PI3K/Akt/mTOR signaling pathway
Phosphatidylinositol-4,5-bisphosphate
3-kinase (PI3K)/protein kinase B (Akt) pathway
The PI3K/Akt pathway is an intracellular signaling
pathway of great importance in the cell cycle process. It is
associated with cellular quiescence, proliferation, cancer and
longevity. PI3K activation phosphorylates and further activates
Akt, which localizes to the cellular membrane (1). Activated Akt fulfills various
biological functions, including activating cAMP-response element
binding protein (2), inhibiting
p27 (3), localizing forkhead box
protein O (FOXO) in the cytoplasm (3), activating phosphatidylinositol
3-phosphate (PtdIns3 ps or PI3P) (4) and activating mammalian target of
rapamycin (mTOR) (3). It has been
identified that the PI3K/Akt pathway can be enhanced by several
biological molecules, including epidermal growth factor (EGF)
(5), sonic hedgehog (2), insulin-like growth factor (IGF)-1
(2), insulin (3) and calmodulin (4). Conversely, this pathway is
antagonized by other molecules, including phosphatase and tensin
homolog (PTEN) (6), glycogen
synthase kinase 3β (2) and
transcription factor HB9 (5).
Structure and function of PI3Ks
The PI3Ks are a family of lipid kinases, which
generate second messengers by specific catalytic 3-hydroxy
phosphorylation of phosphatidylinositol (PI) (7). The PI3K family is divided into three
classes, I–III; these classes share four homologous regions, among
which, the kinase domain is the most conserved.
The substrate for Class I PI3Ks includes PI,
phosphatidylinositol 4-phosphate and phosphatidylinositol (4,5)
bisphosphate [PI(4,5)P2]. Class I PI3Ks are
heterodimeric molecules composed of a regulatory and a catalytic
subunit, which are further divided into IA and IB subsets. Class IA
PI3Ks are composed of a heterodimer between a p110 catalytic
subunit and a p85 regulatory subunit (8,9). The
p85 subunit consists of α, β and γ, which are encoded in mammals by
PI3K regulatory subunit (PIK3R)1, PIK3R2 and PIK3R3, respectively.
The p85 subunit serves different roles in receptor binding, enzyme
activation and localization (10).
The p110 subunit consists of α, β and δ, which are encoded by PI3K
catalytic subunit (PIK3C)A, PIK3CB and PIK3CD, respectively
(11). p110α and p110β mainly
influence cellular proliferation and insulin signaling in various
tissues, whereas p110δ is found only in leukocytes and is involved
in immune function and inflammation (12). Class IB PI3Ks consist of regulatory
p101 and catalytic p110γ, and are encoded by a single gene each
(8). p110γ is mainly expressed in
leukocytes, with reduced expression in the heart, pancreas, liver
and skeletal muscle. It combines with the Gβγ subunit of the G
protein-coupled receptor and activates PI3K (13). The catalytic p110 subunit of Class
IA PI3Ks consists of an N-terminal p85-binding domain (p110γ does
not contain this domain), Ras binding domain, a proline-rich region
[which reacts with proteins containing the SRC Homology (SH)3
domain], C-terminal homologous region (HR)3 (containing a basic
leucine zipper-like domain bZIP), HR2 region (also termed PIK
domain) and a C-terminal HR1 region (catalytic effect domain). HR3,
HR2 and HR1 regions are PI3K HRs, which are responsible for
membrane binding, substrate presentation and kinase domain, and
catalytic inositol lipid 3-hydroxy-phosphorylated (14). The p85 regulatory subunit of Class
IA PI3Ks consists of an SH3 domain, RHO-binding domain/breakpoint
cluster region homology region, C-terminal SH2 domain and a
connecting region (15).
Class II PI3Ks have monomeric catalytic isoforms,
and contain α, β and γ subtypes. They contain a proline-rich
region, Ras binding domain, HR3 region, HR2 region, HR1 region, PX
domain and the C2 domain (16).
Class II PI3Ks catalyze the production of PI(3)P from PI and
phosphatidylinositol (3,4)-bisphosphate [PI(3,4)P2]
from phosphoinositide (PIP). However, little is currently known
about the mechanism.
The sole member of Class III PI3K is Vps34, which is
the human homolog of a yeast gene product. Vps34 is a heterodimer
formed by a p150 regulatory subunit and a p100 myristoyl-flower
catalytic subunit, which phosphorylates PI to PI(3)P. Human Class
III PI3K is a threonine/serine kinase also named as Vps34, which is
the only PI3-kinase expressed in all eukaryotic cells. It was first
identified in a Saccharomyces cerevisiae (budding yeast) screen for
proteins involved in vesicle-mediated vacuolar protein sorting. It
is tightly bound to regulatory subunit p150 and its function is not
only phosphorylation, but also the recruitment of catalytic
subunits to the cell membrane (17). Although PI(3)P is widespread, its
level does not change when cells are stimulated. Therefore, Class
III PI3K may be a housekeeping kinase that does not serve a role in
signal transduction (18).
Akt
Akt (~60 kDa) has 68% homology with protein kinase A
(PKA) and 73% homology with protein kinase C (PKC); therefore, Akt
is also termed PKA and PKC-associated kinase. PKB is a product of
oncogene v-akt encoding the retrovirus Akt-8, which is also known
as Akt (19). Akt is a
serine/threonine kinase, which is the central mediator of the PI3K
pathway that serves a key role in numerous cellular processes,
including glucose metabolism, apoptosis, cell proliferation,
transcription and cell migration. There are three Akt subtypes:
PKBα (Akt1), PKBβ (Akt2) and PKBγ (Akt3). Akt1 is widely expressed
in several tissues, whereas Akt2 is mainly expressed in
insulin-sensitive tissues, with a lower expression in other
tissues. Akt3 is specific to the brain, lung, heart, kidney, testis
and skeletal muscle (20). The
three Akt isoforms share homologous amino acid sequences, including
the N-terminal regulatory region, kinase catalytic domain and
C-terminal regulatory domain. The N-terminal regulatory region is
also termed the Akt homology domains/pleckstrin homology domains
(PHD) domain. The kinase catalytic domain is highly homologous to
enzymatically active regions in PKA and PKC, and its Thr308 site is
necessary for Akt activation. The Ser473 site of C-terminal
regulatory domain is important for complete activation of Akt
(21).
mTOR
mTOR-targeted proteins belong to the
phosphatidylinositol 3-kinase-related kinase family, which are
serine/threonine protein kinases. This family serves a key role in
identifying nutrition signals, cell growth and proliferation. mTOR
is composed of mTOR complex (mTORC)1 and mTORC2, which contain five
domains: TOR1, focal adhesion kinase (FAT), FKBP-rapamycin-binding
(FRB) domain, kinase domain, negative regulatory domain, and FRAP,
ATM, TRRAP C-terminal (FATC) domain (22). It has been confirmed that there is
an amino acid residues kinase domain near the C-terminus of mTOR,
which has a similar structure to the catalytic domain of PI3K.
Upstream of the kinase domain is an FRB domain, which is the
binding site of the FKBP12-rapamycin complex (23). Upstream of the FRB kinase domain is
the FAT domain, which lies in the C-terminal of mTOR. This is also
termed the FATC domain and serves a key role in mTOR stability.
Deletion of one amino acid within its structure can lead to loss of
mTOR activity (24). mTORC1, which
comprises regulatory-associated protein of mTOR and mammalian
ortholog of LST8 (mLST8), mainly regulates cell growth and energy
metabolism, and is sensitive to rapamycin (25–28).
mTORC2, which comprises rapamycin insensitive companion of mTOR,
mitogen-activated protein kinase-associated protein 1 and mLST8, is
mainly involved in cytoskeletal remodeling and cell survival, and
is not sensitive to rapamycin (29,30).
Regulation of the PI3K/Akt signaling
pathway
PI3K activation
PI3KIA kinase is activated while the growth factor
receptor such as insulin receptor is bound to the p85 regulatory
subunit of the SH2 domain of PI3KIA kinase. PI3K is also activated
by other members of the non-receptor tyrosine kinase Src family. It
can be activated by thrombin through focal adhesion kinase (FAK),
which interacts with the SH3 domain of p85 subunits.
