Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
6th most common general malignancy worldwide, and five million new
cases of HNSCC are diagnosed each year (1). Tobacco smoking, alcohol consumption,
and human papilloma virus infections are associated with the
occurrence of HNSCC (2). The
5-year overall survival rate of patients with HNSCC is 40–50%, and
this has remained unchanged over the last four to five decades,
despite substantial advances in multimodal therapies.
MicroRNAs (miRNAs) are small non-coding RNAs that
silence genes by binding to the target mRNAs to either inhibit
their translation or destabilize the mRNA (3). Since the beginning of the 21st
century, research on the relationship between miRNAs and cancer
development has intensified (4).
The Cancer Genome Atlas (TCGA) is a landmark cancer
genomics program, that molecularly characterizes over 20,000
primary cancers and matches normal samples spanning 33 cancer
types. The creation of TCGA and the ubiquity of high-throughput
sequencing data has meant that the sharing of genomic datasets
related to cancer research has become more common (5). For instance, a study by Long et
al (6) reported that four
overall-survival (OS)-related genes sourced from a hepatocellular
carcinoma dataset deposited in the TCGA were predictive of
different prognoses. This result demonstrates that clinically
relevant cancer biomarkers can be identified from next-generation
sequencing (NGS) datasets.
In this study, a TCGA dataset consisting of miRNA
expression data from HNSCC tumor and adjacent normal tissues was
obtained and identification of potential gene biomarkers was
attempted. Based on the results of this analysis, a seven-miRNA
prognostic model was constructed and designed to recognize a miRNA
expression signature to predict the overall survival rate of HNSCC
patients when provided with clinical information and bioinformatic
data as inputs. Functional analysis of the identified miRNAs was
also performed. These results may improve our knowledge concerning
the mechanisms of HNSCC progression and may be useful for guiding
the clinical treatments of HNSCC patients.
Materials and methods
Sources of expression profiles and
clinical data
A dataset containing miRNA expression profiles from
paired samples of HNSCC tissue and adjacent non-tumorous head and
neck tissue was obtained. As of January 1, 2018, this dataset,
which was acquired from the TCGA data portal (https://portal.gdc.cancer.gov/) (7), contained 525 HNSCC tissue samples and
44 samples of adjacent non-tumorous tissue. All sequencing
procedures were performed on an Illumina HiSeq platform.
Accompanying clinical data (‘id’, ‘survival time’, ‘survival
state’, ‘the prognostic model’, ‘T-stage’, ‘N-stage’, ‘perineural
invasion’, ‘lymphovascular invasion’, ‘sex’, ‘age’, ‘histologic
grade’ and ‘tumor site’) were obtained from the University of
California, Santa Cruz Xena browser (UCSC http://xenabrowser.net/datapages/?dataset). All data
used was obtained from the TCGA database or the UCSC browser.
Because the original deposition of the data abided by the ethical
guidelines of the TCGA platform, there was no need to obtain
additional ethical clearance from our local research ethics
committee.
Identification of differentially
expressed miRNAs in HNSCC and adjacent normal tissue samples
Approximately 1,881 miRNA expression profiles of
HNSCC tissue samples were acquired from the TCGA database. Next,
differentially expressed miRNAs (DEMs) were identified using the
edgeR package (8). DEMs
were identified by having an absolute log2 fold change
(FC) >1.5 and a P-value <0.01.
Establishment of a miRNA-related
prognostic model
A prognostic model was constructed by implementing
log-rank tests, LASSO-Cox regression (9), and multivariate Cox regressions to
analyze the DEM dataset. First, all miRNAs were screened using
log-rank tests of the overall survival rates of patients and the
miRNA expression levels. Only DEMs with P-values <0.05 were
retained for further analyses. Second, a LASSO-Cox regression
analysis was used to develop a prognostic miRNA panel for HNSCC
patients. Next, the independency of the prognosis power of the
miRNA panel was assessed using a stepwise multivariate Cox
regression. Finally, a novel seven-miRNA prognostic model was
established by multiplying the expression levels of the constituent
miRNAs with a regression coefficient derived from the multivariate
Cox regression. Every HNSCC patient can obtain a risk score
according to the prognostic model given the correct inputs. Risk
scores were employed to classify patients into low- and high-risk
groups by the median method. Finally, the predictive ability of the
model was evaluated using a time-dependent receiver operating
characteristic curve (ROC) and analysis of Kaplan-Meier survival
estimates (10,11).
Univariate and multivariate Cox
regression analyses
Both univariate and multivariate Cox regression
analyses (12–17) were performed on the HNSCC patient
list to assess the independence of the prognosis from other
clinical information (i.e. age, sex, histologic grade, T stage, N
stage, perineural invasion, and lymphovascular invasion). Hazard
ratios (HRs) and 95% confidence intervals were calculated using
SPSS version 19.0 (IBM Corp.). All tests performed were
two-tailed.
Functional enrichment analysis
The function of the downstream target genes of the
seven miRNAs using TargetScan (18) was predicted. The Gene Ontology (GO)
annotations of predicted target genes were determined and a Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analysis was conducted on them using the clusterProfiler R
package (19).
