The Hayflick limit is a response to cellular
lesions, which are triggered by multiple mechanisms, including
replicative senescence, oncogene activation, telomerase dysfunction
and DNA lesions (1–3). Senescent cells arrested at the
G1 phase demonstrated more properties associated with
dysfunctional cells compared with normal cells (4). Although senescence is an undesirable
stress for normal cells, it is beneficial for the body as it
restrains excessive proliferation of tumor cells. Therefore,
senescence is used as a means of suppressing cancer and is an
important cancer treatment method (5–8).
Extracellular signal-regulated protein kinase
(ERK)1/2 is a mitogen-activated protein kinase (MAPK) family
protein with typical cascade signaling characteristics and serves
an important role in signal transduction pathways and the function
of transcription factors, including activator protein-1,
proto-oncogene c-Fos (c-Fos) and ETS domain-containing protein
Elk-1 (Elk1) (9). The majority of
research has focused on its regulatory effect on cell growth and
differentiation (10–14); however, a number of previous
studies demonstrated that ERK1/2 promotes cell senescence (15–17).
Based on these characteristics, numerous small molecule MAPK/ERK
kinase (MEK) inhibitors were examined in early-phase clinical
trials, including PD098059, U0126, CI-1040, PD0325901 and AZD6244
(18); however, none of them were
approved by The Food and Drug Administration due to adverse side
effects or other toxic reactions (18). Many of these inhibitors negatively
affected normal and abnormal cells. Notably, these effects may have
been the result of the dual roles of ERK1/2 in senescence, as
demonstrated by other previous studies (19,20).
The present review examines the mechanisms regulating the role of
ERK1/2 in cell senescence and suggests that ERK1/2 is a potentially
useful target in treating cancer.
The mammalian MAPKs consist of cytoplasmic
serine/threonine kinases that are involved in the transduction of
signals from the surface to the interior of the cell. This family
includes the ERK family (ERK1-8), the p38 kinase family (p38
α/β/γ/δ) and the c-Jun N-terminal kinase family (JNK1-3,
additionally termed stress-activated protein kinase) (9). With a number of substrate docking and
enzyme recruitment sites (21),
ERK1/2 (MAPK1/3) is a multifunctional serine/threonine kinase that
is able to phosphorylate numerous substrates, including protein
kinases, signal effectors, receptors, cellular scaffold proteins
and nuclear transcriptional regulators (21). At present, five types of ERK
isoforms are known. ERK1 and ERK2 are thought to be the most
important isoforms with 84% homology for the primary sequences and
similar functions (21).
The Ras/Raf/MEK/ERK1/2 signaling pathway is a small
GTPase ligation of activated tyrosine receptors and cytoplasmic
kinase signal transduction cascades. The key point of activation is
to transmit a signal from tyrosine receptors, including epidermal
growth factor receptor (EGFR), which subsequently recruit Son of
sevenless (SOS) through intracellular Shc and Grb2 domains,
ultimately catalyzing the conversion of inactive Ras/guanosine
diphosphate to the active Ras/guanosine triphosphate complex
(22). As an activator of ERK1/2,
MEK1/2 catalyzes the phosphorylation of ERK1/2 at Tyr204/187 and
Thr202/185 by casein kinase 2 (CK2) (23). This enzyme subsequently binds to
importin7 and translocates ERK1/2 from the cytoplasm to the nucleus
(24), where it functions as an
upstream regulator of substrate genes that encode for transcription
factors, including Elk1, c-Myc, signal transducers and activators
of transcription (STATs), c-Jun and c-Fos (21). These transcription factors regulate
their counterpart target genes to alter the expression or activity
of various proteins and are involved in the regulation of a large
variety of processes, including adhesion, cell cycle progression,
migration, survival, differentiation, metabolism, proliferation and
transcription (21). The
Ras/Raf/MEK/ERK cascade is a highly efficient signaling pathway,
aided by scaffold proteins, including kinase suppressor of Ras, MEK
partner 1/p14 complex, β-arrestins, fibroblast growth factor
receptor substrate 2, MAPK organizer 1 and flotillin-1 (25). The function of scaffold proteins is
characterized by combinatorial inhibition, which is the
stoichiometry of a scaffold and its signaling partners; the
expression levels of scaffold proteins should not be too high (the
kinase and its substrate may each bind to an individual scaffold
protein) or too low (the phosphorylation of the cascade is
sub-optimal) (26). With these
different scaffold proteins, the phosphorylation of different
isoforms is accurately regulated; the scaffold protein MEK protein
1 specifically binds ERK1, not ERK2 (27).
Cellular senescence was first observed in cultured
fibrocytes, when the Hayflick limit demonstrated that as the cells
divided, their cell cycle became arrested (replicative senescence)
(28). Senescent cells have
abnormal metabolic activity (3),
accompanied by morphological, biological and genetic alterations.
When β-galactosidase expression (an important senescence marker)
increases (29), the cell cycle is
arrested at the G1/S phase (30). Cell cycle dependent kinase (CDK)
and cyclin A activity additionally decrease with increased activity
of cyclin-dependent kinase inhibitors (p16INK4a and p21) (31). Without the protection of histones,
mitochondrial reactive oxygen species (ROS) may damage
mitochondrial DNA, which induces a series of oxidative stress
reactions (32). Telomere
shortening, which causes DNA to lose protection from the telomeres,
is another feature of senescence and leads to DNA integration and
degradation. Oxidative stress reactions and mitochondrial
dysfunction (33) accelerate the
shortening of telomeres. Cellular senescence is triggered by a
number of signaling pathways and mechanisms, including DNA injury,
telomerase dysfunction, oncogenes, oxidative stress reactions and
mitochondrial dysfunction (34).
ERK1/2 is an important messenger for extracellular
and intracellular signals, which serve a vital role in processes,
including proliferation, differentiation, cytoskeleton construction
and cellular senescence (35). In
the majority of cases, ERK1/2 is a regulator of cellular
proliferation; however, it has been identified that ERK1/2 may
additionally promote senescence. Strong, constitutively active
expression of MEK1 (an upstream activator of ERK1/2) in
non-immortalized intestinal epithelial cells (HIEC cells) promotes
cellular senescence, whereas, in immortalized intestinal cells
(IEC-6 cells), it does not, suggesting that cell type may serve a
role (36). This phenomenon
requires further investigation to improve the clinical use of
ERK1/2-associated reagents, including ERK1/2 inhibitors.
ERK1/2 is associated with cell survival,
proliferation and development. To investigate the role of ERK1/2 in
different cell types and animal models, a number of previous
studies investigating this were reviewed. ERK1/2 functions more
frequently as a cellular proliferation marker than as a dual role
kinase (Table I) (37–54).
Based on these previous studies, cellular proliferation is
primarily regulated by the effects of ERK1/2 on cell cycle entry
and protein synthesis.
As a transcription factor regulator, ERK1/2
transduces signals from the cell membrane to the nucleus. ERK1/2
may additionally regulate carbamoyl phosphate synthetase II
(55), which catalyzes the initial
rate-limiting step in the de novo synthesis of pyrimidine
nucleotides. Furthermore, it was identified that ERK1/2
phosphorylates high motility group boxes of nucleolar transcription
factor 1, an RNA polymerase I factor transcriptional enhancer that
enhances ribosomal RNA genes (56).
In addition to the substrates involved in cell
proliferation, ERK1/2 additionally regulates cellular tumor antigen
p53 (p53) phosphorylation. p53 is a tumor suppressor protein and
functions as a transcription factor by binding to a number of
genes, including cyclin-dependent kinase inhibitor 1A, which
encodes p21. p21 binds and inactivates CDKs, which are crucial for
cell entry into the G1/S phase (67,68).