Phosphorylation of FAK itself promotes the interaction with the SH2
domain of p85 subunits. PI3K is also activated by small GTP enzyme
Ras through interaction with p110α (31). In general, PI3K is mainly activated
directly and indirectly through the FAK pathway. It can be
activated i) through receptor tyrosine protein kinase (RTK)
dimerization, phosphorylation and interaction with the SH2 domain
of p85 subunit by various growth factors, including
platelet-derived growth factor (PDGF), IGF and EGF (32–34).
ii) By RTK recruiting related proteins including Shc, Grb2 and Grb1
and Ras-related small molecules G proteins (Rho, Rac and Cdc42)
(35). iii) By non-receptor
protein tyrosine kinases, including Janus kinase, integrin linked
kinase, FAK, Fyn, Lck, Lyn and Src through p85/p110 (36). iv) By the Gβγ subunit of G
protein-coupled receptors (37).
Akt activation and PI3K/Akt negative
regulation
PI(3,4,5)P3 has been identified as an Akt
activator following the discovery of Akt. It serves an important
role in the process of Akt activation. The head group of the lipid
is directly attached to the N-end PH domain of Akt and is
indirectly regulated through the PH domain of lysis protein
kinase-3-phosphoinositide-dependent kinase 1 (PDK1).
PI(3,4,5)P3 recruits Akt from the cytoplasm to the cell
membrane, which causes Akt conformational alterations and
ring-phosphorylation through a combination of the PH domain of PDK1
(8,38). With the aid of PDK2, the Akt C
hydrophobic terminal is phosphorylated and double-phosphorylated
Akt separates from the membrane, thus resulting in the cellular
reaction with the substrate including phosphoinositide-dependent
kinase 2 (PDK2), integrin-linked kinase (ILK), mechanistic target
of rapamycin complex (mTORC) and DNA-dependent protein kinase
(DNA-PK). It has been confirmed that the PI3K/Akt pathway is
activated by the following steps: i) PI(3,4,5)P3 generation, ii)
Akt conformational alterations and iii) double-phosphorylation of
Akt (20,39,40).
The PI3K/Akt signaling pathway regulates cell proliferation,
migration, differentiation and apoptosis through activation or
inhibition of downstream proteins (40). Phosphatase and tensin homologue
(PTEN) transform PI-3,4,5-P3 into PI-4,5-P2 (41); therefore, Akt and the downstream
signaling pathway is activated when PTEN activity is inhibited
(42). PIP3 expression levels are
higher in various tissue and cells, such as hypothalamus and
pancreatic beta cells in PTEN gene knockout mice compared with in
wild-type mice (43,44). In addition, carboxyl terminal
modulator protein C can block the PI3K/Akt signaling pathway,
acting as a negative regulator by inhibiting Akt phosphorylation
(45).
PI3K/Akt signaling transduction pathway and
HIF-1α
Hypoxia-inducible factor (HIF)-1
structure and regulation
HIF-1 structure
HIF-1 is a heterodimer, which is composed of HIF-1α
and HIF-1β (also known as the aryl hydrocarbon receptor nuclear
transporter, ARNT) (46). The
HIF-1α and HIF-1β subunits (120 kDa and 91–94 kDa, respectively)
belong to the basic helix-loop-helix (bHLH) protein family and
contain the period circadian protein-ARNT-single-minded protein
(PAS) domain (47). The HIF-1α
gene is located at the q21-24 region of human chromosome 14 and is
regulated by hypoxic signaling. The HIF-1β gene is located at the
q21 region of human chromosome 1 and is stably expressed (48,49).
The two subunits contain an N-terminal bHLH/PAS homologous region,
which is essential for dimerization and binding of target genes.
The middle of the HIF-1α subunit is an oxygen-dependent degradation
domain (ODDD), which is rich in proline-serine-threonine. The
C-terminal region contains two transactivation domains (TAD)-N and
TAD-C and an inhibitory domain located between two TADs, which
inhibits transcriptional activation of TAD (50). HIF-1 serves its important role in
the regulation of transcription only when the two subunits form a
dimer (51).
HIF-1 regulation
HIF-1α maintains the balance between synthesis and
decomposition under normoxic conditions. The stability and
transcription of HIF-1α are significantly increased under hypoxia
conditions. HIF-1α is regulated by two oxygen-dependent factors;
factor-inhibiting HIF-1 and prolyl hydroxylase (PH).
Under normoxic conditions, HIF-1 transcription is
inhibited by C-terminal transactivation domain through CBP/p300
(52). However, under hypoxic
conditions, the increase in HIF-1α expression induces transcription
of downstream target genes (53,54).
Under normoxic conditions, HIF-1α is degraded by
ubiquitin-proteasome, which is formed via prolyl hydroxylase and
causes ubiquitination of the HIF-1α subunit (55,56).
Conversely, under hypoxic conditions, HIF-1α degradation is reduced
by von Hippel-Lindau tumor suppressor (57). HIF-1α combines with nuclear pore
protein under the nuclear localization signal and enters the
nucleus to form a dimer with HIF-1β. Subsequently, the dimer
combines with CBP/p300 and initiates the transcription of HIF-1
target genes.
HIF-1α is regulated through the PI3K/Akt pathway,
which is activated by growth factors including PDGF, IGF and EGF,
transforming growth factor (TGF), tumor necrosis factor-α and
interleukin-1β (58). Expression
of HIF-1α is enhanced when the PI3K/Akt pathway is activated
through RTK.
Arrest-defective-1 (ARD1) acetylates the middle ODDD
532 lysine of the HIF-1α subunit. Since the mRNA and protein
expression levels of ARD1 are reduced under hypoxic conditions,
expression of HIF-1α increases due to decreased HIF-1α acetylation
(59). HIF-1α, as a phosphoric
acid protein, can regulate the synthesis and degradation through
the phosphoric process by itself. However, hypoxia-induced HIF-1α
phosphorylation enhances HIF-1 transcriptional activity (60).
PI3K/Akt signaling pathway and HIF-1α
under normoxic conditions
A number of factors such as growth factors and
cytokines indirectly regulate HIF-1α stabilization under normoxic
conditions. Growth factors and cytokines regulate the PI3K/Akt and
mitogen-activated protein kinase (MAPK) signaling pathways.
Phosphorylation of HIF-1α causes activation of its transcription by
the extracellular signal-regulated kinase/MAPK pathway, whereas
HIF-1α protein levels are regulated by PI3K/Akt.
Nitric oxide (NO) increases HIF-1α stability under
normoxic whereas the opposite occurs under hypoxia (61–64).
Co-culture of mouse astrocytes and endothelial cells demonstrated
that vascular endothelial cells increase the stability of HIF-1α in
astrocytes by producing endothelial NO synthase. It also increases
the production of glucose transporter-1 (GLUT1), hexokinase-2,
monocarboxylate transporter-4 and lactate (65). HIF-1α is associated with the
activation of cyclooxygenase-2 via the PI3K/Akt pathway in lung
cancer cells (66).
IGF-1 increases the expression of HIF-1α protein in
UCT116 cells, whereas the inhibitor of PI3K, LY294002, inhibits
induction of HIF-1α (67). It is
unclear whether the PI3K/Akt pathway induces HIF-1α expression or
inhibits HIF-1α degradation under normoxic conditions. The study
have demonstrated that in response to PI3K/Akt/mTOR induction in
the normoxic environment, the synthesis of HIF-1α is increased,
whereas its degradation is not inhibited (68).
The PI3K inhibitors wortmannin and LY294002, or the
mTOR inhibitor rapamycin, suppress expression of HIF-1α and
transcription of HIF-1 target genes which are induced and activated
by EGF and angiotensinII via the PI3K/Akt pathway (69,70).
Isakoff et al (71)
revealed that EGFR can combine with the p85 regulatory subunit of
PI3K through its C-terminal amino acid sequence. EGF binding to
EGFR can indirectly activate expression of HIF-1α protein via the
PI3K/Akt pathway (72).
