potentially
tumor-suppressive miRNAs, including miR-26, miR-34a and miR-96, are
downregulated (23). For example,
the putative onco-miR, miR-155, enhances reactive oxygen
species-induced proliferation by targeting forkhead box O (FoxO)3a
and GTPase KRas, and also promotes tumor growth via tumor protein
p53-inducible nuclear protein 1 in PDAC (24,25).
The overexpression of miR-214 decreases the sensitivity of PDAC
tumor cells to gemcitabine by inhibiting inhibitor of growth family
member 4 (26). Furthermore, the
association of certain aberrantly expressed miRNAs with the
clinical outcomes of patients with PDAC has been reported in
numerous previous studies (27–30).
For example, elevated miR-21 expression levels are significantly
correlated with worse overall survival and progression-free
survival in patients with PDAC (31). Additionally, downregulation of
miR-506 leads to increased sphingosine kinase 1 expression, which
is also associated with shorter survival in patients with PDAC
(32).
Based on the potential of miRNAs to be effective
biomarkers, further in-depth analysis of the association between
miRNAs and PDAC prognosis is important. Traditional biological
research focuses on the specific functions and characteristics of
individual genes, transcripts and proteins, which can only explain
part of a biological system. Weighted gene co-expression network
analysis (WGCNA) is an effective systems biology method used to
assign highly co-expressed genes into modules using unsupervised
hierarchical clustering and assess the association of modules with
clinical features, allowing the identification of key genes in a
module for further analysis according to intra-modular connectivity
and gene significance (33).
In the present study, WGCNA was used to analyze
miRNAs to detect the potential key miRNAs associated with the
prognosis of patients with early-stage PDAC that underwent radical
resection. These results may produce further useful information for
the assessment of PDAC prognosis.
Materials and methods
Data collection and processing
miRNA sequencing data and clinical information,
including survival profiles, of patients with PDAC were obtained
from The Cancer Genome Atlas (TCGA; cancergenome.nih.gov/). Datasets with the following
criteria were used: i) The histological type was pancreatic ductal
adenocarcinoma; ii) the pathological stage was stage I or II,
according to the standard of American Joint Committee on Cancer
(7th edition) (34);
iii) patients underwent radical resection, including the Whipple
procedure, distal or total pancreatectomy; and iv) survival data
were available. Consequently, level three miRNAseq data generated
using the Illumina HiSeq platform (Illumina, Inc., San Diego, CA,
USA) for 124 patients with PDAC and the corresponding clinical data
were available for analysis. The variance in expression of each
miRNA in each sample was calculated and ranked using the quartile
method. miRNAs with expression variance less than the median
variance were removed. Subsequently, the ‘goodSamplesGenes’
function in the R package ‘WGCNA’ (R version 3.3.3) was used to
remove miRNAs with verbose <3 for excessive missing values and
identification of outlier samples (35). Additionally, the clustering
analysis of samples was performed using the ‘hclust’ function
(method ‘average’) in R with an appropriate cut-off value. Further
approval of the data and analysis in the present study was not
required by the TCGA ethics committee. Meanwhile, miRNA sequencing
data with relevant clinical data were searched on the Gene
Expression Ominbus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) to further validate
our results (36).
Weighted gene co-expression network
analysis
Based on the filtered miRNA expression data, the
scale-free gene modules of co-expression were constructed by WGCNA
(35). To ensure the reliability
of the co-expression network, hierarchical clustering was performed
based on Euclidean distance to detect and remove sample outliers.
The Pearson correlation coefficient between all input miRNAs was
calculated for converting expression data into correlation
matrices. An adequate soft-threshold power that met the scale-free
topology criterion was selected for transforming the former
correlation matrix into an adjacency matrix, which was subsequently
converted into a Topological Overlap Matrix (TOM) using the
‘TOMsimilarity’ function in R (33,37).
TOM-based dissimilarity was computed as measure distance, and a
miRNA clustering tree (dendrogram) and module colors were obtained.
In the clustering dendrogram, the minimum module size and cut
height were separately set to 30 and 0.25, respectively; thus,
modules below these values were merged into new modules.
Analysis of association between
modules and clinical characteristics
The association between modules and clinical
characteristics (age, gender, pathological stage, pathological T
stage, pathological N stage, histological grade, type of surgery
performed, radical resection, number of lymph nodes, radiation
therapy, targeted molecular therapy, history of alcohol consumption
and smoking history) were estimated by Pearson's correlation tests
for the phenotype (clinical characteristics) and module eigengene.
P<0.05 was considered to indicate statistical significance
(38). The association of
individual miRNAs with clinical characteristics was quantified by
Gene Significance (GS), while the correlation of the miRNA
expression profile with module eigengenes was weighted by Module
Membership (MM) (39).
Identification of hub miRNAs and
functional annotation
Hub miRNAs are highly connected intra-modular miRNAs
that determine the characteristics of the module to a certain
extent (40,41). The module eigengene function was
used for calculating the eigengene, which represents the whole
module miRNA expression level. Subsequently, the ‘signedKME’
function was used to calculate the distance between a gene and a
module eigengene. Screening based on GS and MM was used to detect
the hub miRNAs in specified modules. The corrected miRNAs with
q-weighted P<0.01 were selected as hub miRNAs (35). For visualizing the network that
consists of the hub miRNAs in the turquoise module, Cytoscape
software (version 3.6.1; http://cytoscape.org) was used, which is an open
source software platform used primarily for visualization of
biological pathways and molecular interactions (42).
For interpreting the biological function of the hub
miRNAs, target genes were predicted using miRWalk 2.0 (zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/index.html),
miRDB (www.mirdb.org), TargetScanHuman 7.2
(www.targetscan.org/vert_71/), miRanda
(34.236.212.39/microrna/home.do), miRTarBase 7.0 (mirtarbase.mbc.nctu.edu.tw/php/index.php) and PicTar
(pictar.mdc-berlin.de) (43–48).
Genes common to four of these six databases were selected as target
genes and used in further analysis. The list of target genes was
then uploaded to the Database For Annotation, Visualization And
Integrated Discovery (DAVID) bioinformatics resource (version 6.8;
http://david.ncifcrf.gov) for Gene Ontology (GO)
and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis
(49).
