Introduction
Proliferating cell nuclear antigen (PCNA) is a clamp
homotrimer that encircles mammalian DNA as a scaffold and is
overexpressed in various types of cancer; thus, PCNA participates
in DNA replication, DNA damage repair and cell cycle regulation and
plays a critical role in anti-apoptosis, chromatin metabolism and
gene expression (1,2). Notably, the importance of PCNA in DNA
repair, as revealed by a subset of methods, including homologous
recombination, nucleotide excision repair, mismatch repair and
translesion DNA synthesis, have revealed the importance of PCNA in
DNA repair, recruiting various repair factors to disrupt the DNA
damage from chemotherapy treatments, such as platinum-based drugs
(2,3). The overexpression of PCNA in tumors
is often associated with the maintenance of aggressiveness and the
stimulation of excessive proliferation by providing the genomic
material, which is necessary for cell division and genomic
integrity (4–6). Given the essential function of PCNA
in cancer or normal cells, whose functions mostly depend on
posttranslational modifications, an intricate network of associated
proteins and signaling molecules may exist, but the key factors or
exact mechanisms that mediate its expression are not understood
(1).
One possible approach that may provide insightful
and valuable information is examining the tumor microenvironment.
Previous studies have established that limited nutrient supply or
rather nutrient stress (NS), a common but distinguished feature, is
also an important stimulus of proliferation, antagonism, survival
and metastasis for different cancer types (7–13).
The excessive nutrient consumption hinders cell division, and the
metabolic phenotype makes cancer cells more sensitive to hypoxia
and nutrient starvation (14,15).
The abnormal metabolism of multiple tumors confers the ability to
utilize other metabolic substances represented by acetate (16,17).
In light of restricted resources resulting in stress, cancer cells
convert acetate to acetyl-CoA and other various biomolecules and
energy sources to support cellular activity, particularly
proliferative or metastatic activity (18–20).
Acetyl-CoA synthetase short-chain family member 2 or acetyl-CoA
synthetase 2 (ACSS2) is responsible for the production of acetyl
CoA from acetate, and recent research has uncovered its
differential and even conflicting roles in proliferation, fatty
acid synthesis, metastasis and chemoresistance (14,20–24).
Moreover, the role of ACSS2 in metabolism reprogramming and further
signal transmission, especially under NS, has only sparsely been
investigated.
New evidence suggests that beyond classical
resistance mechanisms, microenvironment stress also results in
antagonism-independent alterations of the drug target, overactive
DNA repair and survival pathways, enhanced expression of
detoxification proteins and drug efflux (25,26).
In coping with drug-induced double-strand breaks (DSBs), cancer
cells rapidly initiate the DNA damage response (DDR) to ensure the
efficient and accurate repair of damaged DNA and cell survival.
Before initiating repair, the phosphorylation and position of H2AX
with the recruitment of PCNA in the DNA breaks provide a docking
site for other DNA repair factors. Among these factors, ataxia
telangiectasia mutated (ATM) and DNA-dependent protein kinase
catalytic subunit (DNA-PKcs) are two primary kinases that
phosphorylate H2AX at S139 in response to DSBs (1,3,27).
Although multiple signaling transduction pathways are involved in
the activation and transcriptional regulation, targeting
AMP-activated protein kinase (AMPK) activation, yet as a
double-edged sword, is worth exploring (28–31).
Moreover, studies have reported that PCNA expression is affected by
AMPK, a master nutrient sensor that is imbalanced in several types
of cancer, yet the physiological implications of the PCNA and AMPK
interaction, especially under NS, are poorly understood (32–36).
Elucidation of the metabolic alterations during the progression of
cancer may offer potential therapeutic targets for the challenge of
chemoresistance. The present study investigated the impact of ACSS2
on chemoresistance under NS and sought a more complete
understanding of the underlying mechanisms between the AMPK pathway
and PCNA in DNA repair, beyond its role in proliferation.
Materials and methods
Cell lines and reagents
ESCC cell lines TE-1 and ECA-109 were purchased from
The Cell Resource Centre of Shanghai Institutes for Biological
Sciences, Chinese Academy of Science (Shanghai, China) and cultured
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., China)
supplemented with 10% dialyzed fetal bovine serum (FBS; Biological
Industries, Kibbutz Beit-Haemek, Israel), 1% compound antibiotics
(Pen Strep; Gibco; Thermo Fisher Scientific, Inc.). Immortalized
human normal esophageal epithelial cells, Het-1A (purchased from
BeNa Culture Collection, Kunshan, Jiangsu, China), were maintained
in DMEM (Gibco; Thermo Fisher Scientific, Inc., China),
supplemented with 10% FBS as described previously (37). For induction of nutrient
starvation, ESCC cells (ESCCs) or Het-1A cells (confluence of
50–60%) were transferred to serum lacking media (supplemented with
1% FBS) for the indicated periods of time. Cells were tested
negative for mycoplasma or other infectious agents, and maintained
at 37°C in a humidified atmosphere with 5% CO2. ACSS2
(cat. no. sc-398559; Santa Cruz Biotechnology, Sant Cruz, CA, USA),
ATM (product #2873)/p-ATM (product #5883), BRCA1 (product
#9010)/p-BRCA1 (product #9009), Bcl-xL (product #2762), γH2AX
(product #9718), DNA-PKcs (product #4602), ULK1 (product
#8054)/p-ULK1 (product #5869), AMPK (product #5831)/p-AMPK (product
#50081), PCNA (product #2586), Ki-67 (product #9449) and β-actin
(product #58169) (Cell Signaling Technology, Danvers, MA, USA), Bax
(catalog no. AF0120; Affinity, Changzhou, China), Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.), and dorsomorphin
(an inhibitor of the AMPK pathway; APExBIO, USA) were used
following the manufacturer's instructions.
Patients and specimens
Cancer tissues and paired normal tissues were
obtained from 28 patients with ESCC at the Affiliated Hospital of
Jiangsu University (Zhenjiang, China) from 2010 to 2018. The Ethics
Committee of the Affiliated Hospital of Jiangsu University approved
the research. Firstly, the ESCC samples were confirmed by the
Pathology Department, and then serial sections were processed and
stained with anti-ACSS2 (1:200 dilution; cat. #sc-398559; Santa
Cruz Biotechnology), anti-Ki-67 (1:1,000 dilution; product #9449,
Cell Signaling Technology) and anti-PCNA (1:4,000 dilution; product
#2586, Cell Signaling Technology) antibodies for the
immunohistochemistry (IHC) assay. Ki-67 scores ranged from 0 to
100% as the percentage of positive cells within the area of
invasive cells. The evaluation criteria for ACSS2 or PCNA were
based on our previously published study (13). The proportion of positively
staining tumor cells and the staining intensity were examined and
scored by five independent pathologists, who were blinded to the
patient clinical information as previously described (13). All methods, including the
collection and use of patient samples, were performed in accordance
with the relevant guidelines and regulations and all patients
provided signed informed consent.
RNA interference and plasmid
infection
Knockdown of human ACSS2 and PCNA were performed
using gene-specific siRNAs. These gene-specific siRNA and including
control siRNA were purchased from Shanghai GenePharma (Shanghai
GenePharma Co., Ltd., Shanghai, China), which were reconstituted in
sterile DEPC water to a stock concentration of 20 µM, and
transfected into TE-1 and ECA-109 cells using Lipofectamine 2000.
The siRNA sequences were as follows: siRNA-ACSS2: Sense,
5′-UAUGCUUGGUGACAGGCUCAUCUCC-3′ and antisense,
5′-GGAGAUGAGCCUGUCACCAAGCAUA-3′; siRNA-PCNA: Sense,
5′-UAUGGUAACAGCUUCCUCCTT-3′ and antisense,
5′-GGAGGAAGCUGUUACCAUATT-3′. The inhibitory effect of the siRNA
transfection was evaluated with RT-qPCR and western blotting.
Upregulation of ACSS2 expression was performed by cloning ACSS2
full-length complementary DNA into a GV141 carrier plasmid,
including the control plasmid group, which were purchased from
Shanghai Genechem Co., Ltd. (Shanghai, China), and these plasmids
were transfected into TE-1 and ECA-109 cells using Lipofectamine
2000. The upregulation effect of the plasmid was evaluated with
RT-qPCR and western blotting. After transfection, the cells were
subjected to other treatments for the indicated duration and then
used for the subsequent assays.
