Introduction
Spinal tumors are rare and, based on their location,
can be classified into various subtypes, such as intramedullary and
extramedullary tumors (1).
Ependymoma is a type of intramedullary tumor that tends to occur in
the spinal cord in adults (2).
Ependymoma is the most common type of spinal cord tumor and has
significant impacts on the quality of life of patients, and may
cause mortality (3). Classifying
ependymal tumors has represented a challenge, as the current
grading systems do not accurately describe their clinical
characteristics (4,5). However, previous studies have found
that DNA methylation patterns are reliable biomarkers for the
classification of different molecular subtypes of spinal ependymal
tumors (6).
Improved detection methods and the identification of
disease-associated biomarkers have been shown to facilitate the
diagnosis and treatment of patients (7–11).
DNA methylation is one of the most studied epigenetic modifications
in mammals (12,13). Notably, aberrant methylation is
associated with cancer and aging (14,15).
Some DNA methylation markers have been used in commercially
available clinical tests, and most of the sites of methylation are
located in the promoters of genes (16). Despite several previous studies
suggesting that DNA methylation is a reliable molecular biomarker
in ependymal tumors, to the best of our knowledge, the role of
methylation in long non-coding RNA (lncRNA) genes has not been
investigated (4,6). lncRNAs are >200 nucleotides in
length and do not encode proteins. Similarly to mRNAs, lncRNAs have
their own promoters (17), are
transcribed by RNA polymerase II and have a polyadenylated tail
(18). An increasing number of
studies have identified various roles of lncRNAs in multiple
biological processes and diseases through various mechanisms
(19,20). Previous studies have demonstrated
that lncRNAs can serve as biomarkers for various cancer features
(21). Recent studies have
provided insight on the mechanism underlying DNA methylation of
lncRNA genes in carcinogenesis (22,23).
However, to the best of our knowledge, a systematic investigation
of the DNA methylation status and the function of lncRNA genes in
spinal cord ependymal tumor has yet to be reported.
In the present study, the DNA methylation landscape
of lncRNAs in spinal cord ependymal tumors was investigated and
certain lncRNAs were identified to have distinct DNA methylation
states among various tumor subtypes. Additionally, various tumor
subtype-specific lncRNAs were identified to be involved in spinal
cord development. In the present study, a functional
characterization of lncRNAs was performed using lncRNA-protein
interaction data, and a random forest algorithm was used to
identify 30 lncRNAs with high classification efficiency. Notably,
the majority of subtype-specific lncRNAs was identified to be
hypomethylated. A subset of the identified lncRNAs was found to
have potential roles in cancer and nervous system development.
Furthermore, a functional analysis identified the role of
subtype-specific lncRNAs in spinal cord ependymal tumors. To the
best of our knowledge, the present study is the first to
investigate the DNA methylation status of lncRNA genes in order to
identify their clinical significance in various molecular subtypes
of spinal cord ependymal tumors.
Materials and methods
Methylation and lncRNA data
collection
Whole-genome DNA methylation data from spinal cord
ependymal tumors were downloaded from Gene Expression Omnibus
version 1 (24) [accession no.
GSE65362 (4)]. Specifically, these
samples were classified into three classes: i) Spinal ependymoma
(SP-EPN); ii) spinal myxopapillary EPN (SP-MPE); and iii) spinal
subependymoma (SP-SE). lncRNA annotation data were downloaded from
GENCODE (version 28) (25). These
lncRNAs were classified into nine groups based on their location
and features (http://vega.archive.ensembl.org/info/about/gene_and_transcript_types.html)
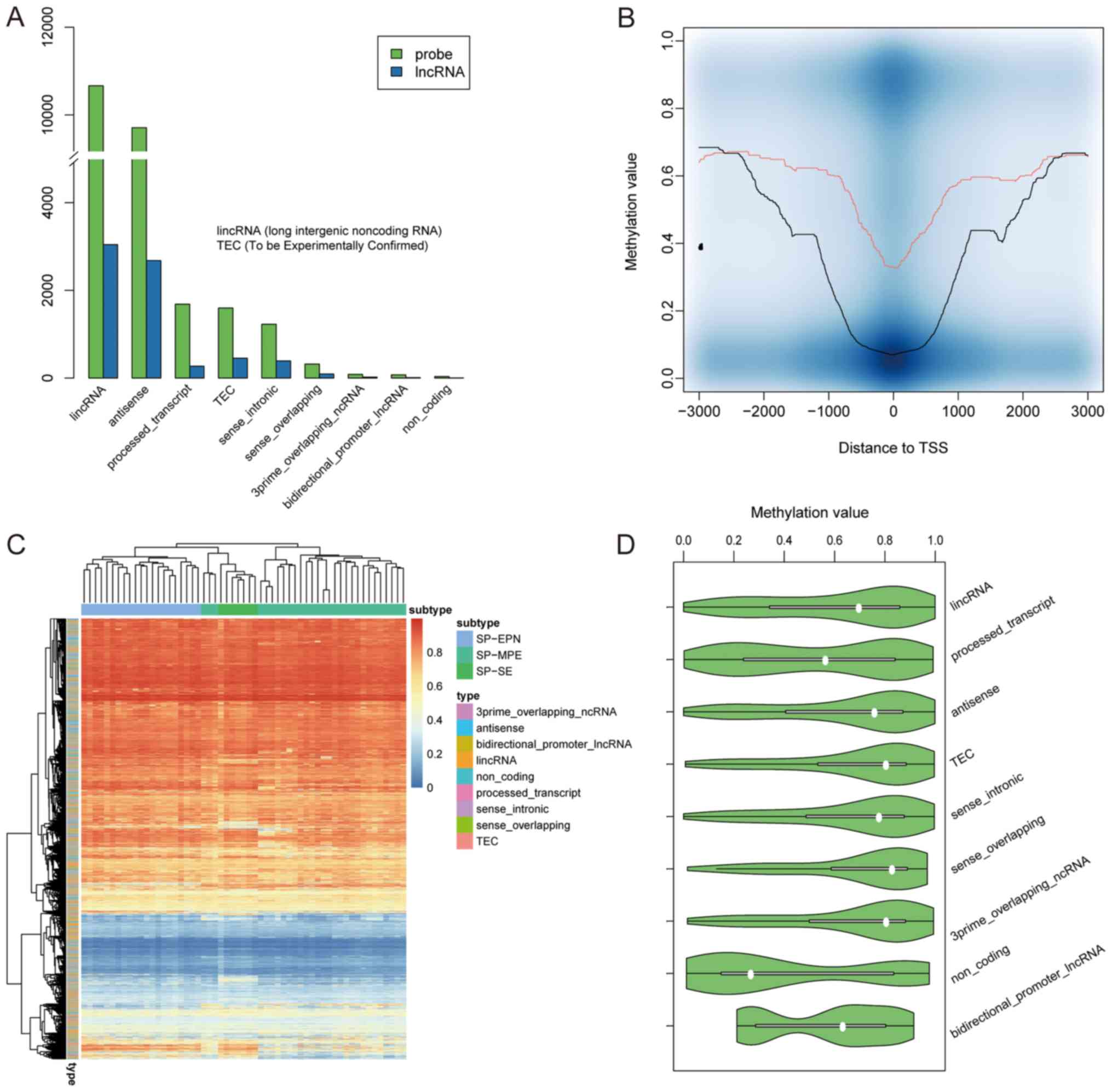
(Fig. 1A). Notably, the To be
Experimentally Confirmed (TEC) category has been specifically
created for the ENCODE project to indicate regions that could
indicate the presence of protein coding genes (PCGs) that require
experimental validation (25).