Prognostic model construction and
survival analysis
After dichotomizing the data around the median
expression of each hub miRNAs, the R packages ‘survival’ and
‘survminer’ (package version 0.4.2) (https://cran.r-project.org/web/packages/survminer/index.html)
were used for the single hub miRNA survival analysis and
constructing the survival curves. Subsequently, the ‘rbsurv’
function in R was applied to develop models that utilized the
partial likelihood of the Cox model to select survival-associated
miRNAs by employing forward selection, generating a series of gene
models. One of these models, which was validated to have
significant importance for survival analysis via the ‘ggsurvplot’
function contained in the ‘survminer’ package, was the optimal
model (50,51). Meanwhile, the expression of the
miRNAs for each sample in this model was visualized with the
‘pheatmap’ package in R (https://cran.r-project.org/web/packages/pheatmap/index.html)
(52). For all the survival
analysis, overall survival (OS) was determined as survival
endpoints, the hazard ratio (HR) was calculated via a Cox
regression model and Kaplan-Meier curves were compared by the
log-rank test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Study population and sequencing
data
The clinical characteristics with survival profiles
of 185 patients with pancreatic adenocarcinoma and a miRNA
sequencing dataset from 178 patients were downloaded from TCGA. A
total of 129 patients diagnosed with early-stage PDAC who underwent
radical resection met the inclusion criteria of the present study
(Table I). It should be noted that
five patients were removed due to detection as sample outliers by
clustering analysis prior to construction of the co-expression
network in the subsequent analysis. Using miRNA sequencing data,
523 miRNAs for each sample were filtered into further analysis
according to the ranked expression variance. Following testing
using the ‘goodSamplesGenes’ function, the expression of these
selected 523 miRNAs in 129 patients with PDAC were implemented into
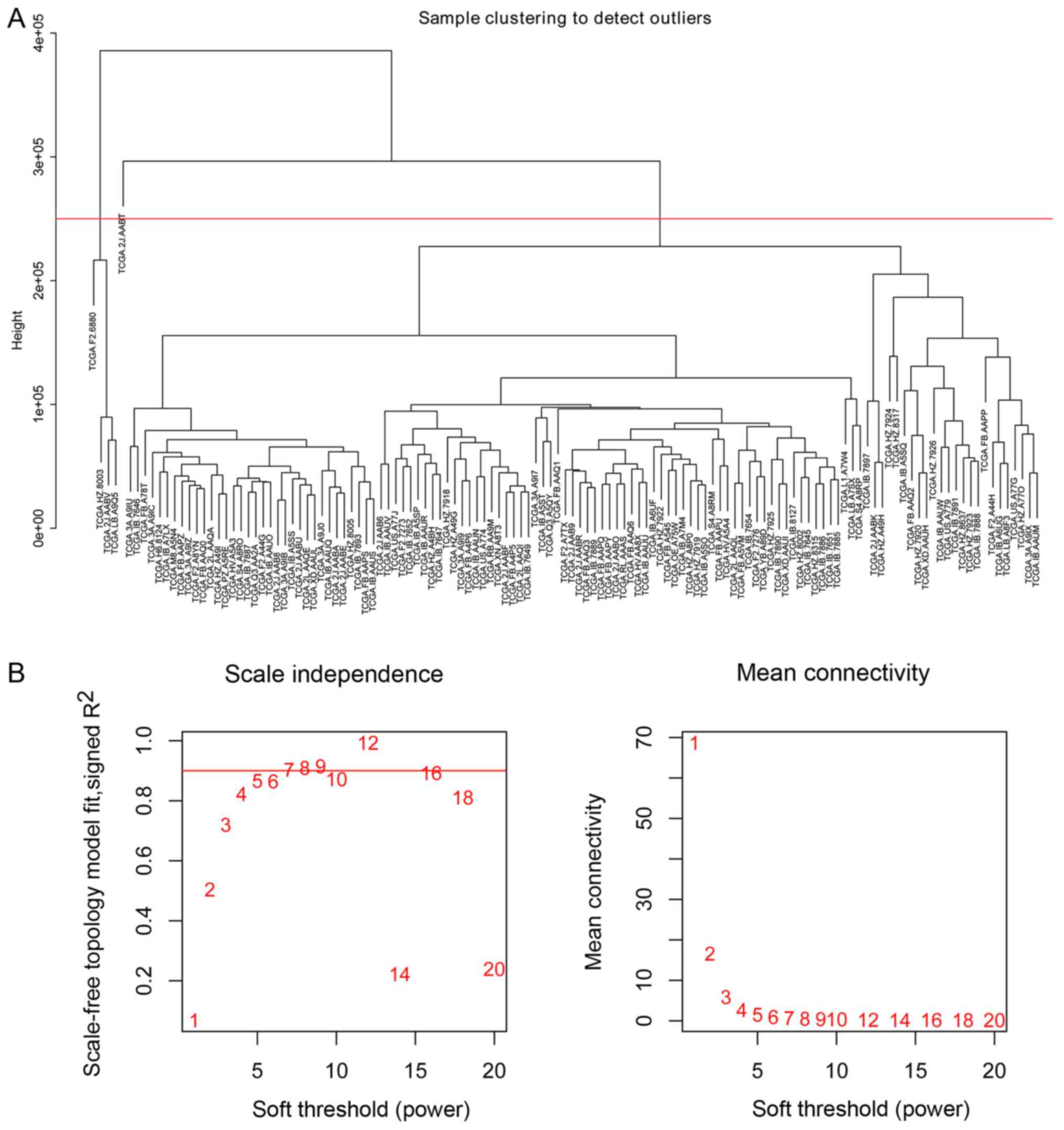
hierarchical clustering analysis (Fig.
1A). Consequently, five outlier samples whose leaf node height
was significantly higher than other samples and deviated from the
main cluster obviously in the dendrogram were identified. To avoid
effects on the subsequent analysis, a height cut at a certain value
ranged from about 2.2e+05 to 2.9e+05 was utilized to remove these
samples automatically. The expression data of 523 miRNAs in 124
patients with PDAC was used for construction of the co-expression
network.
 | Table I.Clinical characteristics of patients
with pancreatic ductal adenocarcinoma (n=124). |
Table I.
Clinical characteristics of patients
with pancreatic ductal adenocarcinoma (n=124).
| Variable | Number of cases
(%) |
|---|
| Age (years) |
|
|
<60 | 40 (32.3) |
|
≥60 | 84 (67.7) |
| Sex |
|
|
Female | 55 (44.4) |
|
Male | 69 (55.6) |
| Pathological
stage |
|
| I | 10 (8.1) |
| II | 114 (91.9) |
| Pathological T
stage |
|
|
T1/T2 | 18 (14.5) |
| T3 | 106 (85.5) |
| Pathological N
stage |
|
| N0 | 28 (22.6) |
| N1 | 95 (76.6) |
| NX | 1 (0.8) |
| Histological
grade |
|
| 1 | 16 (12.9) |
| 2 | 71 (57.2) |
|
3/4 | 37 (29.8) |
| Type of surgery
performed |
|
|
Whipple | 107 (86.3) |
|
Distal/total
pancreatectomy | 17 (13.7) |
| Residual tumor |
|
| R0 | 72 (58.1) |
|
R1/R2 | 44 (35.5) |
| RX | 8 (6.4) |
| Number of lymph
nodes |
|
| 0 | 27 (21.8) |
| ≥1 | 97 (78.2) |
| Radiation
therapy |
|
| No | 80 (64.5) |
|
Yes | 34 (27.4) |
| NA | 10 (8.1) |
| Targeted molecular
therapy |
|
| No | 34 (27.4) |
|
Yes | 61 (49.2) |
| NA | 29 (23.4) |
| Alcohol consumption
history |
|
| No | 46 (37.1) |
|
Yes | 71 (57.3) |
| NA | 7 (5.6) |
| Tobacco smoking
history |
|
|
<3 | 63 (50.8) |
| ≥3 | 38 (30.6) |
| NA | 23 (18.6) |
Co-expression network analysis
The ‘PickSoftThreshold’ function in the WGCNA
package was used to estimate the suitable soft threshold power,
which was set as five (Fig. 1B).
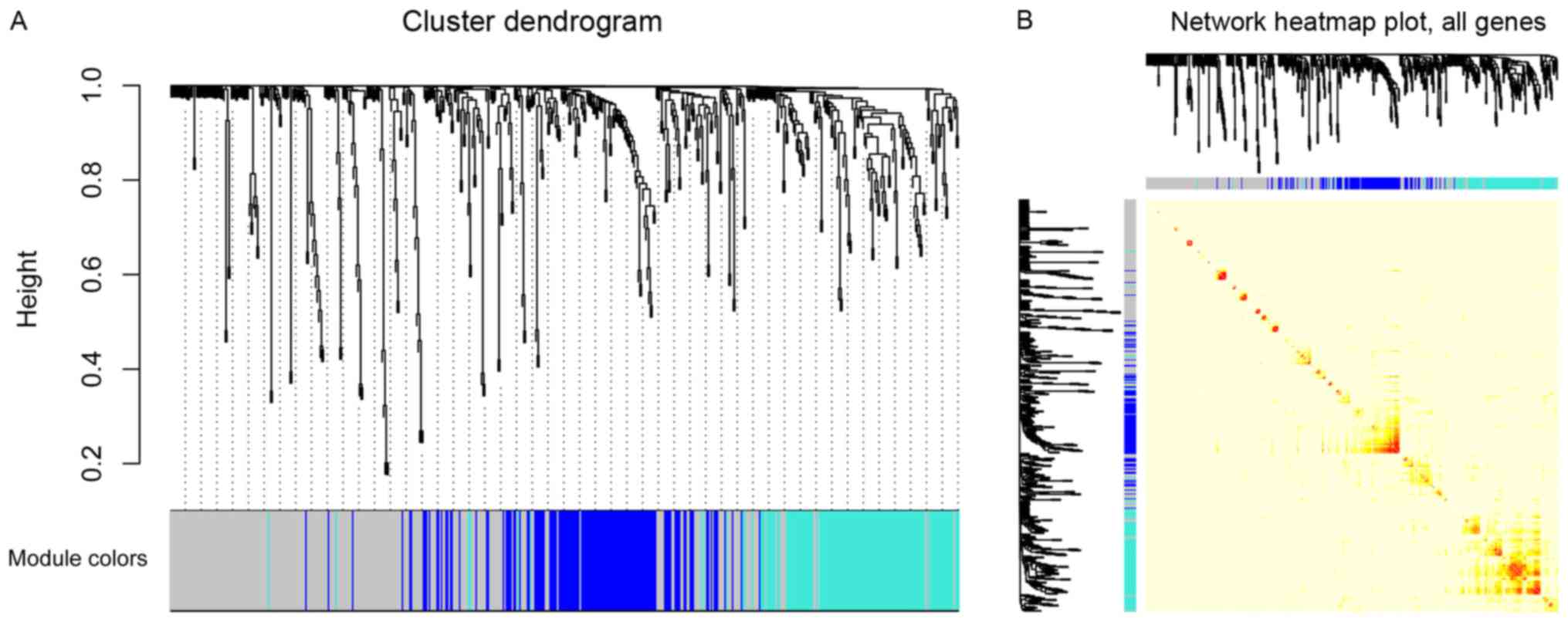
Three modules were identified by a one-step network construction
method using the ‘blockwiseModules’ function in R (minModuleSize,
30; mergeCutHeight, 0.25) containing blue, turquoise and grey
modules. Because module identification does not employ prior
biological knowledge about the miRNA, the biological meaning of
each module is initially unknown and hence the module were assigned
a color label (blue, turquoise and grey). Notably, miRNAs that
failed to be classified into a module were assigned to the grey
module (Fig. 2A). The number of
miRNAs in each module was 131 in blue, 131 in turquoise and 261 in
grey. The co-expression network was visualized as a TOM plot
consisting of a hierarchical clustering dendrogram and TOM matrix
(Fig. 2B).