Quantitative real-time polymerase
chain reaction (qPCR)
RNA was extracted using RNAiso Plus in accordance
with the manufacturer's instructions and reverse transcribed into
cDNA using a reverse transcription reagent kit, according to the
manufacturer's protocol (both from Takara Biotechnology Co., Ltd.,
Dalian, China). The RT-PCR reactions were prepared using a PCR kit
(Takara Biotechnology Co., Ltd.) and performed on an Agilent
Mx3000P™ Real-Time PCR System (Stratagene). The primers used were
synthesized by Invitrogen (Invitrogen; Thermo Fisher Scientific,
Inc.), and the sequences were as follows: ACSS2 primers,
5′-GGATTCCAGCTGCAGTCTTC-3′ (forward) and 5′-CAGCCAGCTCCTTCAGGTT-3′
(reverse); PCNA primers, 5′-CTGAAGCCGAAACCAGCTAGACT-3′ (forward)
and 5′-TCGTTGATGAGGTCCTTGAGTGC-3′ (reverse); β-actin primers,
5′-TCACCCACACTGTGCCCATCTACGA-3′ (forward) and
5′-CAGCGGAACCGCTCATTGCCAATGG-3′ (reverse). Relative expression
levels were calculated using the 2−ΔΔCq method (38) and β-actin was used as an internal
reference gene.
Immunoblotting
Cells of different treatment were collected by
centrifugation with 1200 rpm in 5 min, processed and lysed in RIPA
buffer supplemented with a protease inhibitor cocktail and PMSF
(all purchased from Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China) according to the manufacturer instructions.
Protein concentration was determined by BCA protein assay (CoWin
Biotech Co., Ltd, Beijing, China), and 5 µl of protein were loaded
and separated by 8 to 12% SDS-PAGE. After transfer to PVDF
membranes, 5% BSA in TBST was used to block the membrane at room
temperature for 1 h. The membranes were incubated with primary
antibodies [ACSS2 (at 1:2,000 dilution); ATM, p-ATM, BRCA1,
p-BRCA1, Bcl-xL, γH2AX, DNA-PKcs, ULK1, p-ULK1, AMPK, p-AMPK, PCNA,
and Ki-67 (at 1:1,000 dilution); β-actin (at 1:2,000 dilution); Bax
(at 1:500 dilution)] saturated with 5% BSA in TBST at 4°C
overnight. On the next day, the membranes were incubated with
HRP-conjugated anti-rabbit or -mouse antibody (at 1:5,000 dilution,
product #7074 and #7076; Cell Signaling Technology) for 1 h at room
temperature, and then exposed to enhanced ECL reagent (Nanjing
Vazyme Biotech Co., Ltd., Nanjing, China), imaged by an automatic
ChemisScope-4300 imager (Clinx Science Instruments Co., Ltd.,
Shanghai, China) and data were analyzed with Fluor Chem FC3
software (Protein-Simple, USA).
CCK-8 assay
Cell proliferation was examined using the CCK-8 Cell
Counting Kit (Nanjing Vazyme Biotech Co., Ltd.) according to the
manufacturer's instructions. Briefly, TE-1/ECA-109 and Het-1A cells
were grown in a 96-well plate for 24 h, transfected with
siRNA-ACSS2 or the negative control, and then cultured in normal
medium or under NS. To measure the cell proliferation at 0, 24, 48,
or 72 h after transfection, 10 µl of CCK-8 was added to each well
for 1 h. Absorbance was measured at a wavelength of 450 nm by a
microplate reader (BioTek, USA). Assays were repeated at least
three times.
Flow cytometric analysis
The effect of ACSS2 on cell cycle distribution was
determined by flow cytometry. Briefly, cells subjected to the
indicated treatments were harvested when they reached 80%
confluence, washed with PBS and resuspended in 1 ml of DNA staining
solution and 10 µl of permeabilization solution (MultiSciences
Biotech Co., Ltd., Hangzhou, China). Following incubation for 30
min in the dark at room temperature, the cells were analyzed by
flow cytometry at 72 h after interference. The fractions of cells
in the G0/G1, S, and G2/M phases were analyzed. Cell apoptosis was
detected using an Annexin V-FITC/PI Apoptosis Detection kit
(MultiSciences Biotech Co., Ltd., Hangzhou, China) according to the
supplier's protocol. Cells were seeded in 12-well plates
(6×104/well). The cells were harvested after
transfection with siRNA or NC and with or without DDP (5 µg/ml)
treatment for 24 h, and washed in cold PBS. Then, cells were
resuspended in 500 µl of binding buffer. Next, 5 µl of Annexin
V-FITC and 10 µl of PI working solution were added to each reaction
system at room temperature for 5 min. Flow cytometric analysis was
performed immediately on a flow cytometer (BD FACSCanto II; BD
Biosciences) and data were analyzed with BD FACSDiva software (BD
Biosciences). Each experiment was repeated at least three
times.
TUNEL staining and
immunofluorescence
After pretreatment with siRNA or NC, and with or
without DDP (5 µg/ml) for the indicated time, ESCC monolayers were
spun down on slides, and stained by a TUNEL Apoptosis Detection kit
(#A113-01; Nanjing Vazyme Biotech Co., Ltd., Nanjing, China) for in
situ detection of apoptosis according to the manufacturer's
instructions. In brief, slides were further incubated in the
prescribed mixture of enzyme and label solution, at 37°C for 1 h in
a humidified chamber. The TUNEL-stained cells were counterstained
with PBS containing 4′,6-diamidino-2-phenylindole (DAPI, 5 µg/ml)
(#422801, BioLegend, USA) for 5 min, and counted to calculate the
TUNEL indices. For immunofluorescence, after being washed 3 times
with PBS and fixed with 4% paraformaldehyde, the cells were
permeabilized with 0.5% Triton X-100 in PBS for 10 min. The fixed
cells were blocked in 5% BSA in PBS for 1 h at room temperature and
incubated with primary antibodies against γH2AX (at 1:400 dilution;
product #9718, Cell Signaling Technology), p-ATM (at 1:500
dilution; product #5883, Cell Signaling Technology) and PCNA (at
1:2,000 dilution; product #2586, Cell Signaling Technology)
overnight at 4ºC. FITC-conjugated anti-mouse, Cy3-conjugated
anti-rabbit secondary antibodies (1:100 dilution; cat. #SA00003 and
#SA00009, Proteintech Group, Wuhan, China) were used to detect the
bound primary antibody. Then, the slides were washed 3 times in PBS
and incubated with DAPI for 5 min. Immunofluorescence results are
representatives of at least three independent experiments. Images
were captured on a fluorescence (BX51, Olympus, Japan) or laser
confocal microscope (LSM800, ZEISS, Germany) with fixed settings
between samples and analyzed using ImageJ software (National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All experimental data are presented as the means ±
standard deviation (SD) from three independent experiments.
Survival curves were generated by Kaplan-Meier analysis and tested
for significance using the Breslow test. The χ2-test was
used to explore the relationship between two variables. The
differences between two groups were analyzed using Student's
t-test, and comparisons in datasets containing multiple groups were
analyzed using one-way analysis of variance (ANOVA) followed by the
Newman-Keuls post-test. Significant differences were determined
using a threshold P-value of 0.05.
