Introduction
Heterotopic ossification (HO)
Definition and classification of
HO
HO refers to the appearance of osteoblasts in soft
tissue and the formation of bone tissue under pathological
conditions, including local and systemic massive ossification
(1). In general, HO can be divided
into two categories: Genetic heterotopic ossification (GHO) and
acquired heterotopic ossification (AHO). GHO includes
fibrodysplasia ossificans progressiva (FOP) (2) and progressive osseous heteroplasia
(POH) (3). AHO is characterized by
pain and a limited range of motion in the joints. FOP is a rare
autosomal-dominant disease characterized by progressive HO. A
common mutation (617G>A; R206H) of ACVR1/ALK2 causes >95% of
cases (4,5). Multiple congenital skeletal
malformations are associated with FOP, most frequently including an
abnormal first toe (6),
dysmorphology affecting the digits of the hand (7) and malformations of the cervical
spine. POH is a genetic disease that has a predilection for the
skin and subcutis characterized by plaque-like HO, beginning in
infancy in the dermis and subsequently developing to involve deep
connective tissues, such as the muscles and joints, which sometimes
leads to loss of mobility (8).
However, a loss-of-function mutation in guanine nucleotide binding
protein G subunit α, an imprinted gene, is responsible for POH
(9–11). AHO often occurs after severe
trauma, such as muscle injury, fracture or dislocation, major joint
surgery, myelitis, encephalitis or central nervous system damage
(3,12–15).
The occurrence of HO can seriously affect quality of life,
especially in military service members and veterans. Because these
people often suffer combat-related extremity injuries, the global
incidence of HO in the military is 63–65%, far higher than the rate
of 10–30% reported globally for ordinary citizens (16). In total, 60% of AHO occurs in the
lower extremities and ~40% occurs in the upper extremity joints,
such as the elbow and shoulder (15,17).
Once HO begins to form, it can cause swelling, pain, nerve
compression and joint movement disorders around the joints; if it
forms in the spine, it leads to limited spinal mobility and spinal
cord compression (18). In the
early stage of the formation of HO, the patient may experience
swelling, pain and elevated skin temperature. The patient cannot
undergo rehabilitation training because of fear of pain, which
adversely affects the recovery process (19). As the disease progresses, the HO
gradually matures, wrapping around or blocking joints, hindering
joint activities and reducing the range of motion of the joints,
and thereby further affecting the ability of the patient to sit and
stand, and hindering weight and gait training. As such, HO has a
great impact on the rehabilitation of patients (20,21).
Formation process of HO
The homeostasis of bone formation is maintained by
osteoblasts and osteoclasts, and its formation process is
predominantly divided into intramembranous ossification and
endochondral ossification (22).
Intramembranous ossification begins with mesenchymal
differentiation into an embryonic connective tissue membrane, which
then forms bone in the membrane, while endochondral ossification
mainly refers to the differentiation of mesenchymal precursor cells
into chondrocytes (22,23). The development process of HO is
similar to the development of normal bone tissue, but it is not
strictly in accordance with normal osteogenesis. The ectopic bone
not only remodels (just like the normal skeleton) but also expands,
with expansion leading to the gradual accumulation of ectopic bone
(23). Ectopic bone produced in
FOP is formed by endochondral ossification, while intramembranous
ossification accounts for POH, while intramembranous ossification
and endochondral ossification both contribute to AHO (24). A previous study found that the
formation of endochondral-mediated HO involves the inflammation and
destruction of connective tissue and the formation of
cartilage/bone tissue but not POH (25). Following a variety of stimuli, such
as injury, burns and amputation, a large number of perivascular
lymphocytes in the blood leak out of the blood vessel wall during
inflammation and move into the early HO area within the muscle and
other connective tissues, simultaneously accompanied by damage to
the connective tissue structure. Along with lymphocytic
infiltration and the destruction of connective tissue structure, an
inflammatory reaction, fibrosis and angiogenesis occur, which give
rise to the release of osteogenic factors, inducing bone
morphogenetic proteins (BMPs) (22,23).
These factors induce the formation of mesenchymal cells in the
local microenvironment and promote the differentiation of
mesenchymal precursor cells into osteoblasts and chondrocytes,
eventually forming ectopic bone (Fig.
1) (25,26). However, the cellular origin of the
ectopic bone in HO remains a unclear (27) and there have been different
hypotheses for the origin of HO progenitor cells, including from
muscle (28), vascular endothelia
(29), brown fat (30) and the endoneurium (31). In summary, ectopic bone formation
requires three conditions: Osteogenic-inducing factors, precursor
cells and an appropriate local microenvironment (32).
BMP family and pathway
BMPs belong to the tumor growth factor (TGF)-β
superfamily. There are ~20 types of BMPs, which are divided into
four classes in mammals based on sequence and functional
similarity: i) BMP2 and 4; ii) BMP5, 6, 7, 8a and 8b; iii) BMP9 and
10; and iv) BMP12, 13 and 14. BMP2, 4, 5, 6, 7 and 9 have a high
osteogenic activity (7,33). The BMP signaling pathways are
mediated primarily by the formation of heterotetrameric complexes
of type I and type II BMP receptors (BMPRs). The TGF-β family
receptor members are composed of type I and type II receptors,
among which there are seven type I BMPRs (ALK1-7) and three type II
BMPRs [BMPRII, Activin type II receptor (ActRII) and ActRIIB]
(34).
Among all type I receptors, BMPs are more likely to
bind to ALK1, 2, 3 and 6 (6,35).
BMP signaling pathways regulate a number of cellular activities,
including cell differentiation, proliferation, apoptosis, migration
and stem cell self-renewal (36).
BMPs have the specific ability to induce cartilage formation and
osteogenesis, as well as to promote the formation of endochondral
bone (37). Previous clinical and
basic medical studies have shown that the BMP pathway plays an
important role in HO (36,38). When a ligand binds to a type II
BMPR and phosphorylates it, the type II BMPR phosphorylates and
activates the glycine/serine (GS) domain of the type I receptors,
which in turn activate Smad-dependent (BMP/Smad) and
Smad-independent [BMP/mitogen activated protein kinase (MAPK)]
signaling reactions. The BMP/Smad meditated phosphorylation of type
I receptors leads to the phosphorylation of Smad1/5/8, which
subsequently bind to Smad4 and transport it to the nucleus. In the
nucleus, Smad1/5/8-Smad4 bind to target genes and upregulate the
expression of bone related transcription factors, thereby promoting
bone formation. BMP/MAPK mediated phosphorylation of type I
receptors activates the MAPK pathways, such as Erk, JNK and p38,
thereby regulating transcription factor activity (Fig. 2) (6,35,36,39).
As such, both pathways regulate bone metabolism.