Association of modules with clinical
characteristics
The associations of co-expression network modules
with the clinical characteristics of the patients with PDAC are
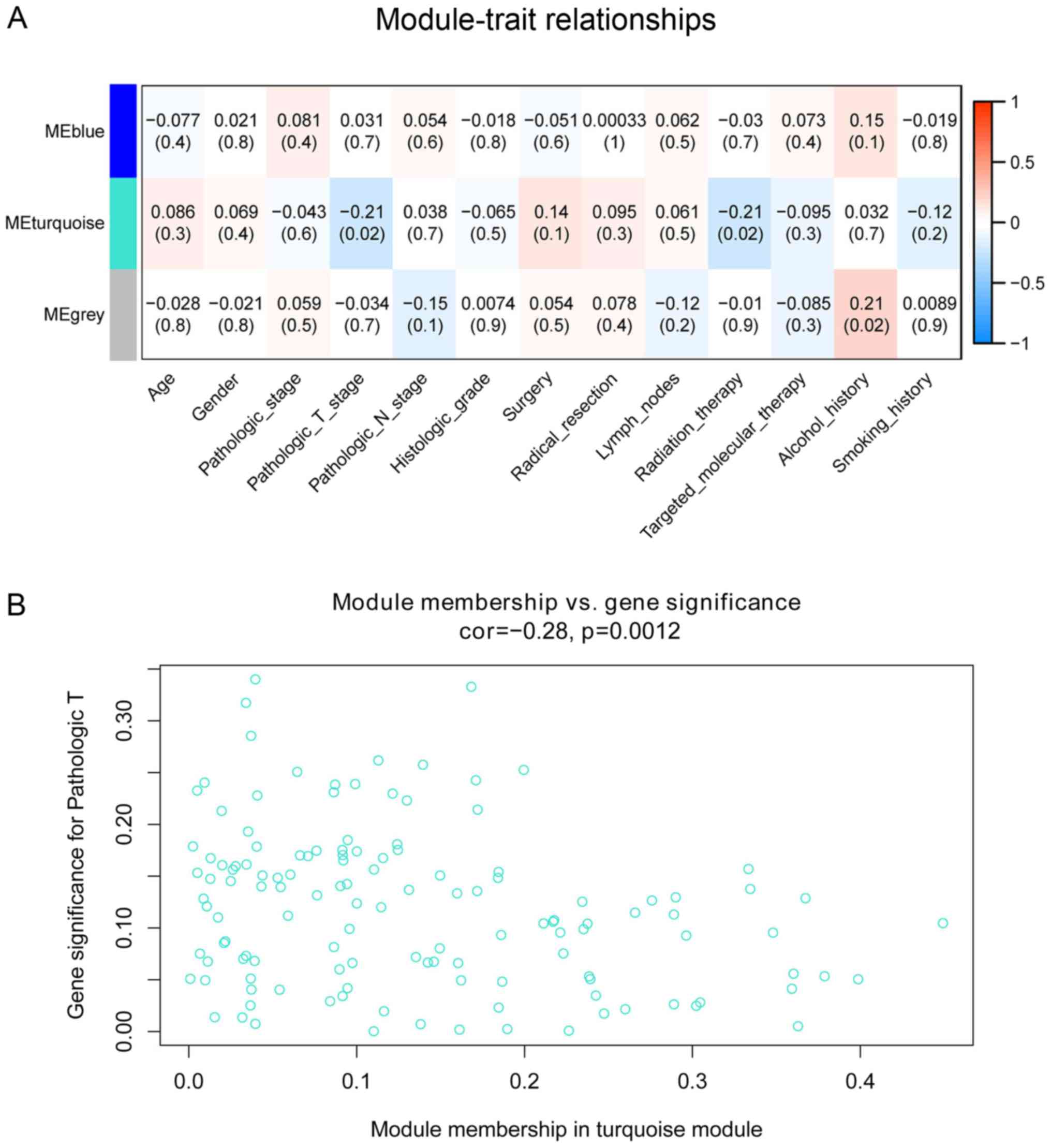
illustrated as heatmaps of module-trait correlation (Fig. 3A). The turquoise module was
significantly associated with pathological T stage (cor=−0.21;
P=0.02) and radiation therapy (cor=−0.21; P=0.02). A scatterplot of
GS vs. MM in the turquoise module was generated. This analysis
revealed a highly significant association between the turquoise
module and pathological T stage (cor=−0.21; P=0.0012; Fig. 3B); however, radiotherapy was not
significantly associated with the turquoise module in this analysis
(cor=0.035; P=0.69).