The association between ERK1/2 and p53 remains unclear. A previous
study suggested that p53 functions upstream of ERK1/2 (69); however, the most widely accepted
hypothesis is that ERK1/2 regulates p53 by activating STAT3
(70) and other transcription
factors. ERK1/2 and p53 have hundreds of substrates, thus, it is
easy for them to engage in crosstalk, as is the case with
dual-specificity phosphatases (DUSPs) (71). The effect of ERK1/2 on its
downstream substrates may accelerate the degradation of p53
(72). ERK1/2 regulates p53
phosphorylation [a form that protects p53 from E3 ubiquitin-protein
ligase Mdm2 (73)] through the
forkhead box M1/c-myc/polycomb complex protein BMI-1 pathway, which
inhibits p19 phosphorylation, attenuating cellular senescence
(74).
Mitochondria not only provide energy to cells;
however, additionally serve a decisive role in cell fate.
Mitochondria within the respiratory chain are responsible for
maintaining the proton gradient and providing various respiratory
enzymes; it was demonstrated that the proton gradient is not just
associated with adenosine triphosphate synthesis. Rasola et
al (75) identified that
ERK1/2 phosphorylates glycogen synthase kinase-3β, inhibiting
permeability transition pore opening by regulating cyclophilin D
and preventing the release of apoptotic substances, including
mitochondrial cytochrome C, ROS, Ca2+ and free radicals.
The number of mitochondria is additionally an important hallmark of
cellular proliferation. A previous study demonstrated that ERK2 may
phosphorylate dynamin-1-like protein (an important regulator of
mitochondrial fission) on serine 616 in several tumor models
(76), resulting in tumor
growth.
Based on extensive investigations in a variety of
cell types, previous studies identified that ERK1/2 may
additionally facilitate cellular senescence under certain
circumstances. In the present review, a number of previous studies
are discussed to gain a better understanding of the role of ERK1/2
during cellular senescence and the underlying mechanisms behind its
control. In contrast, the role of ERK1/2 in cellular proliferation
was only studied in numerous cell types, primarily fibroblasts,
providing limited information. The previous studies investigating
ERK1/2 involvement in cellular senescence (Table II) (77–90)
identified a number of possible mechanisms for the role of ERK1/2
that are associated with abnormal signaling of negative feedback
loops, caused by constitutive and overexpressed ERK1/2 (20,77,78),
and ERK1/2 cellular localization (79).
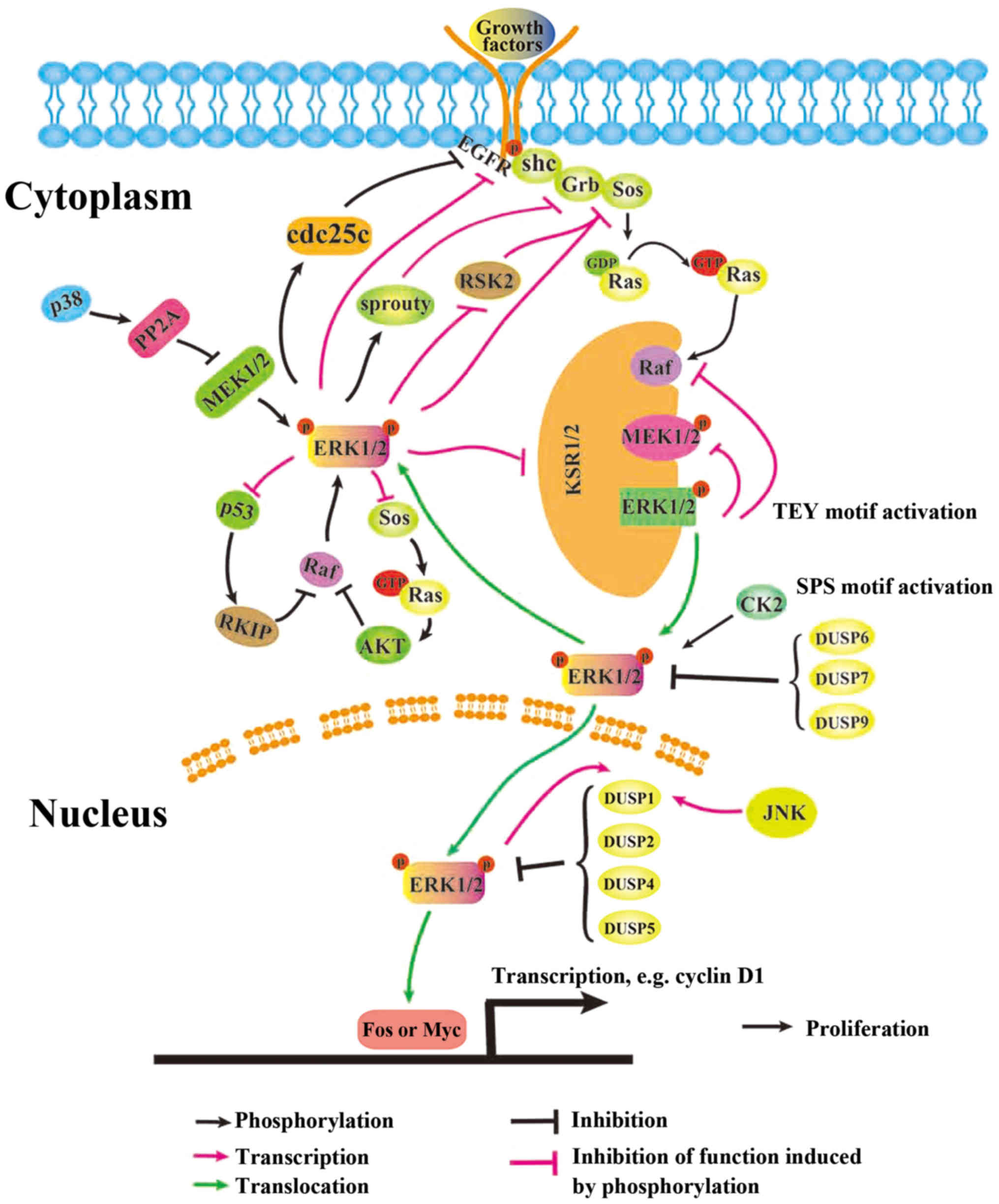
A number of previous studies demonstrated that
negative regulation of ERK1/2 within MAPK signaling cascades
regulate ERK1/2 signaling. ERK1/2 phosphorylates proteins within
this cascade at alternate sites, which interrupts the normal
binding behavior of their respective downstream substrates
(91). ERK1/2 phosphorylates EGFR
at T669 (92) and decreases
constitutive tyrosine phosphorylation activity, decreasing the
ability of the phosphorylated loop to cross-activate other adaptors
(93). ERK1/2 may additionally
phosphorylate dual specificity Cdc25C at T48, which
dephosphorylates EGFR at Y1068 (94). Furthermore, ERK1/2 was identified
to phosphorylate MAPK signaling components, including fibroblast
growth factor receptor (FGFR) at S777 (95); SOS1 at S1132, S1167, S1178 and
S1193 (96); fibroblast growth
factor receptor substrate 2 (FRS2)α at T132, T135, T138, T376,
T452, T455, T458 and T463 (97,98);
RAF proto-oncogene serine/threonine-protein kinase (Raf-1) at S29,
S289, S296, S301 and S642 (99–103); serine/threonine-protein kinase
B-raf (B-Raf) at S151, T401, S750, T753 and S642 (104,105); MEK1 at T292 and T386 (106,107); and kinesin suppressor or Ras 1 at
T260, T274, S320, S443 and S463 (108,109) (Fig.
1). The phosphorylation of all these components results in a
disruption of binding to downstream substrates.
ERK1/2 requires dual phosphorylation of threonine
and tyrosine residues to acquire its biological kinase function.