Results
Some miRNAs are differentially
expressed in HNSCC and adjacent normal tissue samples
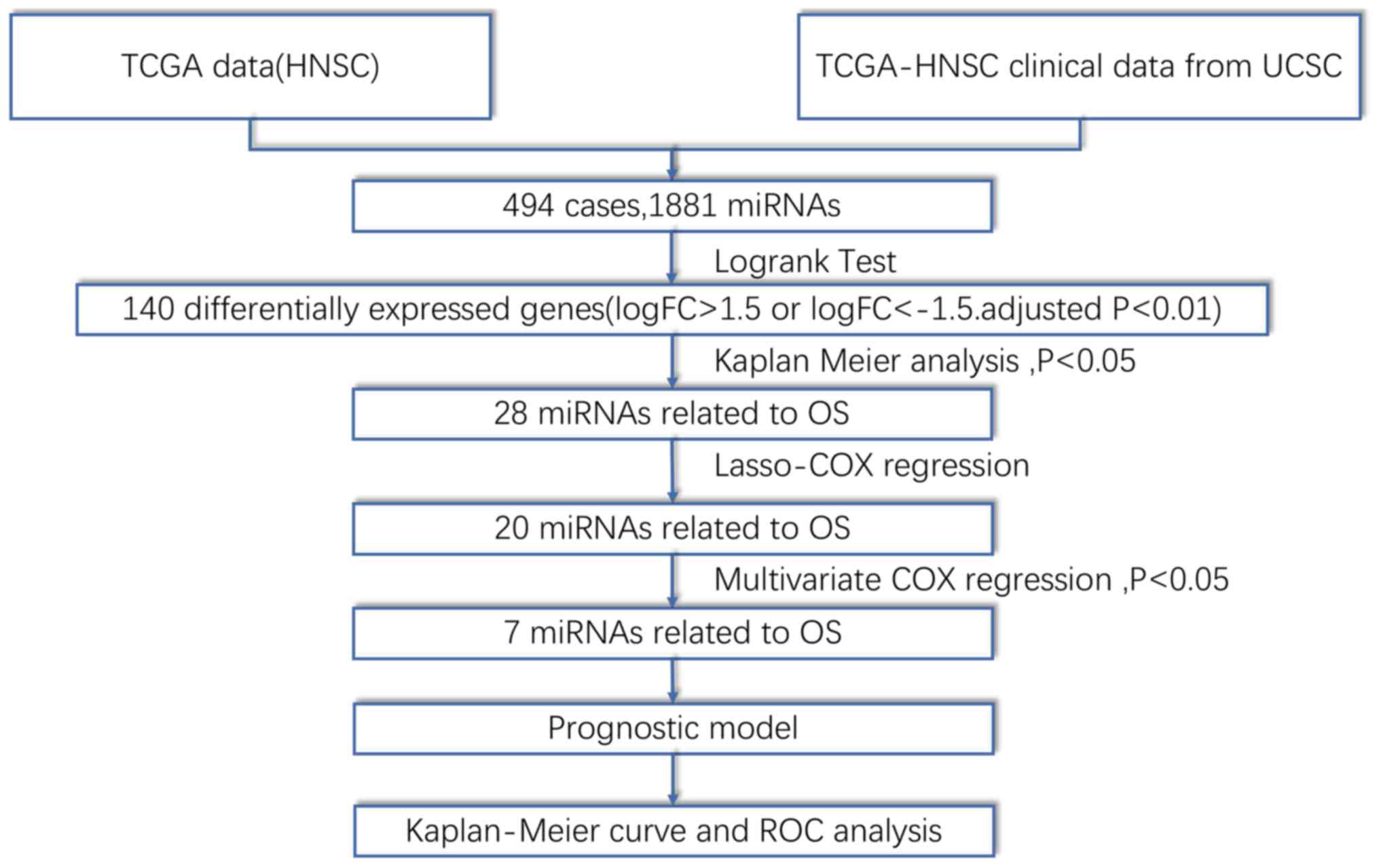
Fig. 1 depicts a
flow chart of the analysis procedure used for this study.
Approximately 140 DEMs (i.e. genes with logFC>1.5 or <-1.5
and adjusted P<0.01) were identified in the miRNA expression
profiles of HNSCC tissue samples (n=525) relative to adjacent
normal tissue samples (n=44). Of these DEMs, 52 miRNAs were found
to be downregulated in HNSCC tissue, while 88 miRNAs were
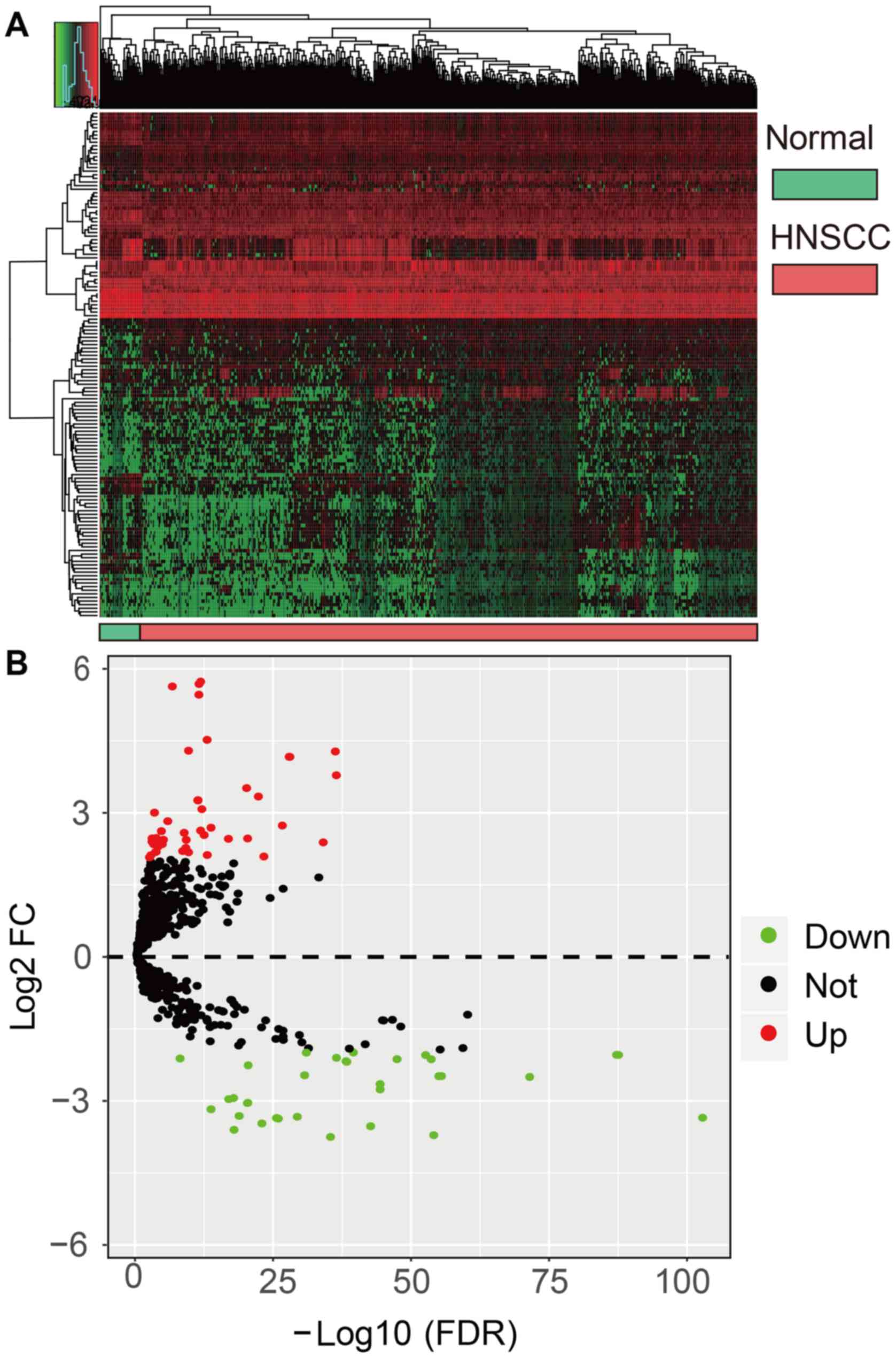
upregulated. Fig. 2 reveals a
volcano plot and heatmap of the 140 DEMs as generated by the
edgeR and ggplot2 R packages, respectively. Normal
and HNSCC tissues are distinguished by red or green labels in
Fig. 2A.