Importantly, Ensembl (https://www.ensembl.org/info/genome/genebuild/biotypes.html)
and Vega (http://vega.archive.ensembl.org/info/about/gene_and_transcript_types.html)
databases classify TEC genes as lncRNAs.
Mapping methylation probes to lncRNA
promoters
The genomic locations of the Infinium Human
Methylation 450 (HM450) BeadChip (Illumina, Inc.) probes based on
GRCh37 were converted to the genomic locations in GRCh38 (The
Genome Reference Consortium; http://www.ncbi.nlm.nih.gov/grc) using the University
of California, Santa Cruz (UCSC) Batch Coordinate Conversion
liftOver tool version 2 (26).
Probes exhibiting single nucleotide polymorphisms (SNPs) >5 bp
from their 3′-end and probes with non-unique 3′-subsequences of 30
bases were excluded, as previously described (27). lncRNA promoter regions were defined
as 3-kb windows from either side of the transcription start site
(TSS), as previously described (22). Subsequently, the methylation probes
were mapped to the lncRNA promoter regions using BEDtools version
2.24.0 (28). The same approach
was used to map the PCG probes. In addition, methylation probes
that simultaneously mapped to lncRNAs and PCGs were excluded.
lncRNA DNA methylation in spinal cord
ependymal tumors
lncRNA promoter methylation values were calculated
as the mean values of all probes located in the corresponding
promoter. DNA methylation patterns around TSSs were calculated in
100 bp windows based on the median methylation value across
samples. The lncRNA methylation value was used to cluster samples
using the pheatmap package (version 1.0.12; http://cran.r-project.org/web/packages/pheatmap/index.html),
using the default parameters, on R (version 3.3.0; http://cran.r-project.org/). The similarity between
tumor samples was measured by Pearson correlation coefficients
using R to determine if tumors within a subtype were more similar
to each other than those from other subtypes. In addition,
principal component analysis was used to investigate the
methylation patterns of different tumor subgroups. Tumor
subtype-specific lncRNAs were identified by ANOVA using R, as
previously described (29). The
RCircos package (version 1.2.1; http://cran.r-project.org/web/packages/RCircos/index.html)
was used with R to display the distribution of methylation levels
and the locations of tumor subtype-specific lncRNAs in the
genome.
Tumor subtype-specific lncRNA-protein
interaction network construction
RNA-protein interaction data were downloaded from
the RAID database (version 2.0) (30), which integrates experimental and
computational prediction interactions from the literature and other
database resources under one common framework. lncRNA-protein
interaction associations were calculated by mapping
subtype-specific lncRNAs to mRNAs.
Tumor subtype-specific lncRNA
signature identification
A random forest algorithm (31) was performed using the DNA
methylation value of the lncRNAs identified in the constructed
network. The lncRNAs with a feature importance value >0,
calculated using the random forest algorithm, were considered
potential tumor subtype-specific signatures (32). All statistical analyses were
performed using R (version 3.3.0; R project).
Functional enrichment analysis of
lncRNAs and PCGs
Tumor subtype-specific lncRNAs were annotated using
their promoter regions by the Genomic Regions Enrichment of
Annotations Tool (GREAT; version 3.3.0) using the default
parameters (33); in addition, the
lncRNA promoter regions were used as ‘background regions’.
Specifically, the genomic coordinates of the lncRNAs used in GREAT
were first converted into GRCh37 coordinates using the UCSC
liftover tool, as aforementioned. Kyoto Encyclopedia of Genes and
Genomes (KEGG; version 73.0) (34)
enrichment analysis was performed on the PCGs using the Enrichr
online tool (version 2.0), using the default parameters (35), and the significantly enriched KEGG
pathways were identified (P<0.01).
Results
Global DNA methylation patterns in
lncRNA promoters in spinal cord ependymal tumors
To characterize lncRNA methylation patterns, a
computational pipeline was used to annotate HM450 probes to lncRNA
promoters. In total, 485,506 HM450 probes were successfully
converted into GRCh38 coordinates, and 433,532 probes were obtained
after filtering for SNPs and copy number-associated probes. The
present analysis resulted in a set of 29,402 probes annotated in
6,967 lncRNA promoter regions (Fig.
1A). In total, 12,668 and 11,711 methylation probes were found
in the promoters of long intergenic noncoding RNAs (lincRNAs) and
antisense lncRNAs, respectively. Additionally, lncRNAs exhibited
methylation states similar to those of PCGs around TSSs; both had
low methylation values in proximity to the TSS and had higher
methylation values as the distance from the TSS increased (Fig. 1B). However, the overall methylation
level in lncRNAs was higher compared with PCGs (Fig. 1B). Cluster analysis suggested that
tumor samples from patients with the same molecular subtype had
similar methylation patterns in lncRNAs and that the methylation
levels of various lncRNAs in the same class were not similar
(Fig. 1C). Additionally, the
majority of lncRNAs identified in the present study had a
methylation value >0.4 (Fig.
1C). Certain lncRNAs, including those classified as
‘processed_transcript’ and ‘bidirectional_promoter_lncRNA’ lncRNAs,
exhibited bimodally distributed methylation levels in tumor
samples, whereas other lncRNAs, including ‘antisense’ and
‘sense_intronic’ lncRNAs, were hypermethylated in the majority of
the patients (Fig. 1D).