Results
ESCCs are more sensitive to NS, and
are associated with increased expression of ACSS2
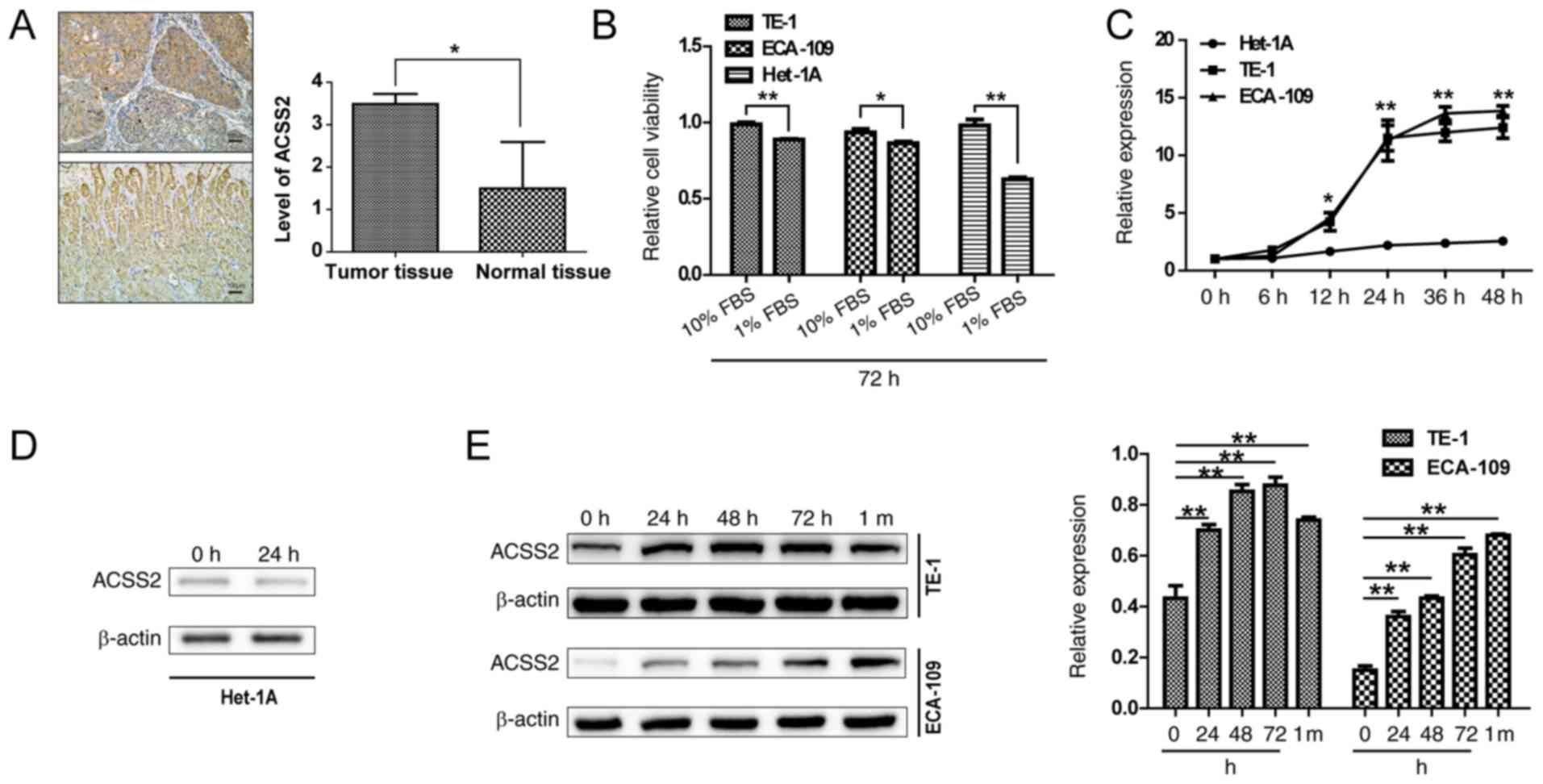
IHC staining analysis was used to measure ACSS2
expression in 28 pairs of human ESCC and adjacent normal tissues,
and the results revealed that ACSS2 exhibited positive cytoplasmic
staining. In cancer tissues, ACSS2 protein was highly expressed in
central zones and hypercellular areas, while ACSS2-positive cells
in adjacent noncancerous tissues were concentrated in stroma cells
and the glandular epithelium at basal and lower levels (Fig. 1A). To investigate the potential
role of NS caused by excessive proliferation or poor nutrient
supply, we first examined the effect of limited serum on cell
viability. As shown in Fig. 1B,
the proliferation of TE-1 and ECA-109 cells was significantly
decreased at 72 h after the replacement of both media with 1% FBS
(NS; P=0.008 and 0.03), while the proliferation of Het-1A cells was
decreased more significantly (P<0.01), suggesting that normal
esophageal epithelial cells are more sensitive to nutrient supply.
To study the role of ACSS2 in the adaptive mechanisms of ESCCs to
NS, the transcription levels of ACSS2 were analyzed at different
times in limited serum medium. ACSS2 expression showed a sharp
increase at 12 h in the TE-1 and ECA-109 cells compared to that in
the Het-1A cells (P=0.03) under NS. The mRNA levels reached 9- to
12-fold at 24 h (P<0.01), and the expression levels of ACSS2
mRNA in the TE-1 and ECA-109 cells tended to reach a plateau after
24 h, but the levels were still at a higher level than those in the
Het-1A cells (P<0.01) (Fig.
1C). However, the mRNA and protein expression of ACSS2 in the
normal esophageal epithelial Het-1A cells did not correlate at all
times or under all conditions (Fig. 1C
and D), thus effectively adapting to stress and effectively
played an extraordinary role, which has been described as a
characteristic of cancer cells. When ACSS2 was analyzed in the ESCC
cell lines, it was found that ACSS2 protein was upregulated with an
increasing period under NS (Fig.
1E). Similarly, ACSS2 was persistently upregulated under NS for
more than 1 month (Fig. 1E). ACSS2
has been shown to be upregulated by glucose, lipid deprivation and
hypoxia in different cancer cell lines (14,39,40).
Together, these data indicate that the proliferation of ESCCs is
less restricted than that of human esophageal squamous epithelial
cells in response to low serum culture and that the upregulation of
ACSS2 may help maintain ESCC cell survival under NS.
ACSS2 contributes to the proliferation
and DNA repair that are regulated by PCNA
To elucidate the role of ACSS2 in ESCC cell
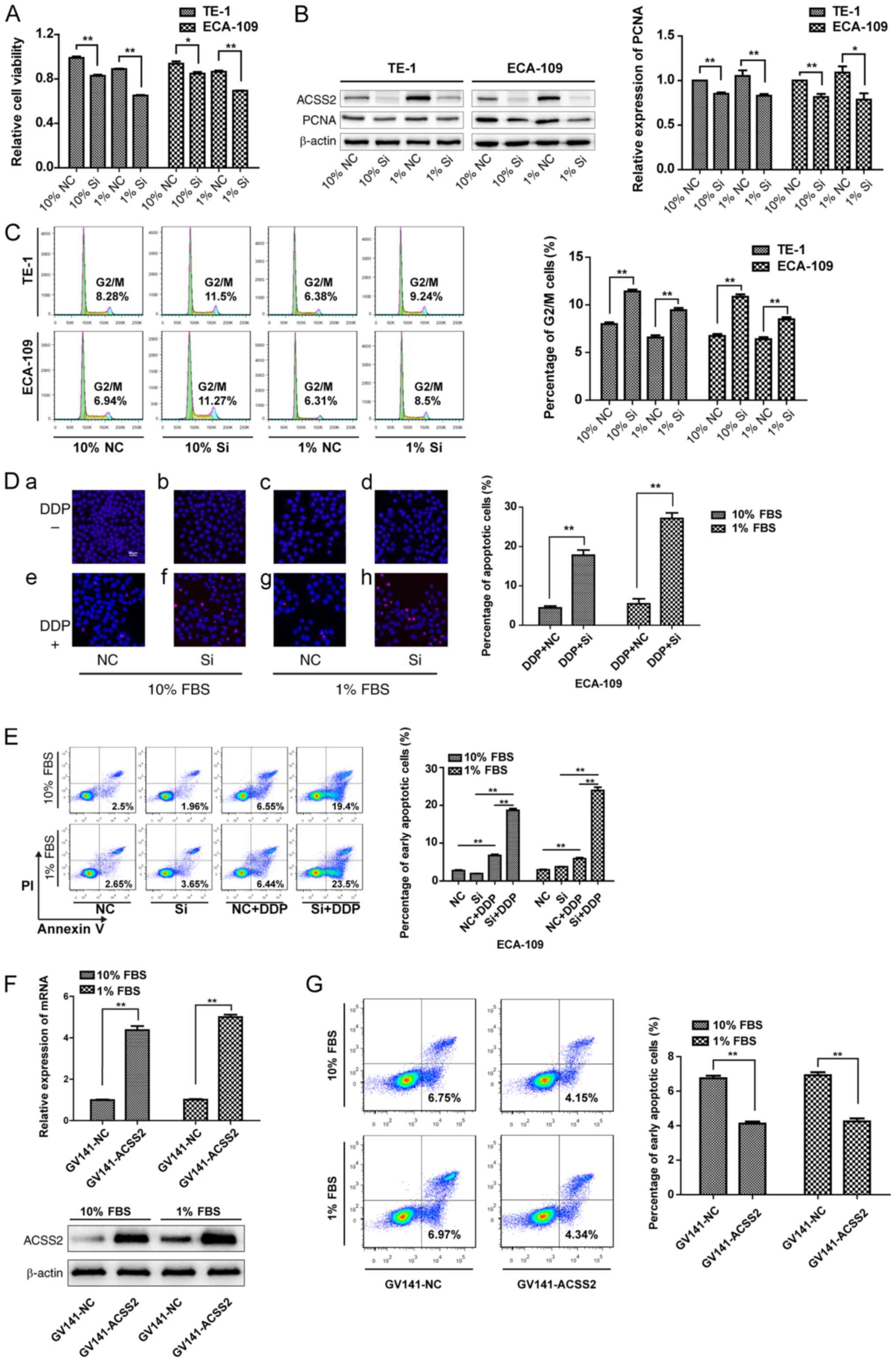
viability, we first performed siRNA experiments. Data from Fig. 2B show that the specific siRNA
treatment observably decreased the protein levels of ACSS2. CCK-8
assays were performed to investigate cell proliferation. The
results showed that suppression of ACSS2 expression significantly
affected the proliferation of TE-1 and ECA-109 cells compared to
that of the control group (NC), regardless of whether the cells
were grown in normal medium or under NS (P<0.05, Fig. 2A). As stated above, PCNA acts as a
scaffold to recruit proteins for replication, recombination and DNA
repair. siRNA-ACSS2 treatment led to a 15 and 18% decrease in PCNA
transcript levels which was further decreased 21 and 28% in
combination with NS in the TE-1 and ECA-109 cells, respectively.