Dual-specificity Thr/Tyr phosphatases [DUSPs; additionally termed
MAPK phosphatases (MKP)] represent a large family that regulates
the activity of MAPKs by dephosphorylating threonine and tyrosine
residues within the activation loop of MAPKs, which in turn
regulates the biologically active form of ERK1/2 in the cytoplasm
and the nucleus (111). DUSP1,
DUSP2, DUSP4 and DUSP5 are located in the nucleus, whereas DUSP6,
DUSP7 and DUSP9 are located in the cytoplasm (112). Their binding to ERK1/2 is
regulated by a conserved motif within the amino-terminal
non-catalytic domain (kinase-interacting motif) of the protein
(113,114) and results in a significant
increase in catalytic activity, which deprives ERK1/2 of the
phosphate group. The interaction between MAPKs and DUSPs is a
two-way regulation; MAPKs are able to upregulate transcription of
DUSPs (113), primarily those in
the cytoplasm (115), in a
delayed manner following MAPK activation, whereas, DUSPs strictly
regulate MAPK signaling.
Sprouty [protein sprout homolog (Spry) 1–4] is
another ERK1/2 regulator family that is not well-studied. A
previous Japanese study demonstrated that Spry1 and Spry2 are
phosphorylated at Y53 and Y55, which creates a docking site for
growth factor receptor-bound protein 2 at the Src homology 2 domain
and consequently disrupts association with the FGFR adaptor FRS2 in
C2C12 cells (116). Other
previous studies suggested that the spry protein inhibits receptor
tyrosine kinase signaling, which suppresses the activation of Ras
in BRAFV600E melanomas (117,118). Another previous study
demonstrated that in 293T cells, Spry1 and Spry2 regulate Raf-1 by
directly binding to it (119).
Lake et al (91) suggested
that Sprouty serves its functions at multiple nodes in a
context-specific manner.
In contrast to signaling that regulates cell
proliferation, AKT/mTOR and ERK1/2 engage in crosstalk that
sustains cell proliferation and survival, which in turn helps cells
escape from either PI3K/AKT or ERK1/2 suppression (120). Crystal structure analysis
demonstrated that astrocytic phosphoprotein PEA-15 (PEA-15) may
efficiently bind to the ERK2 activation loop at the Thr-X-Tyr
region (121), activating
transportation of ERK1/2 from the nucleus. Sinha et al
(122) observed that ERK1/2
decreases phosphorylated (p)-AKT expression levels in mouse renal
proximal tubular cells via Ras/PI3K through a negative feedback
pathway, whereas AKT phosphorylates and stabilizes PEA-15, which
subsequently decreases the nuclear localization of ERK1/2 (123) and induces cellular senescence.
Although the regulatory nodes shared by Ras/MAPK and PI3K/AKT
signaling are complicated, the two signaling pathways are
influenced by co-effectors, including TSC1/2, mTOR, ER and S6, and
regulate each other in concentration- and context-dependent manners
(124).
As a messenger of extracellular and intracellular
proteins, ERK1/2 has many substrates. MEK1/2 functions upstream of
ERK1/2 and serves a vital regulatory role in ERK1/2 activation.
MEK1/ERK signaling promotes cell proliferation, whereas MEK2/ERK
signaling promotes G1/S cell cycle arrest (125). In different tissues and cell
types, including mouse embryos and fibroblasts, cellular senescence
induced by the Ras/Raf/MEK/ERK signaling pathway is dependent on
the integrity of p16/INK4A, p21 and p53; in human primary
fibroblasts, inhibition of either p16 or p53 is not able to reverse
ERK1/2-induced senescence (17,20,78).
However, a previous study demonstrated that the matrix cell protein
G1/S-specific cyclin CCN1 induces senescence through the p53/p21
pathway and inhibits lung cancer growth (36). p53 and ERK1/2 have two-way
regulation, which means there is a negative feedback loop between
ERK1/2 and p53. Lee et al (126) suggested that a novel p53 target
protein, Raf kinase inhibitor protein, inhibits ERK1/2 by affecting
Raf proteins and promoting senescence. Notably, p53 may regulate
the transcription of all nuclear DUSPs (DUSP1/2/4/5) (127–129).
Exposure of cells to certain bioactive chemicals,
including the natural ethanolic Rhus coriaria extract may
lead to activation of ERK1/2 and p21 upregulation (130). The microtubule stabilizing agent
discodermolide was identified to induce cellular senescence due to
overexpressed ERK1/2 in A549 cells (66). Administration of
epigallocatechin-3-gallate leads to cellular senescence during PC-3
prostate cancer cell proliferation via MEK-independent ERK1/2
activation (85). In addition, a
Ras or B-Raf mutation is common in tumors, particularly in
malignant tumors, which may lead to sustained activation of ERK1/2
signaling. In specific cases, downstream proteins are activated by
ERK1/2 aberrantly, resulting in different physiological effects
(103). Expression of proteins
downstream of ERK1/2, including BRAF-induced insulin-like growth
factor-binding protein 7 (IGFBP7) in normal melanoma cells, is low
and primarily controlled by autocrine or paracrine functions that
influence cell proliferation (103). Examples of such phenomenon
include BRAFV600E-positive nevi, which contain high BRAF
expression; the continuously activated RAF/MEK/ERK pathway, which
increases IGFBP7 expression; and high expression levels of IGFBP7,
which inhibit the RAF/MEK/ERK pathway within cells. However, in
melanoma cells, IGFBP7 expression is lost, which results in
uncontrolled proliferation (131).
miRNAs are important for regulating cell biology.
Previous studies demonstrated that miRNA-34a induces persistent
activation of ERK1/2, leading to cellular senescence via inhibition
of p53 signaling (89) and MEK1/2
(132). miR-21 increases the
expression level of p-ERK1/2 by inhibiting Spry1 (116) and Spry2 (133).
A number of previous studies suggest that
ERK1/2-induced cellular senescence may be associated with the
strength of ERK1/2 signals and the duration of its activation
(20,77,78,86,90,134). As ERK1/2 has many substrates,
different biological effects may occur when the signal proteins
compete for the same target protein. As discussed above, many
negative feedback loops and regulatory nodes shape ERK1/2
signaling, and when ERK1/2 is overexpressed, it may activate those
negative loops. However, this regulatory system is dependent on the
cellular context.