DNA methylation in lncRNAs is
correlated with histological characteristics of spinal cord
ependymal tumors
The patterns of DNA methylation in the lncRNA genes
were investigated in different tumor histopathological subtypes. In
total, there were 57 patients with spinal cord tumors, 29 (50.9%)
patients were in the SP-MPE group, whereas 21 (36.8%) and 7 (12.3%)
were in the SP-EPN and SP-SE group, respectively. DNA methylation
correlation values were calculated between each pair of samples,
and tumor samples were clustered using their methylation
correlation values. lncRNA methylation levels were found to be more
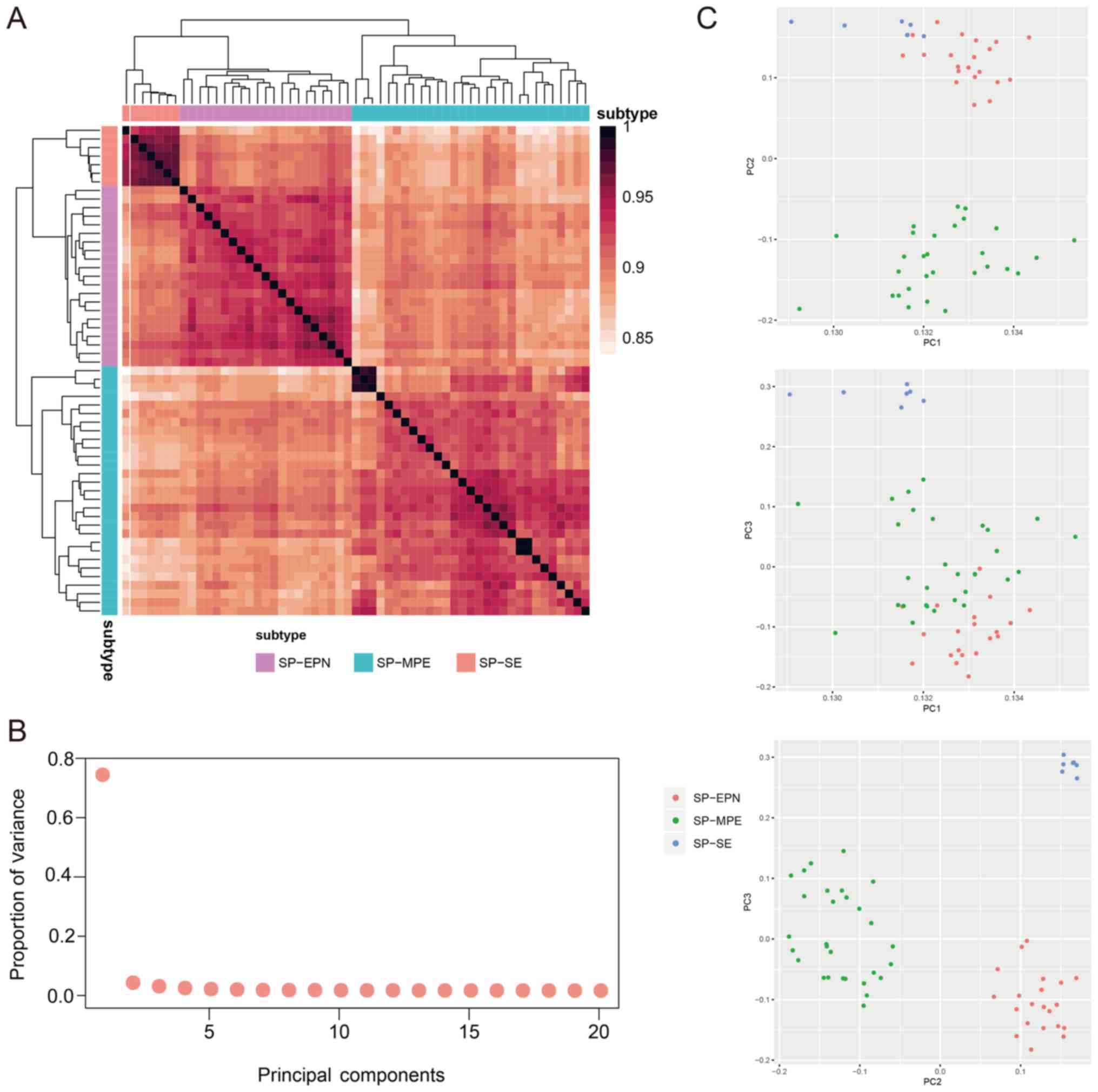
similar within groups than between groups (Fig. 2A). In particular, patients in the
SP-SE group exhibited the most consistent methylation status, with
a mean methylation correlation value of 0.96. The mean methylation
correlation values in the SP-EPN and SP-MPE were 0.93 and 0.92,
respectively. This effect may be due to the little tumor
heterogeneity among SE samples. According to the World Health
Organization classification (36,37),
SP-SE is considered a grade I tumor and its prognosis is more
favorable compared with the majority of ependymal tumors (36). In addition, principal component
analysis was performed to analyze the tumor samples based on the
DNA methylation of the lncRNAs. The first three principal
components had the highest proportion of variance values (Fig. 2B). These three components were used
to characterize the DNA methylation features in these tumor
subtypes, and the results suggested that patients with the same
tumor subtype were likely to have similar lncRNA methylation levels
(Fig. 2C). The present results
suggested that the DNA methylation in lncRNA genes was correlated
with histological characteristics of spinal cord ependymal
tumors.