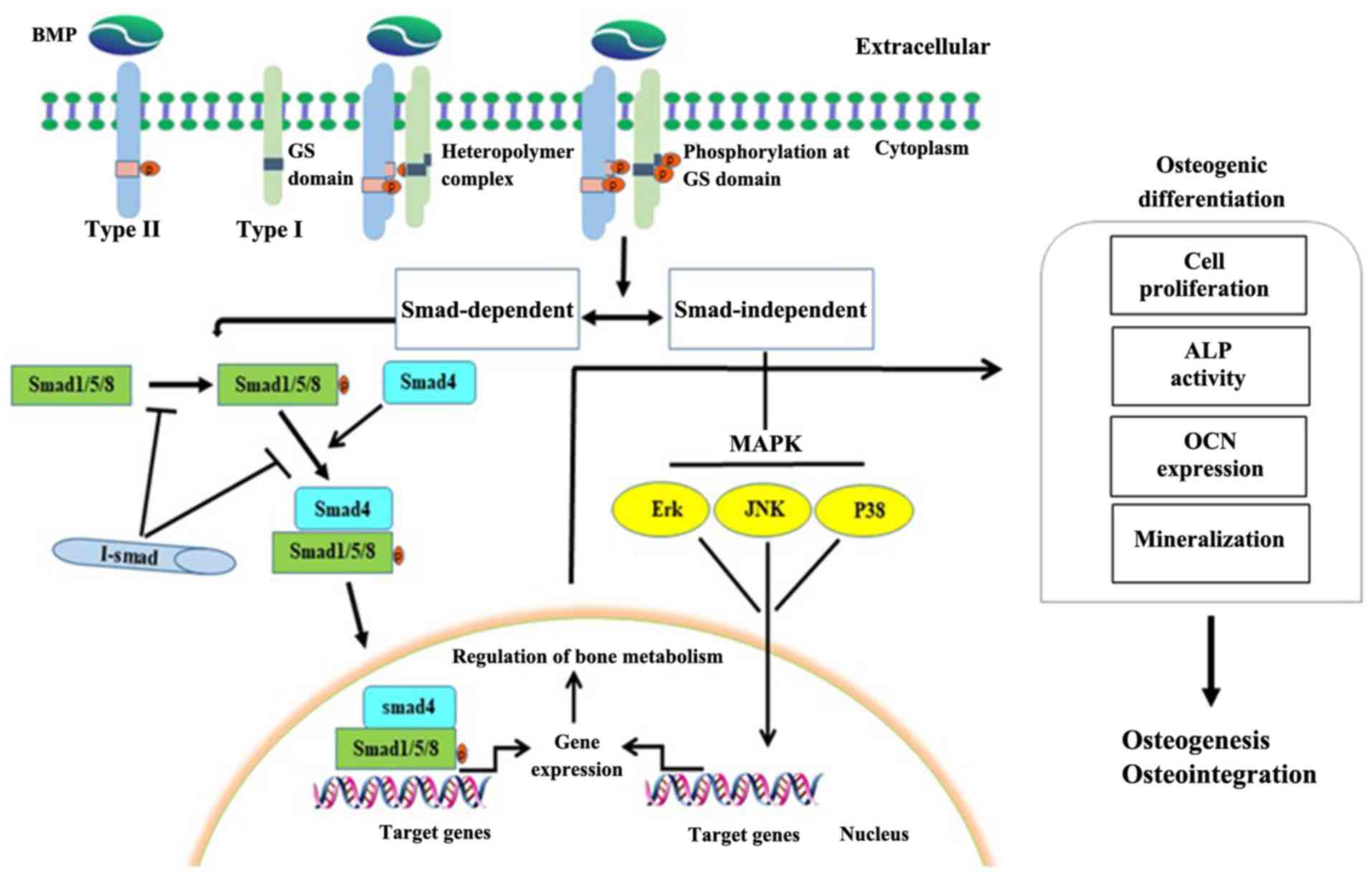 | Figure 2.Regulation of the BMP signaling
pathway. When BMPs bind to type I (ALK1/ALK2/ALK3/ALK6) and type II
(BMP receptors II/Act-II/and Act-IIB) receptors, type II BMP
receptors phosphorylate and activate type I receptors through the
GS domain, which in turn activates BMP/Smad and BMP/MAPK cascade
reactions. The BMP/Smad-mediated phosphorylation of type I
receptors leads to the phosphorylation of Smad1/5/8, which binds to
Smad4. This complex is transported into the nucleus where it binds
to target genes and upregulates the expression of bone related
transcription factors, promoting bone formation. The
BMP/MAPK-mediated phosphorylation of type I receptors activates
MAPK pathways, such as Erk, JNK and P38, thereby regulating
transcription factors. Activation of both pathways promotes bone
formation. BMP, bone morphogenetic proteins; ALK, activin
receptor-like kinase; Act-II, activin type II receptor; GS,
glycine/serine; MAPK, mitogen-activated protein kinase; I-Smad,
inhibitor Smad; ALP, alkaline phosphatase; OCN, osteocalcin. |
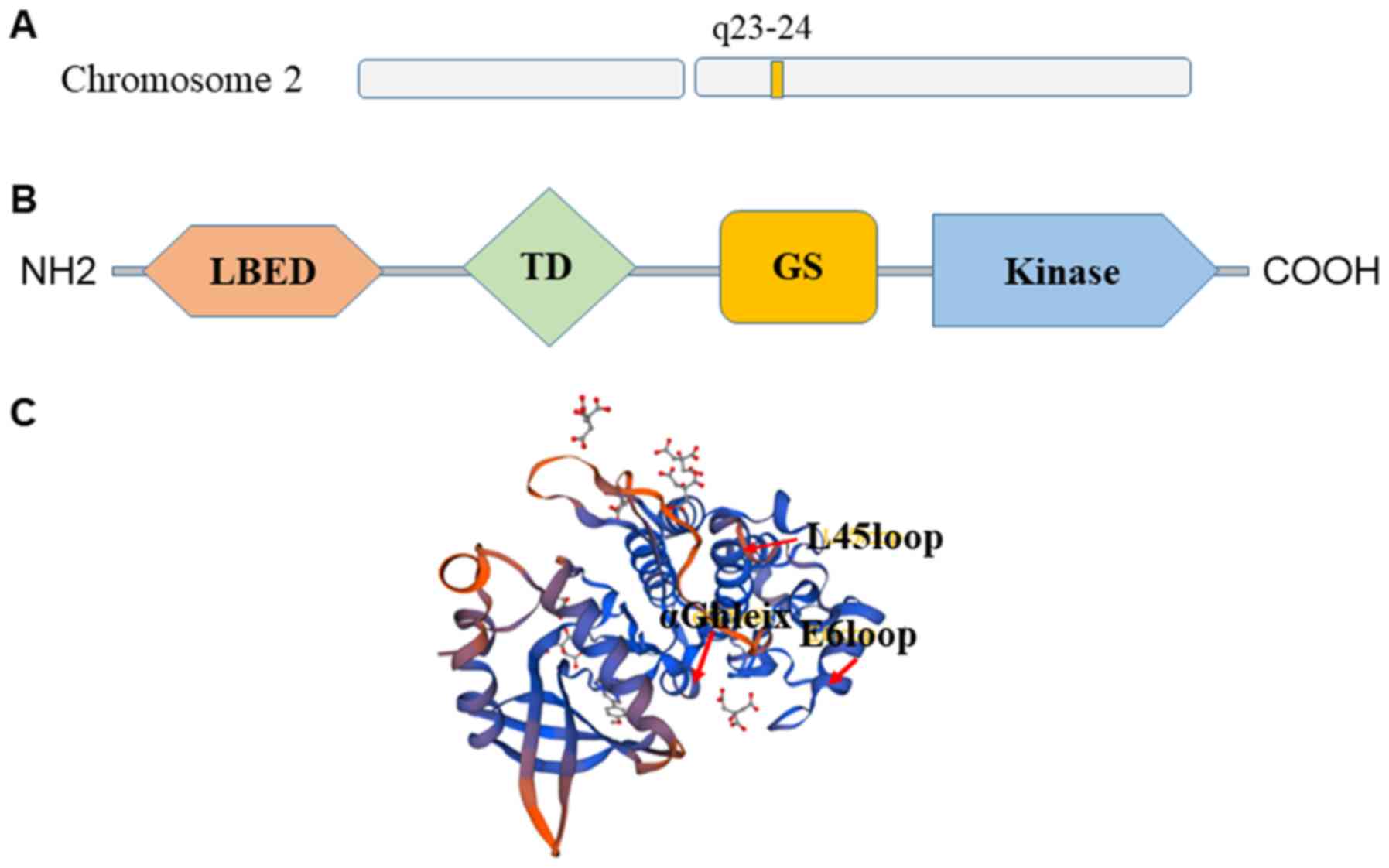
Structure of ALK2
ALK2 belongs to the BMP receptor family (40). In addition, it is a product of the
ACVR1 gene, which is located at 2q23-24 (40) (Fig.
3A). The structure of the cytoplasmic domain of ALK2 was
resolved in complex with the inhibitors 12 kDa FK506 binding
protein (FKBP12) and dorsomorphin (40) which provided a new model for the
structure based lead optimization of BMP inhibitors. ALK2 contains
the typical ALK receptor domains (Fig.
3B), including a ligand binding extracellular domain, a
transmembrane domain, a membrane-associated GS-rich domain and a
large kinase domain (41,42). In addition, ALK2 has a typical
bilobal kinase architecture with a pattern of specific insertions.
These include the L45 loop, the E6 loop, insertions flanking the αF
helix and an insertion in the substrate pocket preceding the αG
helix (41) (Fig. 3C).
Type I receptor kinases are activated by the
phosphorylation of the GS domain by type II receptor kinases in the
tetramer complex (36). It was
shown that the kinase domain has a bifoliate structure, with the GS
domain extending from the N-terminal to the helix motif, which can
bind to the endogenous inhibitor FKBP12 (40,42).
A previous study also showed that FKBP12 prevents entry into the
regulatory GS ring and inhibits the αC movement required for kinase
activation (42). In the absence
of FKBP12, the inactive conformation in this region is relatively
stable, suggesting the conformation and activity of ALK2 are
related (40,42).
ALK2 signaling pathway in HO
ALK2 signaling pathway in GHO
Mutations in ALK2 have not only been associated with
cancer, particularly diffuse intrinsic pontine glioma, but have
also been linked to HO (40,41).