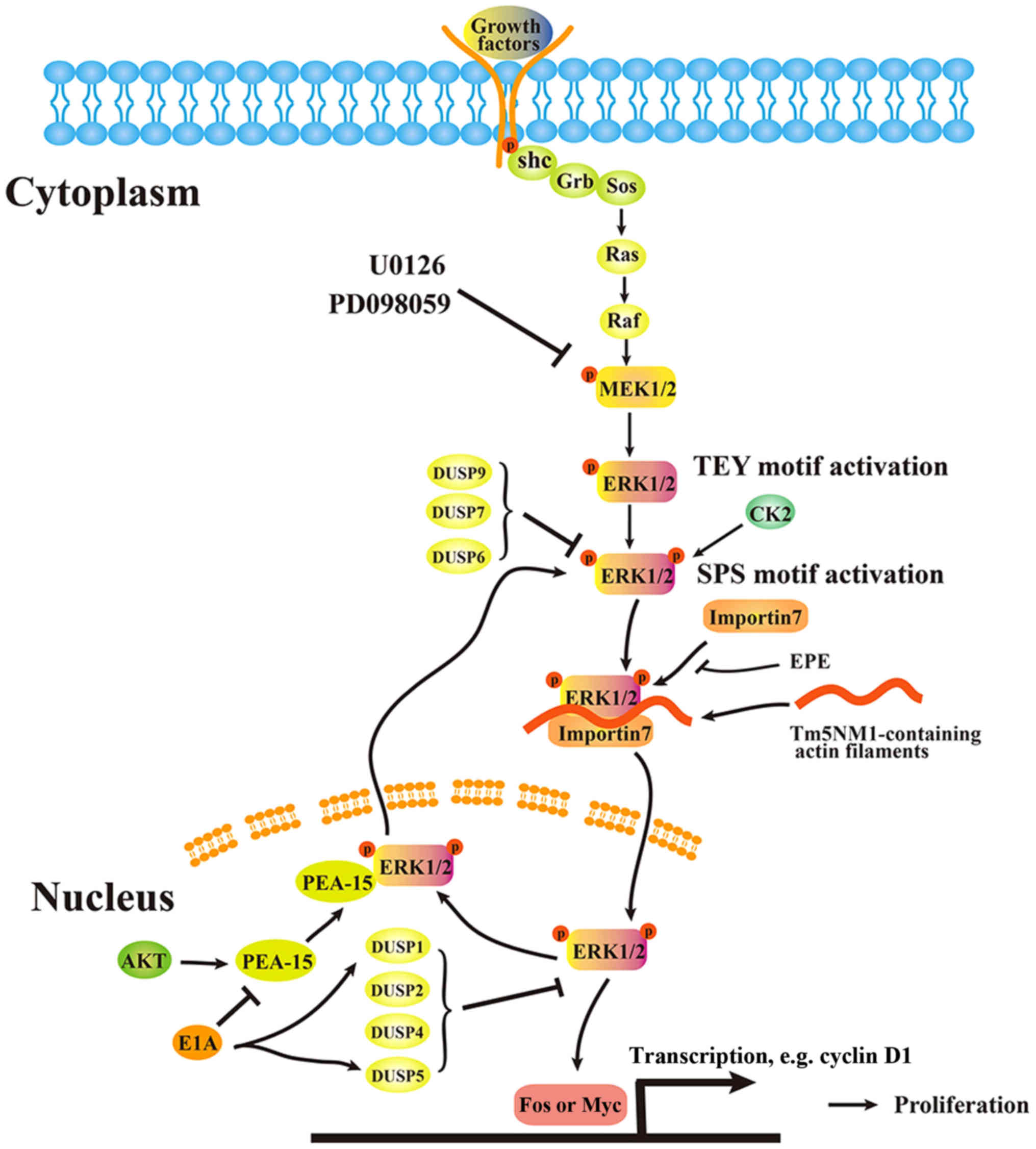
Scaffold proteins facilitate protein translocation,
and translocation of ERK1/2 from the cytoplasm to the nucleus is
essential for regulation of the cell cycle and cellular
proliferation (135). This
process requires dual-phosphorylation of specific residues within
the activation loop. ERK is initially phosphorylated by MAPK/ERK
kinase (MEK) on the Thr-Glu-Tyr motif, with subsequent
phosphorylation on the Ser-244/Pro-245/Ser-246 (SPS) nuclear
translocation sequence (NTS) (108,109). This is achieved primarily by CK2
to generate pSPS-pERK, which binds to the shuttling protein
importin7 (Imp7) (136,137). A previous study investigating
mouse embryo fibroblasts demonstrated that Tm5NM1-containing actin
filaments facilitate the binding of pSPS-pERK
(Ser-244/Pro-245/Ser-246) and Imp7, possibly by functioning as a
scaffold and/or recruiting myosin motors to assist in the physical
transportation of pSPS-pERK from the cytoplasm to the nucleus
(136). Inhibiting the binding of
pSPS-pERK and Imp7 appears to be an effective way of blocking the
translocation of dual-phosphorylated ERK. Plotnikov et al
(135) developed a myristoylated,
NTS-derived phosphomimetic peptide (EPE peptide) that competes with
the binding of Imp7 and blocks this process in a number of cell
lines.
ETS translocation variant 4 (E1A) is additionally a
negative regulator of activated ERK1/2 translocation from the
cytoplasm to the nucleus (79,138). A previous study examining normal
human diploid fibroblast IMR90 cells demonstrated that E1A
decreases expression levels of PEA-15 (139), an ERK1/2 nuclear export
factor, and increases expression levels of MKP1/DUSP1 and
DUSP5 (138). The translocation
regulation model is presented in Fig.
2.
ERK1/2 serves a vital role in cellular outcomes,
which involve numerous substrates, regulators and scaffolding
proteins. If a small dose of a MEK1/2 inhibitor, including U0126 or
PD098059, is applied to a cell, the cell will recover by utilizing
a compensatory pathway to regain its proliferation capacity. Even
in the same type of cell or animal model, ERK1/2 serves a dual role
in cellular senescence under different circumstances, which are
dose- and duration-dependent. Cells subjected to drugs against
ERK1/2 may additionally contain mutations in upstream (AKT, MEK and
Ras/Raf) or downstream (p21 and p53) proteins. Analyzing cells for
Ras/Raf/MAPK mutations and testing cell sensitivity may help to
determine the proper dosage and duration of drug administration. In
terms of drug development, altering the translocation of ERK1/2
from the cytoplasm to the nucleus (135), which is primarily required for
induction of cell proliferation, may help to decrease the
proliferation of cancer cells. However, as cells may shift their
proliferation signals between a number of different proteins, a
combination of numerous anti-proliferation tactics require
consideration.
Not applicable.
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81460374, 31460304
and 81572753), the Natural Science Foundation of Jiangxi Province
of China (grant nos. 20171BAB205062 and 20171BCB23086) and the
Education Department of Jiangxi Province of China (grant no.
160032).
Not applicable.
JZ and TL contributed equally in reviewing the
publications and writing this manuscript. PG and DH drafted the
manuscript. JY, QX, YL and RK designed the schema of ERK1/2
signaling. All authors read and approved the final version of the
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Cristofalo VJ, Lorenzini A, Allen RG,
Torres C and Tresini M: Replicative senescence: A critical review.
Mech Ageing Dev. 125:827–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zeiser R: Trametinib. Recent Results
Cancer Res. 201:241–248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Salama R, Sadaie M, Hoare M and Narita M:
Cellular senescence and its effector programs. Genes Dev.
28:99–114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tominaga K: The emerging role of senescent
cells in tissue homeostasis and pathophysiology. Pathobiol Aging
Age Relat Dis. 5:277432015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gewirtz DA: Autophagy and senescence in
cancer therapy. J Cell Physiol. 229:6–9. 2014.PubMed/NCBI
|
|
6
|
Ohtani N, Mann DJ and Hara E: Cellular
senescence: Its role in tumor suppression and aging. Cancer Sci.
100:792–797. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu CH, van Riggelen J, Yetil A, Fan AC,
Bachireddy P and Felsher DW: Cellular senescence is an important
mechanism of tumor regression upon c-Myc inactivation. Proc Natl
Acad Sci USA. 104:13028–13033. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Plotnikov A, Zehorai E, Procaccia S and
Seger R: The MAPK cascades: Signaling components, nuclear roles and
mechanisms of nuclear translocation. Biochim Biophys Acta.
1813:1619–1633. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tan X, Wang YL, Yang XL and Zhang DD:
Ethyl acetate extract of Artemisia anomala S. Moore displays
potent anti-inflammatory effect. Evid Based Complement Alternat
Med. 2014:6813522014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Montero-Conde C, Ruiz-Llorente S,
Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA,
Rosen N and Fagin JA: Relief of feedback inhibition of HER3
transcription by RAF and MEK inhibitors attenuates their antitumor
effects in BRAF-mutant thyroid carcinomas. Cancer Discov.
3:520–533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bell RM, Kunuthur SP, Hendry C,
Bruce-Hickman D, Davidson S and Yellon DM: Matrix metalloproteinase
inhibition protects CyPD knockout mice independently of RISK/mPTP
signalling: A parallel pathway to protection. Basic Res Cardiol.