DNA methylation-based detection of
tumor subtypes-specific lncRNAs
Tumor subtype-specific lncRNAs were identified based
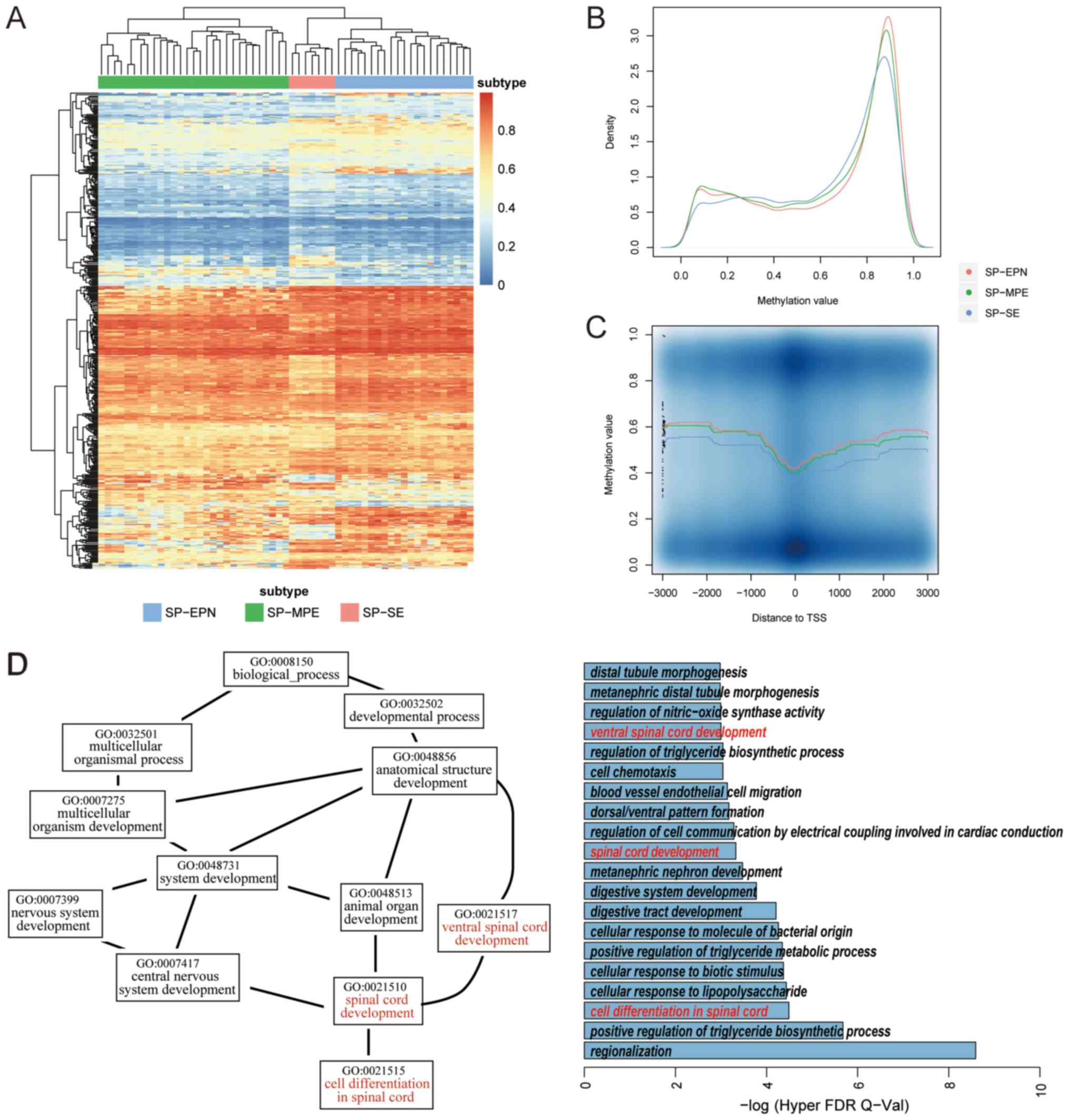
on their DNA methylation values using ANOVA. In total, 1,046
subtype-specific lncRNAs were identified [false discovery rate
(FDR) <0.05; Figs. 3 and
S1]. These lncRNAs tended to have
higher methylation values (Fig.
3A), and the majority had methylation values ~0.9. Furthermore,
the patients in the SP-EPN group had the highest methylation
levels, whereas the patients in the SP-SE had the lowest
methylation levels (Fig. 3B and
C). GREAT was used to investigate the potential mechanisms of
lncRNAs in spinal cord ependymal tumors. Among the significant Gene
Ontology (38) terms (FDR q-value
<0.01), three of them were associated with spinal cord
development (Fig. 3D). ‘Ventral
spinal cord development’ and ‘spinal cord development’ processes
can affect cell differentiation of spinal cord cells, and a
previous study demonstrated that cell differentiation is associated
with tumorigenesis (39).
Therefore, the distinct DNA methylation levels of various lncRNAs
may affect the expression levels of lncRNAs in each tumor subtype,
thus affecting genes associated with the nervous system and spinal
cord development. The present results indicated the role of lncRNAs
in human spinal cord development and suggested that they could be
lncRNA signatures in spinal cord ependymal tumors.
Tumor subtype-specific lncRNA
signature discovery
Biological networks represent the interactions
between molecules in vivo, and can be used to identify
regulatory pathways and processes (40,41).