Log-rank tests and least absolute
shrinkage and selection operator (LASSO) regression
First, the prognostic significance of each miRNA was
evaluated by performing log-rank tests as implemented by the
survival R package. miRNAs with P-values <0.01 are listed
in Table I. Next, LASSO regression
was performed to simplify and regularize the miRNA model for HNSCC
patients. LASSO is a biased estimation method used to process data
with complex patterns of collinearity. The LASSO procedure
generates a relatively concise model by constructing a penalty
function, which forces the sum of the absolute values of the
coefficients to be less than a standardized constraint parameter
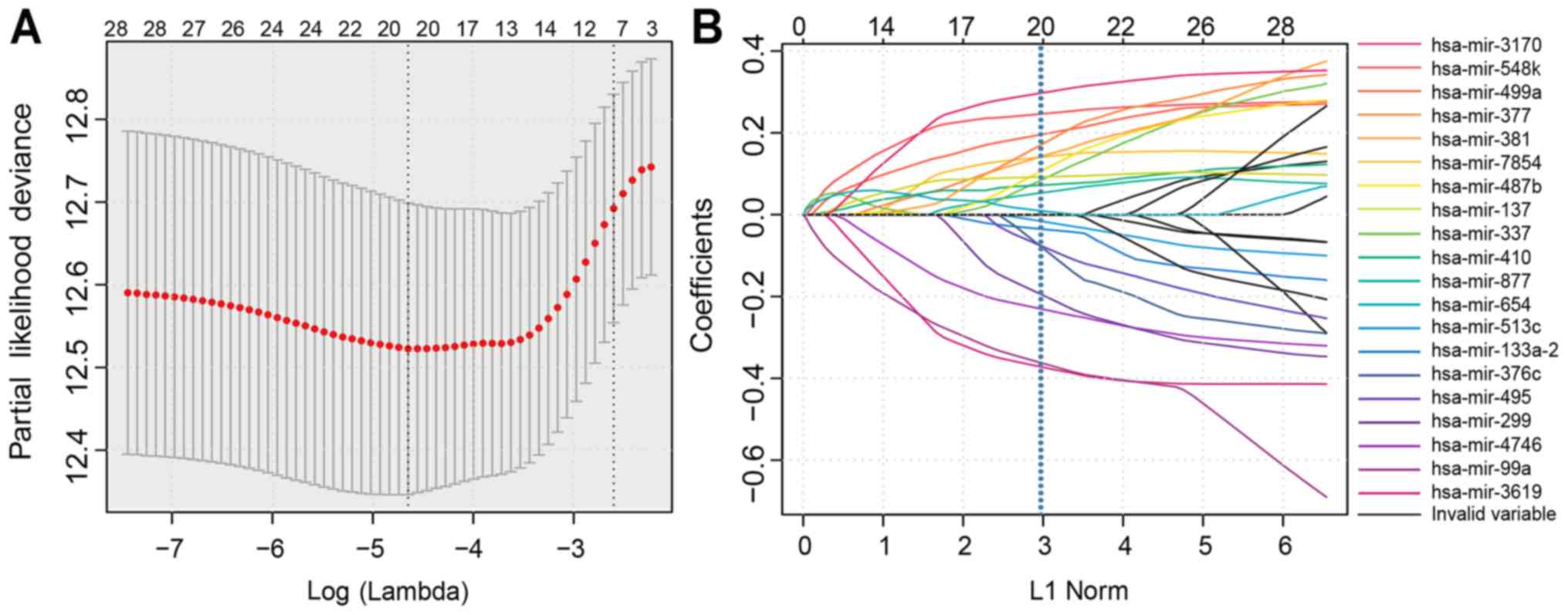
Lambda (λ). Fig. 3A reveals a
generalized cross-validation plot for the HNSCC patient data. The
partial likelihood deviance was plotted against log (λ), where λ is
the tuning parameter. The dotted vertical lines were drawn at the
optimal values by minimum criteria and 1-SE criteria. The suitable
λ occurred when the model included 20 variables. Fig. 3B reveals the estimated coefficients
from the LASSO-derived model and the dotted line indicates the
coefficients selected by cross-validation. Thus, 20 miRNAs were
included in further analyses (20). Selected miRNAs with non-zero
coefficients were deemed to be potential prognostic indicators
(Fig. 3).
 | Table I.miRNAs with P<0.01 assessed by the
log-rank test. |
Table I.
miRNAs with P<0.01 assessed by the
log-rank test.