PI3K/Akt signaling pathway and HIF-1α
under hypoxic conditions
The controversy over the association between the
PI3K/Akt signaling pathway and HIF-1α may be associated with
disease and cell type under hypoxic. Stability of HIF-1α is
associated with the PI3K/Akt signaling pathway, and HIF-1α
expression is reduced by PI3K inhibitors (68,69,73–78).
Under hypoxia (8% O2), hepatocytes exhibit increased
HIF-1α levels (~6-fold), HIF-2α levels (~5-fold) and HIF-3α levels
(~3-fold) compared with under normoxia. The insulin-dependent
increase of the HIF 1α protein is mediated via PI3K, inasmuch as
the PI3K inhibitor, wortmannin, eradicates the insulin-dependent
enhancement of HIF-1α (79). FOXO4
not only reduces HIF-1α protein expression in HeLa cells under
hypoxia or deferoxamine-simulated chemical hypoxia via the PI3K/Akt
pathway, but also downregulate the stability of HIF-1α which cannot
be caused by hydroxylation of proline (80). Blocking the PI3K/Akt/mTOR pathway
inhibits serum-induced HIF-1, but not hypoxia-induced HIF-1
expression, hypoxia-inducible transcription gene activation of
phosphoglycerate kinase (PGK) and GLUT1 (81). It has also been suggested that
hypoxia can not only activate the PI3K/Akt signaling pathway in
HeLa cells but can also promote the expression of HIF-1α. However,
it appears that hypoxia-induced HIF-1α precedes PI3K/Akt activation
(82). Nevertheless, hypoxia does
not cause Akt phosphorylation in HEK293T, PC-3, COS-7, U373 and 3T3
cells. These data demonstrated that PI3K/Akt activity is only
induced in response to hypoxia in certain cell types (82).
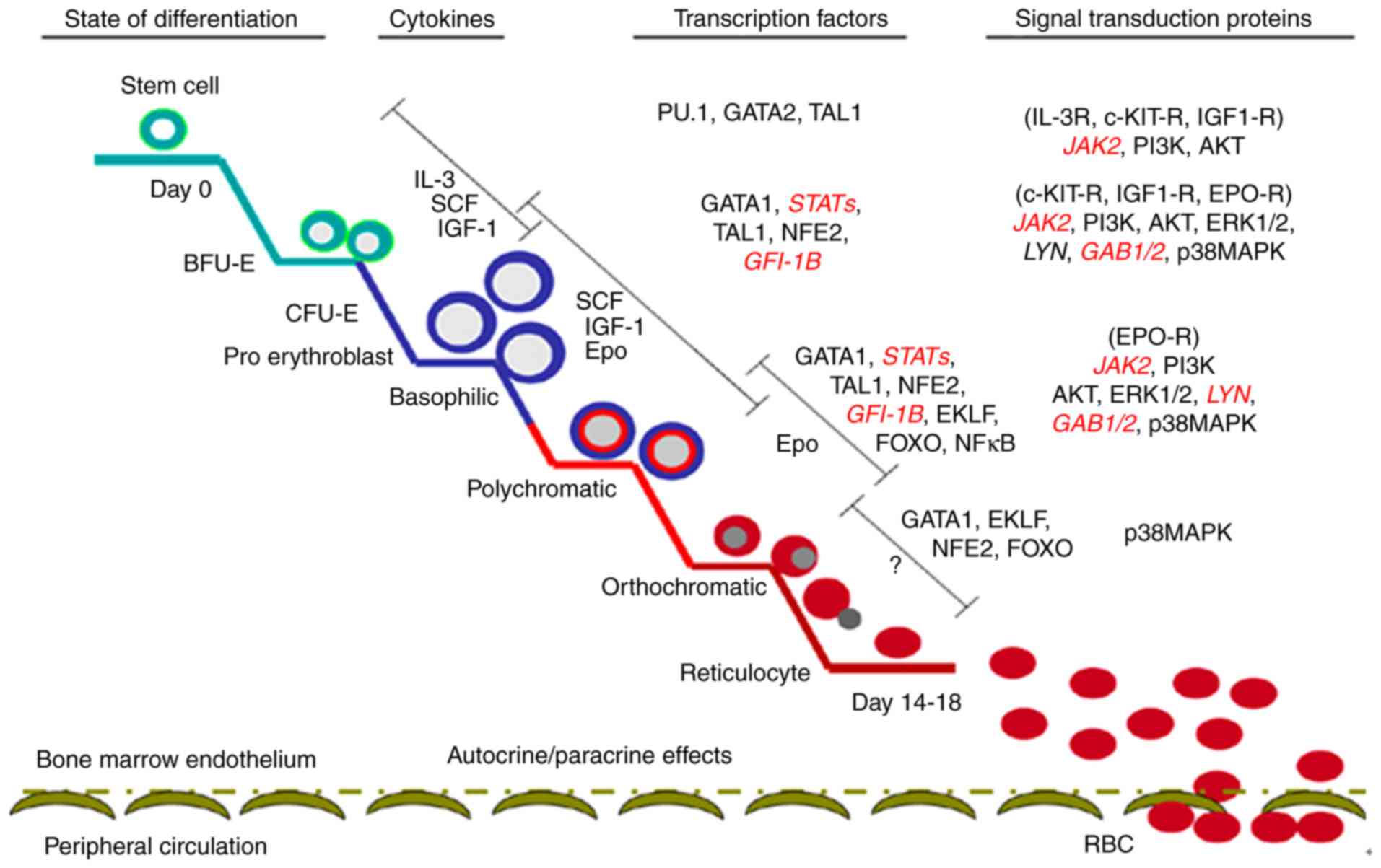
Hypoxia and erythropoiesis
Erythropoiesis is a complex and sophisticated
process, which originates in hematopoietic stem cells (HSCs);
during this process, HSCs sequentially form burst-forming units of
erythroid (BFU-E), colony-forming units of erythroid (CFU-E),
proerythroblast and erythroblast, finally resulting in the
formation of mature erythrocytes in the bone marrow (83–85).
The main regulatory mechanism underlying erythropoiesis includes
external hematopoietic cytokines, hematopoietic cytokine receptors,
transcription factors and signaling molecules (Fig. 1) (86).
HIF-1 regulates numerous downstream genes, including
angiogenesis genes [vascular endothelial growth factor (VEGF) and
endothelin 1], erythropoiesis and energy metabolism genes (GLUT1,
ALDOA, enolase 1, lactate dehydrogenase A, phosphofructokinase,
liver type, PGK1 and HK), cell proliferation and differentiation
genes (fibroblast growth factor, TGF and IGF), and
apoptosis-associated genes (caspase-3 and cytochrome c) (87,88).
HIF-α is divided into three subtypes (HIF-1α, HIF-2α and HIF-3α),
each with specific functions. HIF-1α is involved in the acute
hypoxic response, whereas HIF-2α is associated with the response to
chronic hypoxia (89).
Hematopoietic organs improve the oxygen-carrying capacity of the
blood by increasing the number of red blood cells during oxygen
plateau or strenuous exercise. This process is achieved via the
erythropoietin (EPO) gene, which is mediated by HIF-α in the acute
hypoxic response (89,90). León-Velarde et al (91) reported that EPO levels in the
normal population at oxygen plateau and in patients with high
altitude polycythemia (HAPC) were higher compared with in the
normal population. However, no differences were observed between in
normal population at oxygen plateau and patients with HAPC. The
above findings indicate that EPO is an important regulatory factor
of erythropoiesis in acute hypoxia but not in a chronic hypoxic
environment.