108:3312013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu J, Xu J, Eksioglu EA, Chen X, Zhou J,
Fortenbery N, Wei S and Dong J: Icariside II induces apoptosis of
melanoma cells through the downregulation of survival pathways.
Nutr Cancer. 65:110–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Desar IM, Gilles R, van Herpen CM,
Timmer-Bonte AJ, Cantarini MV, van der Graaf WT and Oyen WJ:
(18)F-FLT-PET for response evaluation of MEK inhibitor selumetinib
(AZD6244, ARRY-142886) in patients with solid tumors. World J Nucl
Med. 11:65–69. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barlin JN, Jelinic P, Olvera N, Bogomolniy
F, Bisogna M, Dao F, Barakat RR, Chi DS and Levine DA: Validated
gene targets associated with curatively treated advanced serous
ovarian carcinoma. Gynecol Oncol. 128:512–517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rybakova Y, Akkuratov E, Kulebyakin K,
Brodskaya O, Dizhevskaya A and Boldyrev A: Receptor-mediated
oxidative stress in murine cerebellar neurons is accompanied by
phosphorylation of MAP (ERK 1/2) kinase. Curr Aging Sci. 5:225–230.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhuang D, Mannava S, Grachtchouk V, Tang
WH, Patil S, Wawrzyniak JA, Berman AE, Giordano TJ, Prochownik EV,
Soengas MS and Nikiforov MA: C-MYC overexpression is required for
continuous suppression of oncogene-induced senescence in melanoma
cells. Oncogene. 27:6623–6634. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang D, Boerner SA, Winkler JD and LoRusso
PM: Clinical experience of MEK inhibitors in cancer therapy.
Biochim Biophys Acta. 1773:1248–1255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang W, Chen JX, Liao R, Deng Q, Zhou JJ,
Huang S and Sun P: Sequential activation of the MEK-extracellular
signal-regulated kinase and MKK3/6-p38 mitogen-activated protein
kinase pathways mediates oncogenic ras-induced premature
senescence. Mol Cell Biol. 22:3389–3403. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin AW, Barradas M, Stone JC, van Aelst L,
Serrano M and Lowe SW: Premature senescence involving p53 and p16
is activated in response to constitutive MEK/MAPK mitogenic
signaling. Genes Dev. 12:3008–3019. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roskoski R Jr: ERK1/2 MAP kinases:
Structure, function, and regulation. Pharmacol Res. 66:105–143.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gureasko J, Galush WJ, Boykevisch S,
Sondermann H, Bar-Sagi D, Groves JT and Kuriyan J:
Membrane-dependent signal integration by the Ras activator Son of
sevenless. Nat Struct Mol Biol. 15:452–461. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chuderland D, Konson A and Seger R:
Identification and characterization of a general nuclear
translocation signal in signaling proteins. Mol Cell. 31:850–861.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Matsubayashi Y, Fukuda M and Nishida E:
Evidence for existence of a nuclear pore complex-mediated,
cytosol-independent pathway of nuclear translocation of ERK MAP
kinase in permeabilized cells. J Biol Chem. 276:41755–41760. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meister M, Tomasovic A, Banning A and
Tikkanen R: Mitogen-activated protein (MAP) kinase scaffolding
proteins: A recount. Int J Mol Sci. 14:4854–4884. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Good MC, Zalatan JG and Lim WA: Scaffold
proteins: Hubs for controlling the flow of cellular information.
Science. 332:680–686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schaeffer HJ, Catling AD, Eblen ST,
Collier LS, Krauss A and Weber MJ: MP1: A MEK binding partner that
enhances enzymatic activation of the MAP kinase cascade. Science.
281:1668–1671. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hayflick L and Moorhead PS: The serial
cultivation of human diploid cell strains. Exp Cell Res.
25:585–621. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang NC and Hu ML: The limitations and
validities of senescence associated-beta-galactosidase activity as
an aging marker for human foreskin fibroblast Hs68 cells. Exp
Gerontol. 40:813–819. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen KY: Transcription factors and the
down-regulation of G1/S boundary genes in human diploid fibroblasts
during senescence. Front Biosci. 2:d417–d426. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hernandez-Segura A, Nehme J and Demaria M:
Hallmarks of cellular senescence. Trends Cell Biol. 28:436–453.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Passos JF and von Zglinicki T:
Mitochondria, telomeres and cell senescence. Exp Gerontol.
40:466–472. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu L, Trimarchi JR, Smith PJ and Keefe
DL: Mitochondrial dysfunction leads to telomere attrition and
genomic instability. Aging cell. 1:40–46. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Campisi J: Cellular senescence as a
tumor-suppressor mechanism. Trends Cell Biol. 11:S27–S31. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun Y, Liu WZ, Liu T, Feng X, Yang N and
Zhou HF: Signaling pathway of MAPK/ERK in cell proliferation,
differentiation, migration, senescence and apoptosis. J Recept
Signal Transduct Res. 35:600–604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Boucher MJ, Jean D, Vézina A and Rivard N:
Dual role of MEK/ERK signaling in senescence and transformation of
intestinal epithelial cells. Am J Physiol Gastrointest Liver
Physiol. 286:G736–G746. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ravasi S, Citro S, Viviani B, Capra V and
Rovati GE: CysLT1 receptor-induced human airway smooth muscle cells
proliferation requires ROS generation, EGF receptor transactivation
and ERK1/2 phosphorylation. Respir Res. 7:422006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gong X, He X, Qi L, Zuo H and Xie Z:
Stromal cell derived factor-1 acutely promotes neural progenitor
cell proliferation in vitro by a mechanism involving the ERK1/2 and
PI-3K signal pathways. Cell Biol Int. 30:466–471. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Iyengar L, Patkunanathan B, Lynch OT,
McAvoy JW, Rasko JE and Lovicu FJ: Aqueous humour- and growth
factor-induced lens cell proliferation is dependent on MAPK/ERK1/2
and Akt/PI3-K signalling. Exp Eye Res. 83:667–678. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang L, Liu T, Nishioka M, Aguirre RL, Win
SS and Okada N: Activation of ERK1/2 and cyclin D1 expression in
oral tongue squamous cell carcinomas: Relationship between
clinicopathological appearances and cell proliferation. Oral Oncol.
42:625–631. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
De Rosa V, Procaccini C, Cali G, Pirozzi
G, Fontana S, Zappacosta S, La Cava A and Matarese G: A key role of
leptin in the control of regulatory T cell proliferation. Immunity.
26:241–255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li M, Feurino LW, Li F, Wang H, Zhai Q,
Fisher WE, Chen C and Yao Q: Thymosinalpha1 stimulates cell
proliferation by activating ERK1/2, JNK, and increasing cytokine
secretion in human pancreatic cancer cells. Cancer Lett. 248:58–67.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li H, Cai X, Fan X, Moquin B, Stoicov C
and Houghton J: Fas Ag-FasL coupling leads to ERK1/2-mediated
proliferation of gastric mucosal cells. Am J Physiol Gastrointest
Liver Physiol. 294:G263–G275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
He Z, Jiang J, Kokkinaki M, Golestaneh N,
Hofmann MC and Dym M: Gdnf upregulates c-Fos transcription via the
Ras/Erk1/2 pathway to promote mouse spermatogonial stem cell
proliferation. Stem Cells. 26:266–278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mancinelli R, Onori P, Gaudio E, DeMorrow
S, Franchitto A, Francis H, Glaser S, Carpino G, Venter J, Alvaro
D, et al: Follicle-stimulating hormone increases cholangiocyte
proliferation by an autocrine mechanism via cAMP-dependent
phosphorylation of ERK1/2 and Elk-1. Am J Physiol Gastrointest
Liver Physiol. 297:G11–G26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sirianni R, Chimento A, De Luca A,
Casaburi I, Rizza P, Onofrio A, Iacopetta D, Puoci F, Andò S,
Maggiolini M and Pezzi V: Oleuropein and hydroxytyrosol inhibit
MCF-7 breast cancer cell proliferation interfering with ERK1/2
activation. Mol Nutr. 54:833–840. 2010. View Article : Google Scholar
|
|
47
|
Yang Y and Han C: GDNF stimulates the
proliferation of cultured mouse immature Sertoli cells via its
receptor subunit NCAM and ERK1/2 signaling pathway. BMC Cell≠≠≠
Bio≠≠.