lncRNAs may contribute to cancer by interacting with proteins
(42), and an increasing number of
studies have investigated lncRNA-protein interactions (43). lncRNA-protein interaction data in
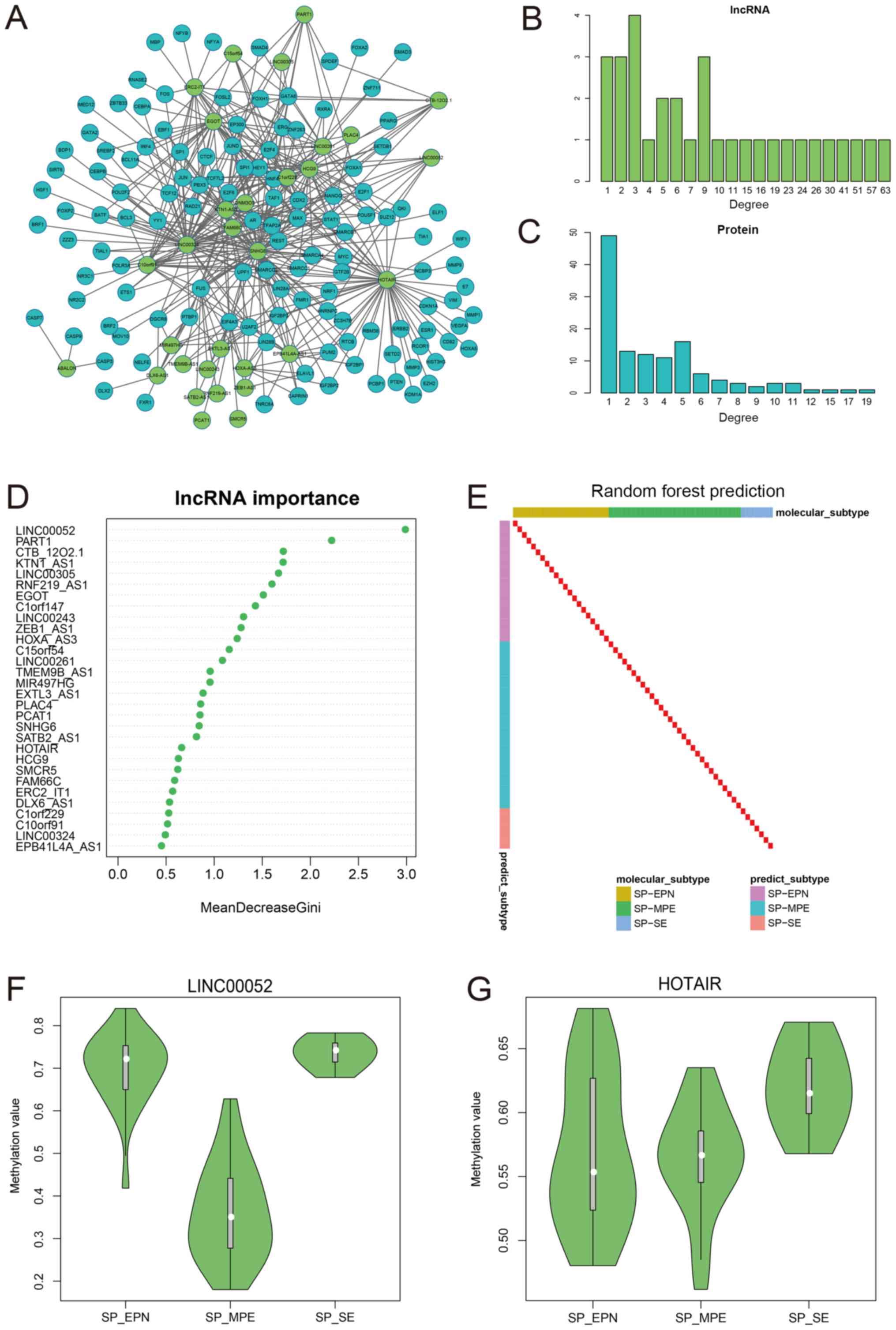
humans have been downloaded from the RAID database (30). In total, 32 subtype-specific
lncRNAs exhibited associations with proteins (Fig. 4A). The network degrees of lncRNAs
and proteins were separately analyzed (Fig. 4B and C). lncRNAs exhibited more
associations compared with proteins. The present results indicated
the important role of lncRNAs in interacting with proteins and
suggested that these 32 lncRNAs may affect the disease status by
affecting protein function in spinal cord ependymal tumors.
Furthermore, in order to identify the lncRNA signatures in
ependymal tumors, a random forest classification method was used.
In total, 30 lncRNAs with importance values >0, ranging between
0.48 and 2.98, were identified (Fig.
4D). Using the methylation values of these 30 lncRNAs, the
three tumor subtypes could be reliably distinguished with
sensitivity and specificity values of 1 (Fig. 4E). These lncRNAs may be potential
biomarkers associated with tumor subtypes, and may facilitate the
diagnosis and treatment of patients with spinal cord ependymal
tumors.
Long intergenic non-coding RNA 52 (LINC00052) is a
lincRNA, and exhibited the highest importance value in the
classification of the three tumor subtypes (Fig. 4D and F). LINC00052 has been
previously shown to promote breast cancer and hepatocarcinoma
development (44,45). HOX transcript antisense RNA
(HOTAIR) is an oncogenic lncRNA in multiple types of cancer,
including breast, gastric, colorectal and cervical cancer (46). Additionally, its DNA methylation
status can serve as a biomarker in primary ovarian cancer (47). HOTAIR has been investigated as a
prognostic factor in mesenchymal glioma, another type of tumor
affecting the nervous system (48). In the network constructed in the
present study, HOTAIR was identified to exhibit the features of a
hub. HOTAIR interacted with 51 proteins and displayed tumor
subtype-specific DNA methylation characteristics (Fig. 4A and G). The lncRNAs listed in
Fig. 4D may be potential novel
biomarkers for treating and diagnosing spinal cord ependymal
tumors. Analyzing the DNA methylation values of these lncRNAs in
patients with ependymal tumors may facilitate the classification of
the tumors and the development of personalized therapies.
Functional and epigenomic
characteristics suggest the roles of lncRNAs in spinal cord
ependymal tumors
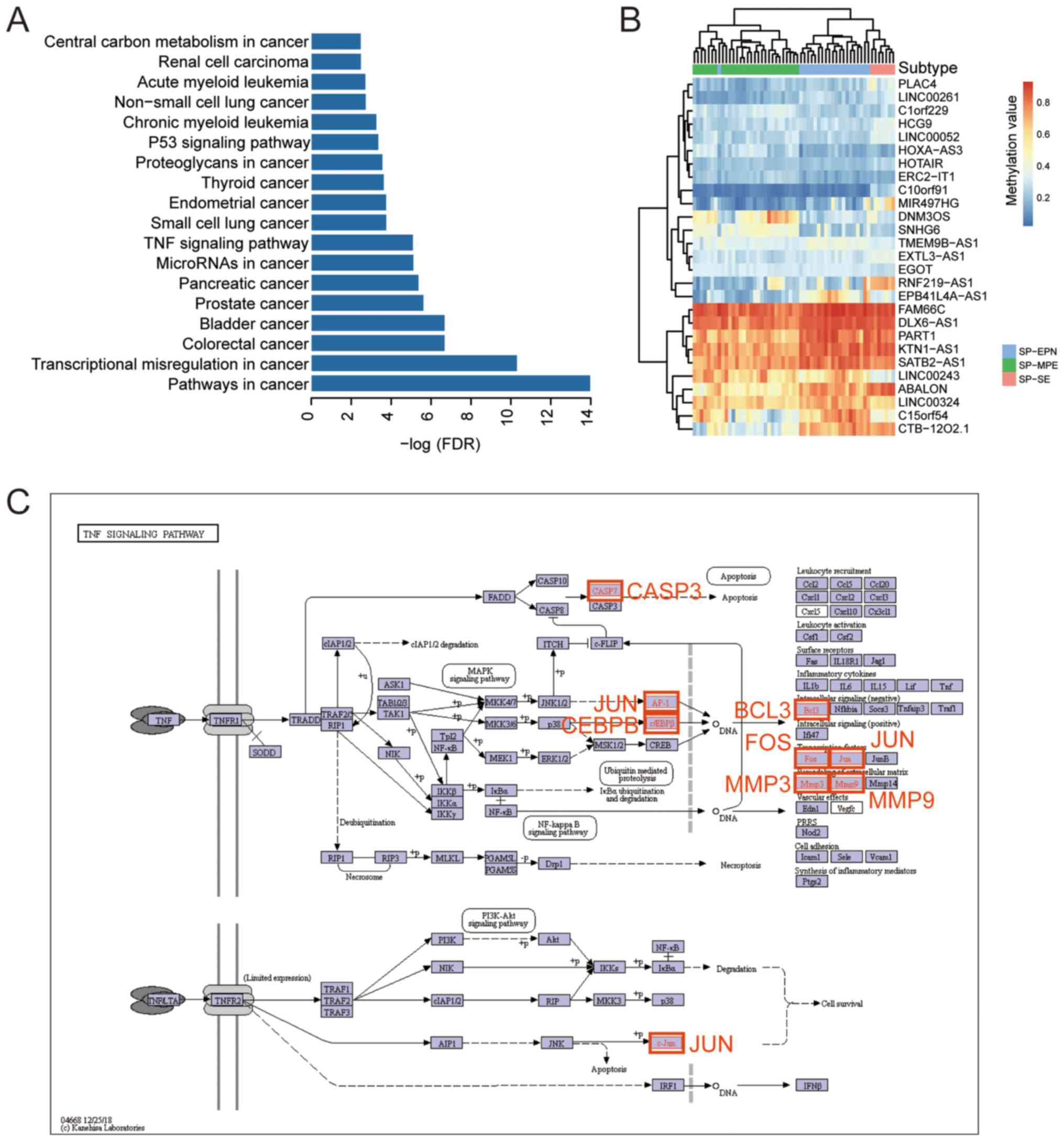
KEGG pathway enrichment analysis was performed in
order to analyze the PCGs interacting with the identified lncRNA
signatures. In total, 128 PCGs were analyzed. A total of 39
significantly enriched pathways (adjusted P<0.01; Fig. 5A) were identified. Out of 39
pathways, 17 were associated with cancer, including
‘transcriptional misregulation in cancer’, ‘p53 signalling
pathway’, ‘colorectal cancer’ and ‘bladder cancer’. Furthermore,
the pathway ‘proteoglycans in cancer’ was identified, which is
involved in central nervous system-associated functions and
diseases (49). Chondroitin
sulphate proteoglycans (CSPGs) are enriched in the nervous system
and contribute to neural cell migration and axon extension
(50). A previous study
demonstrated that CSPGs are upregulated after spinal cord injury
(51). Additionally, the tumor
necrosis factor (TNF) signaling pathway was identified, which is
involved in various diseases (Fig.
5C). Interestingly, most of the lncRNAs identified to exhibit
subtype-specific expression were hypomethylated compared with the
total amount of lncRNAs identified (Figs. 1C and 5B).
Discussion
Ependymal tumor is a rare type of malignant tumor
(52). Ependymal tumors can arise
from both the brain and spinal cord (53). Notably, depending on its origin,
there is a large genetic difference between these two types of
ependymal tumor (54). Previous
studies have investigated ependymal tumor arising from the brain
(6,55), and the current knowledge of the