Type I receptor kinases are typically activated by
phosphorylation of the GS domain by type II receptor kinases in the
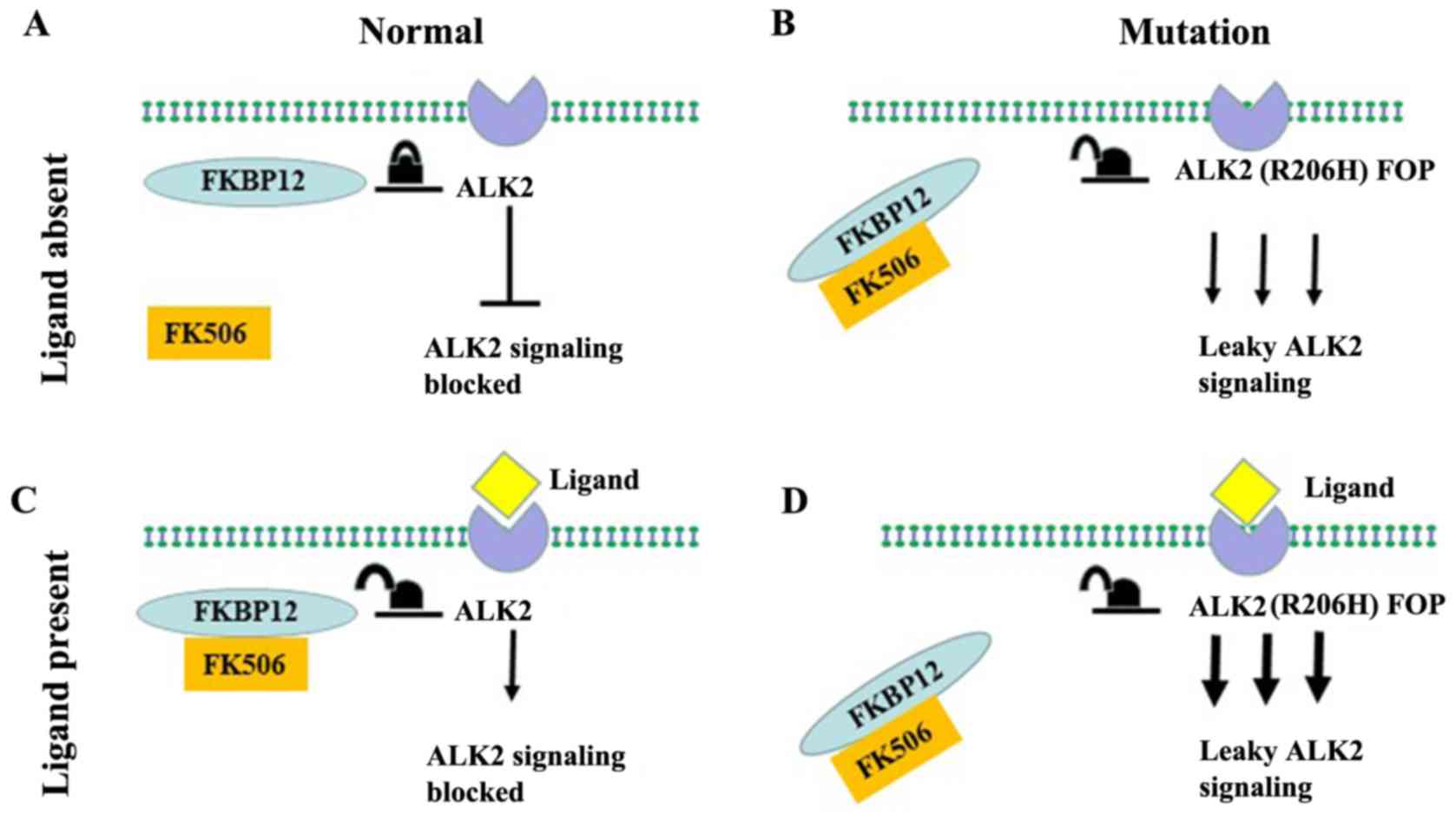
tetramer complex (40,43). However, when ALK2 acquires a
functional mutation, ALK2 has reduced binding to the inhibitor
FKBP12 and promotes leaky signaling, which continues to activate
downstream signals in the absence of the ligand (Fig. 4B) (44). In addition, a loss of function
mutation of ALK2 inhibits cartilage formation in mouse embryonic
fibroblasts (MEFs) in vitro by inhibiting the
phosphorylation of Smad1/5/8, therefore, decreasing ectopic bone
formation (9,15); the R206H mutation is not involved
in other forms of HO, such as acquired forms and POH. Culbert et
al (9) described the function
of ALK2 signaling both in vitro and in vivo in the
presence of the BMP ligand (Fig.
4D). ALK2R206H/+ MEFs were compared with wild-type MEFs in
three-dimensional chondrogenic culture, which were stained with
Alcian blue, and it was found that, at the same BMP4 concentration,
ALK2R206H/+ cells were more sensitive to BMP4 than wild-type cells.
Wild-type or ALK2R206H/+ MEFs with 500 ng BMP4 were injected into
the limb muscles of the mice, and it was found that limbs which had
been injected with wild-type cells developed no measurable
mineralization, while all limbs injected with Alk2R206H/+ cells
developed robust mineralization. In addition, even in the absence
of BMP ligands, the ALK2 signaling pathway was continuously
activated in ALK2R206H/+ cells. The ALK2R206H gain-of-function
mutation enhances both canonical (phospho-Smad1/5/8) and
noncanonical BMP signaling responses in the absence of ligands
(9,45). However, ALK2R206H/+ mice in which
the ALK2R206H mutation was introduced into the second allele
closely replicated key features of classic FOP. Chakkalakal et
al (46) developed
constitutive chimeric ACVR1R206H knock-in mice, as the ALK2R206H
mutation causes perinatal lethality. The deformation of the first
digits in the posterior limb and ectopic bone formation were
observed in the chimeric mice, features also commonly seen in FOP
patients before HO lesions appear; features that are often an
important clinical marker of the disease. By 6–8 weeks of age, the
mice had extensive HO in skeletal muscle. Moreover, HO developed at
injury sites and in the surrounding soft tissues. Other genetic
models of FOP, that conditionally express ALK2R206H in mice, were
developed by Lin et al (42), Hatsell et al (47) and Katagiri (48), and these novel mouse models of FOP
carrying the R206H mutation in ALK2 were created using a
conditional knock-in genetic technique. Yu et al (49) obtained CAGGS-Cre estrogen receptor
mice, which express a tamoxifen-inducible Cre recombinase
ubiquitously expressed under the control of the cytomegalovirus
immediate-early enhancer and the chicken β-actin promoter/enhancer.
After crossing with Cre driver mice or tamoxifen injection, the
ALK2R206H mice developed unprovoked HO in anatomic sites, including
the back, limbs, sternum, ribcage, jaw and hip. In addition, it was
noted the mouse model formed HO in response to skeletal muscle
injury, similar to patients with FOP. A related ALK2R206H FOP mouse
model was also developed by Lees-Shepard et al (5), in which the expression of ALK2R206H
was Cre-dependent.
However, two previous reports demonstrated that the
R206H substitution in ALK2 has novel functions, with the
substitution altering activin-specific signaling (47,50).
Under normal condition, Activin A usually does not have an
osteogenic role. However, in FOP, Activin A leads to activation of
Smad1/5/8 phosphorylation via mutant ALK2 signaling, triggering HO
formation. Activin A is secreted by many cell types, among which
the cells of the immune system play a basic role in the early
phases of FOP lesion formation (47). The binding of activin A to its
cognate receptor molecules activates an intracellular signaling
cascade mediated by Smad2/3 that regulates the expression of target
genes after translocation into the nucleus (47,51).
By contrast, the binding of activin A to the ALK2R206H mutant
receptor activates downstream signaling through the canonical
Smad1/5/8 mediators in several cell types expressing mutated
ALK2R206H, such as in induced pluripotent stem cells (iPSCs)
derived from patients with FOP and mouse models (5,47,50).
The targeted expression of the disease-causing ALK2 (R206H) allele
to fibro/adipogenic progenitors (FAPs) recapitulates all the
features of HO observed in patients with FOP. ALK2R206H-expressing
FAPs activate osteogenic signaling in response to activin ligands,
but wild-type FAPs do not (5). Lin
et al (42), Hatsell et
al (47) and Olsen et
al (51) reported that activin
A can be an antagonist of the BMP/ALK2 signaling pathway by acting
as an activator of the ALK2 signaling pathway, with activin A as a
precursor signal. For patients with FOP, trauma readily induces new
ectopic bone formation, predominantly because activin A activates
the mutated ALK2, and the mutation of ALK2 increases sensitivity to
activin A (23). These findings
have been supported by another previous study (50). Upadhyay et al (23) reported that in the FOP mouse model,
both activin A pharmacological and immunological inhibitors could
reduce the formation of new ectopic bone in the early stages of
ectopic bone formation. It was also reported that, in the case of
activin deficiency, even if the Smad1 receptor is continuously
activated (promoted by ALK2Q207D), new heterotopic bone cannot be
generated (23,50,51).