Since PCNA directly participates in proliferation, these data
suggest that ACSS2 reactivated PCNA in both cell lines, especially
under NS (Fig. 2B). This was
further confirmed by using flow cytometry, demonstrating
substantial increases in G2/M phase arrest in both cell lines upon
siRNA-ACSS2 treatment; the histogram (right) shows that the
percentage of cells in the G2/M phase in the siRNA-ACSS2 group was
approximately 1.4-fold higher than that in the NC group under NS or
not (P<0.01) (Fig. 2C).
Notably, the G2/M DNA damage checkpoint is important for repairing
DNA after replication (6,41–43);
thus, its defect would lead to apoptosis or death. Most cells under
apoptosis preferentially are labeled by TUNEL for fragmented DNA at
later stages. To examine whether DNA damage increased after
siRNA-ACSS2 treatment, TUNEL analysis was performed to determine
apoptosis with or without cisplatin (DDP) treatment. At least five
viewing fields were utilized to obtain each data point and a
representative of three individual dose-dependent experiments is
shown. The results showed that there was no difference in the
TUNEL-positive cell ratio between the NC group and the siRNA-ACSS2
group in TE-1 or ECA-109 cells, suggesting that the downregulation
of ACSS2 did not induce cell death. However, the level of
TUNEL-staining was significantly increased on day 3 posttreatment
with siRNA-ACSS2 compared with the NC when treated with DDP (10%
FBS: 17.78±1.16 vs. 4.09±0.89%; 1% FBS: 27.14±1.23 vs. 5.45±1.07%;
P<0.01) (Fig. 2D).
Additionally, ACSS2 interference also significantly increased the
percentage of early apoptotic cells compared to that in NC groups
after DDP treatment, whether under NS or not (P<0.01), but the
differences between the siRNA-ACSS2 and NC groups were not
statistically significant at 72 h after transfection (Fig. 2E). To further confirm the role of
ACSS2 in chemoresistance, plasmid transfection experiment were
performed, and the data from Fig.
2F shows that specific plasmid treatment significantly
increased the mRNA and protein levels of ACSS2 (P<0.01).
Upregulation of ACSS2 expression reduced the percentage of early
apoptotic cells in comparison to the NC groups after DDP treatment
under NS or not (P<0.01) (Fig.
2G). These preliminary data indicate that ACSS2 is a factor
that maintains DNA stability and may be associated with PCNA
expression.
ACSS2/AMPK signaling stabilizes PCNA
expression and regulates the DNA damage response under NS
To investigate the potential mechanism of
ACSS2-dependent ESCC chemoresistance under NS, the levels of PCNA,
p-ATM, γH2AX and related proteins were assessed. The cooperation of
ATM protein kinase and DNA-PKcs plays critical roles in the
regulation of the DNA damage response and cellular homeostasis
(44,45). As expected, among the siRNA-ACSS2
groups, the expression levels of p-ATM increased 4.5-fold under NS
combined with DDP compared to 2.0-fold following DDP treatment
alone. However, the downregulation of ACSS2 had no significant
effects on DNA-PKcs or p-BRCA1 levels. Importantly, we also
observed that both siRNA-ACSS2 and DDP influenced the accumulation
of γH2AX, exhibiting obvious synergistic effects under NS. B-cell
lymphoma-extra large (Bcl-xL) is a member of the Bcl-2 family, an
anti-apoptotic protein in mitochondria that prevents the release of
cytochrome c. As an apoptosis activator, Bax leads to a loss
in membrane potential and the release of cytochrome c.
Considering that the lower ACSS2 under NS is much more vulnerable
to DNA damage, we further detected variations in Bcl-xL and Bax.
However, the suppression of ACSS2 expression in the ESCCs had no
significant effect on the protein content of Bcl-xL or Bax, whether
under sufficient nutrition or deficiency. Interestingly, despite
its inhibitory effect on Bcl-xL, the presence of DDP had no
influence on Bax expression (Fig.
3A). As shown in Fig. 3B and
C, changes in PCNA and p-ATM and γH2AX foci were detected in
the ESCCs, represented by ECA-109 cells, after treatment with
cisplatin. In regards to the p-ATM and PCNA signals, although the
percentages of p-ATM-positive cells increased steadily after
cisplatin, the frequencies of p-ATM foci peaked at 24 h after
downregulation of the expression of ACSS2; 89 to 95% of ESCCs with
increased p-ATM showed decreased PCNA staining after low-serum
culture for 48 h (Fig. 3B). The
changes in the γH2AX signals were similar to those of the p-ATM
foci, and the percentages of PCNA were steadily depended on the
levels of ACSS2, but the intensity of γH2AX was stronger at 24 h
post damage (Fig. 3C). These data
suggest that nuclear-wide γH2AX expression, p-ATM responses and
PCNA expression are related to ACSS2 expression in ESCCs after
cisplatin treatment. More importantly, the effective interference
of PCNA combined with DDP treatment also increased γH2AX and p-ATM
expression especially during NS (Fig.
3D), indicating that PCNA plays a vital role as a scaffold
associated with ACSS2-regulated DNA repair in ESCCs. The formation
of PCNA, γH2AX, and p-ATM in the DNA damage response is related to
the activation of the AMPK signaling pathway in multiple cancer
cells (1,46,47).
Similarly, our results showed that the significant phosphorylation
of AMPK was induced by culture of TE-1 and ECA-109 cells under NS.
The inhibition of ACSS2 accompanied by the significant
downregulation of p-AMPK showed a decreasing tendency, whereas
there was no significant difference in p-ULK1, which mediates
autophagy (Fig. 3E). Furthermore,
the levels of PCNA and ACSS2 were significantly affected after
dorsomorphin treatment, showing an even stronger inhibition of
ACSS2 (Fig. 3F). These results
indicated that the interaction of ACSS2 with AMPK signaling
mediates the stabilization of PCNA and DNA repair.