Hub miRNA detection and functional
analysis
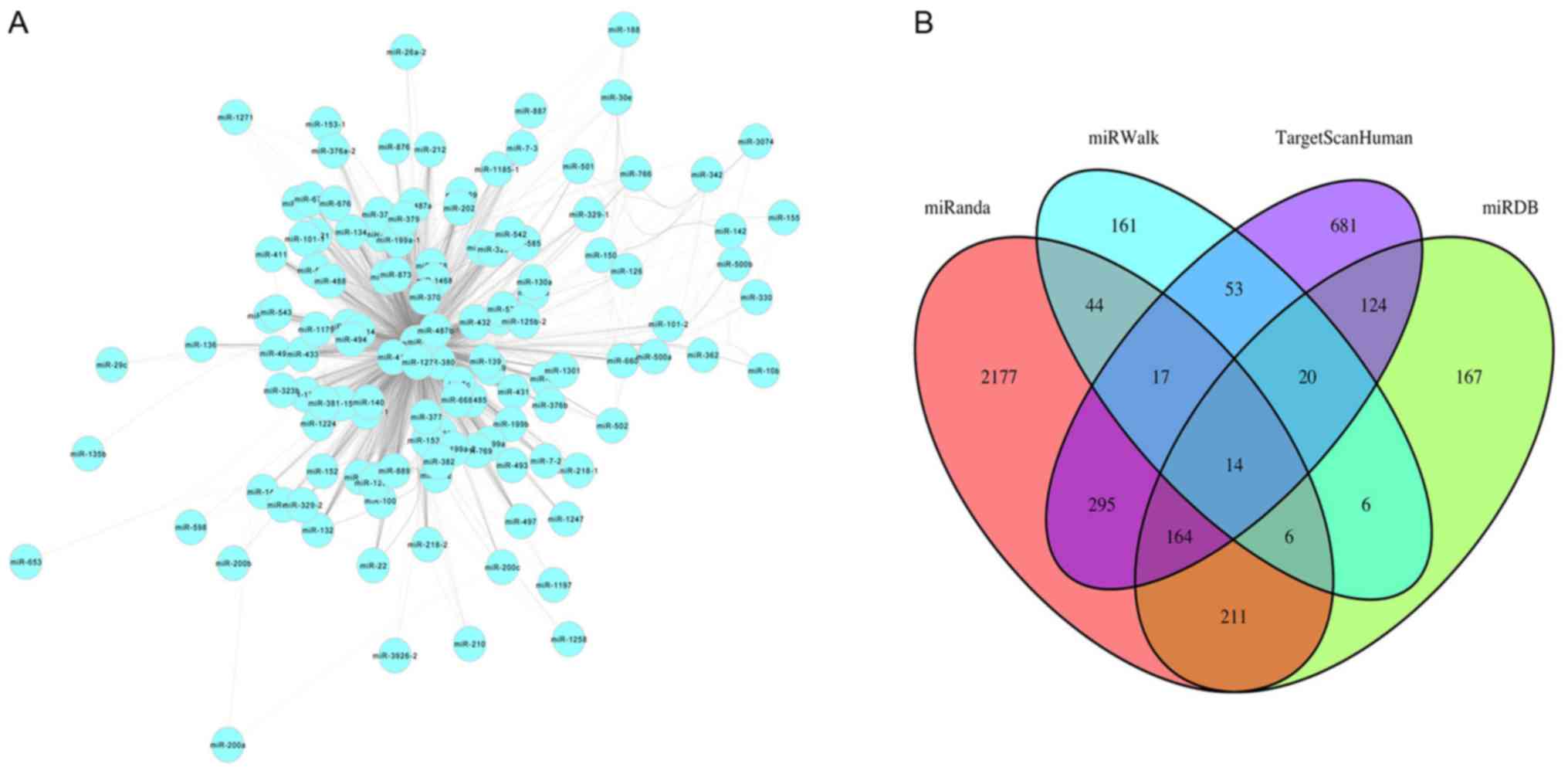
Cytoscape software (version 3.6.1; http://cytoscape.org) was used to visualize the miRNAs
in the co-expression network turquoise module based on topological
overlap of miRNAs (Fig. 4A). Nodes
represent miRNAs and edges represent connectivity. The more edges
the node is connected with, the higher intra-module connectivity
the node possesses, and the more likely it is to be a hub miRNA.
Through function ‘networkScreening’ based on GS and MM, 39 hub
miRNAs were identified in the turquoise module (Table II). TargetScanHuman, miRWalk,
miRDB, miRanda, miRTarBase and PicTar were used to identify target
genes of the miRNAs. Genes common to four of these six databases
were selected as target genes. Overlapping target genes for each
miRNA from four web tools were screened out using Venn diagrams in
order to reduce the false positive rate, such as for miR-140
(Fig. 4B). In total, 404 genes
were identified as targeted genes of the 39 hub miRNAs in the
turquoise module.
| Table II.Hub miRs associated with pathological
T stage in the turquoise module. |
Table II.
Hub miRs associated with pathological
T stage in the turquoise module.
| Module | Hub miRNAs | q-weighted
P-value |
|---|
| Turquoise | miR-376c, miR-379,
miR-654, miR-873, miR-889, miR-487b, miR-323, miR-410, miR-204,
miR-127, miR-758, miR-487a, miR-432, miR-154, miR-381, miR-431,
miR-376a-1, miR-409, miR-377, miR-543, miR-370, miR-433, miR-375,
miR-382, miR-1468, miR-551b, miR-539, miR-129-1, miR-1224,
miR-129-2, miR-369, miR-1179, miR-496, miR-212, miR-1197,
miR-218-2, miR-134, miR-411, miR-140 | <0.01 |
For further insights into the biological relevance
of the target genes, GO and KEGG pathway enrichment analysis were
performed using the data uploaded to DAVID. The GO biological
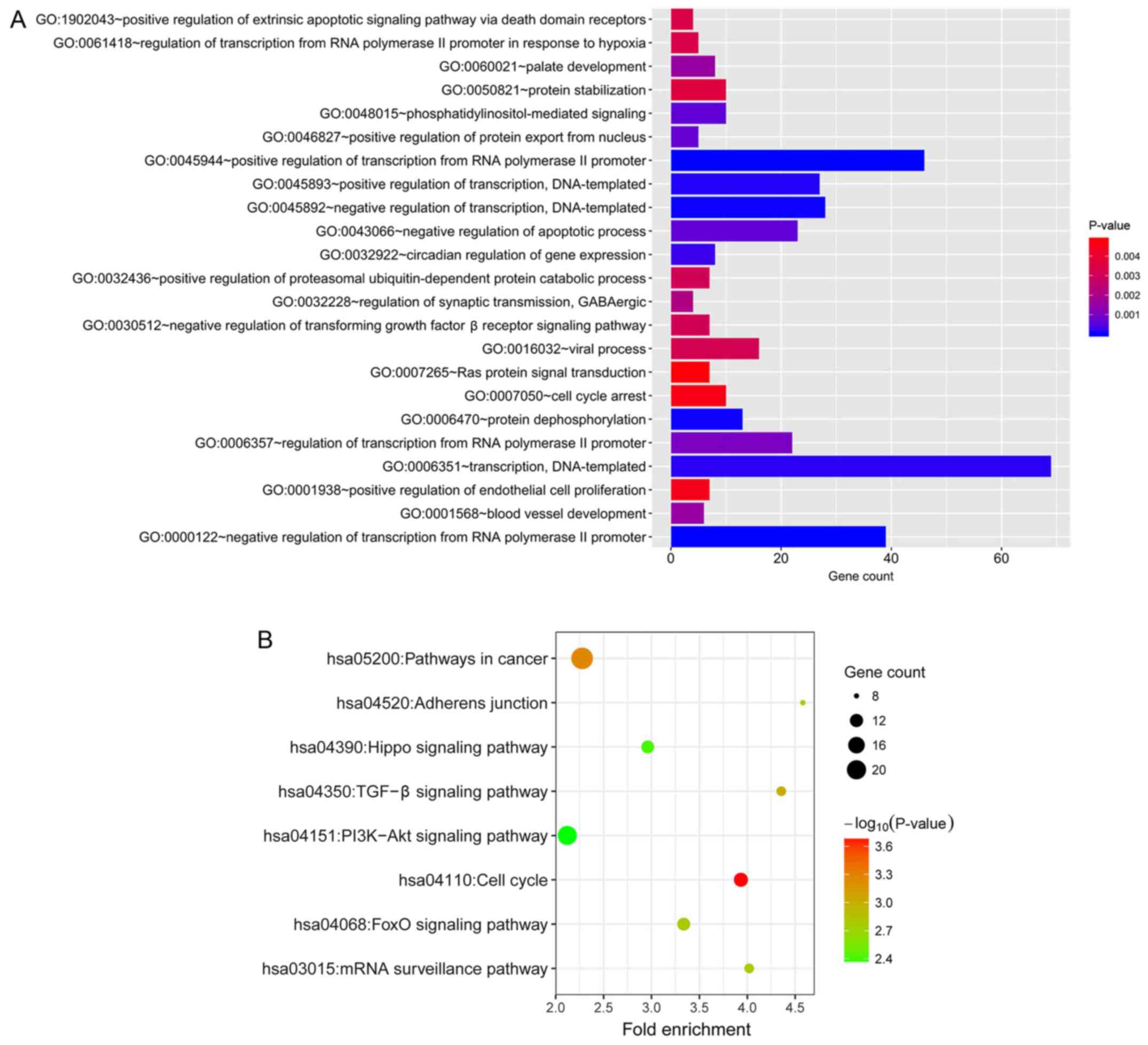
process that was most enriched was ‘negative regulation of
transcription from RNA polymerase II promoter’ (GO:0000122;
Fig. 5A). The KEGG pathways that
were most enriched were ‘cell cycle’ (hsa04110), ‘pathways in
cancer’ (hsa05200), ‘TGF-β signaling pathway’ (hsa04350), ‘FoxO
signaling pathway’ (hsa04068), ‘mRNA surveillance pathway’
(hsa03015), ‘adherens junction’ (hsa04520), ‘Hippo signaling
pathway’ (hsa04390), ‘PI3K-Akt signaling pathway’ (hsa04151;
Fig. 5B).