molecular characteristics of spinal cord-derived ependymal tumor
remains limited.
By investigating the DNA methylation status of
lncRNAs, the present study provided novel insight into the
understanding of ependymal tumors and the possible treatments of
patients with this disease. The present study investigated the DNA
methylation profile of multiple lncRNA promoters and identified
that the majority of lncRNAs in ependymal tumors presented high
methylation levels. The present study suggested that the DNA
methylation level in lncRNA promoters are consistent among samples
belonging to the same tumor subtype. The regions with differential
methylation levels corresponded to lncRNA signatures that may be
involved in spinal cord development.
The present study suggested that tumor
subtype-specific lncRNAs may be involved in spinal cord
development. By integrating lncRNA-PCG interaction data, a total of
30 lncRNAs were identified, and their DNA methylation levels may be
a signature in spinal cord ependymal tumors. Some of these lncRNAs
were identified to be associated with cancer and nervous system
diseases. The PCGs regulated by the lncRNA signatures were
identified to be enriched in many cancer- and nervous
system-associated pathways. Interestingly, most of the lncRNAs
interacting with cancer- and nervous system-associated proteins
were found to be hypomethylated. In addition, the TNF signaling
pathway was identified as being involved in spinal cord ependymal
tumorigenesis. In previous studies, TNF-α was shown to be involved
in cell survival, apoptosis, inflammation and immunity (56–58),
and its role has been investigated in various diseases,
particularly in cancer (59). A
previous study investigating the function of TNF in the nervous
system demonstrated that TNF affects the nervous system, and it is
involved in neurodegenerative diseases (60). Although previous studies have
examined the function of the TNF signalling pathway in the nervous
system, the present study may provide insight for future studies
aimed to investigate its function in spinal cord tumors. In the
present study, lncRNAs with target genes associated with cancer and
the nervous system were identified, and their methylation patterns
in tumor samples were investigated. Therefore, DNA methylation may
be a principal factor in regulating the biological features of
lncRNAs, thus influencing the function of proteins downstream of
these lncRNAs.
Collectively, spinal cord ependymal tumors were
investigated using a novel approach, and the methylation status of
lncRNAs in ependymal tumors was characterized. The present study
may lay the foundations for future studies aimed to investigate
spinal cord tumors. However, the present results require validation
using a high number of tumor samples. The present findings may
contribute to the development of novel strategies for diagnosing
and treating spinal cord ependymal tumors.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW conceived and designed the experiments. LW
performed most of the experiments. CZ and YX performed certain
experiments. JH and SG identified tumor-subtype specific lncRNA
signatures. FX and WJ performed functional enrichment analysis. JH,
SG, FX and WJ aided in the interpretation of the results and wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
lncRNA
|
long non-coding RNA
|
|
PCGs
|
protein coding genes
|
|
SP-EPN
|
spinal ependymoma
|
|
SP-SE
|
spinal subependymoma
|
|
SP-MPE
|
spinal myxopapillary
|
|
TSS
|
transcript start site
|
|
GREAT
|
Genomic Regions Enrichment of
Annotations Tool
|
|
HM450
|
Human Methylation 450
|
|
CSPGs
|
chondroitin sulfate proteoglycans
|
|
SNP
|
single nucleotide polymorphism
|
|
TEC
|
To be Experimentally Confirmed
|
References
|
1
|
Arnautovic K and Arnautovic A:
Extramedullary intradural spinal tumors: A review of modern
diagnostic and treatment options and a report of a series. Bosn J
Basic Med Sci. 9 (Suppl 1):S40–S45. 2009. View Article : Google Scholar :
|
|
2
|
Allen JC, Siffert J and Hukin J: Clinical
manifestations of childhood ependymoma: A multitude of syndromes.
