Introduction
Dilated cardiomyopathy (DCM) is a common type of
non-ischemic cardiomyopathy, which leads to progressive heart
failure and sudden cardiac-associated mortality (1–3). DCM
is characterized by ventricular chamber enlargement and systolic
dysfunction, and has strong associations with cardiac development,
organ morphology, myocardial energy metabolism and abnormal gene
expression (4,5). In patients with DCM, the possible
causes of the condition are often ascribed to coronary artery
disease, hypertension, viral infections and alcohol abuse (6). However, the exact mechanisms involved
in the development of DCM remain unknown.
Long noncoding RNAs (lncRNAs) are a cluster of
transcribed RNAs with a length >200 nucleotides, which lack
protein-coding potential (7,8).
lncRNAs function by regulating their neighboring or distant target
genes through connection with microRNAs and interaction with
proteins (9,10). Previous studies have verified that
lncRNAs serve critical roles in the progression of cardiovascular
diseases by modulating cardiac development, homeostasis and
regeneration (7,11–14).
Nevertheless, determining the genome-wide profile is urgently
required to uncover the expression variation and potential
functions of lncRNAs in the development of DCM.
In the present study, the genome-wide profile of
circulating lncRNAs between patients with DCM and controls was
detected by RNA sequencing, which is considered as a credible
method to define lncRNA expression (15–17).
Gene Ontology (GO) analysis, Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway analysis and lncRNA-messenger RNA (mRNA)
co-expression network analysis were performed to explore the
potential functions of differentially expressed lncRNAs in DCM.
Materials and methods
Ethical statement
All procedures in the present study were approved by
the Ethics Committee of the Liaocheng People's Hospital (Liaocheng,
China). All subjects provided written informed consent prior to
enrollment into the study.
Study subjects
The 8 control patients who admitted for routine
testing and 8 patients with DCM underwent internal medical
treatment were enrolled in the present study at Liaocheng People's
Hospital between July 2015 and May 2016 (Table I). The inclusion criteria for
patients with DCM were as follows: i) Left ventricular
end-diastolic diameter (LVED) of >50 mm (females) or >55 mm
(males); and ii) left ventricular ejection fraction (LVEF) of
<45% and/or left ventricular fraction shortening of <25%.
Patients with clear causes of idiopathic DCM, including
hypertension, coronary artery diseases, valvular disease,
congenital defect, alcoholic cardiomyopathy, tachycardia-induced
cardiomyopathy and peripartum cardiomyopathy, were excluded from
the present study. Patients with a positive family history of DCM
were also excluded. The following data were gathered for the DCM
and control subjects: Sex, age, blood pressure on admission, heart
rate, New York Heart Association classes (18), LVED, LVEF, N-terminal pro b-type
natriuretic peptide, medical history and diagnostic testing
(Table I).
 | Table I.Clinical characteristics of control
subjects and DCM patients. |
Table I.
Clinical characteristics of control
subjects and DCM patients.
|
| Controls (n=8) | DCM patients
(n=8) |
|---|
|
|
|
|
|---|
| Group | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|
| Sex | M | M | M | M | F | M | M | M | M | M | M | M | F | M | M | M |
| Age (years) | 32 | 38 | 35 | 35 | 61 | 60 | 46 | 35 | 33 | 40 | 33 | 36 | 63 | 61 | 47 | 34 |
| SBP (mmHg) | 110 | 120 | 112 | 118 | 124 | 122 | 126 | 118 | 140 | 120 | 110 | 108 | 104 | 102 | 102 | 124 |
| DBP (mmHg) | 72 | 74 | 70 | 74 | 74 | 80 | 82 | 72 | 90 | 80 | 70 | 72 | 76 | 70 | 86 | 78 |
| Heart rate
(rpm) | 66 | 68 | 72 | 65 | 73 | 67 | 76 | 74 | 105 | 86 | 98 | 85 | 72 | 105 | 78 | 90 |
| LVED (mm) | 46 | 44 | 41 | 46 | 44 | 47 | 43 | 44 | 56 | 63 | 67 | 78 | 84 | 62 | 68 | 75 |
| LVEF (%) | 68 | 61 | 68 | 66 | 65 | 73 | 77 | 68 | 42 | 26 | 20 | 16 | 19 | 25 | 30 | 27 |
| NT-proBNP
(pg/ml) | 70 | 82 | 60 | 84 | 96 | 90 | 88 | 78 | 5,400 | 6,630 | 1,470 | 2,450 | 2,980 | 1,760 | 2,530 | 2,228 |
| NYHA class | – | – | – | – | – | – | – | – | IV | III | III | III | IV | IV | IV | III |
Specimen collection
Blood samples for RNA sequencing were collected from
8 typical patients with DCM and 8 healthy controls. In addition,
blood samples from another 20 patients with DCM (16 males and 4
females, 43.2±8.7 years old) and 20 healthy controls (16 males and
4 females, 45.3±7.9 years old) were collected to identify the
expression levels of the top 10 lncRNAs with the highest degree via
Reverse transcription-polymerase chain reaction (RT-PCR), which was
conducted as described below. All the patients were diagnosed with
DCM by two experienced clinicians and underwent internal medical
treatment at Liaocheng People's Hospital based on the American
Heart Association guidelines (19,20).