Prognostic model construction and
survival analysis
To explore the association of the 39 hub miRNAs with
the prognosis of the patients with PDAC and construct a prognostic
model, the expression data of hub miRNAs and corresponding survival
profiles were analyzed in R software using the ‘rbsurv’ function. A
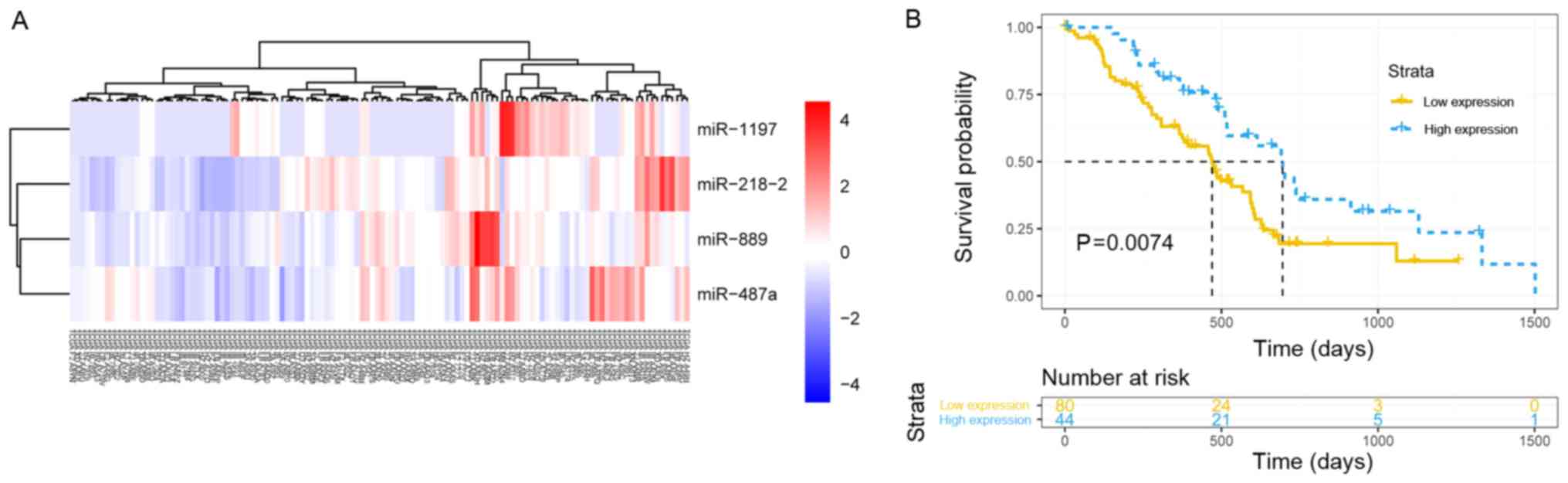
model consisting of four hub miRNAs (miR-1197, miR-218-2, miR-889
and miR-487a) associated with pathological T staging was built and
validated by survival analysis. The patients with PDAC were
successfully stratified into high and low signature expression
groups using the ‘pheatmap’ function (agglomeration method, ward.
D2) in R (Fig. 6A). The median
overall survival was 695 vs. 470 days in the high and low miR
signature expression groups, respectively (P=0.0074; Fig. 6B). The results demonstrated that
the higher expression of the signature, the longer the OS of the
patients with early-stage PDAC who underwent radical resection.
Discussion
As one of the most fatal cancers worldwide,
treatment of PDAC faces therapeutic challenges due to an increasing
incidence rate and insufficient diagnosis at the early stage
(3). Although patients who receive
radical resection have a relatively good clinical outcome, <20%
of patients are eligible for surgery (53). Furthermore, low sensitivity to
adjuvant therapy and substantial toxicity are major problems
(4,54). Identification of reliable
biomarkers to screen for patients who will benefit from surgery is
ongoing (55). Advancements in
algorithms have allowed the use of systems biology methods to
explore the mechanisms of tumorigenesis and development in depth
(56).
In the present study, WGCNA was performed using
miRNA expression data and corresponding clinical information of 124
patients with PDAC. An association between the turquoise module and
pathological T stage was detected in the co-expression network. The
pathological T stage indicates whether the adjacent tissues have
been invaded by the tumor and what the original tumor size was, and
was previously identified as an independent prognostic factor for
patients with PDAC who underwent resection in previous studies
(57,58); however, the mechanisms by which
miRNAs affect the pathological T stage remains unknown. In the
present study, 39 hub miRNAs that represented the features of the
turquoise module were selected to predict target genes for
enrichment analysis. In GO enrichment analysis, ‘negative
regulation of transcription from RNA polymerase II promoter’ was
the most significantly enriched biological process, while the other
enriched processes included ‘regulation of transcription’,
‘negative regulation of apoptotic process’, ‘TGF-β receptor
signaling pathway’, ‘positive regulation of endothelial cell
proliferation’, ‘cell cycle arrest’ and ‘Ras protein signal
transduction’.
The proliferation and growth of tumor cells is
regulated by various factors, including abnormalities in growth
promotion and inhibitory signaling, increased telomerase activity,
reduced apoptosis, continuous tumor angiogenesis and self-renewal
of cancer stem cells (59). The
functions of the majority of oncogenes and tumor suppressor genes
are associated with the cell cycle. Gene mutations may cause
dysregulation of the cell cycle, resulting in uncontrolled growth
via excessive proliferation and decreased apoptosis (60). In addition, tumor growth requires
delivery of oxygen and nutrients via blood vessels. Vascular
endothelial growth factor (VEGF) is secreted by tumor cells and
acts directly on receptors on the surface of endothelial cells to
induce their proliferation and activation to form new blood vessels
(61). Transforming growth
factor-β (TGF-β) is a multifunctional cytokine. In early-stage
pancreatic cancer, cell growth is inhibited by activation of TGF-β
receptor signaling, which increases Smad3 phosphorylation and
nuclear translocation; in advanced tumors, the activation of TGF-β
signaling can promote tumor vascularization and metastasis by
targeting VEGFA, and increasing fibrosis in the pancreatic tissue
(62–64). Ras-Raf-MEK 1-mitogen activated
protein kinase signaling, a receptor tyrosine kinase-mediated
signaling pathway, serves an important role in the regulation of
cell proliferation and survival. As an upstream mediator of this
pathway, abnormal activation of Ras signaling may lead to
unrestrained cell proliferation, and is associated with poor
prognosis in PDAC (65).
In KEGG pathway analysis, the most significantly
enriched pathway was ‘cell cycle’, and other significant
cancer-associated pathways included ‘TGF-β signaling pathway’,
‘FoxO signaling pathway’, ‘mRNA surveillance pathway’, ‘adherens
junction’, ‘Hippo signaling pathway’ and ‘PI3K-Akt signaling
pathway’. It is well established that cell cycle disorder can cause
tumorigenesis (66). Additionally,
TGF-β signaling promotes cell proliferation, angiogenesis and the
self-renewal of tumor stem cells. FoxOs, as a subfamily of the fork
head transcription factor family, have confirmed roles in tumor
cell differentiation, proliferation, apoptosis, DNA repair and
damage, and also act as mediators of oxidative stress (67). The translation of mRNA transcripts
into proteins is an essential part of the central dogma of
molecular biology. However, fidelity errors are occasionally
present in the mRNA molecules, which may result in errors in the
translated protein. The mRNA surveillance pathway is a quality
control mechanism that detects and degrades abnormal mRNA molecules
(68).