| miRNAs | P-value |
|---|
| hsa-let-7c |
4.363×10−2 |
| hsa-mir-127 |
4.794×10−4 |
| hsa-mir-133a-1 |
2.701×10−2 |
| hsa-mir-133a-2 |
3.285×10−2 |
| hsa-mir-137 |
1.177×10−2 |
| hsa-mir-206 |
2.287×10−3 |
| hsa-mir-299 |
2.963×10−2 |
| hsa-mir-3170 |
4.739×10−2 |
| hsa-mir-337 |
1.329×10−4 |
| hsa-mir-3619 |
9.871×10−3 |
| hsa-mir-369 |
1.572×10−2 |
| hsa-mir-376c |
8.053×10−3 |
| hsa-mir-377 |
4.575×10−4 |
| hsa-mir-99a |
2.318×10−4 |
| hsa-mir-379 |
2.769×10−4 |
| hsa-mir-381 |
3.687×10−2 |
| hsa-mir-410 |
1.425×10−2 |
| hsa-mir-411 |
1.950×10−3 |
| hsa-mir-433 |
3.721×10−2 |
| hsa-mir-4746 |
9.439×10−3 |
| hsa-mir-487b |
3.156×10−3 |
| hsa-mir-495 |
8.206×10−3 |
| hsa-mir-499a |
2.196×10−3 |
| hsa-mir-513c |
4.539×10−2 |
| hsa-mir-548k |
4.249×10−2 |
| hsa-mir-654 |
1.788×10−4 |
| hsa-mir-7854 |
2.614×10−3 |
| hsa-mir-877 |
7.084×10−3 |
Multivariate Cox regression analysis and predictive
model building. Table II shows
the results of a stepwise multivariate Cox regression analysis as
implemented by SPSS, which established seven significant OS-related
miRNAs. Then, the seven-miRNA-based prognostic model was
established using the risk score method according to the following
formula: Risk score=(0.21 × hsa-miR-499a expression) + (0.264 ×
hsa-miR-548k expression)-(0.404 × hsa-miR-3619 expression)-(0.367 ×
hsa-miR-99a expression) + (0.123 × hsa-miR-137 expression) + (0.298
× hsa-miR-3170 expression) + (0.222 × hsa-miR-654 expression). The
median risk score (−0.9560) was used as a threshold to divide
patients into the high-risk and low-risk groups.
| Table II.Correlation between the seven miRNAs
and the overall survival of HNSCC patients based on TCGA dataset
using stepwise multivariate Cox analysis. |
Table II.
Correlation between the seven miRNAs
and the overall survival of HNSCC patients based on TCGA dataset
using stepwise multivariate Cox analysis.
|
| Multivariate
Cox |
|---|
|
|
|
|---|
| Gene | P-value | Exp(B) | 95.0% CI
[Exp(B)] |
|---|
| hsa-mir-499a | 0.001 | 1.234 | 1.090–1.397 |
| hsa-mir-548k | 0.004 | 1.302 | 1.087–1.559 |
| hsa-mir-3619 | 0.001 | 0.667 | 0.523–0.852 |
| hsa-mir-99a | <0.001 | 0.693 | 0.597–0.805 |
| hsa-mir-137 | 0.024 | 1.130 | 1.016–1.258 |
| hsa-mir-3170 | 0.003 | 1.347 | 1.107–1.640 |
| hsa-mir-654 | 0.007 | 1.248 | 1.063–1.466 |
Kaplan-Meier curve and time-dependent
ROC curve validation of the performance of the prognostic
model
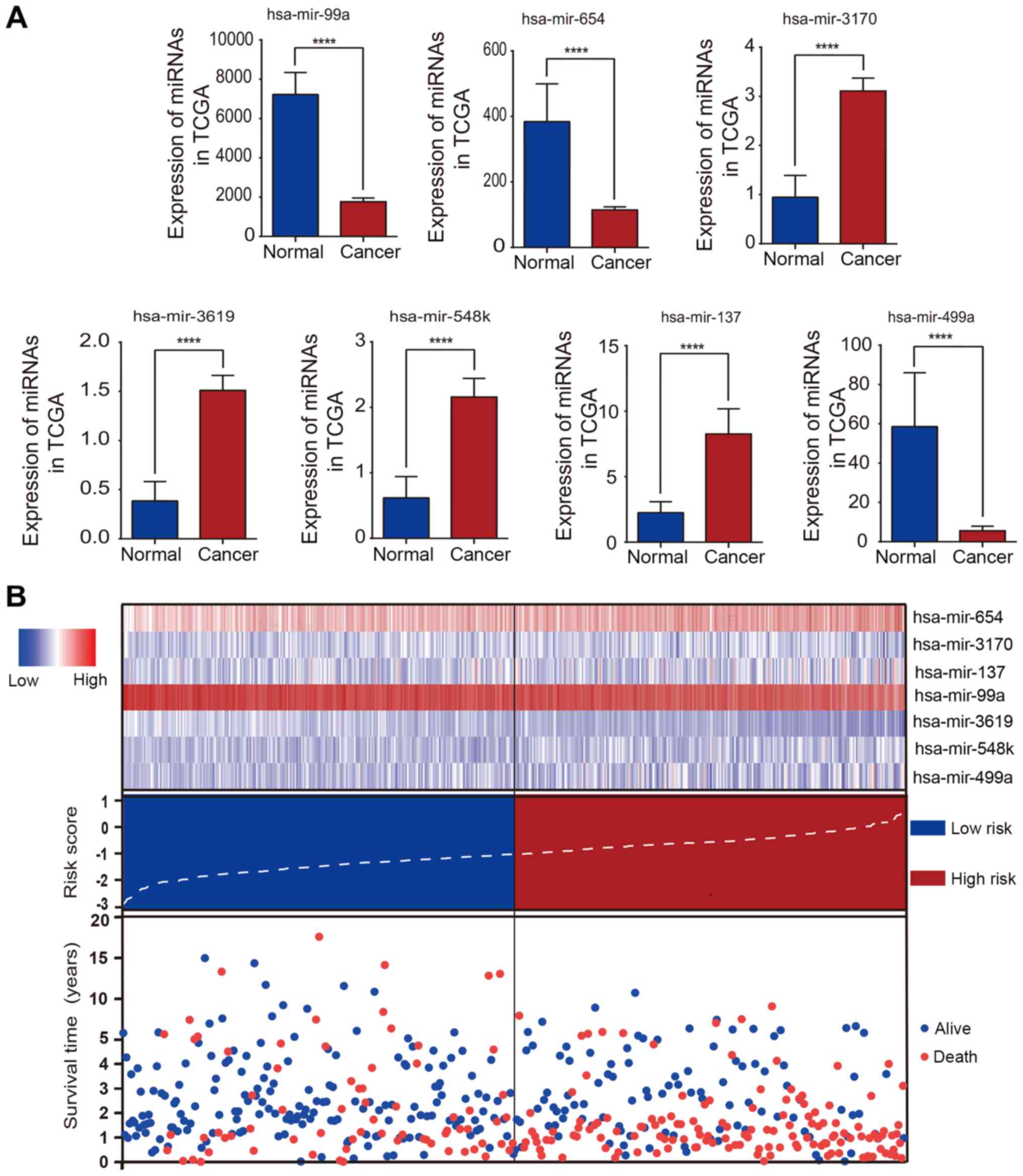
Fig. 4A reveals a
bar chart of the expression levels of every selected miRNA. The
patients were divided into high- and low-risk groups by the risk
scores presented in Fig. 4B. In
the image, the x-axis represents for the distribution of the HNSCC
patients from low-risk to high-risk. The image in Fig. 4B reveals that HNSCC patients in
whom hsa-miR-654, hsa-miR-548k, and hsa-miR-499a were highly
expressed often had a high risk of poor prognosis. As revealed in
Fig. 5, risk scores based on the
seven OS-related miRNAs divided patients into two groups that
significantly differed in overall survival time (P<0.0001). In
addition, the prognostic capacity of the seven-miRNA model was
assessed by computing the AUC of a time-dependent ROC curve. The
higher time-dependent AUC represented better performance and
stability for the prognostic model. The AUCs of the seven-miRNA
prognostic model were 0.64, 0.67, 0.68, 0.68 and 0.64 for 1-, 2-,
3-, 4- and 5-year survival times. Collectively, these measures
indicated that the prognostic model possessed good specificity,
sensitivity, and time-dependent stability for predicting the
overall survival of HNSCC patients.