Excess iron causes oxidative stress via the Fenton
reaction and inhibits the interaction of HIF-2α with the EPO
promoter in the kidneys of mice (92), however, an antioxidant compound,
such as tempol, can restore this process. It has also been
demonstrated that iron supplementation reduces EPO gene expression
via an oxidative stress-HIF-2α-dependent signaling pathway
(92). HIF prolyl hydroxylase
enzyme inhibitors serve their roles by stabilizing the HIF complex
and stimulating endogenous EPO production. They also improve iron
mobilization to the bone marrow in patients with end-stage kidney
disease (93). Classically,
erythrocytosis is classified as either primary, caused by intrinsic
defects in erythroid progenitor cells in the presence of normal or
low serum EPO levels, or secondary. The causes of primary
erythrocytosis include mutations in the Janus kinase 2 (JAK2) and
EPOR genes, which can lead to EPO-independent proliferation of
erythroid precursors or hypersensitivity to EPO (94). Secondary erythrocytosis is due to
defects in the oxygen-sensing pathway, including mutations in the
genes for HIF prolyl 4-hydroxylase 2 (HIF-P4H-2), HIF-2α, and von
Hippel Lindau (VHL) protein, and impaired oxygen delivery or tissue
hypoxia, all associated with the activation of the EPO pathway and
elevated serum EPO levels (95,96).
Large-spectrum conditional inactivation of HIF-P4H-2 in mice leads
to severe erythrocytosis (97).
HIF stabilization can thus mediate non-erythropoietin-driven
splenic erythropoiesis via altered Notch signaling pathway
(97). Genes associated with
hypoxic environments have been identified in Tibetan populations
living at high altitude; these populations are adapted to the
plateau environment, in order to protect from polycythemia. The
encoded PHD2-specific EGLN1 haplotypes can reduce HIF accumulation
under hypoxic conditions. The effects of the EGLN1 haplotype on
hemoglobin at low altitude are age-dependent, whereas EPAS1
rs142764723 C/C alleles exist to maintain low levels of hemoglobin
at high altitude (98).
PI3K/Akt signaling pathway and
glycolysis
Glycogen synthase kinase 3β (GSK-3) is an important
downstream molecule regulated by Akt. It consists of Axis
inhibition protein, β-catenin and adenomatous polyposis coli
protein, and belongs to the serine/threonine protein kinase family.
There are two subtypes: GSK-3α and GSK-3β, and 97% of amino acid
sequences in the catalytic region of these two subtypes is
homologous. They are both expressed widely in numerous cells and
tissues, and possess similar biological properties. Previous
studies have identified that GSK-3β can phosphorylate various
endogenous substrates, including proteins associated with
metabolism and transcription factors. GSK-3β serves an important
role in cell growth, development, tumorigenesis and the regulation
of glucose homeostasis (99–102). PI3K-dependent Akt is activated by
insulin and growth factors that cause GSK-3β N terminal serine
phosphorylation to inhibit GSK-3β activity. The decreased glycogen
synthesis and increased accumulation of cyclin D1 result in cell
cycle progression and proliferation (103). HIF-1α can activate the expression
of associated glycolytic enzymes (87,88).
HIF-1α is regulated by growth factors and cytokines via the
PI3K/Akt/mTOR pathway and through this pathway HIF-1α can activate
the expression of VEGF (104,105). Furthermore, PI3K/Akt regulates
fructose 2,6-bisphosphatase (PFKFB2) expression and strengthens
glycolysis. The activity of PFKFB2 is regulated by phosphorylation
of C-terminal Ser466 and Ser483 residues (106–109). By pretreating LNCaP cells with
R1881 (methyltrienolone) for 72 h, followed by LY294002 PI3K
inhibition, Moon et al (110) revealed that phosphorylation of
Ser466 and Ser483 was reduced and reversed by R1881. The
association between glycolysis target genes regulated by HIF-1α and
PI3K/Akt remains to be elucidated. HIF-1α is regulated by
downstream mTOR of the PI3K/Akt signaling pathway (111). A previous study suggested that
HIF-1α regulates glycolysis via the PI3K/Akt pathway (111). PI3K/Akt can regulate the
reduction of glycogen synthesis and strengthen glycolysis.
N-terminal serine phosphorylation of GSK-3β initiates cell cycle
progression and proliferation by inhibiting GSK-3β activity and
increasing the accumulation of cyclin D1. Therefore, reduction of
glycogen synthesis and glycolysis may be a key point in cell
proliferation regulation.
PDGF activates HIF-1α and c-Myc to reduce
mitochondrial complex IV activity by activating the PI3K/Akt
pathway in vitro. Furthermore, PDGF stimulation can reverse
the decrease in glucose uptake and lactate production resulting
from GLUT1 inhibitors in colon cancer cells (112). These findings suggested that PDGF
may lead to reduced mitochondrial activity and increased glycolysis
(112). Silencing cluster of
differentiation 147 by specific small interfering RNA can
downregulate GLUT1 levels by inhibiting PI3K/Akt signaling and
decreasing glucose uptake in A375 cells (113). Under hypoxic conditions, the
expression of HIF-1α and associated glycolytic enzymes, including
GLUT1, hexokinase II, phosphofructokinase 2 and lactate
dehydrogenase A, are increased, and extracellular lactate
concentration is increased in the esophageal carcinoma cell lines
Eca109 and TE13. However, the use of the PI3K/Akt inhibitor
Wortmannin can reverse the alterations in glycolytic enzymes and
the secretion of lactic acid (114). Primary exudative lymphoma (PEL)
is a rare subtype of B-cell non-Hodgkin lymphoma, in which the dual
PI3K and mTOR inhibitor PF-04691502 and Akt inhibitor (Akti-½) can
reduce lactate production and extracellular acidification rate.
PF-04691502 and Akti-½ can alter the metabolism of PEL cells from
aerobic glycolysis towards oxidative respiration in combination
with the glycolysis inhibitor 2-deoxyglucose (115). Serum stimulation can induce a
slight accumulation of HIF-1α protein in a PI3K/Akt
pathway-dependent manner (81).
However, hypoxia induces much higher levels of HIF-1α protein and
HIF-1 DNA binding activity independent of PI3K and mTOR activity.
In addition, it has been identified that the effects of active Akt
on HIF-1 activity are cell-type specific. High levels of Akt
signaling can modestly increase HIF-1α protein, but not affect
HIF-1 target gene expression (81). Therefore, the PI3K/Akt pathway may
not be necessary for hypoxic induction of HIF-1 subunits or
activity, and constitutively active Akt is not sufficient to induce
HIF-1 activity by itself (81).