In addition, tissue hypoxia and inflammation promote BMP signaling,
chondrogenesis and ectopic bone formation through mutant ALK2
(30,37,52).
ALK2 signaling pathway in AHO
The incidence of acquired forms of HO may be due to
multiple factors, including injury to soft tissues or the central
nervous system, burns, and amputations (53). A previous study found that BMP2 is
highly expressed in the cerebrospinal fluid of patients with
traumatic brain injury (54). The
overexpression of various osteogenic transcription factors,
including BMP2 and BMP3, has been reported in mouse models of
traumatic injury (55).
Furthermore, HO can be successfully induced by the injection of
BMPs into mice, especially BMP2, BMP4 and BMP9 (39). Therefore, BMPs play an important
role in the development of HO. However, along with the BMPs, the
receptors also play important roles. It has been reported that the
expression of ALK is upregulated in an ectopic bone model of spinal
cord injury, and that the phosphorylation of Smad and its
subsequent translocation into the nucleus are also increased
(56). In a tendon burn model,
BMP-Smad signaling continued to be activated during the immature
phase of ectopic bone formation and fibroproliferation (55). LDN-193189, an inhibitor of BMP type
I receptors, effectively reduced the differentiation of mesenchymal
stem cells to osteogenic cells in a mouse model of HO following by
hip surgery (49). These previous
studies have shown that the ALK2 signaling pathway plays an
important role in the process of AHO (Fig. 4A and C).
In summary, both AHO and GHO have some
commonalities, including the formation of bone tissue in the wrong
location and that both are likely regulated by the ALK2 signaling
pathway. FOP provides an important model for studying the mechanism
of occurrence and development of HO. ALK2 is predominantly
expressed in undifferentiated cells, such as mesenchymal stem
cells, which have the potential for multifunctional differentiation
(9). These undifferentiated cells
can differentiate into osteoblast (57). However, the expression of other BMP
type I receptors are not necessarily the same. For example, ALK3 is
stably expressed throughout the differentiation process, while ALK6
is highly expressed during the differentiation stage compared with
the undifferentiated stage (41).
The prevention and treatment of diseases should target the
underlying causes of the early stages of the disease, therefore, it
is imperative to study drug targets for the prevention and
treatment of HO.
Development of treatments for HO by
targeting ALK2
Targeted therapies for HO are being explored, these
therapies include the inhibition of BMP receptors (including type I
and type II) to block the continued activation of the BMP signaling
pathway and thereby inhibit HO (55) (Table
I).
 | Table I.Inhibition of ALK2. |
Table I.
Inhibition of ALK2.
| Target | Type | Molecule | Clinical trial
phase |
|---|
| ALK2 kinase | Small molecule
inhibitor (49,69–74) | Dorsomorphin |
|
|
|
| LDN-193189 |
|
|
|
| LDN-212854 |
|
|
|
| K02288 |
|
|
|
| DMH-1 |
|
|
|
| ML347 |
|
|
|
| VU465350 |
|
| ALK2 mRNA | Compound (67) | Dipyridamole |
|
|
| Gene (78–85) | microRNA |
|
|
|
| AON |
|
|
|
| ASP-RNAi |
|
| ALK2 signaling | FDA approved drugs
(88,89) | Fendiline |
|
|
|
| Perhexiline |
|
|
|
| Metformin (42) |
|
| Ligand | Antibody
(66,91) | Anti-activin-A
antibody (REGN2477) | Phase II |
|
|
| Therapeutic
monoclonal antibodies |
|
|
|
| specific for
ALK2 |
|
|
| Ligand traps
(66,90,92) | sActR-IIA-Fc |
|
|
|
| sActR-IIB-Fc |
|
The prevention and treatment of HO in the clinic
predominantly includes nonsteroidal anti-inflammatory drugs
(NSAIDs), bisphosphonates and other drug treatments, low-dose local
radiation therapy and surgical resection, rehabilitation treatment
and physical therapy (58).
However, most of these therapies have additional clinical benefits.
For instance, the use of non-selective NSAIDs, such as naproxen,
after hip surgery significantly reduced the incidence of HO,
despite the primary use being to relieve inflammation and joint
stiffness (59). In addition,
indomethacin, which is primarily used to relieve inflammation, is
the most effective drug for the inhibition of osteoblast survival
in vitro and can promote wound healing in (60). However, NSAIDs may cause
gastrointestinal bleeding. Therefore, the topical application of
NSAIDs may be a safe and effective method to prevent HO, while
avoiding various gastrointestinal complications (61). Radiotherapy may induce malignant
tumors and promote the early closure of the epiphysis (62). Bisphosphonates mainly reduce the
bone mineralization process by blocking the conversion of amorphous
calcium phosphate into hydroxyapatite, however, they do not affect
the synthesis of bone matrix. Bisphosphonates delay rather than
block the mineralization of HO, therefore, it is easy to resume
treatment after stopping administration (63). Surgical resection may cause new
ectopic bone formation (64).
These methods can only prevent the occurrence of HO but are not
therapies specifically targeted at the pathogenesis of HO, as such,
they do not tackle the root cause of the occurrence of HO (61,62,64,65).
Therefore, scholars have been working to develop targeted therapies
based on the pathogenesis of HO.
Several previous studies have suggested that
blocking the ALK2 signaling pathway may inhibit HO (49,66,67).
There are three possible mechanisms to inhibit the ALK2 signaling
pathway: i) Block the binding of ligands; ii) inhibit the
phosphorylation of smad1/5/8 by inhibiting the activity of the ALK2
kinase, and iii) to upregulate the expression of ACVR1, the gene
encoding ALK2, at the transcriptional level. Many endogenous
protein antagonists that block the binding of ligands to receptors
have been discovered, including noggin, follistatin, chordin and
gremlin (68). The strategy of
targeting ALK2 activity for inhibition has attracted much
attention.
Targeting the ALK2 kinase
Targeting the ALK2 kinase in AHO
An early example of a targeted inhibitor of a BMP
type I receptor is dorsomorphin, which inhibits BMP signaling by
targeting ALK2, ALK3 and ALK6 (69). However, this inhibitor has poor
selectivity and may cause a series of side effects, therefore,
optimization is required. At a similar time, the highly selective
dorsomorphin derivative LDN-193819 (targeting ALK2 and ALK3) was
introduced (70). Yu et al
(49) found that HO in a rat model
was effectively treated with the high-efficiency ALK2 inhibitor
LDN-193819 7 days after the initiation of treatment. At 30 days
after the initiation of treatment, ~66% of the rats had effective
inhibition of the symptoms of HO and symptoms in the other rats
were also reduced. Furthermore, HO did not recur after the
withdrawal of treatment. Further analysis showed that LDN-193819
significantly inhibited the phosphorylation of acetyl-CoA
carboxylase, thereby inhibiting the BMP receptor-mediated signal
transduction pathway, suggesting that BMP type I receptors can be
used as targets for the treatment of HO. Other derivatives have
also been developed, including LDN-212854, K02288, DMH-1, ML347 and
VU465350 (49,71–74).
Targeting ALK2 kinase in FOP
Among the aforementioned inhibitors, LDN-212854 has
a much higher selectivity for ALK2 than for ALK3 or ALK5 compared
with other derivatives (75).
LDN-212854 showed significant efficacy in the ALK2Q207D transgenic
mouse (71) and in a mouse model
with the ALK2R206H disease allele knocked in (76), which highly mimics human FOP.
Therefore, the interaction of LDN-212854 with ALK2 is suitable for
targeting ALK2R206H. The breakthrough in the identification of the
structure of this complex provides a template for the future
development of targeted ALK2 inhibitors for the treatment of FOP
and other related HO disorders (70).
Targeting ALK2 mRNA
ALK2 is encoded by the gene ACVR1. The
aforementioned inhibition of the ALK2 kinase can effectively
inhibit HO, therefore, it is important to determine whether
directly downregulating the expression of ALK2 at the
transcriptional level has a similar effect. The 2.9 kb sequence
located at the 5′end of the ACVR1 gene is an active promoter
(77). Cappato et al
(67) found dipyridamole, which
was identified using high-throughput screening, inhibited the
production of ectopic bone by downregulating the expression of
ACVR1 transcription in a BMP-induced mouse model of HO. Previous
studies have also shown that micro (mi)RNAs may regulate the
expression of ACVR1 (78–82). Specifically, miRNAs that target the
sequence at the 3′untranslated region of the ALK2 gene have been
shown to downregulate the expression of ACVR1/ALK2, these miRNAs
include miR-365, miR-148b, miR-148a, miR-30-C and miR-130a
(78–85).