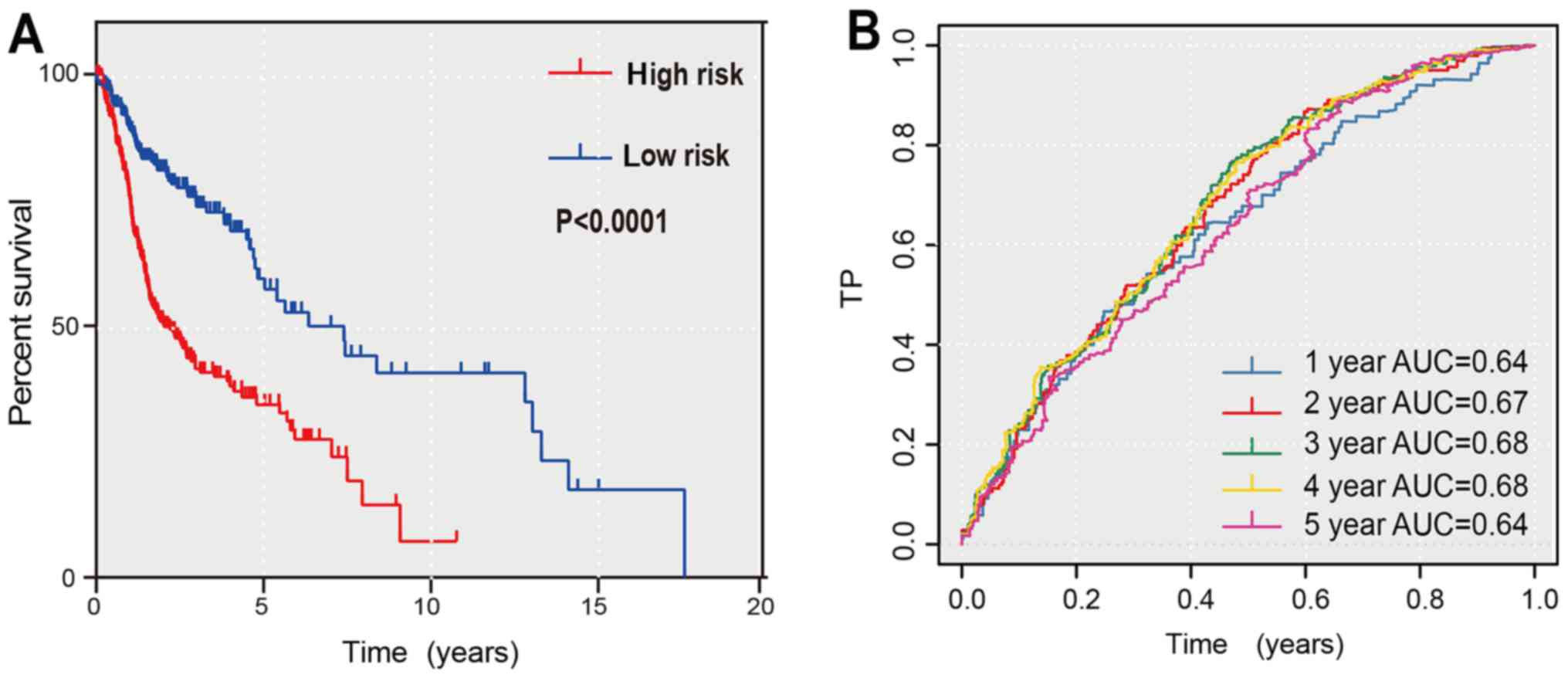 | Figure 5.Kaplan Meier curves and time-dependent
ROC curves for the prognostic model in the TCGA HNSCC cohort. (A)
The Kaplan Meier survival curves revealed the overall survival in
low- and high-risk groups divided by the median risk score. (B) The
time-dependent ROC curves evaluated the stability for predicting
the overall survival of HNSCC patients at different time-points (1,
2, 3, 4, 5 years). The area under the ROC curve was 0.64, 0.67,
0.68, 0.68 and 0.64 for the prognostic model at 1, 2, 3, 4 and 5
years, respectively. ROC, receiver operating characteristic; TCGA,
The Cancer Genome Atlas; HNSCC, head and neck squamous cell
carcinoma; AUC, area under the curve. |
Independence of the prognostic model
is verified by multivariate Cox analysis
The complete clinical information of 279 HNSCC
patients from the TCGA HNSCC cohort was obtained. Univariate and
multivariate Cox regression analysis was then performed on this
dataset to assess the independent predictive ability of the
prognostic model. The univariate Cox regression results revealed
significant differences between surviving and non-surviving HNSCC
patients when grouped by ‘the prognostic model’, ‘T-stage’,
‘N-stage’, ‘perineural invasion’ and ‘lymphovascular invasion’
predictors (P<0.05). However, sex, age, and histologic grade did
not correlate with OS (P>0.05). In addition, the primary tumor
site had no influence on the overall survival (Fig. S1) (P>0.05). Then all factors
were included into a multivariate Cox regression analysis (Table III), which indicated that the
‘T-stage’, ‘perineural invasion’ and ‘prognostic model’ variables
were independent prognostic factors related to OS (P<0.05).
| Table III.Correlations of the seven miRNAs with
the overall survival of HNSCC patients based on TCGA dataset using
univariate and multivariate Cox analysis. |
Table III.