|
|
48
|
Lee JG and Kay EP: PI 3-kinase/Rac1 and
ERK1/2 regulate FGF-2-mediated cell proliferation through
phosphorylation of p27 at Ser10 by KIS and at Thr187 by
Cdc25A/Cdk2. Invest Ophthalmol Vis Sci. 52:417–426. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gao M, Zhan YQ, Yu M, Ge CH, Li CY, Zhang
JH, Wang XH, Ge ZQ and Yang XM: Hepassocin activates the EGFR/ERK
cascade and induces proliferation of L02 cells through the
Src-dependent pathway. Cell Signal. 26:2161–2166. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tocharus C, Puriboriboon Y, Junmanee T,
Tocharus J, Ekthuwapranee K and Govitrapong P: Melatonin enhances
adult rat hippocampal progenitor cell proliferation via ERK
signaling pathway through melatonin receptor. Neuroscience.
275:314–321. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wu Z, Uchi H, Morino-Koga S, Shi W and
Furue M: Resveratrol inhibition of human keratinocyte proliferation
via SIRT1/ARNT/ERK dependent downregulation of aquaporin 3. J
Dermatol Sci. 75:16–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu H, Wu Y, Zhu S, Liang W, Wang Z, Wang
Y, Lv T, Yao Y, Yuan D and Song Y: PTP1B promotes cell
proliferation and metastasis through activating src and ERK1/2 in
non-small cell lung cancer. Cancer Lett. 359:218–225. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang J, He C, Zhou T, Huang Z, Zhou L and
Liu X: NGF increases VEGF expression and promotes cell
proliferation via ERK1/2 and AKT signaling in Müller cells. Mol
Vis. 22:254–263. 2016.PubMed/NCBI
|
|
54
|
Kim SH, Pei QM, Jiang P, Yang M, Qian XJ
and Liu JB: Effect of active vitamin D3 on VEGF-induced ADAM33
expression and proliferation in human airway smooth muscle cells:
Implications for asthma treatment. Respir Res. 18:72017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Graves LM, Guy HI, Kozlowski P, Huang M,
Lazarowski E, Pope RM, Collins MA, Dahlstrand EN, Earp HS III and
Evans DR: Regulation of carbamoyl phosphate synthetase by MAP
kinase. Nature. 403:328–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Stefanovsky V, Langlois F, Gagnon-Kugler
T, Rothblum LI and Moss T: Growth factor signaling regulates
elongation of RNA polymerase I transcription in mammals via UBF
phosphorylation and r-chromatin remodeling. Mol Cell. 21:629–639.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mendoza MC, Er EE and Blenis J: The
Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends
Biochem Sci. 36:320–328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang Y, Zhu L, Kuokkanen S and Pollard JW:
Activation of protein synthesis in mouse uterine epithelial cells
by estradiol-17β is mediated by a PKC-ERK1/2-mTOR signaling
pathway. Proc Natl Acad Sci USA. 112:E1382–E1391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hoang B, Benavides A, Shi Y, Yang Y, Frost
P, Gera J and Lichtenstein A: The PP242 mammalian target of
rapamycin (mTOR) inhibitor activates extracellular signal-regulated
kinase (ERK) in multiple myeloma cells via a target of rapamycin
complex 1 (TORC1)/eukaryotic translation initiation factor 4E
(eIF-4E)/RAF pathway and activation is a mechanism of resistance. J
Biol Chem. 287:21796–21805. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ma L, Chen Z, Erdjument-Bromage H, Tempst
P and Pandolfi PP: Phosphorylation and functional inactivation of
TSC2 by Erk implications for tuberous sclerosis and cancer
pathogenesis. Cell. 121:179–193. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chambard JC, Lefloch R, Pouysségur J and
Lenormand P: ERK implication in cell cycle regulation. Biochim
Biophys Acta. 1773:1299–1310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lavoie JN, L'Allemain G, Brunet A, Muller
R and Pouysségur J: Cyclin D1 expression is regulated positively by
the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol
Chem. 271:20608–20616. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Seth A, Alvarez E, Gupta S and Davis RJ: A
phosphorylation site located in the NH2-terminal domain of c-Myc
increases transactivation of gene expression. J Biol Chem.
266:23521–23524. 1991.PubMed/NCBI
|
|
64
|
Daksis JI, Lu RY, Facchini LM, Marhin WW
and Penn LJ: Myc induces cyclin D1 expression in the absence of de
novo protein synthesis and links mitogen-stimulated signal
transduction to the cell cycle. Oncogene. 9:3635–3645.
1994.PubMed/NCBI
|
|
65
|
Walsh S, Margolis SS and Kornbluth S:
Phosphorylation of the cyclin B1 cytoplasmic retention sequence by
mitogen-activated protein kinase and Plx. Mol Cancer Res.
1:280–289. 2003.PubMed/NCBI
|
|
66
|
Palmer A, Gavin AC and Nebreda AR: A link
between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation:
p90(rsk) phosphorylates and inactivates the p34(cdc2) inhibitory
kinase Myt1. EMBO J. 17:5037–5047. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shaw PH: The role of p53 in cell cycle
regulation. Pathol Res Pract. 192:669–675. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wesierska-Gadek J, Wojciechowski J,
Ranftler C and Schmid G: Role of p53 tumor suppressor in ageing:
Regulation of transient cell cycle arrest and terminal senescence.
J Physiol Pharmacol. 56:15–28. 2005.PubMed/NCBI
|
|
69
|
Lee SY, Choi HC, Choe YJ, Shin SJ, Lee SH
and Kim HS: Nutlin-3 induces BCL2A1 expression by activating ELK1
through the mitochondrial p53-ROS-ERK1/2 pathway. Int J Oncol.