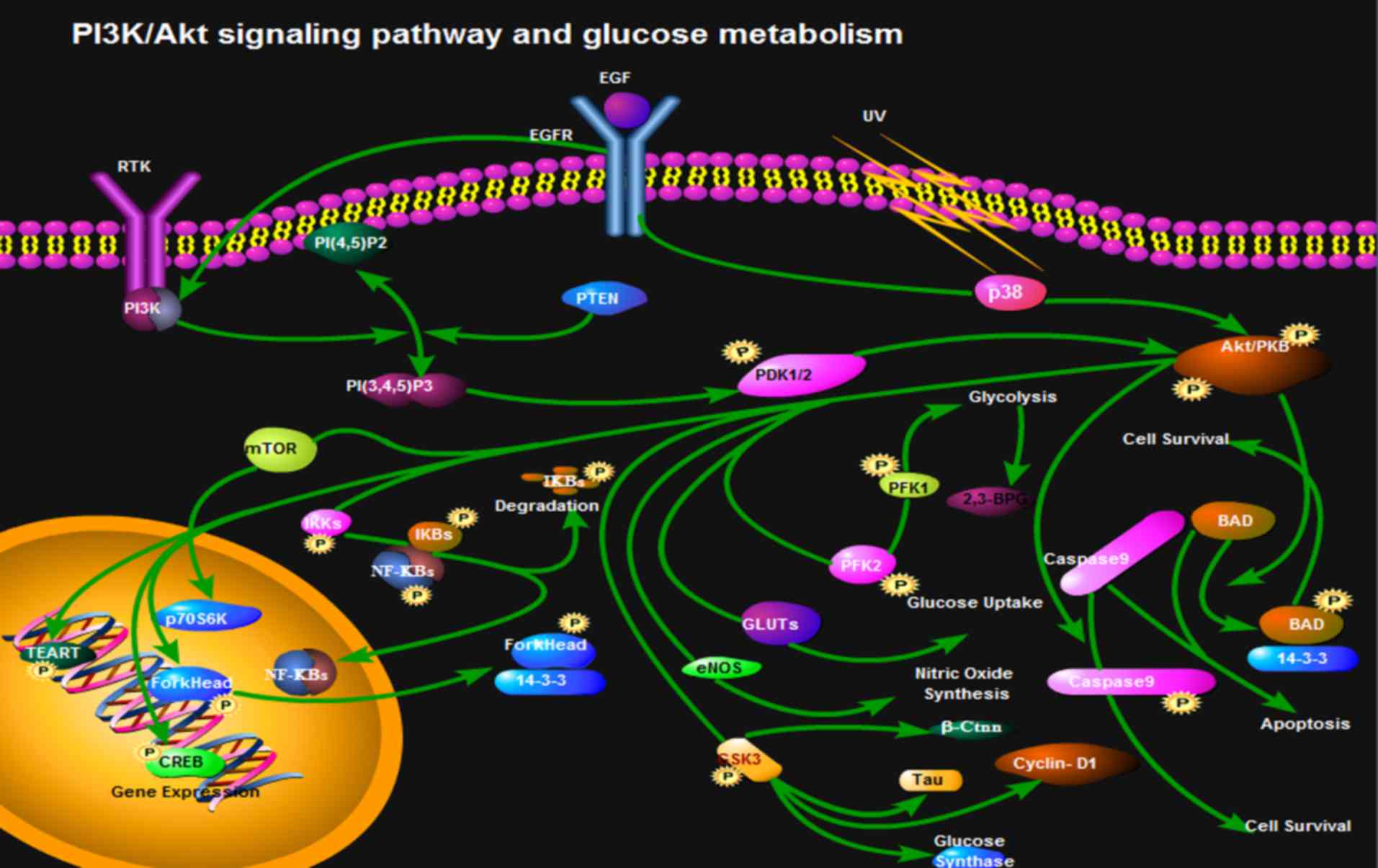
In cell glycolysis, l, 3-bisphosphoglycerate (BPG)
catalyzed by PGK generates 3-phosphoglyceric acid. However, 1,3-BPG
catalyzed by BPG mutase (BPGM) generates 2,3-BPG in the red blood
cells. Under normal circumstances, the 2,3-BPG branch is only 19%
of the glycolytic pathway (116);
however, 2,3-BPG accounts for 50% of the glycolytic pathway under
hypoxic conditions. Benesch et al (117) hypothesized and confirmed that
2,3-BPG and hemoglobin mainly regulate the affinity of hemoglobin
and O2. The mRNA expression of BPGM is distributed
mainly during development from bone marrow erythroid cells to
mature erythrocytes. Low levels of BPGM mRNA can be detected in
non-red blood cells, whereas the synthesis of 2,3-BPG only occurs
in red blood cells (118). The
Creteil I mutation, which results in inactivation of BPGM, or the
Creteil II mutation, which is a reading frame shift, results in the
inability of cells to express BPGM correctly, which then leads to a
low content of 2,3-BPG in human peripheral blood. As a result, the
bone marrow produces more red blood cells to transport more
O2 into tissues (119). Nevertheless, it remains to be
elucidated as to whether the PI3K/Akt signaling pathway regulates
synthesis of BPGM (Fig. 2).
Summary and perspective
The PI3K/Akt signaling pathway is involved in
numerous biological processes including cell cycle, cell apoptosis,
angiogenesis and glucose metabolism. The PI3K/Akt pathway is
indispensable to cell proliferation and apoptosis, and serves an
important role in the occurrence and development of tumors
(120). It has been confirmed
that inhibition of PI3K/Akt can suppress tumor cell proliferation
and induce apoptosis in vitro and in vivo. Certain
inhibitors of PI3K/Akt have been applied in clinical trials
(121). The efficacy of PI3K
inhibition can also derive from interfering with the ability of
cancer cells to respond to stromal signals, as illustrated by the
approved PI3Kδ inhibitor Idelalisib in B-cell malignancies
(121). Inhibition of the
leukocyte-enriched PI3Kδ or PI3Kγ may unleash more potent antitumor
T-cell responses by inhibiting regulatory T cells and
immune-suppressive myeloid cells. In addition, tumor angiogenesis
may be targeted by PI3K inhibitors to enhance cancer therapy.
Chronic mountain sickness is characterized by erythrocytosis with
or without pulmonary hypertension (122). Whether the PI3K/Akt signaling
pathway is involved in erythrocytosis and chronic hypoxia-induced
glucose metabolism requires further study, in order to provide
novel directions for the diagnosis or prevention of chronic
mountain sickness. Inhibitors of the PI3K/Akt signaling pathway may
prevent overproduction of red blood cells (123,124).
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (grant no. 81360084), the project of
2014 Qinghai Talent ‘Xiao Gao Di’ and the National Key Disciplines
(hematology) of Qinghai Provincial People's Hospital (grant no.
Ministry of Finance and Public Health 2011-170), the guiding
project of Qinghai Provincial Health and Family Planning Commission
(2018-wjzdx-21).
Availability of data and materials
Not applicable.
Authors' contributions
JL and YG designed the study. YX, XS, KS, GH, WL,
QZ, BJ and JF wrote the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
Authors declare that they have no competing
interests.
References
|
1
|
King D, Yeomanson D and Bryant HE: PI3King
the lock: Targeting the PI3K/Akt/mTOR pathway as a novel
therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol.
37:245–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peltier J, O'Neill A and Schaffer DV:
PI3K/Akt and CREB regulate adult neural hippocampal progenitor
proliferation and differentiation. Dev Neurobiol. 67:1348–1361.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rafalski VA and Brunet A: Energy
metabolism in adult neural stem cell fate. Prog Neurobiol.
93:182–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Man HY, Wang Q, Lu WY, Ju W, Ahmadian G,
Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, et al: Activation
of PI3-kinase is required for AMPA receptor insertion during LTP of
mEPSCs in cultured hippocampal neurons. Neuron. 38:611–624. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ojeda L, Gao J, Hooten KG, Wang E,
Thonhoff JR, Dunn TJ, Gao T and Wu P: Critical role of
PI3K/Akt/GSK3β in motoneuron specification from human neural stem
cells in response to FGF2 and EGF. PLoS One. 6:e234142011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wyatt LA, Filbin MT and Keirstead HS: PTEN
inhibition enhances neurite outgrowth in human embryonic stem
cell-derived neuronal progenitor cells. J Comp Neurol.
522:2741–2755. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fruman DA, Meyers RE and Cantley LC:
Phosphoinositide kinases. Annu Rev Biochem. 67:481–507. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Courtney KD, Corcoran RB and Engelman JA:
The PI3K pathway as drug target in human cancer. J Clin Oncol.
28:1075–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Breitkopf SB, Yang X, Begley MJ, Kulkarni
M, Chiu YH, Turke AB, Lauriol J, Yuan M, Qi J, Engelman JA, et al:
A cross-species study of PI3K protein-protein interactions reveals
the direct interaction of P85 and SHP2. Sci Rep. 6:204712016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: Variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Katso R, Okkenhaug K, Ahmadi K, White S,
Timms J and Waterfield MD: Cellular function of phosphoinositide
3-kinases: Implications for development, homeostasis, and cancer.
Annu Rev Cell Dev Biol. 17:615–675. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Amzel LM, Huang CH, Mandelker D, Lengauer
C, Gabelli SB and Vogelstein B: Structural comparisons of class I
phosphoinositide 3-kinases. Nat Rev Cancer. 8:665–669. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schauder C, Ma LC, Krug RM, Montelione GT
and Guan R: Structure of the iSH2 domain of human
phosphatidylinositol 3-kinase p85β subunit reveals conformational
plasticity in the interhelical turn region. Acta Crystallogr Sect F
Struct Biol Cryst Commun. 66:1567–1571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Falasca M and Maffucci T: Role of class II
phosphoinositide 3-kinase in cell signalling. Biochem Soc Trans.