Pediatr Neurosurg. 28:49–55. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thuppal S, Propp JM and McCarthy BJ:
Average years of potential life lost in those who have died from
brain and CNS tumors in the USA. Neuroepidemiology. 27:22–27. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pajtler KW, Witt H, Sill M, Jones DT,
Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C,
Johann P, et al: Molecular classification of ependymal tumors
across all CNS compartments, histopathological grades, and age
groups. Cancer Cell. 27:728–743. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pfeffer C and Olsen BR: Editorial: Journal
of negative results in biomedicine. J Negative Results Biomed.
1:22002. View Article : Google Scholar
|
|
6
|
Witt H, Gramatzki D, Hentschel B, Pajtler
KW, Felsberg J, Schackert G, Löffler M, Capper D, Sahm F, Sill M,
et al: DNA methylation-based classification of ependymomas in
adulthood: Implications for diagnosis and treatment. Neuro
Oncology. 20:1616–1624. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma DL, Lin S, Leung KH, Zhong HJ, Liu LJ,
Chan DS, Bourdoncle A, Mergny JL, Wang HM and Leung CH: An
oligonucleotide-based label-free luminescent switch-on probe for
RNA detection utilizing a G-quadruplex-selective iridium(III)
complex. Nanoscale. 6:8489–8494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He HZ, Chan DS, Leung CH and Ma DL: A
highly selective G-quadruplex-based luminescent switch-on probe for
the detection of gene deletion. Chem Commun (Camb). 48:9462–9464.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leung KH, He HZ, Chan DS, Fu WC, Leungb CH
and Maa DL: An oligonucleotide-based switch-on luminescent probe
for the detection of kanamycin in aqueous solution. Sensors
Actuators B: Chemical. 177:487–492. 2013. View Article : Google Scholar
|
|
10
|
Wu C, Wu KJ, Kang TS, Wang HD, Leung CH,
Liu JB and Ma DL: Iridium-based probe for luminescent nitric oxide
monitoring in live cells. Sci Rep. 8:124672018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kiltschewskij D and Cairns MJ:
Temporospatial guidance of activity-dependent gene expression by
microRNA: Mechanisms and functional implications for neural
plasticity. Nucleic Acids Res. 47:533–545. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kulis M and Esteller M: DNA methylation
and cancer. Adv Genet. 70:27–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiao Y, Yu F, Pang L, Zhao H, Liu L, Zhang
G, Liu T, Zhang H, Fan H, Zhang Y, et al: MeSiC: A model-based
method for estimating 5 mC levels at Single-CpG resolution from
MeDIP-seq. Sci Rep. 5:146992015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Klutstein M, Nejman D, Greenfield R and
Cedar H: DNA methylation in cancer and aging. Cancer Res.
76:3446–3450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu F, Quan F, Xu J, Zhang Y, Xie Y, Zhang
J, Lan Y, Yuan H, Zhang H, Cheng S, et al: Breast cancer prognosis
signature: Linking risk stratification to disease subtypes. Brief
Bioinform. Sep 3–2018.doi: 10.1093/bib/bby073 (Epub ahead of
print). View Article : Google Scholar
|
|
16
|
Koch A, Joosten SC, Feng Z, de Ruijter TC,
Draht MX, Melotte V, Smits KM, Veeck J, Herman JG, Van Neste L, et
al: Analysis of DNA methylation in cancer: Location revisited. Nat
Rev Clin Oncol. 15:459–466. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guttman M, Amit I, Garber M, French C, Lin
MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al:
Chromatin signature reveals over a thousand highly conserved large
non-coding RNAs in mammals. Nature. 458:223–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu Q, Kim YC, Lu J, Xuan Z, Chen J, Zheng
Y, Zhou T, Zhang MQ, Wu CI and Wang SM: Poly A-transcripts
expressed in HeLa cells. PLoS One. 3:e28032008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu F, Zhang G, Shi A, Hu J, Li F, Zhang X,
Zhang Y, Huang J, Xiao Y, Li X and Cheng S: LnChrom: A resource of
experimentally validated lncRNA-chromatin interactions in human and
mouse. Database (Oxford). 20182018.doi:
10.1093/database/bay039.
|
|
20
|
Zhang Y, Li X, Zhou D, Zhi H, Wang P, Gao
Y, Guo M, Yue M, Wang Y, Shen W, et al: Inferences of individual
drug responses across diverse cancer types using a novel competing
endogenous RNA network. Mol Oncol. 12:1429–1446. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bolha L, Ravnik-Glavac M and Glavac D:
Long noncoding RNAs as biomarkers in cancer. Dis Markers.
2017:72439682017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Z, Yang B, Zhang M, Guo W, Wu Z, Wang
Y, Jia L, Li S; Cancer Genome Atlas Research Network, ; Xie W and
Yang D: lncRNA epigenetic landscape analysis identifies EPIC1 as an
oncogenic lncRNA that interacts with MYC and promotes cell-cycle
progression in cancer. Cancer Cell. 33:706–720.e9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Heilmann K, Toth R, Bossmann C, Klimo K,
Plass C and Gerhauser C: Genome-wide screen for differentially
methylated long noncoding RNAs identifies Esrp2 and lncRNA Esrp2-as
regulated by enhancer DNA methylation with prognostic relevance for
human breast cancer. Oncogene. 36:6446–6461. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Clough E and Barrett T: The gene
expression omnibus database. Methods Mol Biol. 1418:93–110. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harrow J, Frankish A, Gonzalez JM,
Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa
A, Searle S, et al: GENCODE: The reference human genome annotation
for The ENCODE Project. Genome Res. 22:1760–1774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou W, Laird PW and Shen H: Comprehensive
characterization, annotation and innovative use of Infinium DNA
methylation BeadChip probes. Nucleic Acids Res.