Correlations of the seven miRNAs with
the overall survival of HNSCC patients based on TCGA dataset using
univariate and multivariate Cox analysis.
|
| Univariate Cox | Multivariate
Cox |
|---|
|
|
|
|
|---|
| Variable | P-value | HR (95% CI) | P-value | HR (95% CI) |
|---|
| Sex (male vs.
female) | 0.627 | 1.114
(0.722–1.718) | – | – |
| Age (>60 vs.
≤60) | 0.590 | 1.112
(0.755–1.638) | – | – |
| Grade (G3-4 vs.
G1-2) | 0.839 | 0.968
(0.708–1.324) | – | – |
| T stage (T3-4 vs.
T1-2) | <0.001 | 2.665
(1.583–4.489) | 0.002 | 2.262
(1.336–3.830) |
| N stage (N1-2 vs.
N0) | 0.001 | 2.111
(1.380–3.230) | – | – |
| Perineural invasion
(positive vs. negative) | <0.001 | 2.772
(1.844–4.168) | <0.001 | 2.209
(1.455–3.353) |
| Lymphovascular
invasion (positive vs. negative) | 0.001 | 1.943
(1.320–2.861) | – | – |
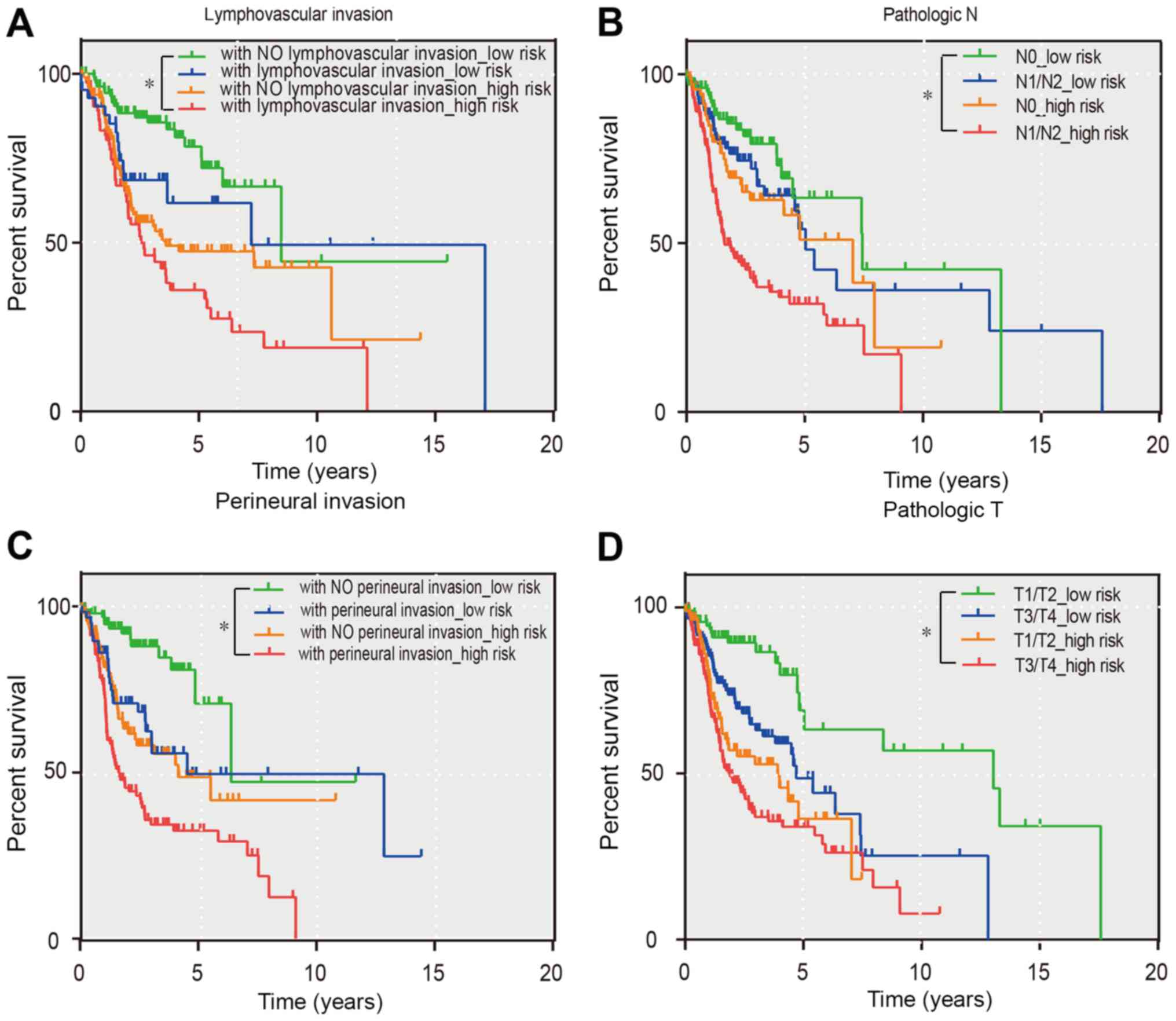
Combination of the prognostic model
and pathological characteristics
Since the T-stage, N stage, perineural invasion, and
lymphovascular invasion variables had non-zero prognostic value
according to the univariate Cox regression analysis, the prognostic
model with the aforementioned four pathological features were
separately combined to refine its predictive capacity using a
Kaplan-Meier curve. As revealed in Fig. 6, all groups revealed that the
patients belonging to the green curve possessed a markedly good
prognosis. This information may, therefore, help clinicians
distinguish between different prognoses for their patients. In
addition, patients with high-risk scores and poor pathological
characteristics can be identified earlier by clinicians. These
patients should also have health checks more frequently than the
other groups.