35:211–214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Backer JM: The regulation and function of
Class III PI3Ks: Novel roles for Vps34. Biochem J. 410:1–17. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bohdanowicz M, Cosío G, Backer JM and
Grinstein S: Class I and class III phosphoinositide 3-kinases are
required for actin polymerization that propels phagosomes. J Cell
Biol. 191:999–1012. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Staal SP and Hartley JW: Thymic lymphoma
induction by the AKT8 murine retrovirus. J Exp Med. 167:1259–1264.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coffer PJ, Jin J and Woodgett JR: Protein
kinase B (c-Akt): A multifunctional mediator of
phosphatidylinositol 3-kinase activation. Biochem J. 335:1–13.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Woodgett JR: Recent advances in the
protein kinase B signaling pathway. Curr Opin Cell Biol.
17:150–157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Andrade MA and Bork P: HEAT repeats in the
Huntington's disease protein. Nat Genet. 11:115–116. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jacinto E and Hall MN: Tor signalling in
bugs, brain and brawn. Nat Rev Mol Cell Biol. 4:117–126. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peterson RT, Beal PA, Comb MJ and
Schreiber SL: FKBP12-rapamycin-associated protein (FRAP)
autophosphorylates at serine 2481 under translationally repressive
conditions. J Biol Chem. 275:7416–7423. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Du K and Tsichlis PN: Regulation of the
Akt kinase by interacting proteins. Oncogene. 24:7401–7409. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carnero A, Blanco-Aparicio C, Renner O,
Link W and Leal JF: The PTEN/PI3K/AKT signalling pathway in cancer,
therapeutic implications. Curr Cancer Drug Targets. 8:187–198.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tokunaga E, Oki E, Egashira A, Sadanaga N,
Morita M, Kakeji Y and Maehara Y: Deregulation of the Akt pathway
in human cancer. Curr Cancer Drug Targets. 8:27–36. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Manning BD and Toker A: AKT/PKB Signaling:
Navigating the network. Cell. 169:381–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yip CK, Murata K, Walz T, Sabatini DM and
Kang SA: Structure of the human mTOR complex I and its implications
for rapamycin inhibition. Mol Cell. 38:768–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ward SG and Finan P: Isoform-specific
phosphoinositide 3-kinase inhibitors as therapeutic agents. Curr
Opin Pharmacol. 3:426–434. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yokota J, Chosa N, Sawada S, Okubo N,
Takahashi N, Hasegawa T, Kondo H and Ishisaki A: PDGF-induced
PI3K-mediated signaling enhances the TGF-β-induced osteogenic
differentiation of human mesenchymal stem cells in a
TGF-β-activated MEK-dependent manner. Int J Mol Med. 33:534–542.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma X and Bai Y: IGF-1 activates the
P13K/AKT signaling pathway via upregulation of secretory clusterin.
Mol Med Rep. 6:1433–1437. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dudu V, Able RA Jr, Rotari V, Kong Q and
Vazquez M: Role of epidermal growth factor-triggered PI3K/Akt
signaling in the migration of medulloblastoma-derived cells. Cell
Mol Bioeng. 5:413–502. 2012. View Article : Google Scholar
|
|
35
|
Osaki M, Oshimura M and Ito H: PI3K-Akt
pathway: Its functions and alterations in human cancer. Apoptosis.
9:667–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Geltz NR and Augustine JA: The p85 and
p110 subunits of phosphatidylinositol 3-kinase-alpha are
substrates, in vitro, for a constitutively associated protein
tyrosine kinase in platelets. Blood. 91:930–939. 1998.PubMed/NCBI
|
|
37
|
Kang BH, Shim YJ, Tae YK, Song JA, Choi
BK, Park IS and Min BH: Clusterin stimulates the chemotactic
migration of macrophages through a pertussis toxin sensitive
G-protein-coupled receptor and Gβγ-dependent pathways. Biochem
Biophys Res Commun. 445:645–650. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hresko RC and Mueckler M: mTOR RICTOR is
the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J
Biol Chem. 280:40406–40416. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tang JM, He QY, Guo RX and Chang XJ:
Phosphorylated Akt overexpression and loss of PTEN expression in
non-small cell lung cancer confers poor prognosis. Lung Cancer.
51:181–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wishart MJ and Dixon JE: PTEN and
myotubularin phosphatases: From 3-phosphoinositide
dephosphorylation to disease. Trends Cell Biol. 12:579–585. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stiles BL, Kuralwalla-Martinez C, Guo W,
Gregorian C, Wang Y, Tian J, Magnuson MA and Wu H: Selective
deletion of Pten in pancreatic beta cells leads to increased islet
mass and resistance to STZ-induced diabetes. Mol Cell Biol.
26:2772–2781. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nguyen KT, Tajmir P, Lin CH, Liadis N, Zhu
XD, Eweida M, Tolasa-Karaman G, Cai F, Wang R, Kitamura T, et al:
Essential role of Pten in body size determination and pancreatic
beta-cell homeostasis in vivo. Mol Cell Biol. 26:4511–4518. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao S, Fu J, Liu F, Rastogi R, Zhang J
and Zhao Y: Small interfering RNA directed against CTMP reduces
acute traumatic brain injury in a mouse model by activating Akt.
Neurol Res. 36:483–490. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang GL and Semenza GL: Purification and
characterization of hypoxia-inducible factor 1. J Biol Chem.
270:1230–1237. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci
USA. 92:5510–5514. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Semenza GL: Hypoxia-inducible factor 1 and
cancer pathogenesis. IUBMB Life. 60:591–597. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Loor G and Schumacker PT: Role of
hypoxia-inducible factor in cell survival during myocardial
ischemia-reperfusion. Cell Death Differ. 15:686–690. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jiang BH, Zheng JZ, Leung SW, Roe R and
Semenza GL: Transactivation and inhibitory domains of
hypoxia-inducible factor 1alpha. Modulation of transcriptional
activity by oxygen tension. J Biol Chem. 272:19253–19260. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Adams JM, Difazio LT, Rolandelli RH, Luján
JJ, Haskó G, Csóka B, Selmeczy Z and Németh ZH: HIF-1: A key
mediator in hypoxia. Acta Physiol Hung. 96:19–28. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lendahl U, Lee KL, Yang H and Poellinger
L: Generating specificity and diversity in the transcriptional
response to hypoxia. Nat Rev Genet. 10:821–832. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kaelin WG Jr and Ratcliffe PJ: Oxygen
sensing by metazoans: The central role of the HIF hydroxylase
pathway. Mol Cell. 30:393–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Peet DJ, Lando D, Whelan DA, Whitelaw ML
and Gorman JJ: Oxygen-dependent asparagine hydroxylation. Methods
Enzymol. 381:467–487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kaelin WG: Proline hydroxylation and gene
expression. Annu Rev Biochem. 74:115–128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kondo K and Kaelin WG Jr: The von
Hippel-Lindau tumor suppressor gene. Exp Cell Res. 264:117–125.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Arjumand W and Sultana S: Role of VHL gene
mutation in human renal cell carcinoma. Tumour Biol. 33:9–16. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Niu G, Briggs J, Deng J, Ma Y, Lee H,
Kortylewski M, Kujawski M, Kay H, Cress WD, Jove R and Yu H: Signal
transducer and activator of transcription 3 is required for
hypoxia-inducible factor-1alpha RNA expression in both tumor cells
and tumor-associated myeloid cells. Mol Cancer Res. 6:1099–1105.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fisher TS, Etages SD, Hayes L, Crimin K
and Li B: Analysis of ARD1 function in hypoxia response using
retroviral RNA interference. J Biol Chem. 280:17749–17757. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sandau KB, Fandrey J and Brüne B:
Accumulation of HIF-1alpha under the influence of nitric oxide.
Blood. 97:1009–1015. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kasuno K, Takabuchi S, Fukuda K,
Kizaka-Kondoh S, Yodoi J, Adachi T, Semenza GL and Hirota K: Nitric
oxide induces hypoxia-inducible factor 1 activation that is
dependent on MAPK and phosphatidylinositol 3-kinase signaling. J
Biol Chem. 279:2550–2558. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Park YK, Ahn DR, Oh M, Lee T, Yang EG, Son
M and Park H: Nitric oxide donor,
(+/-)-S-nitroso-N-acetylpenicillamine, stabilizes transactive
hypoxia-inducible factor-1alpha by inhibiting von Hippel-Lindau
recruitment and asparagine hydroxylation. Mol Pharmacol.