Adherens junctions are created by homotypic
interactions between the extracellular domains of cadherin proteins
to join epithelial cells together, and alterations in cell adhesion
through adherens junctions may mediate tumor cell invasion and
migration in pancreatic cancer (57). Hippo signaling is a highly
conserved serine/threonine kinase cascade involved in the control
of cell proliferation and apoptosis to regulate organ size
(69). Phosphoinositide 3-kinase
(PI3K)-protein kinase B (Akt) signaling, which is one of the most
commonly dysregulated signaling pathways in cancer, can be
triggered to cause continuous activation of Akt, which ultimately
inhibits tumor cell apoptosis. In PDAC, mechanistic target of
rapamycin kinase (mTOR), a key kinase downstream of PI3K-Akt, plays
key roles in the renewal of cancer stem cells, and resistance to
radiotherapy or chemotherapy (70).
Based on the current enrichment analysis, 39 hub
miRNAs in the turquoise module were identified to be enriched in
the biological processes and pathways involved in tumor development
and progression, which may be involved in the negative correlation
detected between miRNA expression and pathological T stage.
By utilizing prognostic biomarkers, PDAC patients
could be divided into different subgroups and received
individualized adjuvant therapy, which might improve clinical
outcomes (71). However, a single
miRNA is less specific and sensitive than a panel of miRNAs
(72,73). With data on miRNAs as prognostic
biomarkers accumulating, inconsistencies among study results have
arisen. Though factors might ascribe to discrepancies of lab
protocols, measurement platforms, samples size and ethnicity, the
credibility of the predictive value of a single miRNA is dubious.
Besides, focusing on the target protein of a single miRNA typically
results in researchers overlooking the pathways involved in complex
mechanisms such as drug metabolism and treatment resistance, which
could be simultaneously affected by certain prognosis-associated
miRNAs (74). Furthermore, some
studies have proven that it is more effective to predict prognosis
with a panel than a single miRNA by statistical analysis (73,75).
To explore the association between the 39 hub miRNAs
and PDAC prognosis, a predictive model was constructed to identify
a prognostic signature composed of miR-218-2, miR-889, miR-487a and
miR-1197. The previous study by Guan et al (76) reported that miR-218 acts as an
inhibitor of tumor angiogenesis by targeting rapamycin-insensitive
companion of mTOR, an mTOR component, in prostate cancer (76). In a study by Zhu et al
(77), miR-218-5p was demonstrated
to suppress the cell proliferation and migration of non-small-cell
lung cancer via epidermal growth factor receptor (77). Although miR-218, which is
transcribed from two loci located at chromosome 5q35.1 (miR-218-2)
and 4p15.31 (miR-218-1), has been considered as an anti-oncogene in
various types of cancer, miR-218-2 has opposing roles in different
types of cancer. In glioblastoma, miR-218-2 promotes cell
proliferation, migration and invasion by targeting cell division
cycle 27 (78); whereas in thyroid
cancer, co-expression of miR-218-2 with its host gene, slit
guidance ligand 3, exerts a tumor-suppressive effect (79). Xie et al (80) reported that miR-889 downregulation
by histone deacetylase enhances the cytotoxicity of natural killer
cells in hepatocellular carcinoma. Xu et al (81) observed that miR-889 promotes the
transformation of esophageal cancer cells from the G1 to S phase;
furthermore, miR-889 may induce epithelial-to-mesenchymal
transition (EMT) via the Akt-mTOR and Wnt pathways (81). In a study of advanced colorectal
cancer, Molina-Pinelo et al (82) reported that the reduced expression
of miR-889 is significantly associated with improved overall
survival and progression-free survival in patients who received
chemotherapy (82). Chang et
al (83) revealed that
miR-487a has the ability to promote hepatocellular carcinoma
metastasis and proliferation by targeting phosphoinositide-3-kinase
regulatory subunit 1 and sprouty-related EVH1 domain-containing 2
mRNA (83). Additionally, miR-487a
was demonstrated to induce cell invasion and migration of lung
adenocarcinoma by regulating migration-associated genes AXL
receptor tyrosine kinase and ovo-like zinc finger 2 (84). In breast cancer cells, EMT induced
by TGF-β1 may be restricted by overexpression of the
membrane-associated guanylate kinase WW and PDZ domain-containing 2
gene, which is regulated by miR-487a (85). Only a limited number of studies
have investigated miR-1197 (86,87).
The effects of these miRNAs on tumor development and progression
are inconsistent with each other and vary among different types of
cancer (78,79). The role of each miRNA in biological
processes and the expression level should be considered when
interpreting study results among varying cancer types. Thus,
comprehensive consideration and further study are required.
Certain limitations of the present study should be
considered. As the results were based on single-data source, Gene
Expression Omnibus datasets were searched for further validation.
Unfortunately, no data with full survival profiles were available
for analysis. Additionally, the conclusions made are based on
bioinformatics data only, lacking of independent external
validation with experimental and clinical data. Although the
accuracy of this bioinformatics method has been proven, a realistic
assessment of the performance of the panel will be warranted in the
future.
Despite these limitations, the present study
identified 39 hub miRNAs associated with pathological T stage via
WGCNA. A four-miRNA signature associated with pathological T stage
was identified and validated for association with the prognosis of
PDAC, which may serve as a novel biomarker for prognosis prediction
and to improve clinical outcome in patients with PDAC. The
prognostic signature consisted of four hub miRNAs (miR-1197,
miR-218-2, miR-889 and miR-487a) associated with pathological T
stage, and expression of the signature miRs was significantly
associated with survival. These findings suggest that the increased
expression of the signature in early-stage PDAC patients who
underwent radical resection surgery may be an indicator of improved
overall survival.
Acknowledgements
Not applicable.
Funding
The present study was supported by People's
Liberation Army General Hospital Medical Big Data R&D Project
(grant no. 2017MBD-013).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LY and JW performed data analyses and wrote the
manuscript. FZ, JZ, HT and XZ contributed significantly in data
analyses and manuscript revision. YH conceived and designed the
study. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kamposioras K and Papadopoulos V:
Pancreatic adenocarcinoma treatment. Anticipating better results.
European Society for Medical Oncology, Lugano. 2012, https://www.esmo.org/Career-Development/Young-Oncologists-Corner/Journal-Club/Pancreatic-Adenocarcinoma-Treatment.-Anticipating-Better-ResultsNovember
27–2012
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bakkevold KE, Arnesjø B, Dahl O and
Kambestad B: Adjuvant combination chemotherapy (AMF) following
radical resection of carcinoma of the pancreas and papilla of
Vater-results of a controlled, prospective, randomised multicentre
study. Eur J Cancer. 29:698–703. 1993. View Article : Google Scholar
|
|
6
|
Kuhlmann KF, de Castro SM, Wesseling JG,
ten Kate FJ, Offerhaus GJ, Busch OR, van Gulik TM, Obertop H and
Gouma DJ: Surgical treatment of pancreatic adenocarcinoma: Actual
survival and prognostic factors in 343 patients. Eur J Cancer.
40:549–558. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsao JI, Rossi RL and Lowell JA:
Pylorus-preserving pancreatoduodenectomy. Is it an adequate cancer
operation. Arch Surg. 129:405. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yeo CJ, Cameron JL, Lillemoe KD, Sitzmann
JV, Hruban RH, Goodman SN, Dooley WC, Coleman J and Pitt HA:
Pancreaticoduodenectomy for cancer of the head of the pancreas. 201
patients. Ann Surg. 221:721–731. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yeo CJ, Cameron JL, Sohn TA, Lillemoe KD,
Pitt HA, Talamini MA, Hruban RH, Ord SE, Sauter PK, Coleman J, et
al: Six hundred fifty consecutive pancreaticoduodenectomies in the
1990s: Pathology, complications, and outcomes. Ann Surg.
226:248–257. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Millikan KW, Deziel DJ, Silverstein JC,
Kanjo TM, Christein JD, Doolas A and Prinz RA: Prognostic factors
associated with resectable adenocarcinoma of the head of the
pancreas. Am Surg. 65:618–623. 1999.PubMed/NCBI
|
|
11
|
Benassai G, Mastrorilli M, Quarto G,
Cappiello A, Giani U and Mosella G: Survival after
pancreaticoduodenectomy for ductal adenocarcinoma of the head of
the pancreas. Chir Ital. 52:263–270. 2000.PubMed/NCBI
|
|
12
|
Dalton RR, Sarr MG, van Heerden JA and
Colby TV: Carcinoma of the body and tail of the pancreas: Is
curative resection justified. Surgery. 111:489–494. 1992.PubMed/NCBI
|
|
13
|
Johnson CD, Schwall G, Flechtenmacher J
and Trede M: Resection for adenocarcinoma of the body and tail of
the pancreas. Br J Surg. 80:1177–1179. 2010. View Article : Google Scholar
|
|
14
|
Khorana AA, Mangu PB and Katz MHG:
Potentially curable pancreatic cancer: American society of clinical
oncology clinical practice guideline. J Oncol Prac. 13:388–391.