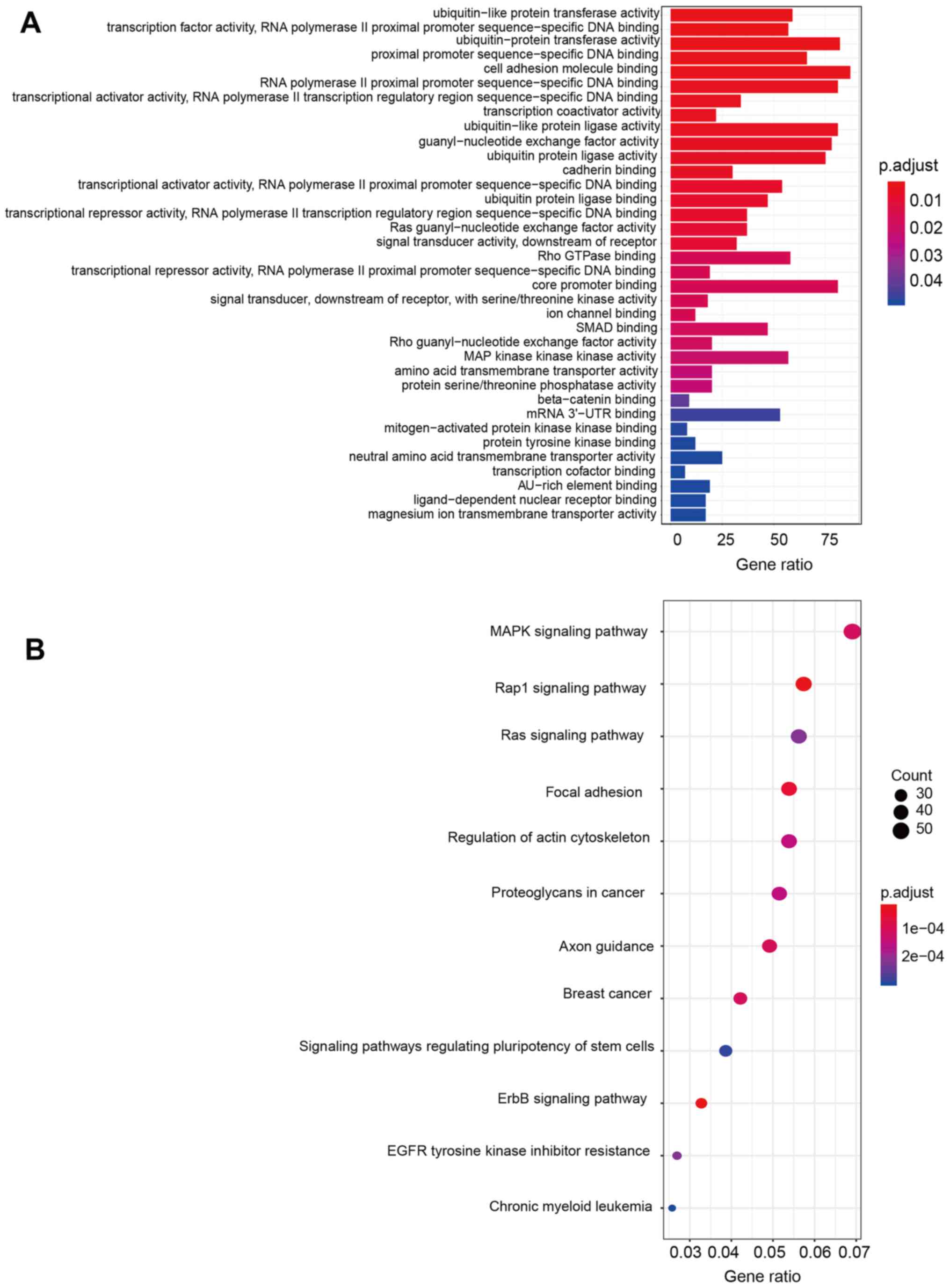
Functional enrichment analysis
To clarify the functional characteristics of the
miRNAs used for this prognostic model, the target genes of all
seven miRNAs were predicted using TargetScan. Next, enrichment
analysis of GO annotations and KEGG pathways with the predicted
target genes were carried out. The results revealed that these
target genes were strongly associated with the ErbB pathway, the
Rap1 signaling pathway, and DNA binding activities. More detailed
information on the predicted functions of target genes based on GO
and KEGG analyses is presented in Fig.
7.
Discussion
In the present study, RNA-seq data was used from the
TCGA database to identify the differences in miRNA expression
between HNSCC and normal tissue samples. Additional analyses were
performed to construct a prognostic model that included the
following miRNAs: Hsa-miR-499a, hsa-miR-548k, hsa-miR-3619,
hsa-miR-99a, hsa-miR-137, hsa-miR-3170 and hsa-miR-654. It was then
revealed that high- and low-risk groups identified by the median
model score exhibited significant differences in mean patient OS
(P<0.0001). Patients with high-risk scores were predisposed to
poor prognoses. If this model was used in clinical practice,
clinicians could adjust patient treatment plans according to its
results to produce individualized and comprehensive therapies for
HNSCC patients. In addition, it is also important to develop
strategies to detect HNSCC early in high-risk populations, who
should be followed closely. Finally, by performing pathway
enrichment and functional analyses, it was determined that the GO
terms associated with the target genes of the seven miRNAs were
related to DNA binding activities. Similarly, the KEGG pathways
associated with the target genes were the ErbB and Rap1 signaling
pathways.
In the present study, seven miRNAs that were
differentially expressed in HNSCC and adjacent normal tissue
samples were identified. Using this information along with clinical
data from HNSCC patients, a miRNA expression-based prognostic model
was developed to forecast the clinical prognoses of HNSCC patients.
Other studies have also reported miR-499a (21) as a prognostic indicator for early
gastric cancer patients. In addition, Zhang et al (22) have reported that hsa-miR-548k plays
a role in promoting lymphatic metastasis of esophageal squamous
cell carcinoma; this miRNA is, therefore, a potentially useful
diagnostic marker for esophageal squamous cell carcinoma.
Furthermore, hsa-miR-137 (23) may
promote the invasion ability of certain lung cancer cell types. Lu
et al (24) have also
reported that miR-654 targets GRB2-related adaptor protein (GRAP)
via the Ras/MAPK signaling pathway to promote proliferation,
metastasis, and resistance to chemotherapy in oral squamous cell
carcinoma. In addition, hsa-mir-3170 (25) has been validated to be
significantly correlated with the OS of patients with uterine
corpus endometrial carcinoma in the latest research. As per our
results, hsa-miR-499a, hsa-miR-548k, hsa-miR-137, hsa-miR-654 and
hsa-miR-3170 are probable risk factors for HNSCC patients. Patients
with high levels of expression of these miRNAs tend to have poor
prognoses.