74:236–245. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sogawa K, Numayama-Tsuruta K, Ema M, Abe
M, Abe H and Fujii-Kuriyama Y: Inhibition of hypoxia-inducible
factor 1 activity by nitric oxide donors in hypoxia. Proc Natl Acad
Sci USA. 95:7368–7373. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Brix B, Mesters JR, Pellerin L and Jöhren
O: Endothelial cell-derived nitric oxide enhances aerobic
glycolysis in astrocytes via HIF-1α-mediated target gene
activation. J Neurosci. 32:9727–9735. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jung YJ, Isaacs JS, Lee S, Trepel J and
Neckers L: IL-1beta-mediated up-regulation of HIF-1alpha via an
NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between
inflammation and oncogenesis. FASEB J. 17:2115–2117. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bárdos JI, Chau NM and Ashcroft M: Growth
factor-mediated induction of HDM2 positively regulates
hypoxia-inducible factor 1alpha expression. Mol Cell Biol.
24:2905–2914. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Laughner E, Taghavi P, Chiles K, Mahon PC
and Semenza GL: HER2 (neu) signaling increases the rate of
hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel
mechanism for HIF-1-mediated vascular endothelial growth factor
expression. Mol Cell Biol. 21:3995–4004. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhong H, Chiles K, Feldser D, Laughner E,
Hanrahan C, Georgescu MM, Simons JW and Semenza GL: Modulation of
hypoxia-inducible factor 1alpha expression by the epidermal growth
factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human
prostate cancer cells: implications for tumor angiogenesis and
therapeutics. Cancer Res. 60:1541–1545. 2000.PubMed/NCBI
|
|
70
|
Pagé EL, Robitaille GA, Pouysségur J and
Richard DE: Induction of hypoxia-inducible factor-1alpha by
transcriptional and translational mechanisms. J Biol Chem.
277:48403–48409. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Isakoff SJ, Cardozo T, Andreev J, Li Z,
Ferguson KM, Abagyan R, Lemmon MA, Aronheim A and Skolnik EY:
Identification and analysis of PH domain-containing targets of
phosphatidylinositol 3-kinase using a novel in vivo assay in yeast.
EMBO J. 17:5374–5387. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Karar J and Maity A: Modulating the tumor
microenvironment to increase radiation responsiveness. Cancer Biol
Ther. 8:1994–2001. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zundel W, Schindler C, Haas-Kogan D, Koong
A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson
AB, et al: Loss of PTEN facilitates HIF-1-mediated gene expression.
Genes Dev. 14:391–396. 2000.PubMed/NCBI
|
|
74
|
Jiang BH, Zheng JZ, Aoki M and Vogt PK:
Phosphatidylinositol 3-kinase signaling mediates angiogenesis and
expression of vascular endothelial growth factor in endothelial
cells. Proc Natl Acad Sci USA. 97:1749–1753. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter
T and Vogt PK: Phosphatidylinositol 3-kinase signaling controls
levels of hypoxia-inducible factor 1. Cell Growth Differ.
12:363–369. 2001.PubMed/NCBI
|
|
76
|
Mazure NM, Chen EY, Laderoute KR and
Giaccia AJ: Induction of vascular endothelial growth factor by
hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt
signaling pathway in Ha-ras-transformed cells through a hypoxia
inducible factor-1 transcriptional element. Blood. 90:3322–3331.
1997.PubMed/NCBI
|
|
77
|
Blancher C, Moore JW, Robertson N and
Harris AL: Effects of ras and von Hippel-Lindau (VHL) gene
mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and
vascular endothelial growth factor expression and their regulation
by the phosphatidylinositol 3′-kinase/Akt signaling pathway. Cancer
Res. 61:7349–7355. 2001.PubMed/NCBI
|
|
78
|
Chen EY, Mazure NM, Cooper JA and Giaccia
AJ: Hypoxia activates a platelet-derived growth factor
receptor/phosphatidylinositol 3-kinase/Akt pathway that results in
glycogen synthase kinase-3 inactivation. Cancer Res. 61:2429–2433.
2001.PubMed/NCBI
|
|
79
|
Kietzmann T, Samoylenko A, Roth U and
Jungermann K: Hypoxia-inducible factor-1 and hypoxia response
elements mediate the induction of plasminogen activator inhibitor-1
gene expression by insulin in primary rat hepatocytes. Blood.
101:907–914. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tang TT and Lasky LA: The forkhead
transcription factor FOXO4 induces the down-regulation of
hypoxia-inducible factor 1 alpha by a von Hippel-Lindau
protein-independent mechanism. J Biol Chem. 278:30125–30135. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Arsham AM, Plas DR, Thompson CB and Simon
MC: Phosphatidylinositol 3-kinase/Akt signaling is neither required
for hypoxic stabilization of HIF-1 alpha nor sufficient for
HIF-1-dependent target gene transcription. J Biol Chem.
277:15162–15170. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Alvarez-Tejado M, Alfranca A, Aragonés J,
Vara A, Landázuri MO and del Peso L: Lack of evidence for the
involvement of the phosphoinositide 3-kinase/Akt pathway in the
activation of hypoxia-inducible factors by low oxygen tension. J
Biol Chem. 277:13508–13517. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Heath DS, Axelrad AA, McLeod DL and
Shreeve MM: Separation of the erythropoietin-responsive progenitors
BFU-E and CFU-E in mouse bone marrow by unit gravity sedimentation.
Blood. 47:777–792. 1976.PubMed/NCBI
|
|
84
|
Fader CM and Colombo MI: Multivesicular
bodies and autophagy in erythrocyte maturation. Autophagy.
2:122–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Swiers G, Patient R and Loose M: Genetic
regulatory networks programming hematopoietic stem cells and
erythroid lineage specification. Dev Biol. 294:525–540. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wickrema A and Crispino JD: Erythroid and
megakaryocytic transformation. Oncogene. 26:6803–6815. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Brahimi-Horn C and Pouysségur J: The role
of the hypoxia-inducible factor in tumor metabolism growth and
invasion. Bull Cancer. 93:E73–E80. 2006.PubMed/NCBI
|
|
88
|
Lee JW, Bae SH, Jeong JW, Kim SH and Kim
KW: Hypoxia-inducible factor (HIF-1)alpha: Its protein stability
and biological functions. Exp Mol Med. 36:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Holmquist-Mengelbier L, Fredlund E,
Löfstedt T, Noguera R, Navarro S, Nilsson H, Pietras A,
Vallon-Christersson J, Borg A, Gradin K, et al: Recruitment of
HIF-1alpha and HIF-2alpha to common target genes is differentially
regulated in neuroblastoma: HIF-2alpha promotes an aggressive
phenotype. Cancer Cell. 10:413–423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lee FS: Genetic causes of erythrocytosis
and the oxygen-sensing pathway. Blood Rev. 22:321–332. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
León-Velarde F, Monge CC, Vidal A,
Carcagno M, Criscuolo M and Bozzini CE: Serum immunoreactive
erythropoietin in high altitude natives with and without excessive
erythrocytosis. Exp Hematol. 19:257–260. 1991.PubMed/NCBI
|
|
92
|
Oshima K, Ikeda Y, Horinouchi Y, Watanabe
H, Hamano H, Kihira Y, Kishi S, Izawa-Ishizawa Y, Miyamoto L,
Hirayama T, et al: Iron suppresses erythropoietin expression via
oxidative stress-dependent hypoxia-inducible factor-2 alpha
inactivation. Lab Invest. 97:555–566. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Gupta N and Wish JB: Hypoxia-inducible
factor prolyl hydroxylase inhibitors: A potential new treatment for
anemia in patients with CKD. Am J Kidney Dis. 69:815–826. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Lee FS and Percy MJ: The HIF pathway and
erythrocytosis. Annu Rev Pathol. 6:165–192. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Prchal JT and Sokol L: ‘Benign
erythrocytosis’ and other familial and congenital polycythemias.