2017. View Article : Google Scholar
|
|
15
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suárez Y and Sessa WC: microRNAs as novel
regulators of angiogenesis. Circ Res. 104:442–454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu P, Guo M and Hay BA: MicroRNAs and the
regulation of cell death. Trends Genet. 20:617–624. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bartel DP and Chen CZ: Micromanagers of
gene expression: The potentially widespread influence of metazoan
microRNAs. Nature Rev Genet. 5:396–400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mcguire A, Brown JAL and Kerin MJ:
Metastatic breast cancer: The potential of miRNA for diagnosis and
treatment monitoring. Cancer Metastasis Rev. 34:145–155. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maura F, Cutrona G, Mosca L, Matis S,
Lionetti M, Fabris S, Agnelli L, Colombo M, Massucco C, Ferracin M,
et al: Association between gene and miRNA expression profiles and
stereotyped subset #4 B-cell receptor in chronic lymphocytic
leukemia. Leuk Lymphoma. 56:3150–3158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song Q, Xu Y, Yang C, Chen Z, Jia C, Chen
J, Zhang Y, Lai P, Fan X, Zhou X, et al: miR-483-5p promotes
invasion and metastasis of lung adenocarcinoma by targeting RhoGDI1
and ALCAM. Cancer Res. 74:3031–3042. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh S, Chitkara D, Kumar V, Behrman SW
and Mahato RI: miRNA profiling in pancreatic cancer and restoration
of chemosensitivity. Cancer Lett. 334:211–220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wald P, Liu XS, Pettit C, Dillhoff M,
Manilchuk A, Schmidt C, Wuthrick E, Chen W and Williams TM:
Prognostic value of microRNA expression levels in pancreatic
adenocarcinoma: A review of the literature. Oncotarget.
8:73345–73361. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang P, Zhu CF, Ma MZ, Chen G, Song M,
Zeng ZL, Lu WH, Yang J, Wen S, Chiao PJ, et al: Micro-RNA-155 is
induced by K-Ras oncogenic signal and promotes ROS stress in
pancreatic cancer. Oncotarget. 6:21148–21158. 2015.PubMed/NCBI
|
|
25
|
Gironella M, Seux M, Xie MJ, Cano C,
Tomasini R, Gommeaux J, Garcia S, Nowak J, Yeung ML, Jeang KT, et
al: Tumor protein 53-induced nuclear protein 1 expression is
repressed by miR-155, and its restoration inhibits pancreatic tumor
development. Proc Natl Acad Sci USA. 104:16170–16175. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang XJ, Ye H, Zeng CW, He B, Zhang H and
Chen YQ: Dysregulation of miR-15a and miR-214 in human pancreatic
cancer. J Hematol Oncol. 3:462010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Giulietti M, Occhipinti G, Principato G
and Piva F: Identification of candidate miRNA biomarkers for
pancreatic ductal adenocarcinoma by weighted gene co-expression
network analysis. Cell Oncol (Dordr). 40:1–12. 2017.PubMed/NCBI
|
|
28
|
Dhayat SA, Abdeen B, Köhler G, Senninger
N, Haier J and Mardin WA: MicroRNA-100 and microRNA-21 as markers
of survival and chemotherapy response in pancreatic ductal
adenocarcinoma UICC stage II. Clin Epigenetics. 7:1322015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hwang JH, Voortman J, Giovannetti E,
Steinberg SM, Leon LG, Kim YT, Funel N, Park JK, Kim MA, Kang GH,
et al: Identification of MicroRNA-21 as a biomarker for
chemoresistance and clinical outcome following adjuvant therapy in
resectable pancreatic cancer. PLoS One. 5:e106302010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang C, Yu XJ, Guo XZ, Sun MH, Wang Z,
Song Y, Ni QX, Li HY, Mukaida N and Li YY: MicroRNA-33a-mediated
downregulation of Pim-3 kinase expression renders human pancreatic
cancer cells sensitivity to gemcitabine. Oncotarget. 6:14440–14455.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Khan K, Cunningham D, Peckitt C, Barton S,
Tait D, Hawkins M, Watkins D, Starling N, Rao S, Begum R, et al:
miR-21 expression and clinical outcome in locally advanced
pancreatic cancer: Exploratory analysis of the pancreatic cancer
Erbitux, radiotherapy and UFT (PERU) trial. Oncotarget.
7:12672–12681. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li J, Wu H, Li W, Yin L, Guo S, Xu X,
Ouyang Y, Zhao Z, Liu S, Tian Y, et al: Downregulated miR-506
expression facilitates pancreatic cancer progression and
chemoresistance via SPHK1/Akt/NF-κB signaling. Oncogene.
35:5501–5514. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang B and Horvath S: A general framework
for weighted gene co-expression network analysis. Stat Appl Genet
Mol Biol. 4:Article 17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cuccurullo V: AJCC cancer staging
handbook: From the AJCC cancer staging manual (7th edition).
European J Nuclear Med Mol Imag. 38:4082011. View Article : Google Scholar
|
|
35
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Barrett T, Troup DB, Wilhite SE, Ledoux P,
Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M,
Marshall KA, et al: NCBI GEO: Archive for high-throughput
functional genomic data. Nucleic Acids Res. 37:D885–D890. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yip AM and Horvath S: Gene network
interconnectedness and the generalized topological overlap measure.
BMC Bioinformatics. 8:222007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Williams S: Pearson's correlation
coefficient. N Z Med J. 109:381996.PubMed/NCBI
|
|
39
|
Shi K, Bing ZT, Cao GQ, Guo L, Cao YN,
Jiang HO and Zhang MX: Identify the signature genes for diagnose of
uveal melanoma by weight gene co-expression network analysis. Int J
Ophthalmol. 8:269–274. 2015.PubMed/NCBI
|
|
40
|
Jeong H, Mason SP, Barabási AL and Oltvai
ZN: Lethality and centrality in protein networks. Nature.
411:41–42. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liang H and Li WH: Gene essentiality, gene
duplicability and protein connectivity in human and mouse. Trends
Genet. 23:375–378. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk-database: Prediction of possible miRNA binding sites by
‘walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wong N and Wang X: miRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43:D146–D152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
ELife. 4:2015. View Article : Google Scholar
|
|
46
|
Doron B, Manda W, Aaron G, Marks DS and
Chris S: The microRNA.org resource: Targets and expression. Nucleic
Acids Res. 36:149–153. 2008.
|
|
47
|
Chou CH, Shrestha S, Yang CD, Chang NW,
Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, et al: miRTarBase
update 2018: A resource for experimentally validated
microRNA-target interactions. Nucleic Acids Res. 46:D296–D302.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Krek A, Grün D, Poy MN, Wolf R, Rosenberg
L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M
and Rajewsky N: Combinatorial microRNA target predictions. Nat
Genet. 37:495–500. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jiao X, Sherman BT, Huang DW, Stephens R,
Baseler MW, Lane HC and Lempicki RA: DAVID-WS: A stateful web
service to facilitate gene/protein list analysis. Bioinformatics.
28:1805–1806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Freije WA, Castrovargas FE, Fang Z,
Horvath S, Cloughesy T, Liau LM, Mischel PS and Nelson SF: Gene
expression profiling of gliomas strongly predicts survival. Cancer
Res. 64:6503–6510. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cho HJ, Yu A, Kim S, Kang J and Hong S-M:
Robust likelihood-based survival modeling with microarray sata. J
Stat Softw. 29:1–16. 2009. View Article : Google Scholar
|
|
52
|
Kolde R: Pheatmap: Pretty Heatmaps.