Another method for downregulating the expression of
ACVR1 uses of antisense oligonucleotides (AON), which bind to
specific exons of genes in the primary mRNA transcript, thereby
preventing splicing and facilitating the skipping of specific exons
(86). Shi et al (83) reported the design of AONs to
knockdown the expression of mouse ALK2 by means of exon skipping.
The ALK2 AON induced exon skipping in cells, which was accompanied
by decreased ALK2 mRNA levels and impaired ALK2 signaling in C2C12
cells and endothelial cells. C2C12 cells are mouse myoblasts that
are often used to study induced differentiation in vitro,
with the cells differentiating into myotubes in vitro under
low serum conditions (87).
However, these strategies for modulating the expression of ALK2
affect both the wild-type and the mutated allele of the
disease-causing gene. From this perspective, RNA interference
(RNAi) is a good tool to develop allele-specific therapies for
genetic diseases. The design of allele-specific RNAi molecules to
target the expression of mutant ALK2 alleles indicated that the
RNAi approach may be successful in FOP (84,85).
Targeting ALK2 signaling
Screening for compounds that can treat HO using
drugs already in clinical use will greatly reduce the cost of drug
development, and the side effects of these drugs are already clear,
therefore, they have high safety profiles. Recently, Yamamoto et
al (88) screened drugs that
have been approved by the Food and Drug Administration (FDA) using
R206H-mutated ALK2-transfected C2C12 cells. It was found that two
drugs for preventing angina, fendiline hydrochloride and
perhexiline maleate, effectively inhibited the aberrantly activated
signaling. Both drugs were also tested in vivo in a mouse
model, animals treated with perhexiline maleate or fendiline
hydrochloride showed a substantial reduction in the volume of newly
formed bone (88). A clinical
trial was performed in which perhexiline maleate was given to five
patients with FOP to determine the effect of this drug in the
control of the disease (89).
While the drug was well tolerated, the study did not allow a
conclusion to be drawn as the serum levels of alkaline phosphatase
(ALP) and bone specific ALP were significantly and synchronously
elevated during flare-ups in the patients. Therefore, the trial did
not provide good evidence for the clinical utility of perhexiline
maleate (89). Dipyridamole, used
as a platelet anti-aggregant, was able to downregulate signaling
and reduce ectopic bone formation in a BMP-induced mouse model of
HO by decreasing the expression of the ACVR1 gene (67). Moreover, it has been reported that
the activation of the 5′AMP-activated protein kinase (AMPK) can
downregulate ALK2 expression by increasing the interaction of the
E3 protein ubiquitin-protein ligase Smurf1 with Smad6, leading to
the inhibition of osteogenic differentiation in MC3T3 cells and
iPSCs derived from patients with FOP (42). Metformin, a first-line drug for the
treatment of diabetes, is also a pharmacological activator of AMPK.
Our team has constructed a trauma-induced HO mouse model and found
that metformin significantly reduced the production of ectopic
bone. In addition, our lab also uses other clinically used AMPK
activators, including berberine and aspirin, to explore the effects
of these drugs on HO and the underlying mechanisms, and have found
them to have significant effects. Thus far, these existing drugs
are thought to inhibit the process of ectopic bone formation by
inhibiting ALK2/ACVR1; however, there are many other signaling
pathways involved in ectopic bone formation. These drugs may also
inhibit the formation of ectopic bone through other signaling
pathway molecules, and this remains to be explored.
Targeting ligand
Therapeutic monoclonal antibodies specific for ALK2
and activin are under development for in vitro and in
vivo studies (66,90). A specific antibody should block the
activity of the target protein and preserve the activation of the
target protein by its ligands.
Targeting activin A
In several cell types expressing mutated ALK2, the
binding of activin A to the R206H mutant receptor activates
downstream signaling through canonical Smad1/5/8 mediators
(5). To research the applicability
of these findings in vivo, Hatsell et al (47) developed a physiologically relevant,
genetically conditional knock-in mouse model of FOP. Upon
pharmacological induction of ALK2R206H expression, mice
spontaneously form HO that completely simulates the phenotype of
FOP, including joint function, premature mortality and the
development of ectopic bone. The injection of activin A in this
mouse model effectively induced ectopic bone formation; moreover,
the phenomenon was also observed in wild-type mice. Importantly,
the formation of new ectopic bone was prevented by treatment with
anti-activin A antibodies (47).
These data were consistent with another study (5). The discovery of antibodies to treat
HO is undoubtedly a major breakthrough in the field. A phase II
trial has recently been approved and is currently recruiting
patients to verify the effects on abnormal bone formation with an
anti-activin A antibody (REGN2477) (91).
Targeting ALK2
Mutant ALK2 demonstrates leaky Smad 1/5/8 signaling
and ligand hyperresponsiveness, providing a rationale for using
antibodies to target ALK2 in the prevention and treatment of FOP.
Therapeutic monoclonal antibodies specific for ALK2 are under
development (66).
An ALK2-Fc fusion protein, which consists of the
human ALK2 extracellular domain (residues 21–123) fused to the Fc
portion of the human immunoglobulin gamma 1 constant region
(residues 99–330), has been produced as a ligand trap for ALK2
(66,90). A previous in vitro study
showed that the ALK2 fusion protein binds to BMP-5/6 with high
affinity, had a weak binding capacity for Activin A and inhibited
the mutant ALK2-induced phosphorylation of Smad1/5/8. Moreover, the
Fc-fusion protein blocked osteoblast differentiation in HUVECs that
stably expressed ALK2R206H (92).
Conclusion and prospects
This review describes HO and the ALK2 signaling
pathways, including the formation of HO, the role of the BMP type I
receptor ALK2/ACVR1 in this process, and the structure and
regulation of ALK2. Furthermore, the current research into the
prevention and treatment of FOP-like HO was summarized and it was
proposed that ALK2 is a potential target for the treatment of
HO.
HO undoubtedly severely limits the ability of a
patient to move and affects quality of life. In the absence of
obvious symptoms and side effects, the ideal treatment is to give
the patient a drug on a chronic basis to prevent the acute phase
and disease progression. The drug should effectively block the
formation of lesions at the very early stages, therefore,
strategies that focus on targeting ligand or mutant receptor
activity may be more promising than others. However, due to the
complexity of the pathogenesis of HO, and in order to improve
treatment tolerance and safety, the use of a combination of drugs
with different targets and synergistic or additive effects may
yield better results, and may reduce the dose of each drug
required. Another key aspect is the possibility that a patient may
need to stop taking the drug for a variety of reasons. In these
cases, it must be ensured that any effects of drug withdrawal will
not occur.
Although basic and translational research on HO has
made great progress, some important factors in the pathogenesis of
the disease deserve further study, such as the role of the innate
and adaptive immune systems, the cross-correlation between
different transduction pathways, and the identification of
biomarkers suitable for monitoring disease and treatment efficacy.
As aforementioned, cells involved in inflammation secrete factors
that induce HO, including BMPs. It is important to identify which
cells are secreting BMPs and to understand how activin A affects
mutated ALK2. These studies will not only help to better understand
the disease mechanism, but will also help to provide new drug
targets and expand the treatment options available for patients
with HO. In addition to FOP, understanding the pathogenesis and
determining treatment goals can benefit patients with other more
common forms of HO, and those without underlying genetic
causes.
Both small molecule inhibitors and FDA-approved
drugs for the treatment of other diseases, as well as noncoding
RNAs and RNAi, are reported to reduce ectopic bone formation at the
cellular level and in animal models. However, all of these
strategies are at the stage of basic research, and it will take a
time for them to enter clinical use. Therefore, more research to
provide an increased number of possibilities for drug developers is
required, as is reducing the distance between clinical medicine and
basic research. In addition, the dysregulation of the BMP signaling
pathway is associated with a variety of diseases, and
pharmacological strategies to modulate BMP signaling are an
effective strategy to elucidate the specific functions, and
multiple biological effects of BMPs. However, the development of
small molecule kinase inhibitors faces a huge challenge. The degree
of homology between BMP type I receptors is very high, and it is
easy to cause off-target effects. With respect to diseases,
especially those requiring long-term treatment, off-target effects
must be reduced or eliminated.
Acknowledgements
Not applicable.
Funding
The present review was supported in part by grants
from the National Natural Science Foundation of China (grant nos.