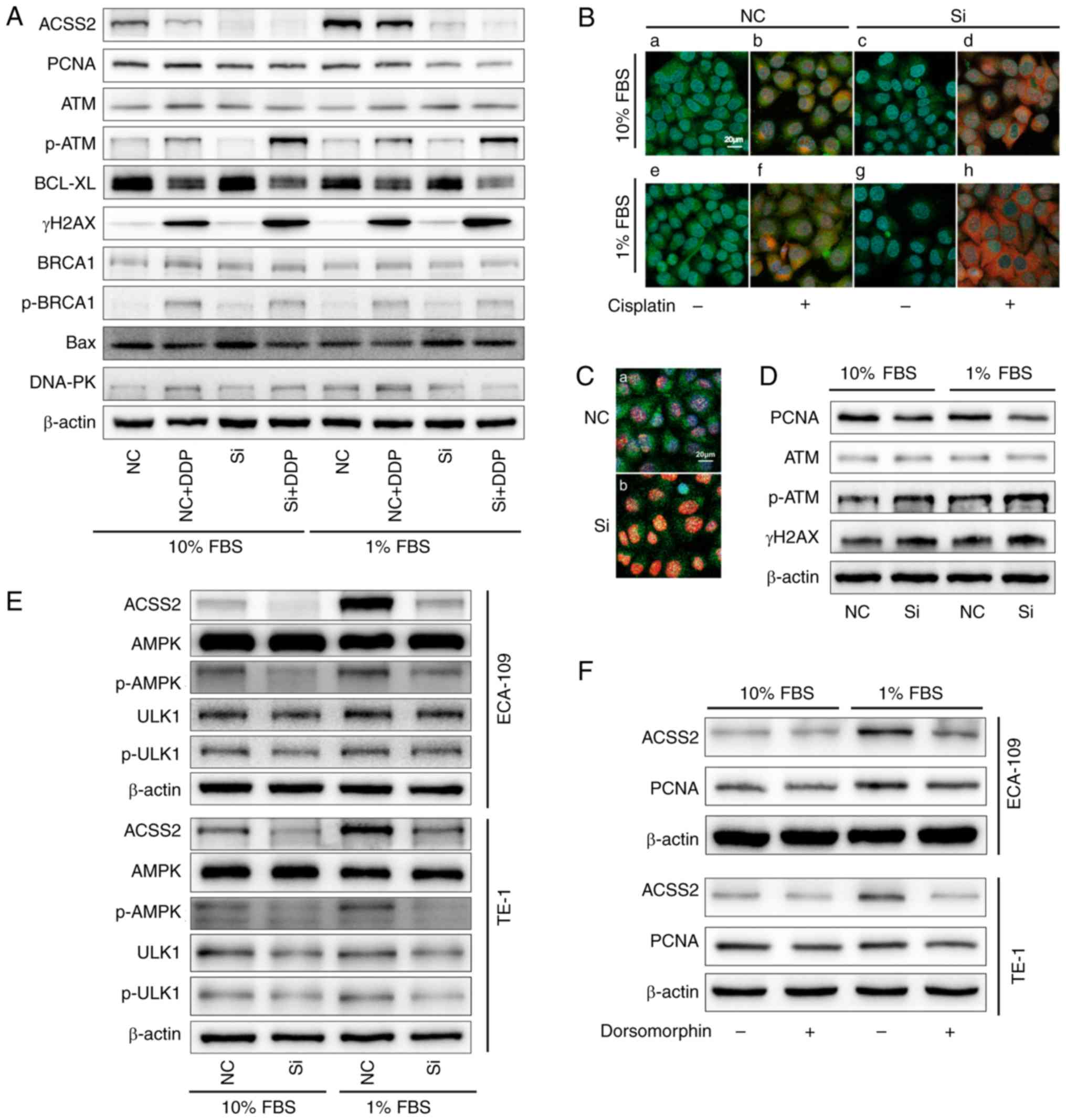 | Figure 3.ACSS2/AMPK/PCNA signaling regulates
DNA damage response under nutrient stress. (A) ECA-109 cells were
grown in different media containing 10% FBS or 1% FBS for 48 h
after being transfected with siRNA-ACSS2 (Si) or negative control
(NC), then stimulated with cisplatin (DDP) (5 µg/ml) for 24 h or
replacement, subjected to immunoblotting analysis for the indicated
proteins. (B and C) Immunofluorescence. Magnification, ×400.
ECA-109 cells were treated with cisplatin (5 µg/ml) for 24 h after
interference for 48 h under basal and stress conditions, then
triple-labeled with anti-PCNA (green), anti-phosphorylated ATM
(p-ATM) (red in B) or anti-γH2AX (red in C), and DAPI (blue,
nuclei) as shown in the figures. (D) ECA-109 cells were treated
with cisplatin for 24 h after being transfected with siRNA against
PCNA (Si) for 24 h under normal or nutrient deficiencies.
Expression of ATM/p-ATM and γH2AX was detected by immunoblotting.
(E) Contribution of ACSS2 to the AMPK pathway for cells incubated
using complete medium or low-serum medium, as assessed by
siRNA-mediated knockdown. (F) Western blot analysis was performed
to show the changes in ACSS2 and PCNA in the dorsomorphin-treated
groups compared with the NC group. ACSS2, acetyl-CoA synthetase
short-chain family member 2; AMPK, AMP-activated protein kinase;
PCNA, proliferating cell nuclear antigen; ATM, ataxia
telangiectasia mutated; Bcl-xL, B-cell lymphoma-extra large; BRCA1,
breast cancer susceptibility gene 1; Bax, Bcl-2 associated X
protein; DNA-PK, DNA-dependent protein kinase catalytic subunit;
γH2AX, phosphorylate H2AX. |
Combination of ACSS2 with PCNA
expression generates a better predictive model for overall
survival
To explore the association between ACSS2 and ESCC
development, we further examined the association of ACSS2
expression with the clinicopathological characteristics of ESCC
patients. The results indicated that the level of ACSS2 was higher
in ESCC tissues, than that in the adjacent normal tissues
(3.23±0.55 vs. 1.56±1.36; P<0.05, Figs. 1A and 4A). To determine the correlation of ACSS2
and PCNA expression, we analyzed samples from two independent
cohorts of ESCC patients. An ACSS2 staining score >3 in the
cohort was considered high, while a score £3 in the cohort was
considered low, and cohorts 1 and 2 included 15 and 13 patients,
respectively. We found that the strong intensity of ACSS2 increased
the expression of PCNA in ESCC tumors (Fig. 4B). However, the clinicopathological
correlation analysis revealed no positive correlation between
elevated ACSS2 levels and age, gender, tumor diameter, lymph node
metastasis, TNM stage and differentiation. Notably, the expression
of ACSS2 was higher in tumors with high PCNA expression compared to
those with low PCNA expression (P=0.0163) and was higher in tumors
with Ki-67 overexpression (P<0.01, Fig. 4C, Table I). Furthermore, to establish a more
sensitive model for predicting the outcomes of patients with ESCC,
we combined ACSS2 expression and PCNA or Ki-67 intensity to create
a prognostic scoring system. Based on the IHC scores, the patients
were stratified into two subgroups (Ki-67 and PCNA, respectively):
A low subgroup for low IHC-positive rates or scores (<50% or ≤3)
and a high subgroup for high IHC-positive rates or scores (≥ 50% or
>3). The analysis showed that the predictive value of ACSS2
alone was higher than that of PCNA (P=0.0311 vs. P=0.4012; Fig. 4D and E), and a combination of ACSS2
and PCNA revealed a better prognostic value (P=0.0446; Fig. 4G). In addition, the combination of
Ki-67 and ACSS2 was better than that for Ki-67 or ACSS2 alone
(P=0.0166; Fig. 4F and H). These
results suggest that the combination of ACSS2 and PCNA expression
could establish a better predictive model for the overall survival
of ESCC patients.