45:675–682. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Murase S, Kim E, Lin L, Hoffman DA and
McKay RD: Loss of signal transducer and activator of transcription
3 (STAT3) signaling during elevated activity causes vulnerability
in hippocampal neurons. J Neurosci. 32:15511–15520. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Carlos AR, Escandell JM, Kotsantis P,
Suwaki N, Bouwman P, Badie S, Folio C, Benitez J, Gomez-Lopez G,
Pisano DG, et al: ARF triggers senescence in Brca2-deficient cells
by altering the spectrum of p53 transcriptional targets. Nat
Commun. 4:26972013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tang D, Wu D, Hirao A, Lahti JM, Liu L,
Mazza B, Kidd VJ, Mak TW and Ingram AJ: ERK activation mediates
cell cycle arrest and apoptosis after DNA damage independently of
p53. J Biol Chem. 277:12710–12717. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ashcroft M and Vousden KH: Regulation of
p53 stability. Oncogene. 18:7637–7643. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ling Q, Meng C, Chen Q and Xing D:
Activated ERK/FOXM1 pathway by low-power laser irradiation inhibits
UVB-induced senescence through down-regulating p21 expression. J
Cell Physiol. 229:108–116. 2014.PubMed/NCBI
|
|
75
|
Rasola A, Sciacovelli M, Pantic B and
Bernardi P: Signal transduction to the permeability transition
pore. FEBS Lett. 584:1989–1996. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kashatus JA, Nascimento A, Myers LJ, Sher
A, Byrne FL, Hoehn KL, Counter CM and Kashatus DF: Erk2
phosphorylation of Drp1 promotes mitochondrial fission and
MAPK-driven tumor growth. Mol Cell. 57:537–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhu J, Woods D, McMahon M and Bishop JM:
Senescence of human fibroblasts induced by oncogenic Raf. Genes
Dev. 12:2997–3007. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cammarano MS, Nekrasova T, Noel B and
Minden A: Pak4 induces premature senescence via a pathway requiring
p16INK4/p19ARF and mitogen-activated protein kinase signaling. Mol
Cell Biol. 25:9532–9542. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kim-Kaneyama, Nose K and Shibanuma M:
Significance of nuclear relocalization of ERK1/2 in reactivation of
c-fos transcription and DNA synthesis in senescent fibroblasts. J
Biol Chem. 275:20685–20692. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lim IK, Won Hong K, Kwak IH, Yoon G and
Park SC: Cytoplasmic retention of p-Erk1/2 and nuclear accumulation
of actin proteins during cellular senescence in human diploid
fibroblasts. Mech Ageing Dev. 119:113–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chaturvedi V, Cesnjaj M, Bacon P, Panella
J, Choubey D, Diaz MO and Nickoloff BJ: Role of INK4a/Arf
locus-encoded senescent checkpoints activated in normal and
psoriatic keratinocytes. Am J Pathol. 162:161–170. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kim HS, Song MC, Kwak IH, Park TJ and Lim
IK: Constitutive induction of p-Erk1/2 accompanied by reduced
activities of protein phosphatases 1 and 2A and MKP3 due to
reactive oxygen species during cellular senescence. J Biol Chem.
278:37497–37510. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Todd DE, Densham RM, Molton SA, Balmanno
K, Newson C, Weston CR, Garner AP, Scott L and Cook SJ: ERK1/2 and
p38 cooperate to induce a p21CIP1-dependent G1 cell cycle arrest.
Oncogene. 23:3284–3295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Klein LE, Freeze BS, Smith AB III and
Horwitz SB: The microtubule stabilizing agent discodermolide is a
potent inducer of accelerated cell senescence. Cell Cycle.
4:501–507. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Albrecht DS, Clubbs EA, Ferruzzi M and
Bomser JA: Epigallocatechin-3-gallate (EGCG) inhibits PC-3 prostate
cancer cell proliferation via MEK-independent ERK1/2 activation.
Chem Biol Interact. 171:89–95. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Deschênes-Simard X, Gaumont-Leclerc MF,
Bourdeau V, Lessard F, Moiseeva O, Forest V, Igelmann S, Mallette
FA, Saba-El-Leil MK, Meloche S, et al: Tumor suppressor activity of
the ERK/MAPK pathway by promoting selective protein degradation.
Genes Dev. 27:900–915. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhu B, Ferry CH, Blazanin N, Bility MT,
Khozoie C, Kang BH, Glick AB, Gonzalez FJ and Peters JM: PPARβ/δ
promotes HRAS-induced senescence and tumor suppression by
potentiating p-ERK and repressing p-AKT signaling. Oncogene.
33:5348–5359. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wang Z, Liu Y, Takahashi M, Van Hook K,
Kampa-Schittenhelm KM, Sheppard BC, Sears RC, Stork PJ and Lopez
CD: N terminus of ASPP2 binds to Ras and enhances Ras/Raf/MEK/ERK
activation to promote oncogene-induced senescence. Proc Natl Acad
Sci USA. 110:312–317. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
El Bezawy R, De Cesare M, Pennati M,
Deraco M, Gandellini P, Zuco V and Zaffaroni N: Antitumor activity
of miR-34a in peritoneal mesothelioma relies on c-MET and AXL
inhibition: Persistent activation of ERK and AKT signaling as a
possible cytoprotective mechanism. J Hematol Oncol. 10:192017.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
del Nogal M, Troyano N, Calleros L, Griera
M, Rodriguez-Puyol M, Rodriguez-Puyol D and Ruiz-Torres MP:
Hyperosmolarity induced by high glucose promotes senescence in
human glomerular mesangial cells. Int J Biochem Cell Biol.
54:98–110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lake D, Correa SA and Müller J: Negative
feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci.
73:4397–4413. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Northwood IC, Gonzalez FA, Wartmann M,
Raden DL and Davis RJ: Isolation and characterization of two growth
factor-stimulated protein kinases that phosphorylate the epidermal
growth factor receptor at threonine 669. J Biol Chem.
266:15266–15276. 1991.PubMed/NCBI
|
|
93
|
Sato K, Shin MS, Sakimura A, Zhou Y,
Tanaka T, Kawanishi M, Kawasaki Y, Yokoyama S, Koizumi K, Saiki I
and Sakurai H: Inverse correlation between Thr-669 and constitutive
tyrosine phosphorylation in the asymmetric epidermal growth factor
receptor dimer conformation. Cancer Sci. 104:1315–1322. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Prahallad A, Sun C, Huang S, Di
Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A
and Bernards R: Unresponsiveness of colon cancer to BRAF(V600E)
inhibition through feedback activation of EGFR. Nature.
483:100–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zakrzewska M, Haugsten EM,
Nadratowska-Wesolowska B, Oppelt A, Hausott B, Jin Y, Otlewski J,
Wesche J and Wiedlocha A: ERK-mediated phosphorylation of
fibroblast growth factor receptor 1 on Ser777 inhibits signaling.
Sci Signal. 6:ra112013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kamioka Y, Yasuda S, Fujita Y, Aoki K and
Matsuda M: Multiple decisive phosphorylation sites for the negative
feedback regulation of SOS1 via ERK. J Biol Chem. 285:33540–33548.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Lax I, Wong A, Lamothe B, Lee A, Frost A,
Hawes J and Schlessinger J: The docking protein FRS2alpha controls
a MAP kinase-mediated negative feedback mechanism for signaling by
FGF receptors. Mol Cell. 10:709–719. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wu YJ, Chen ZJ and Ullrich A: EGFR and
FGFR signaling through FRS2 is subject to negative feedback control
by ERK1/2. Biol Chem. 384:1215–1226. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wartmann M, Hofer P, Turowski P, Saltiel
AR and Hynes NE: Negative modulation of membrane localization of
the Raf-1 protein kinase by hyperphosphorylation. J Biol Chem.
272:3915–3923. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Weiss RH, Maga EA and Ramirez A: MEK
inhibition augments Raf activity, but has variable effects on
mitogenesis, in vascular smooth muscle cells. Am J Physiol.
274:C1521–C1529. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Dougherty MK, Müller J, Ritt DA, Zhou M,
Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP and Morrison
DK: Regulation of Raf-1 by direct feedback phosphorylation. Mol
Cell. 17:215–224. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Hekman M, Fischer A, Wennogle LP, Wang YK,
Campbell SL and Rapp UR: Novel C-Raf phosphorylation sites: Serine
296 and 301 participate in Raf regulation. FEBS Lett. 579:464–468.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Balan V, Leicht DT, Zhu J, Balan K, Kaplun
A, Singh-Gupta V, Qin J, Ruan H, Comb MJ and Tzivion G:
Identification of novel in vivo Raf-1 phosphorylation sites
mediating positive feedback Raf-1 regulation by extracellular
signal-regulated kinase. Mol Biol Cell. 17:1141–1153. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Brummer T, Naegele H, Reth M and Misawa Y:
Identification of novel ERK-mediated feedback phosphorylation sites
at the C-terminus of B-Raf. Oncogene. 22:8823–8834. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ritt DA, Monson DM, Specht SI and Morrison
DK: Impact of feedback phosphorylation and Raf heterodimerization
on normal and mutant B-Raf signaling. Mol Cell Biol. 30:806–819.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Eblen ST, Slack-Davis JK, Tarcsafalvi A,
Parsons JT, Weber MJ and Catling AD: Mitogen-activated protein
kinase feedback phosphorylation regulates MEK1 complex formation
and activation during cellular adhesion. Mol Cell Biol.