81272926, 81572753, and 31660332). HL was supported by the Doctoral
Scientific Research Foundation from Nanchang University and Young
Teachers Research and Development Fund Project from Jiangxi medical
college of Nanchang University (grant no. PY201801) and the Natural
science Foundation of Jiangxi Province (grant no.
2018BAB215012).
Availability of data and materials
Not applicable.
Authors' contributions
FS, JG, JZ, YY and HL were involved in the
conception and design of the review. FS and HL drafted the review.
FS, JZ, YY, JG and HL revised the manuscript. All authors gave
final approval of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ALK2
|
activin receptor-like kinase 2
|
|
HO
|
heterotopic ossification
|
|
BMPs
|
bone morphogenetic proteins
|
|
AHO
|
acquired heterotopic ossification
|
|
HO
|
genetic heterotopic ossification
|
|
FOP
|
fibrodysplasia ossificans
progressive
|
|
POH
|
progressive osseous heteroplasia
|
|
MAPK
|
mitogen-activated protein kinase
|
|
FKBP12
|
12 kDa FK506 binding protein
|
|
MEFs
|
mouse embryonic fibroblasts
|
|
iPSCs
|
induced pluripotent stem cells
|
|
NSAIDs
|
nonsteroidal anti-inflammatory
drugs
|
|
AON
|
antisense oligonucleotides
|
|
AMPK
|
adenosine monophosphate activated
protein kinase
|
|
OCN
|
osteocalcin
|
References
|
1
|
Edwards DS, Kuhn KM, Potter BK and
Forsberg JA: Heterotopic ossification: A review of current
understanding, treatment, and future. J Orthop Trauma. 30 (Suppl
3):S27–S30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hildebrand L, Rossbach B, Kühnen P, Gossen
M, Kurtz A, Reinke P, Seemann P and Stachelscheid H: Generation of
integration free induced pluripotent stem cells from fibrodysplasia
ossificans progressiva (FOP) patients from urine samples. Stem Cell
Res. 16:54–58. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shore EM, Ahn J, Jan de Beur S, Li M, Xu
M, Gardner RJ, Zasloff MA, Whyte MP, Levine MA and Kaplan FS:
Paternally inherited inactivating mutations of the GNAS1 gene in
progressive osseous heteroplasia. N Engl J Med. 346:99–106. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maruyama R and Yokota T:
Morpholino-mediated exon skipping targeting human ACVR1/ALK2 for
fibrodysplasia ossificans progressiva. Methods Mol Biol.
1828:497–502. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lees-Shepard JB, Yamamoto M, Biswas AA,
Stoessel SJ, Nicholas SE, Cogswell CA, Devarakonda PM, Schneider MJ
Jr, Cummins SM, Legendre NP, et al: Activin-dependent signaling in
fibro/adipogenic progenitors causes fibrodysplasia ossificans
progressiva. Nat Commun. 9:4712018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu M, Chen G and Li YP: TGF-β and BMP
signaling in osteoblast, skeletal development, and bone formation,
homeostasis and disease. Bone Res. 4:160092016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abula K, Muneta T, Miyatake K, Yamada J,
Matsukura Y, Inoue M, Sekiya I, Graf D, Economides AN, Rosen V and
Tsuji K: Elimination of BMP7 from the developing limb mesenchyme
leads to articular cartilage degeneration and synovial inflammation
with increased age. FEBS Lett. 589:1240–1248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pereda A, Martos-Tello JM, Garin I,
Errea-Dorronsoro J and Perez de Nanclares G: Progressive osseous
heteroplasia caused by a mosaic GNAS mutation. Clin Endocrinol
(Oxf). 88:993–955. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Culbert AL, Chakkalakal SA, Theosmy EG,
Brennan TA, Kaplan FS and Shore EM: Alk2 regulates early
chondrogenic fate in fibrodysplasia ossificans progressiva
heterotopic endochondral ossification. Stem Cells. 32:1289–300.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feldman G, Li M, Martin S, Urbanek M,
Urtizberea JA, Fardeau M, LeMerrer M, Connor JM, Triffitt J, Smith
R, et al: Fibrodysplasia ossificans progressiva, a heritable
disorder of severe heterotopic ossification, maps to human
chromosome 4q27-31. Am J Hum Genet. 66:128–135. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Regard JB, Malhotra D, Gvozdenovic-Jeremic
J, Josey M, Chen M, Weinstein LS, Lu J, Shore EM, Kaplan FS and
Yang Y: Activation of Hedgehog signaling by loss of GNAS causes
heterotopic ossification. Nat Med. 19:1505–1512. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Forsberg JA, Pepek JM, Wagner S, Wilson K,
Flint J, Andersen RC, Tadaki D, Gage FA, Stojadinovic A and Elster
EA: Heterotopic ossification in high-energy wartime extremity
injuries: Prevalence and risk factors. J Bone Joint Surg Am.
91:1084–1091. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaplan FS, Le Merrer M, Glaser DL, Pignolo
RJ, Goldsby RE, Kitterman JA, Groppe J and Shore EM: Fibrodysplasia
ossificans progressiva. Best Pract Res Clin Rheumatol. 22:191–205.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kaplan FS, Xu M, Glaser DL, Collins F,
Connor M, Kitterman J, Sillence D, Zackai E, Ravitsky V, Zasloff M,
et al: Early diagnosis of fibrodysplasia ossificans progressiva.
Pediatrics. 121:e1295–e1300. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Potter BK, Forsberg JA, Davis TA, Evans
KN, Hawksworth JS, Tadaki D, Brown TS, Crane NJ, Burns TC, O'Brien
FP and Elster EA: Heterotopic ossification following combat-related
trauma. J Bone Joint Surg Am. 92 (Suppl 2):S74–S89. 2010.
View Article : Google Scholar
|
|
16
|
Kan L and Kessler JA: Animal models of
typical heterotopic ossification. J Biomed Biotechnol.
2011:3092872011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Alfieri KA, Forsberg JA and Potter BK:
Blast injuries and heterotopic ossification. Bone Joint Res.
1:192–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shehab D, Elgazzar AH and Collier BD:
Heterotopic ossification. J Nucl Med. 43:346–353. 2002.PubMed/NCBI
|
|
19
|
Pavey GJ, Polfer EM, Nappo KE, Tintle SM,
Forsberg JA and Potter BK: What risk factors predict recurrence of
heterotopic ossification after excision in combat-related
amputations? Clin Orthop Relat Res. 473:2814–2824. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gugala Z, Olmsted-Davis EA, Xiong Y, Davis
EL and Davis AR: Trauma-induced heterotopic ossification regulates
the blood-nerve barrier. Front Neurol. 9:4082018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Juarez JK, Wenke JC and Rivera JC:
Treatments and preventative measures for trauma-induced heterotopic
ossification: A review. Clin Transl Sci. 11:365–370. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carroll SF, Buckley CT and Kelly DJ:
Cyclic tensile strain can play a role in directing both
intramembranous and endochondral ossification of mesenchymal stem
cells. Front Bioeng Biotechnol. 5:732017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Upadhyay J, Xie L, Huang L, Das N, Stewart
RC, Lyon MC, Palmer K, Rajamani S, Graul C, Lobo M, et al: The
expansion of heterotopic bone in fibrodysplasia ossificans
progressiva is activin A-dependent. J Bone Miner Res. 32:2489–2499.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu R, Hu J, Zhou X and Yang Y: Heterotopic
ossification: Mechanistic insights and clinical challenges. Bone.
109:134–142. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lounev VY, Ramachandran R, Wosczyna MN,
Yamamoto M, Maidment AD, Shore EM, Glaser DL, Goldhamer DJ and
Kaplan FS: Identification of progenitor cells that contribute to
heterotopic skeletogenesis. J Bone Joint Surg Am. 91:652–663. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Glaser DL, Economides AN, Wang L, Liu X,
Kimble RD, Fandl JP, Wilson JM, Stahl N, Kaplan FS and Shore EM: In
vivo somatic cell gene transfer of an engineered Noggin mutein
prevents BMP4-induced heterotopic ossification. J Bone Joint Surg
Am. 85:2332–2342. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kan L and Kessler JA: Evaluation of the
cellular origins of heterotopic ossification. Orthopedics.