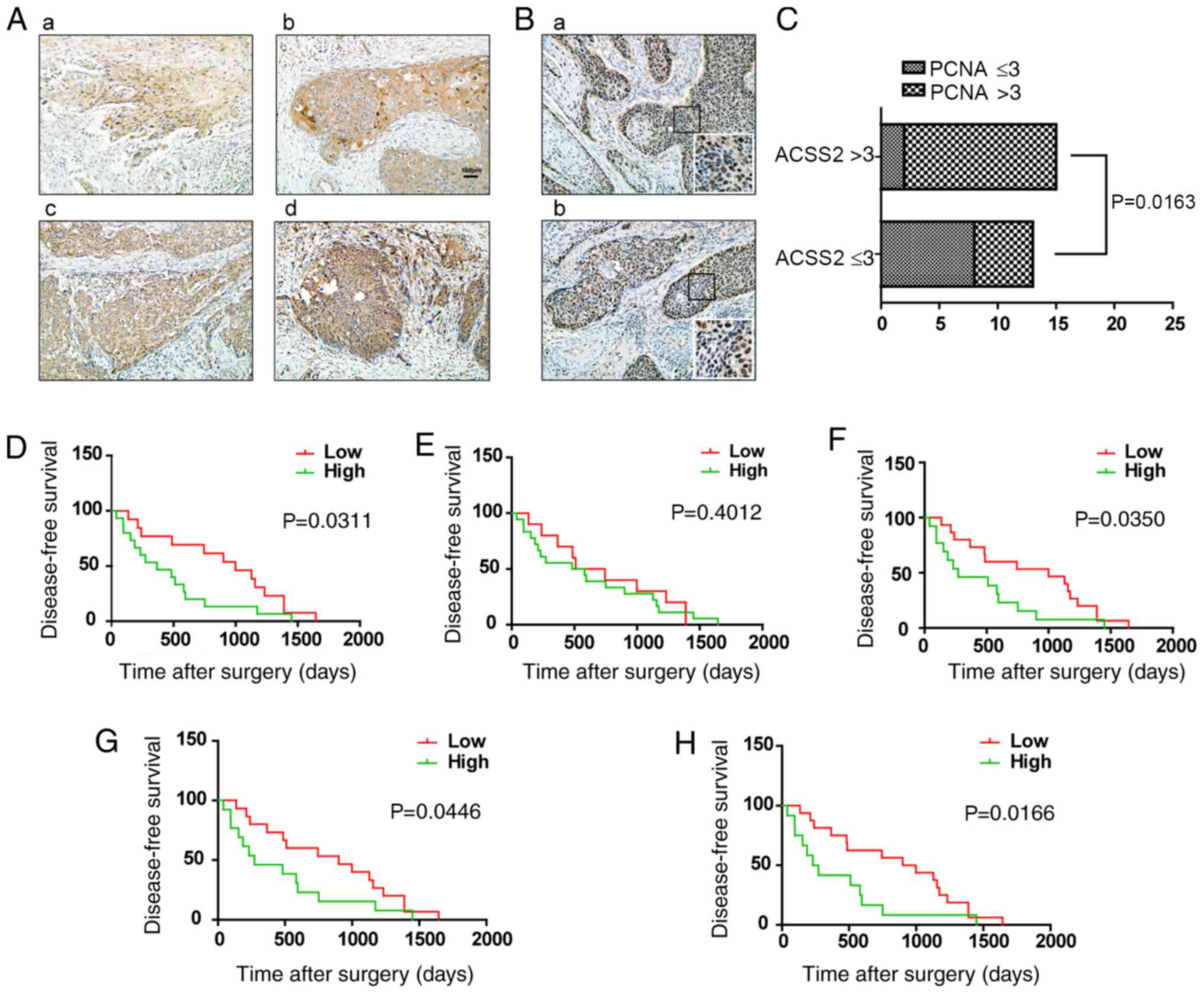 | Figure 4.The prognostic value of ACSS2 and
PCNA expression for patients with ESCC. (A) Representative images
of IHC staining for ACSS2 in 28 pairs of human esophageal cancer
tissues. Magnification, ×200. (a) Weak ACSS2 expression in cancer
tissue is scored as one point. (b) Moderate ACSS2 expression in
cancer tissue is scored as two points. (c) Strong ACSS2 expression
in cancer tissue is scored as three points. (d) Very strong ACSS2
expression in cancer tissue is scored as four points. (B)
Representative images of IHC staining for PCNA in tumors.
Magnification, ×200. (a) High expression of PCNA. (b) Relatively
low level of PCNA. The expression intensity of PCNA is shown in the
magnified image (insert). Magnification, ×400. (C) The association
between ACSS2 and PCNA IHC staining intensity is presented as a
histogram (n=28, P<0.05). (D-F) Kaplan-Meier analysis of
disease-free survival (DFS) of patients with esophageal cancer
(n=28) based on ACSS2, PCNA and Ki-67 expression. (G and H)
Kaplan-Meier estimates of DFS according to the combinations of
ACSS2 and PCNA, ACSS2 and Ki-67 respectively. IHC,
immunohistochemistry; ACSS2, acetyl-CoA synthetase short-chain
family member 2; AMPK, AMP-activated protein kinase; PCNA,
proliferating cell nuclear antigen. |
 | Table I.Association of the expression of
ACSS2 with clinicopathological characteristics of the ESCC
patients. |
Table I.
Association of the expression of
ACSS2 with clinicopathological characteristics of the ESCC
patients.
|
| ACSS2
expression |
|
|---|
|
|
|
|
|---|
| Parameters | Low (13) | High (15) | P-value |
|---|
| Age (years) |
|
|
|
|
<60 | 5 | 6 | 0.9337 |
|
≥60 | 8 | 9 |
|
| Sex |
|
|
|
|
Male | 9 | 14 | 0.0968 |
|
Female | 4 | 1 |
|
| Degree of
differentiation |
|
| 0.1565 |
|
Well | 3 | 5 |
|
|
Moderate | 2 | 6 |
|
|
Poor | 8 | 4 |
|
| Tumor diameter
(cm) |
|
| 0.3903 |
|
<5 | 9 | 8 |
|
| ≥5 | 4 | 7 |
|
| Lymph node
metastasis |
|
| 0.1939 |
|
Yes | 3 | 7 |
|
| No | 10 | 8 |
|
| Tumor grade |
|
| 0.2556 |
|
I–II | 8 | 6 |
|
|
III–IV | 5 | 9 |
|
| PCNA
expression |
|
| 0.0163 |
|
Low | 8 | 2 |
|
|
High | 5 | 13 |
|
| Ki-67
expression |
|
|
<0.01 |
|
≤50% | 12 | 3 |
|
|
>50% | 1 | 12 |
|
Discussion
Due to its immortalized nature, tumor progression is
restricted by an irregular and deficient vascular network that is
characterized by hypoxia and nutrient deficiency. While a majority
of tumor cells remain viable, these cells continue to cautiously
proliferate and tend to develop resistance to current therapies,
thereby permitting survival, repopulation and metastasis (48–50).
Except for the limited blood supply in tumor-restricting drug
distribution, the tumor cells in regions of limited nutrition are
likely to be resistant to drugs. Moreover, increasing evidence
reveals that nutrient stress can stimulate transcriptional
amplification, leading to the increased expression of genes
encoding proteins that cause drug resistance, including autophagy,
DNA repair, cell cycle progression and multidrug-resistant
transporters, through diverse pathways (51–53).
An improved understanding of the mechanisms that provide resistance
to nutrient stress may clarify the reasons for treatment failure in
patients with cancer. Tumor cells located far from functional blood
vessels may receive a low supply, thus serum starvation is a
well-established approach for inducing a broad range of cellular
stresses. In the present study, we observed the upregulation of
ACSS2 when esophageal squamous cell carcinoma cells (ESCCs), but
not normal cells, were exposed to limited serum for a short or long
period of time. This finding led us to consider the underlying
roles for ACSS2 in the coping strategy. In this study, ACSS2
expression was assessed in ESCC patients and higher expression of
ACSS2 was found in tumors focused in core and cell-rich areas.
Thus, identification of whether and how ACSS2 contributes to
malignant behavior may provide new therapeutic opportunities for
ESCC.
In the present study, it was demonstrated that ACSS2
knockdown caused less tolerance to serum deprivation in ESCCs.
First, we observed ACSS2-dependent viability, and the expression of
ACSS2 was more important when cells were cultured in vitro
under limited nutrients. Notably, the level of ACSS2 may stabilize
the transcription and translation of PCNA, especially under
nutrient stress. PCNA executes its major function as a processivity
factor in DNA replication by tethering replicative polymerases to a
genomic template and as a central factor to control genome
stability (2,54–57).
In general, inhibition of proliferation also involves changes in
the cell cycle. Our findings indicated that cells were arrested in
the G2/M phase upon siRNA-ACSS2 treatment. The DNA damage
checkpoint at G2/M is important for DNA repair; thus, its
dysfunction leads to apoptosis or death. In combination with
cisplatin, pretreatment with siRNA-ACSS2 could cause apoptosis and
death, which have marked synergistic effects. The expression of
ACSS2 links nutrient intake and stress signaling with autophagy,
tumor growth and metastasis (14,21,39,58).
Other groups have demonstrated higher ACSS2 expression in the
bladder cancer patients with cisplatin resistance, suggesting that
ACSS2 is involved in lipid metabolic alterations (24,59).
Together with our findings, these data support the proposition that
nutrient stress-induced ACSS2 upregulation may function
protectively to maintain proliferation and prevent apoptosis.
Many tumors exhibit deregulated AMPK activity, which
regulates energy homeostasis and autophagy and in turn requires
increased amounts of carbon sources, such as acetate, glucose and
fatty acids, to meet the needs of reprogrammed anabolic metabolism
(28–30,39).
Indeed, reprogramming energy metabolism is an emerging hallmark of
many cancers as adjusting energy metabolism is essential to fuel
cell growth and division. Acetyl-CoA represents a central node of
carbon metabolism that plays a particular role in the
bioenergetics, cell proliferation, and the regulation of gene
expression. ACSS2 is the key factor that enables cells to maximally
utilize acetate and produce acetyl-CoA for the synthesis of fatty
acids and sterols and the modification of histones (14,39).