The present results also revealed that some miRNAs
are associated with cancer suppression. In another study,
overexpression of hsa-miR-99a (26) could inhibit cell proliferation and
induce apoptosis by downregulating the expression of insulin-like
growth factor 1 receptor (IGFIR) and mechanistic target of
rapamycin kinase (mTOR) genes in oral cancer cells. Moreover,
hsa-miR-3619 (27) is known to
suppress the expression of β-catenin to mediate cancer invasion and
growth in non-small cell lung cancers. In the present prognostic
model, the coefficients of hsa-miR-3619 and hsa-miR-99a were
negative. Hence, it is speculated that hsa-miR-3619 and hsa-miR-99a
are factors that protect against HNSCC.
Several limitations of the present study should be
considered. Patient records lacking clinical information were
excluded, which may cause systematic errors. In addition, the model
did not validate the present results using another database (e.g.
GEO). Moreover, since the type of treatment patients received was
not included in the clinical information, to some extent the
results may contain systematic errors. Collectively, the mechanisms
by which the OS-related miRNAs affect HNSCC require further
investigation in future studies.
In conclusion, in the present study a seven-miRNA
prognostic model that is a reliable predictor of the OS of HNSCC
patients was developed. This model can distinguish between low- and
high-risk patients, thus enabling better clinical management of
patients with HNSCC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Key Research and
Development Project of Jiangsu Province (grant no. BE2016760), the
National Natural Science Foundation of China (grant no. 81470748),
and the Priority Academic Program Development of Jiangsu Higher
Education Institutions (grant no. PAPD 2018–87).
Availability of data and materials
The datasets downloaded and analyzed during the
current study are available from the TCGA data portal (https://portal.gdc.cancer.gov/projects/TCGA-HNSC);
the data contains 525 HNSCC tissue samples and 44 samples of
adjacent non-tumorous tissue as of January 1, 2018.
Authors' contributions
YF and XX contributed to the conception and design
of the work. LL and YW contributed to the acquisition, analysis,
interpretation of data and drafting the manuscript. MF contributed
to designing the work, drafting the manuscript, and revising the
manuscript and figures critically for important intellectual
content. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Petti S, Masood M and Scully C: The
magnitude of tobacco smoking-betel quid chewing-alcohol drinking
interaction effect on oral cancer in South-East Asia. A
meta-analysis of observational studies. PLoS One. 8:e789992013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tomczak K, Czerwinska P and Wiznerowicz M:
The cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
6
|
Long J, Zhang L, Wan X, Lin J, Bai Y, Xu
W, Xiong J and Zhao H: A four-gene-based prognostic model predicts
overall survival in patients with hepatocellular carcinoma. J Cell
Mol Med. 22:5928–5938. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Research Network, ;
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tibshirani R: The lasso method for
variable selection in the Cox model. Stat Med. 16:385–395. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Z, Yamada S, Wu Y, Wang KY, Liu YP,
Uramoto H, Kohno K and Sasaguri Y: Polypeptide
N-acetylgalactosaminyltransferase-6 expression independently
predicts poor overall survival in patients with lung adenocarcinoma
after curative resection. Oncotarget. 7:54463–54473.
2016.PubMed/NCBI
|
|
11
|
Simon RM, Subramanian J, Li MC and Menezes
S: Using cross-validation to evaluate predictive accuracy of
survival risk classifiers based on high-dimensional data. Brief
Bioinform. 12:203–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu JH, Chang WH, Fu HW, Shu WQ, Yuan T and
Chen P: Upregulated long non-coding RNA LOC90784 promotes cell
proliferation and invasion and is associated with poor clinical
features in HCC. Biochem Biophys Res Commun. 490:920–926. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai M, Chen S, Wei X, Zhu X, Lan F, Dai S
and Qin X: Diagnosis, prognosis and bioinformatics analysis of
lncRNAs in hepatocellular carcinoma. Oncotarget. 8:95799–95809.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Niu ZS, Niu XJ and Wang WH: Long
non-coding RNAs in hepatocellular carcinoma: Potential roles and
clinical implications. World J Gastroenterol. 23:5860–5874. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma Y, Luo T, Dong D, Wu X and Wang Y:
Characterization of long non-coding RNAs to reveal potential
prognostic biomarkers in hepatocellular carcinoma. Gene.
663:148–156. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cui H, Zhang Y, Zhang Q, Chen W, Zhao H
and Liang J: A comprehensive genome-wide analysis of long noncoding
RNA expression profile in hepatocellular carcinoma. Cancer Med.
6:2932–2941. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Wu Q, Feng S, Zhao Y and Tao C:
Identification of four prognostic LncRNAs for survival prediction
of patients with hepatocellular carcinoma. PeerJ. 5:e35752017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
ELife. 4:e050052015. View Article : Google Scholar :
|
|
19
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. Omics. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Softw. 33:1–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi H, Yang X, Zhen Y, Huo S, Xiao R and
Xu Z: MicroRNA499 rs3746444 A/G polymorphism functions as a
biomarker to predict recurrence following endoscopic submucosal
dissection in primary early gastric cancer. Mol Med Rep.
15:3245–3251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang W, Hong R, Li L, Wang Y, Du P, Ou Y,
Zhao Z, Liu X, Xiao W, Dong D, et al: The chromosome 11q13.3
amplification associated lymph node metastasis is driven by
miR-548k through modulating tumor microenvironment. Mol Cancer.
17:1252018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu SL, Chen HY, Chang GC, Chen CY, Chen
HW, Singh S, Cheng CL, Yu CJ, Lee YC, Chen HS, et al: MicroRNA
signature predicts survival and relapse in lung cancer. Cancer
Cell. 13:48–57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu M, Wang C, Chen W, Mao C and Wang J:
miR-654-5p targets GRAP to promote proliferation, metastasis, and
chemoresistance of oral squamous cell carcinoma through ras/MAPK
signaling. DNA Cell Boil. 37:381–388. 2018. View Article : Google Scholar
|
|
25
|
Wang Y, Xu M and Yang Q: A six-microRNA
signature predicts survival of patients with uterine corpus
endometrial carcinoma. Curr Probl Cancer. 43:167–176. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen YT, Yao JN, Qin YT, Hu K, Wu F and
Fang YY: Biological role and clinical value of miR-99a-5p in head
and neck squamous cell carcinoma (HNSCC): A bioinformatics-based
study. FEBS Open Bio. 8:1280–1298. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Niu X, Liu S, Jia L and Chen J: Role of
miR-3619-5p in β-catenin-mediated non-small cell lung cancer growth
and invasion. Cell Physiol Biochem. 37:1527–1536. 2015. View Article : Google Scholar : PubMed/NCBI
|