37:329–340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ji Y, Christopherson GT, Kluk MW, Amrani
O, Jackson WM and Nesti LJ: Heterotopic ossification following
musculoskeletal trauma: Modeling stem and progenitor cells in their
microenvironment. Adv Exp Med Biol. 720:39–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Medici D, Shore EM, Lounev VY, Kaplan FS,
Kalluri R and Olsen BR: Conversion of vascular endothelial cells
into multipotent stem-like cells. Nat Med. 16:1400–1406. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Olmsted-Davis E, Gannon FH, Ozen M,
Ittmann MM, Gugala Z, Hipp JA, Moran KM, Fouletier-Dilling CM,
Schumara-Martin S, Lindsey RW, et al: Hypoxic adipocytes pattern
early heterotopic bone formation. Am J Pathol. 170:620–632. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olmsted-Davis EA, Salisbury EA, Hoang D,
Davis EL, Lazard Z, Sonnet C, Davis TA, Forsberg JA and Davis AR:
Progenitors in peripheral nerves launch heterotopic ossification.
Stem Cells Transl Med. 6:1109–1119. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gurkan UA, Golden R, Kishore V, Riley CP,
Adamec J and Akkus O: Immune and inflammatory pathways are involved
in inherent bone marrow ossification. Clin Orthop Relat Res.
470:2528–2540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luu HH, Song WX, Luo X, Manning D, Luo J,
Deng ZL, Montag AG, Haydon RC and He TC: Distinct roles of bone
morphogenetic proteins in osteogenic differentiation of mesenchymal
stem cells. J Orthop Res. 25:665–677. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen D, Zhao M and Mundy GR: Bone
morphogenetic proteins. Growth Factors. 22:233–241. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rahman MS, Akhtar N, Jamil HM, Banik RS
and Asaduzzaman SM: TGF-β/BMP signaling and other molecular events:
Regulation of osteoblastogenesis and bone formation. Bone Res.
15005015.
|
|
36
|
Sánchez-Duffhues G, Hiepen C, Knaus P and
Ten Dijke P: Bone morphogenetic protein signaling in bone
homeostasis. Bone. 80:43–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shore EM and Kaplan FS: Role of altered
signal transduction in heterotopic ossification and fibrodysplasia
ossificans progressiva. Curr Osteoporos Rep. 9:83–88. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bouvard B, Masson C, Legrand E and Audran
M: Fibrodysplasia ossificans progressiva. A case report and focus
on the BMP signaling pathway. Morphologie. 100:250–255. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kan C, Chen L, Hu Y, Ding N, Lu H, Li Y,
Kessler JA and Kan L: Conserved signaling pathways underlying
heterotopic ossification. Bone. 109:43–48. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chaikuad A, Alfano I, Kerr G, Sanvitale
CE, Boergermann JH, Triffitt JT, von Delft F, Knapp S, Knaus P and
Bullock AN: Structure of the bone morphogenetic protein receptor
ALK2 and implications for fibrodysplasia ossificans progressiva. J
Biol Chem. 287:36990–36998. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dudas M, Sridurongrit S, Nagy A, Okazaki K
and Kaartinen V: Craniofacial defects in mice lacking BMP type I
receptor Alk2 in neural crest cells. Mech Dev. 121:173–182. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lin H, Ying Y, Wang YY, Wang G, Jiang SS,
Huang D, Luo L, Chen YG, Gerstenfeld LC and Luo Z: AMPK
downregulates ALK2 via increasing the interaction between Smurf1
and Smad6, leading to inhibition of osteogenic differentiation.
Biochim Biophys Acta Mol Cell Res. 1864:2369–2377. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Engers DW, Frist AY, Lindsley CW, Hong CC
and Hopkins CR: Synthesis and structure-activity relationships of a
novel and selective bone morphogenetic protein receptor (BMP)
inhibitor derived from the pyrazolo[1.5-a]pyrimidine scaffold of
dorsomorphin: The discovery of ML347 as an ALK2 versus ALK3
selective MLPCN probe. Bioorg Med Chem Lett. 23:3248–3252. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Machiya A, Tsukamoto S, Ohte S, Kuratani
M, Fujimoto M, Kumagai K, Osawa K, Suda N, Bullock AN and Katagiri
T: Effects of FKBP12 and type II BMP receptors on signal
transduction by ALK2 activating mutations associated with genetic
disorders. Bone. 111:101–108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
van Dinther M, Visser N, de Gorter DJ,
Doorn J, Goumans MJ, de Boer J and ten Dijke P: ALK2 R206H mutation
linked to fibrodysplasia ossificans progressiva confers
constitutive activity to the BMP type I receptor and sensitizes
mesenchymal cells to BMP-induced osteoblast differentiation and
bone formation. J Bone Miner Res. 25:1208–1215. 2010.PubMed/NCBI
|
|
46
|
Chakkalakal SA, Zhang D, Culbert AL,
Convente MR, Caron RJ, Wright AC, Maidment AD, Kaplan FS and Shore
EM: An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans
progressiva. J Bone Miner Res. 27:1746–1756. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hatsell SJ, Idone V, Wolken DM, Huang L,
Kim HJ, Wang L, Wen X, Nannuru KC, Jimenez J, Xie L, et al:
ACVR1R206H receptor mutation causes fibrodysplasia ossificans
progressiva by imparting responsiveness to activin A. Sci Transl
Med. 7:303ra1372015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Katagiri T: A door opens for
fibrodysplasia ossificans progressiva. Trends Biochem Sci.
41:119–121. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD,
Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, et
al: BMP type I receptor inhibition reduces heterotopic [corrected]
ossification. Nat Med. 14:1363–1369. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hino K, Ikeya M, Horigome K, Matsumoto Y,
Ebise H, Nishio M, Sekiguchi K, Shibata M, Nagata S, Matsuda S and
Toguchida J: Neofunction of ACVR1 in fibrodysplasia ossificans
progressiva. Proc Natl Acad Sci USA. 112:15438–15443. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Olsen OE, Wader KF, Hella H, Mylin AK,
Turesson I, Nesthus I, Waage A, Sundan A and Holien T: Activin A
inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun
Signal. 13:272015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang H, Lindborg C, Lounev V, Kim JH,
McCarrick-Walmsley R, Xu M, Mangiavini L, Groppe JC, Shore EM,
Schipani E, et al: Cellular hypoxia promotes heterotopic
ossification by amplifying BMP signaling. J Bone Miner Res.
31:1652–1665. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kent WT, Shelton TJ and Eastman J:
Heterotopic ossification around the knee after tibial nailing and
ipsilateral antegrade and retrograde femoral nailing in the
treatment of floating knee injuries. Int Orthop. 42:1379–1385.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang YK, Sun WF, Liu XG, Deng J, Yan BE,
Jiang WY and Lin XB: Comparative study of serum levels of BMP-2 and
heterotopic ossification in traumatic brain injury and fractures
patients. Zhongguo Gu Shang. 24:399–403. 2011.(In Chinese).
PubMed/NCBI
|
|
55
|
Peterson JR, De La Rosa S, Eboda O, Cilwa
KE, Agarwal S, Buchman SR, Cederna PS, Xi C, Morris MD, Herndon DN,
et al: Treatment of heterotopic ossification through remote ATP
hydrolysis. Sci Transl Med. 6:255ra1322014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kang H, Dang AB, Joshi SK, Halloran B,
Nissenson R, Zhang X, Li J, Kim HT and Liu X: Novel mouse model of
spinal cord injury-induced heterotopic ossification. J Rehabil Res
Dev. 51:1109–1118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lengner CJ, Lepper C, van Wijnen AJ, Stein
JL, Stein GS and Lian JB: Primary mouse embryonic fibroblasts: A
model of mesenchymal cartilage formation. J Cell Physiol.
200:327–333. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sun E and Hanyu-Deutmeyer AA: Heterotopic
Ossification. StatPearlsTreasure Island (FL): StatPearls Publishing
StatPearls Publishing LLC; 2018
|
|
59
|
Beckmann JT, Wylie JD, Potter MQ, Maak TG,
Greene TH and Aoki SK: Effect of naproxen prophylaxis on
heterotopic ossification following hip arthroscopy: A double-blind
randomized placebo-controlled trial. J Bone Joint Surg Am.