24:2308–2317. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Rossomando AJ, Dent P, Sturgill TW and
Marshak DR: Mitogen-activated protein kinase kinase 1 (MKK1) is
negatively regulated by threonine phosphorylation. Mol Cell Biol.
14:1594–1602. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Canal F, Palygin O, Pankratov Y, Corrêa SA
and Müller J: Compartmentalization of the MAPK scaffold protein
KSR1 modulates synaptic plasticity in hippocampal neurons. FASEB J.
25:2362–2372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
McKay MM, Ritt DA and Morrison DK:
Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci
USA. 106:11022–11027. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Fey D, Croucher DR, Kolch W and Kholodenko
BN: Crosstalk and signaling switches in mitogen-activated protein
kinase cascades. Front Physiol. 3:3552012. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Caunt CJ, Finch AR, Sedgley KR and McArdle
CA: Seven-transmembrane receptor signalling and ERK
compartmentalization. Trends Endocrinol Metab. 17:276–283. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Owens DM and Keyse SM: Differential
regulation of MAP kinase signalling by dual-specificity protein
phosphatases. Oncogene. 26:3203–3213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Huang CY and Tan TH: DUSPs, to MAP kinases
and beyond. Cell Biosci. 2:242012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Peti W and Page R: Molecular basis of MAP
kinase regulation. Protein Sci. 22:1698–1710. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Tanzola MB and Kersh GJ: The dual
specificity phosphatase transcriptome of the murine thymus. Mol
Immunol. 43:754–762. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Hanafusa H, Torii S, Yasunaga T and
Nishida E: Sprouty1 and Sprouty2 provide a control mechanism for
the Ras/MAPK signalling pathway. Nat Cell Biol. 4:850–858. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lito P, Pratilas CA, Joseph EW, Tadi M,
Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub
T, et al: Relief of profound feedback inhibition of mitogenic
signaling by RAF inhibitors attenuates their activity in BRAFV600E
melanomas. Cancer Cell. 22:668–682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Lito P, Rosen N and Solit DB: Tumor
adaptation and resistance to RAF inhibitors. Nat Med. 19:1401–1409.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Yusoff P, Lao DH, Ong SH, Wong ES, Lim J,
Lo TL, Leong HF, Fong CW and Guy GR: Sprouty2 inhibits the Ras/MAP
kinase pathway by inhibiting the activation of Raf. J Biol Chem.
277:3195–3201. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Dent P: Crosstalk between ERK, AKT, and
cell survival. Cancer Biol Ther. 15:245–246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Mace PD, Wallez Y, Egger MF, Dobaczewska
MK, Robinson H, Pasquale EB and Riedl SJ: Structure of ERK2 bound
to PEA-15 reveals a mechanism for rapid release of activated MAPK.
Nat Commun. 4:16812013. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Sinha D, Bannergee S, Schwartz JH,
Lieberthal W and Levine JS: Inhibition of ligand-independent ERK1/2
activity in kidney proximal tubular cells deprived of soluble
survival factors up-regulates Akt and prevents apoptosis. J Biol
Chem. 279:10962–10972. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Trencia A, Perfetti A, Cassese A,
Vigliotta G, Miele C, Oriente F, Santopietro S, Giacco F,
Condorelli G, Formisano P and Beguinot F: Protein kinase B/Akt
binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic
action. Mol Cell Biol. 23:4511–4521. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Aksamitiene E, Kiyatkin A and Kholodenko
BN: Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt
pathways: A fine balance. Biochem Soc Trans. 40:139–146. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Ussar S and Voss T: MEK1 and MEK2,
different regulators of the G1/S transition. J Biol Chem.
279:43861–43869. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Lee SJ, Lee SH, Yoon MH and Park BJ: A new
p53 target gene, RKIP, is essential for DNA damage-induced cellular
senescence and suppression of ERK activation. Neoplasia.
15:727–737. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Li M, Zhou JY, Ge Y, Matherly LH and Wu
GS: The phosphatase MKP1 is a transcriptional target of p53
involved in cell cycle regulation. J Biol Chem. 278:41059–41068.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Shen WH, Wang J, Wu J, Zhurkin VB and Yin
Y: Mitogen-activated protein kinase phosphatase 2: A novel
transcription target of p53 in apoptosis. Cancer Res. 66:6033–6039.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Ueda K, Arakawa H and Nakamura Y:
Dual-specificity phosphatase 5 (DUSP5) as a direct transcriptional
target of tumor suppressor p53. Oncogene. 22:5586–5591. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
El Hasasna H, Athamneh K, Al Samri H,
Karuvantevida N, Al Dhaheri Y, Hisaindee S, Ramadan G, Al Tamimi N,
AbuQamar S, Eid A and Iratni R: Rhus coriaria induces
senescence and autophagic cell death in breast cancer cells through
a mechanism involving p38 and ERK1/2 activation. Sci Rep.
5:130132015. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wajapeyee N, Serra RW, Zhu X, Mahalingam M
and Green MR: Oncogenic BRAF induces senescence and apoptosis
through pathways mediated by the secreted protein IGFBP7. Cell.
132:363–374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ichimura A, Ruike Y, Terasawa K, Shimizu K
and Tsujimoto G: MicroRNA-34a inhibits cell proliferation by
repressing mitogen-activated protein kinase kinase 1 during
megakaryocytic differentiation of K562 cells. Mol Pharmacol.
77:1016–1024. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Sayed D, Rane S, Lypowy J, He M, Chen IY,
Vashistha H, Yan L, Malhotra A, Vatner D and Abdellatif M:
MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Mol
Biol Cell. 19:3272–3282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Deschênes-Simard X, Kottakis F, Lessard F,
Saint-Germain E, Bourdeau VBardeesy N and Ferbeyre G: Tumor
suppressor activity of the ERK/MAPK signaling: Inhibition of cell
reprogramming by degradation of specific proteins. Cancer Res.
74:38952014. View Article : Google Scholar
|
|
135
|
Plotnikov A, Flores K, Maik-Rachline G,
Zehorai E, Kapri-Pardes E, Berti DA, Hanoch T, Besser MJ and Seger
R: The nuclear translocation of ERK1/2 as an anticancer target. Nat
Commun. 6:66852015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Schevzov G, Kee AJ, Wang B, Sequeira VB,
Hook J, Coombes JD, Lucas CA, Stehn JR, Musgrove EA, Cretu A, et
al: Regulation of cell proliferation by ERK and signal-dependent
nuclear translocation of ERK is dependent on Tm5NM1-containing
actin filaments. Mol Biol Cell. 26:2475–2490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Wainstein E and Seger R: The dynamic
subcellular localization of ERK: Mechanisms of translocation and
role in various organelles. Curr Opin Cell Biol. 39:15–20. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Callejas-Valera JL, Guinea-Viniegra J,
Ramirez-Castillejo C, Recio JA, Galan-Moya E, Martinez N, Rojas JM,
Ramón y Cajal S and Sánchez-Prieto R: E1a gene expression blocks
the ERK1/2 signaling pathway by promoting nuclear localization and
MKP up-regulation: Implication in v-H-Ras-induced senescence. J
Biol Chem. 283:13450–13458. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Gaumont-Leclerc MF, Mukhopadhyay UK,
Goumard S and Ferbeyre G: PEA-15 is inhibited by adenovirus E1A and
plays a role in ERK nuclear export and Ras-induced senescence. J
Biol Chem. 279:46802–46809. 2004. View Article : Google Scholar : PubMed/NCBI
|