Our results demonstrated that ACSS2 sufficiently protects cancer
cells from cisplatin, specifically, which directly regulates the
stabilization and expression of PCNA during DNA repair. Another
group demonstrated that glucose deprivation results in
AMPK-mediated ACSS2 phosphorylation and nuclear translocation,
which promote lysosomal biogenesis, autophagy, survival, and
tumorigenesis (39). We also
observed that under nutrient stress, ESCCs exhibited an
accumulation of p-AMPK. In particular, previous studies have shown
that ACSS2 also promotes carcinogenesis by increasing the
expression of autophagy-related factors, such as LAMP1, LC3B, and
ATG3 (39,60). Furthermore, activated AMPK induced
by nutrient stress directly phosphorylates ULK1, a serine/threonine
kinase, which is necessary for the formation of autophagosomes
(61). In contrast to the
activation of AMPK, our results found no significant changes in
either p-ULK1 or ULK by siRNA-ACSS2 or nutrient stress treatment in
ESCCs. Interestingly, the inhibition of AMPK signaling with
dorsomorphin also led to the downregulation of ACSS2, which was
even more obvious than that of PCNA, suggesting that an interaction
between ACSS2 and AMPK signaling ensures the survival and further
resistance of ESCCs. The clinical significance of ACSS2 in ESCC has
been presented in our study, although known prognostic factors,
such as tumor grade, differentiation and lymph node metastasis,
fail to be significant in the analysis. However, we found a
positive correlation between the expression of ACSS2 and PCNA or
Ki-67, suggesting its potential prognostic value. Although the
prognostic relevance of ACSS2 in ESCC needs to be confirmed with
larger-scale clinicopathologic analyses, at the least, our study
has identified aberrant expression of ACSS2 as an independent
prognostic factor in our small samples, and ACSS2 expression could
be integrated with PCNA to generate a better risk stratification
for ESCC patients.
Altogether, PCNA plays a dual role in replication
and DNA repair under nutrient stress in ESCC. By creating an
environment of nutrient stress, we showed here that PCNA is crucial
to manage DNA damage, to complete efficient DSB repair, and to
survive. Furthermore, ACSS2 cooperated with AMPK pathway activity
to regulate the expression of PCNA, especially under nutrient
stress. Finally, it was demonstrated that the combination of ACSS2
and PCNA expression could establish a better predictive model for
the survival of ESCC patients.
Acknowledgements
Not applicable.
Funding
The current study was supported by the National
Natural Science Foundation of China (grant no. 81572956), the
Jiangsu Provincial Science and Technology Supporting Program (grant
no. BE2017096), the Medical Innovation Team of Jiangsu Province
(grant no. CXTDC2016009) and the Student Innovation Training
Program Projects of Jiangsu University (grant no.
201810299262W).
Availability of data and materials
The datasets used and/or during the present study
are available from the corresponding author on reasonable
request.
Authors' contributions
YZ, DC and CM conceived and designed the study. LM,
YZ, DW, QT, XW, HZ, XG, JW, RL and JD performed the experiments. YZ
and DC wrote the paper. YZ, CM, XW and DC reviewed and edited the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The Ethics Committee of the Affiliated Hospital of
Jiangsu University approved the research. All methods, including
the collection and use of patient samples, were performed in
accordance with the relevant guidelines and regulations and all
patients provided signed informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ESCC
|
esophageal squamous cell carcinoma
|
|
NS
|
nutrient stress
|
|
ACSS2
|
acetyl-CoA synthetase short-chain
family member 2
|
|
PCNA
|
proliferating cell nuclear antigen
|
References
|
1
|
Choe KN and Moldovan GL: Forging ahead
through darkness: PCNA, Still the principal conductor at the
replication fork. Mol Cell. 65:380–392. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
De March M and De Biasio A: The dark side
of the ring: Role of the DNA sliding surface of PCNA. Crit Rev
Biochem Mol Biol. 52:663–673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gu L, Lingeman R, Yakushijin F, Sun E, Cui
Q, Chao J, Hu W, Li H, Hickey RJ, Stark JM, et al: The anticancer
activity of a First-in-class Small-molecule targeting PCNA. Clin
Cancer Res. 24:6053–6065. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shiomi Y and Nishitani H: Control of
genome integrity by RFC complexes; conductors of PCNA loading onto
and unloading from chromatin during DNA Replication. Genes. 8(pii):
E522017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tan Z, Wortman M, Dillehay KL, Seibel WL,
Evelyn CR, Smith SJ, Malkas LH, Zheng Y, Lu S and Dong Z:
Small-molecule targeting of proliferating cell nuclear antigen
chromatin association inhibits tumor cell growth. Mol Pharmacol.
81:811–819. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bartová E, Suchanková J, Legartová S,
Malyšková B, Hornáček M, Skalníková M, Mašata M, Raška I and
Kozubek S: PCNA is recruited to irradiated chromatin in late
S-phase and is most pronounced in G2 phase of the cell cycle.
Protoplasma. 254:2035–2043. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Recouvreux MV and Commisso C:
Macropinocytosis: A metabolic adaptation to nutrient stress in
cancer. Front Endocrinol (Lausanne). 8:2612017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
White E, Mehnert JM and Chan CS:
Autophagy, metabolism, and cancer. Clin Cancer Res. 21:5037–5046.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peck B, Ferber EC and Schulze A:
Antagonism between FOXO and MYC regulates cellular powerhouse.
Front Oncol. 3:962013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Singh D, Arora R, Kaur P, Singh B, Mannan
R and Arora S: Overexpression of hypoxia-inducible factor and
metabolic pathways: Possible targets of cancer. Cell Biosci.
7:622017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
El Hout M, Dos Santos L, Hamaï A and
Mehrpour M: A promising new approach to cancer therapy: Targeting
iron metabolism in cancer stem cells. Semin Cancer Biol.
53:125–138. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Conacci-Sorrell M, Ngouenet C, Anderson S,
Brabletz T and Eisenman RN: Stress-induced cleavage of Myc promotes
cancer cell survival. Genes Dev. 28:689–707. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu G, Zhou Y, Zhu Y, Zhou L, Ling R, Wu D,
Mi L, Wang X, Dai D, Mao C and Chen D: Novel transduction of
nutrient stress to Notch pathway by RasGRP3 promotes malignant
aggressiveness in human esophageal squamous cell carcinoma. Oncol
Rep. 38:2975–2984. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen R, Xu M, Nagati J and Garcia JA:
Coordinate regulation of stress signaling and epigenetic events by
Acss2 and HIF-2 in cancer cells. PLoS One. 12:e01902412017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Keenan MM and Chi JT: Alternative fuels
for cancer cells. Cancer J. 21:49–55. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lakhter AJ, Hamilton J, Konger RL,
Brustovetsky N, Broxmeyer HE and Naidu SR: Glucose-independent
acetate metabolism promotes melanoma cell survival and tumor
growth. J Biol Chem. 291:21869–21879. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vysochan A, Sengupta A, Weljie AM, Alwine
JC and Yu Y: ACSS2-mediated acetyl-CoA synthesis from acetate is
necessary for human cytomegalovirus infection. Proc Natl Acad Sci
USA. 114:E1528–E1535. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao TT, Lin SH, Fu L, Tang Z, Che CM,
Zhang LY, Ming XY, Liu TF, Tang XM, Tan BB, et al: Eukaryotic
translation initiation factor 5A2 promotes metabolic reprogramming
in hepatocellular carcinoma cells. Carcinogenesis. 38:94–104. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Comerford SA, Huang Z, Du X, Wang Y, Cai
L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al:
Acetate dependence of tumors. Cell. 159:1591–1602. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schug ZT, Vande Voorde J and Gottlieb E:
The metabolic fate of acetate in cancer. Nat Rev Cancer.
16:708–717. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang S, He J, Jia Z, Yan Z and Yang J:
Acetyl-CoA synthetase 2 enhances tumorigenesis and is indicative of
a poor prognosis for patients with renal cell carcinoma. Urol
Oncol. 36:243.e9–243.e20. 2018. View Article : Google Scholar
|
|
22
|
Sun L, Kong Y, Cao M, Zhou H, Li H, Cui Y,
Fang F, Zhang W, Li J, Zhu X, et al: Decreased expression of
acetyl-CoA synthase 2 promotes metastasis and predicts poor
prognosis in hepatocellular carcinoma. Cancer Sci. 108:1338–1346.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bae JM, Kim JH, Oh HJ, Park HE, Lee TH,
Cho NY and Kang GH: Downregulation of acetyl-CoA synthetase 2 is a
metabolic hallmark of tumor progression and aggressiveness in
colorectal carcinoma. Mod Pathol. 30:267–277. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee MY, Yeon A, Shahid M, Cho E, Sairam V,
Figlin R, Kim KH and Kim J: Reprogrammed lipid metabolism in
bladder cancer with cisplatin resistance. Oncotarget.