97:2032–2037. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rivera JC, Hsu JR, Noel SP, Wenke JC and
Rathbone CR: Locally delivered nonsteroidal antiinflammatory drug:
A potential option for heterotopic ossification prevention. Clin
Transl Sci. 8:591–593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rath E, Warschawski Y, Maman E, Dolkart O,
Sharfman ZT, Salai M and Amar E: Selective COX-2 inhibitors
significantly reduce the occurrence of heterotopic ossification
after Hip arthroscopic surgery. Am J Sports Med. 44:677–681. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Beckmann JT, Wylie JD, Kapron AL, Hanson
JA, Maak TG and Aoki SK: The effect of NSAID prophylaxis and
operative variables on heterotopic ossification after Hip
arthroscopy. Am J Sports Med. 42:1359–1364. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Haran M, Bhuta T and Lee B:
Pharmacological interventions for treating acute heterotopic
ossification. Cochrane Database Syst Rev. CD0033212004.PubMed/NCBI
|
|
64
|
Salazar D, Golz A, Israel H and Marra G:
Heterotopic ossification of the elbow treated with surgical
resection: Risk factors, bony ankylosis, and complications. Clin
Orthop Relat Res. 472:2269–2275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sheybani A, TenNapel MJ, Lack WD, Clerkin
P, Hyer DE, Sun W and Jacobson GM: Risk of radiation-induced
malignancy with heterotopic ossification prophylaxis: A
case-control analysis. Int J Radiat Oncol Biol Phys. 89:584–589.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kaplan FS, Pignolo RJ, Al Mukaddam MM and
Shore EM: Hard targets for a second skeleton: Therapeutic horizons
for fibrodysplasia ossificans progressiva (FOP). Expert Opin Orphan
Drugs. 5:291–294. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Cappato S, Tonachini L, Giacopelli F,
Tirone M, Galietta LJ, Sormani M, Giovenzana A, Spinelli AE,
Canciani B, Brunelli S, et al: High-throughput screening for
modulators of ACVR1 transcription: Discovery of potential
therapeutics for fibrodysplasia ossificans progressiva. Dis Model
Mech. 9:685–696. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Glister C, Regan SL, Samir M and Knight P:
Gremlin, Noggin, Chordin and follistatin differentially modulate
BMP induced suppression of androgen secretion by bovine ovarian
theca cells. J Mol Endocrinol. Oct 1–2018.(Epub ahead of print).
PubMed/NCBI
|
|
69
|
Yu PB, Hong CC, Sachidanandan C, Babitt
JL, Deng DY, Hoyng SA, Lin HY, Bloch KD and Peterson RT:
Dorsomorphin inhibits BMP signals required for embryogenesis and
iron metabolism. Nat Chem Biol. 4:33–41. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Cuny GD, Yu PB, Laha JK, Xing X, Liu JF,
Lai CS, Deng DY, Sachidanandan C, Bloch KD and Peterson RT:
Structure-activity relationship study of bone morphogenetic protein
(BMP) signaling inhibitors. Bioorg Med Chem Lett. 18:4388–4392.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mohedas AH, Xing X, Armstrong KA, Bullock
AN, Cuny GD and Yu PB: Development of an ALK2-biased BMP type I
receptor kinase inhibitor. ACS Chem Biol. 8:1291–1302. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hao J, Ho JN, Lewis JA, Karim KA, Daniels
RN, Gentry PR, Hopkins CR, Lindsley CW and Hong CC: In vivo
structure-activity relationship study of dorsomorphin analogues
identifies selective VEGF and BMP inhibitors. ACS Chem Biol.
5:245–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tsugawa D, Oya Y, Masuzaki R, Ray K,
Engers DW, Dib M, Do N, Kuramitsu K, Ho K, Frist A, et al: Specific
activin receptor-like kinase 3 inhibitors enhance liver
regeneration. J Pharmacol Exp Ther. 351:549–558. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mohedas AH, Wang Y, Sanvitale CE, Canning
P, Choi S, Xing X, Bullock AN, Cuny GD and Yu PB:
Structure-activity relationship of 3,5-diaryl-2-aminopyridine ALK2
inhibitors reveals unaltered binding affinity for fibrodysplasia
ossificans progressiva causing mutants. J Med Chem. 57:7900–7915.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Williams E and Bullock AN: Structural
basis for the potent and selective binding of LDN-212854 to the BMP
receptor kinase ALK2. Bone. 109:251–258. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Dey D, Bagarova J, Hatsell SJ, Armstrong
KA, Huang L, Ermann J, Vonner AJ, Shen Y, Mohedas AH, Lee A, et al:
Two tissue-resident progenitor lineages drive distinct phenotypes
of heterotopic ossification. Sci Transl Med. 8:366ra1632016.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Giacopelli F, Cappato S, Tonachini L, Mura
M, Di Lascio S, Fornasari D, Ravazzolo R and Bocciardi R:
Identification and characterization of regulatory elements in the
promoter of ACVR1, the gene mutated in Fibrodysplasia Ossificans
Progressiva. Orphanet J Rare Dis. 8:1452013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Li L, Liu Y, Guo Y, Liu B, Zhao Y, Li P,
Song F, Zheng H, Yu J, Song T, et al: Regulatory MiR-148a-ACVR1/BMP
circuit defines a cancer stem cell-like aggressive subtype of
hepatocellular carcinoma. Hepatology. 61:574–584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zumbrennen-Bullough KB, Wu Q, Core AB,
Canali S, Chen W, Theurl I, Meynard D and Babitt JL: MicroRNA-130a
is up-regulated in mouse liver by iron deficiency and targets the
bone morphogenetic protein (BMP) receptor ALK2 to attenuate BMP
signaling and hepcidin transcription. J Biol Chem. 289:23796–23808.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Song H, Wang Q, Wen J, Liu S, Gao X, Cheng
J and Zhang D: ACVR1, a therapeutic target of fibrodysplasia
ossificans progressiva, is negatively regulated by miR-148a. Int J
Mol Sci. 13:2063–2077. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Mura M, Cappato S, Giacopelli F, Ravazzolo
R and Bocciardi R: The role of the 3′UTR region in the regulation
of the ACVR1/Alk-2 gene expression. PLoS One. 7:e509582012.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Karbiener M, Neuhold C, Opriessnig P,
Prokesch A, Bogner-Strauss JG and Scheideler M: MicroRNA-30c
promotes human adipocyte differentiation and co-represses PAI-1 and
ALK2. RNA Biol. 8:850–860. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Shi S, Cai J, de Gorter DJ,
Sanchez-Duffhues G, Kemaladewi DU, Hoogaars WM, Aartsma-Rus A, 't
Hoen PA and ten Dijke P: Antisense-oligonucleotide mediated exon
skipping in activin-receptor-like kinase 2: Inhibiting the receptor
that is overactive in fibrodysplasia ossificans progressiva. PLoS
One. 8:e690962013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Takahashi M, Katagiri T, Furuya H and
Hohjoh H: Disease-causing allele-specific silencing against the
ALK2 mutants, R206H and G356D, in fibrodysplasia ossificans
progressiva. Gene Ther. 19:781–785. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kaplan J, Kaplan FS and Shore EM:
Restoration of normal BMP signaling levels and osteogenic
differentiation in FOP mesenchymal progenitor cells by mutant
allele-specific targeting. Gene Ther. 19:786–790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Aartsma-Rus A, Fokkema I, Verschuuren J,
Ginjaar I, van Deutekom J, van Ommen GJ and den Dunnen JT:
Theoretic applicability of antisense-mediated exon skipping for
Duchenne muscular dystrophy mutations. Hum Mutat. 30:293–299. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Miki Y, Morioka T, Shioi A, Fujimoto K,
Sakura T, Uedono H, Kakutani Y, Ochi A, Mori K, Shoji T, et al:
Oncostatin M induces C2C12 myotube atrophy by modulating muscle
differentiation and degradation. Biochem Biophys Res Commun.
516:951–956. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yamamoto R, Matsushita M, Kitoh H, Masuda
A, Ito M, Katagiri T, Kawai T, Ishiguro N and Ohno K: Clinically
applicable antianginal agents suppress osteoblastic transformation
of myogenic cells and heterotopic ossifications in mice. J Bone
Miner Metab. 31:26–33. 2013.PubMed/NCBI
|
|
89
|
Kitoh H, Achiwa M, Kaneko H, Mishima K,
Matsushita M, Kadono I, Horowitz JD, Sallustio BC, Ohno K and
Ishiguro N: Perhexiline maleate in the treatment of fibrodysplasia
ossificans progressiva: An open-labeled clinical trial. Orphanet J
Rare Dis. 8:1632013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kaplan FS, Pignolo RJ and Shore EM: From
mysteries to medicines: Drug development for fibrodysplasia
ossificans progressive. Expert Opin Orphan Drugs. 1:637–649. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Cappato S, Giacopelli F, Ravazzolo R and
Bocciardi R: The horizon of a therapy for rare genetic diseases: A
‘Druggable’ future for fibrodysplasia ossificans progressiva. Int J
Mol Sci. 19(pii): E9892018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Pang J, Zuo Y, Chen Y, Song L, Zhu Q, Yu
J, Shan C, Cai Z, Hao J, Kaplan FS, et al: ACVR1-Fc suppresses BMP
signaling and chondro-osseous differentiation in an in vitro model
of Fibrodysplasia ossificans progressiva. Bone. 92:29–36. 2016.
View Article : Google Scholar : PubMed/NCBI
|