Version 1.0.10. https://cran.r-project.org/web/packages/pheatmap/index.htmlMay
19–2018
|
|
53
|
Bukki J: Pancreatic adenocarcinoma. N Engl
J Med. 371:2139–2140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Guo S, Fesler A, Wang H and Ju J: microRNA
based prognostic biomarkers in pancreatic Cancer. Biomark Res.
6:182018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu IS and Cheung WY: A Contemporary review
of the treatment landscape and the role of predictive and
prognostic biomarkers in pancreatic adenocarcinoma. Can J
Gastroenterol Hepatol. 2018:18635352018.PubMed/NCBI
|
|
56
|
Li J, Li YX and Li YY: Differential
rebgulatory analysis based on coexpression network in cancer
research. Biomed Res Int. 2016:42412932016.PubMed/NCBI
|
|
57
|
Chiang KC, Yeh CN, Ueng SH, Hsu JT, Yeh
TS, Jan YY, Hwang TL and Chen MF: Clinicodemographic aspect of
resectable pancreatic cancer and prognostic factors for resectable
cancer. World J Surg Oncol. 10:772012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yamamoto T, Yagi S, Kinoshita H, Sakamoto
Y, Okada K, Uryuhara K, Morimoto T, Kaihara S and Hosotani R:
Long-term survival after resection of pancreatic cancer: A
single-center retrospective analysis. World J Gastroenterol.
21:262–268. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hanahan D and Robert RA: Weinberg:
Hallmarks of cancer: The next generation. Cell. 144:644–674. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ho A and Dowdy SF: Regulation of G(1)
cell-cycle progression by oncogenes and tumor suppressor genes.
Curr Opin Genet Dev. 12:47–52. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: Structure,
function, intracellular signalling and therapeutic inhibition. Cell
Signal. 19:2003–2012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Massagué J, Blain SW and Lo RS: TGFbeta
signaling in growth control, cancer, and heritable disorders. Cell.
103:295–309. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhang H, Liu C, Kong Y, Huang H, Wang C
and Zhang H: TGFβ signaling in pancreatic ductal adenocarcinoma.
Tumor Biol. 36:1613–1618. 2015. View Article : Google Scholar
|
|
64
|
Katz LH, Likhter M, Jogunoori W, Belkin M,
Ohshiro K and Mishra L: TGF-β signaling in liver and
gastrointestinal cancers. Cancer Lett. 379:166–172. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jonckheere N, Vasseur R and Van SI: The
cornerstone K-RAS mutation in pancreatic adenocarcinoma: From cell
signaling network, target genes, biological processes to
therapeutic targeting. Crit Rev Oncol Hematol. 111:7–19. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Mikhail V: Blagosklonny: Cell immortality
and hallmarks of cancer. Cell Cycle. 2:296–299. 2003.PubMed/NCBI
|
|
67
|
Farhan M, Wang H, Gaur U, Little PJ, Xu J
and Zheng W: FOXO signaling pathways as therapeutic targets in
cancer. Int J Biol Sci. 13:815–827. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Moore MJ: From birth to death: The complex
lives of eukaryotic mRNAs. Science. 309:1514–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dan I, Watanabe NM and Kusumi A: The Ste20
group kinases as regulators of MAP kinase cascades. Trends Cell
Biol. 11:220–230. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Dogan F and Biray AC: Correlation between
telomerase and mTOR pathway in cancer stem cells. Gene.
641:235–239. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Frampton AE, Krell J, Jamieson NB, Gall
TMH, Giovannetti E, Funel N, Mato Prado M, Krell D, Habib NA,
Castellano L, et al: microRNAs with prognostic significance in
pancreatic ductal adenocarcinoma: A meta-analysis. Eur J Cancer.
51:1389–1404. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhou X, Wang X, Huang Z, Xu L, Zhu W and
Liu P: An ER-associated miRNA signature predicts prognosis in
ER-positive breast cancer. J Exp Clin Cancer Res. 33:942014.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Li X, Zhang Y, Zhang Y, Ding J, Wu K and
Fan D: Survival prediction of gastric cancer by a seven-microRNA
signature. Gut. 59:579–585. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhou X, Huang Z, Xu L, Zhu M, Zhang L,
Zhang H, Wang X, Li H, Zhu W, Shu Y and Liu P: A panel of 13-miRNA
signature as a potential biomarker for predicting survival in
pancreatic cancer. Oncotarget. 7:69616–69624. 2016.PubMed/NCBI
|
|
75
|
Greither T, Grochola LF, Udelnow A,
Lautenschläger C, Würl P and Taubert H: Elevated expression of
microRNAs 155: 203, 210 and 222 in pancreatic tumors is associated
with poorer survival. Int J Cancer. 126:73–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Guan B, Wu K, Zeng J, Xu S, Mu L, Yang G,
Wang K, Ma Z, Tian J, Shi Q, et al: Tumor-suppressive microRNA-218
inhibits tumor angiogenesis via targeting the mTOR component RICTOR
in prostate cancer. Oncotarget. 8:8162–8172. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhu K, Ding H, Wang W, Liao Z, Fu Z, Hong
Y, Zhou Y, Zhang CY and Chen X: Tumor-suppressive miR-218-5p
inhibits cancer cell proliferation and migration via EGFR in
non-small cell lung cancer. Oncotarget. 7:28075–28085.
2016.PubMed/NCBI
|
|
78
|
Feng Z, Zhang L, Zhou J, Zhou S, Li L, Guo
X, Feng G, Ma Z, Huang W and Huang F: mir-218-2 promotes
glioblastomas growth, invasion and drug resistance by targeting
CDC27. Oncotarget. 8:6304–6318. 2017.PubMed/NCBI
|
|
79
|
Guan H, Wei G, Wu J, Fang D, Liao Z, Xiao
H, Li M and Li Y: Down-regulation of miR-218-2 and its host gene
SLIT3 cooperate to promote invasion and progression of thyroid
cancer. J Clin Endocrinol Metab. 98:E1334–E1344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Xie H, Zhang Q, Zhou H, Zhou J, Zhang J,
Jiang Y, Wang J, Meng X, Zeng L and Jiang X: microRNA-889 is
downregulated by histone deacetylase inhibitors and confers
resistance to natural killer cytotoxicity in hepatocellular
carcinoma cells. Cytotechnology. 70:513–521. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Xu Y, He J, Wang Y, Zhu X, Pan Q, Xie Q
and Sun F: miR-889 promotes proliferation of esophageal squamous
cell carcinomas through DAB2IP. FEBS Lett. 589:1127–1135. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Molina-Pinelo S, Carnero A, Rivera F,
Estevez-Garcia P, Bozada JM, Limon ML, Benavent M, Gomez J, Pastor
MD, Chaves M, et al: MiR-107 and miR-99a-3p predict chemotherapy
response in patients with advanced colorectal cancer. BMC Cancer.
14:6562014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chang RM, Xiao S, Lei X, Yang H, Fang F
and Yang LY: miRNA-487a promotes proliferation and metastasis in
hepatocellular carcinoma. Clin Cancer Res. 23:2593–2604. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gonzalez-Vallinas M, Rodriguez-Paredes M,
Albrecht M, Sticht C, Stichel D, Gutekunst J, Pitea A, Sass S,
Sánchez-Rivera FJ, Bermejo JL, et al: Epigenetically regulated
chromosome 14q32 miRNA cluster induces metastasis and predicts poor
prognosis in lung adenocarcinoma patients. Mol Cancer Res.
16:390–402. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ma M, He M, Jiang Q, Yan Y, Guan S, Zhang
J, Yu Z, Chen Q, Sun M, Yao W, et al: MiR-487a promotes
TGF-β1-induced EMT, the migration and invasion of breast cancer
cells by directly targeting MAGI2. Int J Biol Sci. 12:397–408.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Stylli SS, Adamides AA, Koldej RM, Luwor
RB, Ritchie DS, Ziogas J and Kaye AH: miRNA expression profiling of
cerebrospinal fluid in patients with aneurysmal subarachnoid
hemorrhage. J Neurosurg. 126:1131–1139. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fisher AJ, Cipolla E, Varre A, Gu H,
Mickler EA and Vittal R: Potential mechanisms underlying
TGF-β-mediated complement activation in lung fibrosis. Cell Mol Med
Open Access. 3:142017. View Article : Google Scholar : PubMed/NCBI
|