Eur J Haematol. 57:263–268. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Patnaik MM and Tefferi A: The complete
evaluation of erythrocytosis: Congenital and acquired. Leukemia.
23:834–844. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Myllymäki MN, Määttä J, Dimova EY, Izzi V,
Väisänen T, Myllyharju J, Koivunen P and Serpi R: Notch
downregulation and extramedullary erythrocytosis in
hypoxia-inducible factor prolyl 4-hydroxylase 2-deficient mice. Mol
Cell Biol. 37(pii): e00529–16. 2017.PubMed/NCBI
|
|
98
|
Tashi T, Scott Reading N, Wuren T, Zhang
X, Moore LG, Hu H, Tang F, Shestakova A, Lorenzo F, Burjanivova T,
et al: Gain-of-function EGLN1 prolyl hydroxylase (PHD2 D4E:C127S)
in combination with EPAS1 (HIF-2α) polymorphism lowers hemoglobin
concentration in Tibetan highlanders. J Mol Med (Berl). 95:665–670.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Inkster B, Zai G, Lewis G and Miskowiak
KW: GSK3β: A plausible mechanism of cognitive and hippocampal
changes induced by erythropoietin treatment in mood disorders.
Transl Psychiatry. 8:2162018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
van der Vaart A, Meng X, Bowers MS, Batman
AM, Aliev F, Farris SP, Hill JS, Green TA, Dick D; COGA Consortium,
; et al: Glycogen synthase kinase 3 beta regulates ethanol
consumption and is a risk factor for alcohol dependence.
Neuropsychopharmacology. 2018.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Sopjani M, Millaku L, Nebija D, Emini M,
Dermaku-Sopjani M and Rifati-Nixha A: The glycogen synthase
kinase-3 in the regulation of ion channels and cellular carriers.
Curr Med Chem. Oct 9–2018.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Frame S and Cohen P: GSK3 takes centre
stage more than 20 years after its discovery. Biochem J. 359:1–16.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Dokken BB, Sloniger JA and Henriksen EJ:
Acute selective glycogen synthase kinase-3 inhibition enhances
insulin signaling in prediabetic insulin-resistant rat skeletal
muscle. Am J Physiol Endocrinol Metab. 288:E1188–E1194. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Secades P, de Santa-María IS, Merlo A,
Suarez C and Chiara MD: In vitro study of normoxic epidermal growth
factor receptor-induced hypoxia-inducible factor-1-alpha, vascular
endothelial growth factor, and BNIP3 expression in head and neck
squamous cell carcinoma cell lines: Implications for anti-epidermal
growth factor receptor therapy. Head Neck. 37:1150–1162. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Park ST, Kim BR, Park SH, Lee JH, Lee EJ,
Lee SH and Rho SB: Suppression of VEGF expression through
interruption of the HIF-1α and Akt signaling cascade modulates the
anti-angiogenic activity of DAPK in ovarian carcinoma cells. Oncol
Rep. 31:1021–1029. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kitamura K, Kangawa K, Matsuo H and Uyeda
K: Phosphorylation of myocardial fructose-6-phosphate,2-kinase:
fructose-2,6-bisphosphatase by cAMP-dependent protein kinase and
protein kinase C. Activation by phosphorylation and amino acid
sequences of the phosphorylation sites. J Biol Chem.
263:16796–16801. 1988.PubMed/NCBI
|
|
107
|
Deprez J, Vertommen D, Alessi DR, Hue L
and Rider MH: Phosphorylation and activation of heart
6-phosphofructo-2-kinase by protein kinase B and other protein
kinases of the insulin signaling cascades. J Biol Chem.
272:17269–17275. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bertrand L, Alessi DR, Deprez J, Deak M,
Viaene E, Rider MH and Hue L: Heart 6-phosphofructo-2-kinase
activation by insulin results from Ser-466 and Ser-483
phosphorylation and requires 3-phosphoinositide-dependent kinase-1,
but not protein kinase B. J Biol Chem. 274:30927–30933. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Depre C, Rider MH, Veitch K and Hue L:
Role of fructose 2,6-bisphosphate in the control of heart
glycolysis. J Biol Chem. 268:13274–13279. 1993.PubMed/NCBI
|
|
110
|
Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ,
Kim JW, Park SW and Kim KS: Androgen stimulates glycolysis for de
novo lipid synthesis by increasing the activities of hexokinase 2
and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in
prostate cancer cells. Biochem J. 433:225–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Agani F and Jiang BH: Oxygen-independent
regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in
cancer. Curr Cancer Drug Targets. 13:245–251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Moench R, Grimmig T, Kannen V, Tripathi S,
Faber M, Moll EM, Chandraker A, Lissner R, Germer CT, Waaga-Gasser
AM and Gasser M: Exclusive inhibition of PI3K/Akt/mTOR signaling is
not sufficient to prevent PDGF-mediated effects on glycolysis and
proliferation in colorectal cancer. Oncotarget. 7:68749–68767.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Su J, Gao T, Jiang M, Wu L, Zeng W, Zhao
S, Peng C and Chen X: CD147 silencing inhibits tumor growth by
suppressing glucose transport in melanoma. Oncotarget.
7:64778–64784. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zeng L, Zhou HY, Tang NN, Zhang WF, He GJ,
Hao B, Feng YD and Zhu H: Wortmannin influences hypoxia-inducible
factor-1 alpha expression and glycolysis in esophageal carcinoma
cells. World J Gastroenterol. 22:4868–4880. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Mediani L, Gibellini F, Bertacchini J,
Frasson C, Bosco R, Accordi B, Basso G, Bonora M, Calabrò ML,
Mattiolo A, et al: Reversal of the glycolytic phenotype of primary
effusion lymphoma cells by combined targeting of cellular
metabolism and PI3K/Akt/ mTOR signaling. Oncotarget. 7:5521–5537.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mulquiney PJ, Bubb WA and Kuchel PW: Model
of 2,3-bisphosphoglycerate metabolism in the human erythrocyte
based on detailed enzyme kinetic equations: In vivo kinetic
characterization of 2,3-bisphosphoglycerate synthase/phosphatase
using 13C and 31P NMR. Biochem J 342 Pt. 3:567–580. 1999.
View Article : Google Scholar
|
|
117
|
Benesch R, Benesch RE and Yu CI:
Reciprocal binding of oxygen and diphosphoglycerate by human
hemoglobin. Proc Natl Acad Sci USA. 59:526–532. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Narita H, Yanagawa S, Sasaki R and Chiba
H: Synthesis of 2,3-bisphosphoglycerate synthase in erythroid
cells. J Biol Chem. 256:7059–7063. 1981.PubMed/NCBI
|
|
119
|
Lemarchandel V, Joulin V, Valentin C, Rosa
R, Galactéros F, Rosa J and Cohen-Solal M: Compound heterozygosity
in a complete erythrocyte bisphosphoglycerate mutase deficiency.
Blood. 80:2643–2649. 1992.PubMed/NCBI
|
|
120
|
Spangle JM, Roberts TM and Zhao JJ: The
emerging role of PI3K/AKT-mediated epigenetic regulation in cancer.
Biochim Biophys Acta Rev Cancer. 1868:123–131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Okkenhaug K, Graupera M and Vanhaesebroeck
B: Targeting PI3K in cancer: Impact on tumor cells, their
protective stroma, angiogenesis, and immunotherapy. Cancer Discov.
6:1090–1105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Villafuerte FC and Corante N: Chronic
mountain sickness: Clinical aspects, etiology, management, and
treatment. High Alt Med Biol. 17:61–69. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Hermida MA, Dinesh Kumar J and Leslie NR:
GSK3 and its interactions with the PI3K/AKT/mTOR signalling
network. Adv Biol Regul. 65:5–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Li N, Zhou H and Tang Q: miR-133: A
suppressor of cardiac remodeling? Front Pharmacol. 9:9032018.
View Article : Google Scholar : PubMed/NCBI
|