9:13231–13243. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dauer P, Nomura A, Saluja A and Banerjee
S: Microenvironment in determining chemo-resistance in pancreatic
cancer: Neighborhood matters. Pancreatology. 17:7–12. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chou CW, Wang CC, Wu CP, Lin YJ, Lee YC,
Cheng YW and Hsieh CH: Tumor cycling hypoxia induces
chemoresistance in glioblastoma multiforme by upregulating the
expression and function of ABCB1. Neuro Oncol. 14:1227–1238. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Iyama T and Wilson DM III: DNA repair
mechanisms in dividing and non-dividing cells. DNA Repair (Amst).
12:620–636. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yung MM, Ngan HY and Chan DW: Targeting
AMPK signaling in combating ovarian cancers: Opportunities and
challenges. Acta Biochim Biophys Sin (Shanghai). 48:301–317. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeng J, Liu W, Fan YZ, He DL and Li L:
PrLZ increases prostate cancer docetaxel resistance by inhibiting
LKB1/AMPK-mediated autophagy. Theranostics. 8:109–123. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oh TI, Lee JH, Kim S, Nam TJ, Kim YS, Kim
BM, Yim WJ and Lim JH: Fascaplysin sensitizes anti-cancer effects
of drugs targeting AKT and AMPK. Molecules. 23(pii): E422017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pan Y, Zhang F, Zhao Y, Shao D, Zheng X,
Chen Y, He K, Li J and Chen L: Berberine enhances chemosensitivity
and induces apoptosis through Dose-orchestrated AMPK signaling in
breast cancer. J Cancer. 8:1679–1689. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lam TG, Jeong YS, Kim SA and Ahn SG: New
metformin derivative HL156A prevents oral cancer progression by
inhibiting the insulin-like growth factor/AKT/mammalian target of
rapamycin pathways. Cancer Sci. 109:699–709. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pan Y, Liu L, Li S, Wang K, Ke R, Shi W,
Wang J, Yan X, Zhang Q, Wang Q, et al: Activation of AMPK inhibits
TGF-β1-induced airway smooth muscle cells proliferation and its
potential mechanisms. Sci Rep. 8:36242018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu L, Pan Y, Song Y, Su X, Ke R, Yang L,
Gao L and Li M: Activation of AMPK α2 inhibits airway smooth muscle
cells proliferation. Eur J Pharmacol. 791:235–243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao Y, Liu Y, Jing Z, Peng L, Jin P, Lin
Y, Zhou Y, Yang L, Ren J, Xie Q and Jin X: N-oleoylethanolamide
suppresses intimal hyperplasia after balloon injury in rats through
AMPK/PPARα pathway. Biochem Biophys Res Commun. 496:415–421. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang T, Guo P, Zhang Y, Xiong H, Yu X, Xu
S, Wang X, He D and Jin X: The antidiabetic drug metformin inhibits
the proliferation of bladder cancer cells in vitro and in vivo. Int
J Mol Sci. 14:24603–24618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Feng J, Qi B, Guo L, Chen LY, Wei XF, Liu
YZ and Zhao BS: miR-382 functions as a tumor suppressor against
esophageal squamous cell carcinoma. World J Gastroenterol.
23:4243–4251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li X, Yu W, Qian X, Xia Y, Zheng Y, Lee
JH, Li W, Lyu J, Rao G, Zhang X, et al: Nucleus-translocated ACSS2
promotes gene transcription for lysosomal biogenesis and autophagy.
Mol Cell. 66:684–697.e9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schug ZT, Peck B, Jones DT, Zhang Q,
Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K,
et al: Acetyl-CoA synthetase 2 promotes acetate utilization and
maintains cancer cell growth under metabolic stress. Cancer Cell.
27:57–71. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang L, Cheng X, Gao Y, Bao J, Guan H, Lu
R, Yu H, Xu Q and Sun Y: Induction of ROS-independent DNA damage by
curcumin leads to G2/M cell cycle arrest and apoptosis in human
papillary thyroid carcinoma BCPAP cells. Food Funct. 7:315–325.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang HW, Tang JY, Ou-Yang F, Wang HR,
Guan PY, Huang CY, Chen CY, Hou MF, Sheu JH and Chang HW: Sinularin
selectively kills breast cancer cells showing G2/M arrest,
apoptosis, and oxidative DNA damage. Molecules. 23(pii): E8492018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hegde M, Vartak SV, Kavitha CV, Ananda H,
Prasanna DS, Gopalakrishnan V, Choudhary B, Rangappa KS and
Raghavan SC: A benzothiazole derivative (5g) induces DNA damage and
potent G2/M arrest in cancer cells. Sci Rep. 7:25332017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Finzel A, Grybowski A, Strasen J,
Cristiano E and Loewer A: Hyperactivation of ATM upon DNA-PKcs
inhibition modulates p53 dynamics and cell fate in response to DNA
damage. Mol Biol Cell. 27:2360–2367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tomimatsu N, Mukherjee B and Burma S:
Distinct roles of ATR and DNA-PKcs in triggering DNA damage
responses in ATM-deficient cells. EMBO Rep. 10:629–635. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shen B, He PJ and Shao CL: Norcantharidin
induced DU145 cell apoptosis through ROS-mediated mitochondrial
dysfunction and energy depletion. PLoS One. 8:e846102013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bártová E, Malyšková B, Komůrková D,
Legartová S, Suchánková J, Krejčí J and Kozubek S: Function of
heterochromatin protein 1 during DNA repair. Protoplasma.
254:1233–1240. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Saggar JK, Yu M, Tan Q and Tannock IF: The
tumor microenvironment and strategies to improve drug distribution.
Front Oncol. 3:1542013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tan Q, Saggar JK, Yu M, Wang M and Tannock
IF: Mechanisms of drug resistance related to the microenvironment
of solid tumors and possible strategies to inhibit them. Cancer J.
21:254–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rohwer N and Cramer T: Hypoxia-mediated
drug resistance: Novel insights on the functional interaction of
HIFs and cell death pathways. Drug Resist Updat. 14:191–201. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ojha R, Bhattacharyya S and Singh SK:
Autophagy in cancer stem cells: A potential link between
chemoresistance, recurrence, and metastasis. Biores Open Access.
4:97–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hua Y, Gorshkov K, Yang Y, Wang W, Zhang N
and Hughes DP: Slow down to stay alive: HER4 protects against
cellular stress and confers chemoresistance in neuroblastoma.
Cancer. 118:5140–5154. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Salaroglio IC, Panada E, Moiso E,
Buondonno I, Provero P, Rubinstein M, Kopecka J and Riganti C: PERK
induces resistance to cell death elicited by endoplasmic reticulum
stress and chemotherapy. Mol Cancer. 16:912017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Baserga R: Growth regulation of the PCNA
gene. J Cell Sci. 98:433–436. 1991.PubMed/NCBI
|
|
55
|
Feng W, Guo Y, Huang J, Deng Y, Zang J and
Huen MS: TRAIP regulates replication fork recovery and progression
via PCNA. Cell Discov. 2:160162016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fox JT, Lee KY and Myung K: Dynamic
regulation of PCNA ubiquitylation/deubiquitylation. FEBS Lett.
585:2780–2785. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Slade D: Maneuvers on PCNA rings during
DNA replication and repair. Genes (Basel). 9(pii): E4162018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bidkhori G, Benfeitas R, Klevstig M, Zhang
C, Nielsen J, Uhlen M, Boren J and Mardinoglu A: Metabolic
network-based stratification of hepatocellular carcinoma reveals
three distinct tumor subtypes. Proc Natl Acad Sci USA.
115:E11874–E11883. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wen H, Lee S, Zhu WG, Lee OJ, Yun SJ, Kim
J and Park S: Glucose-derived acetate and ACSS2 as key players in
cisplatin resistance in bladder cancer. Biochim Biophys Acta Mol
Cell Biol Lipids. 1864:413–421. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yao L, Guo X and Gui Y: Acetyl-CoA
synthetase 2 promotes cell migration and invasion of renal cell
carcinoma by upregulating lysosomal-associated membrane protein 1
expression. Cell Physiol Biochem. 45:984–992. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Puente C, Hendrickson RC and Jiang X:
Nutrient-regulated phosphorylation of ATG13 inhibits
starvation-induced autophagy. J Biol Chem. 291:6026–6035. 2016.
View Article : Google Scholar : PubMed/NCBI
|