45:e222017.PubMed/NCBI
|
|
28
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aine M, Sjödahl G, Eriksson P, Veerla S,
Lindgren D, Ringnér M and Höglund M: Integrative epigenomic
analysis of differential DNA methylation in urothelial carcinoma.
Genome Med. 7:232015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yi Y, Zhao Y, Li C, Zhang L, Huang H, Li
Y, Liu L, Hou P, Cui T, Tan P, et al: RAID v2.0: An updated
resource of RNA-associated interactions across organisms. Nucleic
Acids Res. 45:D115–D118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen X and Ishwaran H: Random forests for
genomic data analysis. Genomics. 99:323–329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nguyen TT, Huang JZ and Nguyen TT:
Unbiased feature selection in learning random forests for
high-dimensional data. TheScientificWorldJournal. 2015:4713712015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McLean CY, Bristor D, Hiller M, Clarke SL,
Schaar BT, Lowe CB, Wenger AM and Bejerano G: GREAT improves
functional interpretation of cis-regulatory regions. Nat
Biotechnol. 28:495–501. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kuleshov MV, Jones MR, Rouillard AD,
Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM,
Lachmann A, et al: Enrichr: A comprehensive gene set enrichment
analysis web server 2016 update. Nucleic Acids Res. 44:W90–W97.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Prayson RA and Suh JH: Subependymomas:
Clinicopathologic study of 14 tumors, including comparative MIB-1
immunohistochemical analysis with other ependymal neoplasms. Arch
Pathol Lab Med. 123:306–309. 1999.PubMed/NCBI
|
|
37
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yao J, Zhang L, Hu L, Guo B, Hu X,
Borjigin U, Wei Z, Chen Y, Lv M, Lau JT, et al: Tumorigenic
potential is restored during differentiation in fusion-reprogrammed
cancer cells. Cell Death Dis. 7:e23142016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Y, Liu D, Wang L, Wang S, Yu X, Dai
E, Liu X, Luo S and Jiang W: Integrated systems approach identifies
risk regulatory pathways and key regulators in coronary artery
disease. J Mol Med (Berl). 93:1381–1390. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiang W, Zhang Y, Meng F, Lian B, Chen X,
Yu X, Dai E, Wang S, Liu X, Li X, et al: Identification of active
transcription factor and miRNA regulatory pathways in Alzheimer's
disease. Bioinformatics. 29:2596–2602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schmitt AM and Chang HY: Long noncoding
RNAs in cancer pathways. Cancer Cell. 29:452–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ferre F, Colantoni A and Helmer-Citterich
M: Revealing protein-lncRNA interaction. Brief Bioinform.
17:106–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xiong D, Sheng Y, Ding S, Chen J, Tan X,
Zeng T, Qin D, Zhu L, Huang A and Tang H: LINC00052 regulates the
expression of NTRK3 by miR-128 and miR-485-3p to strengthen HCC
cells invasion and migration. Oncotarget. 7:47593–47608. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Salameh A, Fan X, Choi BK, Zhang S, Zhang
N and An Z: HER3 and LINC00052 interplay promotes tumor growth in
breast cancer. Oncotarget. 8:6526–6539. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hajjari M and Salavaty A: HOTAIR: An
oncogenic long non-coding RNA in different cancers. Cancer Biol
Med. 12:1–9. 2015.PubMed/NCBI
|
|
47
|
Teschendorff AE, Lee SH, Jones A, Fiegl H,
Kalwa M, Wagner W, Chindera K, Evans I, Dubeau L, Orjalo A, et al:
HOTAIR and its surrogate DNA methylation signature indicate
carboplatin resistance in ovarian cancer. Genome Med. 7:1082015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang JX, Han L, Bao ZS, Wang YY, Chen LY,
Yan W, Yu SZ, Pu PY, Liu N, You YP, et al: HOTAIR, a cell
cycle-associated long noncoding RNA and a strong predictor of
survival, is preferentially expressed in classical and mesenchymal
glioma. Neuro Oncol. 15:1595–1603. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Heindryckx F and Li JP: Role of
proteoglycans in neuro-inflammation and central nervous system
fibrosis. Matrix Biol. 68-69:589–601. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Silver DJ and Silver J: Contributions of
chondroitin sulfate proteoglycans to neurodevelopment, injury, and
cancer. Curr Opin Neurobiol. 27:171–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rolls A, Shechter R, London A, Segev Y,
Jacob-Hirsch J, Amariglio N, Rechavi G and Schwartz M: Two faces of
chondroitin sulfate proteoglycan in spinal cord repair: A role in
microglia/macrophage activation. PLoS Med. 5:e1712008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Villano JL, Parker CK and Dolecek TA:
Descriptive epidemiology of ependymal tumours in the United States.
Br J Cancer. 108:2367–2371. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Armstrong TS, Vera-Bolanos E, Bekele BN,
Aldape K and Gilbert MR: Adult ependymal tumors: Prognosis and the
M. D. Anderson Cancer Center experience. Neuro Oncol. 12:862–870.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee CH, Chung CK and Kim CH: Genetic
differences on intracranial versus spinal cord ependymal tumors: A
meta-analysis of genetic researches. Eur Spine J. 25:3942–3951.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gittleman H, Boscia A, Ostrom QT, Truitt
G, Fritz Y, Kruchko C and Barnholtz-Sloan JS: Survivorship in
adults with malignant brain and other central nervous system tumor
from 2000–2014. Neuro Oncol. 20 (Suppl 7):vii6–vii16. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Olmos G and Lladó J: Tumor necrosis factor
alpha: A link between neuroinflammation and excitotoxicity.
Mediators Inflamm. 2014:8612312014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mak TW and Yeh WC: Signaling for survival
and apoptosis in the immune system. Arthritis Res. 4 (Suppl
3):S243–S252. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
58
|
Parameswaran N and Patial S: Tumor
necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot
Gene Expr. 20:87–103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
van Horssen R, Ten Hagen TL and Eggermont
AM: TNF-alpha in cancer treatment: Molecular insights, antitumor
effects, and clinical utility. Oncologist. 11:397–408. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Probert L: TNF and its receptors in the
CNS: The essential, the desirable and the deleterious effects.
Neuroscience. 302:2–22. 2015. View Article : Google Scholar : PubMed/NCBI
|