Peripheral venous blood (5 ml) samples were collected from all
participants in sterile tubes with ethylenediaminetetraacetic
acid.
RNA isolation, library construction
and sequencing
Total RNA was isolated and enriched using
TRIzol® reagent (cat. no. 15596; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Next, ribosomal RNA was
removed using the Ribo-Zero rRNA Removal kit (cat. no. MRZB12424;
Illumina, Inc., San Diego, CA, USA). Ribosomal RNA-depleted RNAs
were used to construct RNA libraries with the TruSeq Stranded Total
RNA Library Prep kit (cat. no. RS-122-2301; Illumina, Inc.).
Libraries were controlled for quality and quantified using the
Agilent BioAnalyzer 2100 system (cat. no. 5067-1505; Agilent
Technologies Inc., Santa Clara, CA, USA). Libraries (10 pM) were
denatured as single-stranded DNA molecules, captured on Illumina
flow cells, amplified in situ as clusters and finally
sequenced for 150 cycles on the Illumina HiSeq sequencer (HiSeq
4000; Illumina, Inc.). All these procedures were conducted
according to the manufacturer's protocol.
Analysis of RNA sequencing data
Paired-end reads were harvested from Illumina HiSeq
sequencer and quality controlled by Q30 (P<0.001). Subsequent to
removing the 3′ adaptor-trimming and low-quality reads with
cutadapt software (version 1.9.3, http://cutadapt.readthedocs.io/en/stable/index.html),
the high-quality trimmed reads were aligned to the reference genome
(UCSC hg19) guided by the Ensembl Gff gene annotation file with
HISAT2 software (version 2.0.4, http://ccb.jhu.edu/software/hisat2/index.shtml). The
cuffdiff software (version 2.2.1; part of Cufflinks, http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/index.html)
was then used to obtain the expression profiles of lncRNAs and
mRNAs in terms of the fragments per kilobase of transcript per
million fragments mapped (FPKM). Next, the fold-change (FC) and
P-values were calculated based on the FPKM, and differentially
expressed lncRNAs and mRNAs were identified between the DCM and
control groups. The criteria were set as an FC of ≥2 or ≤-2, and a
P-value of <0.05 between two groups. P-values were corrected
with the false discovery rate method. Hierarchical clustering with
average linkage was used to calculate the distinguishable
expression patterns. RNA sequencing data were uploaded to the Gene
Expression Omnibus database (accession no. GSE101585; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE101585).
RT-PCR
Total RNA from the blood samples was isolated and
enriched with TRIzol® reagent (Thermo Fisher Scientific,
Inc.), and then the concentration was measured with a NanoDrop
ND-1000 spectrophotometer (Nanodrop Technologies; Thermo Fisher
Scientific, Inc., Wilmington, MA, USA). Total RNA was reverse
transcribed into cDNA with the SuperScript III reverse
transcriptase (cat. no. 18080044; Invitrogen; Thermo Fisher
Scientific, Inc.). Subsequently, 2X PCR master mix (CloudSeq
Biotech, Inc., Shanghai, China) was applied to perform the PCR with
the target RNAs and internal reference, β-actin (ACTB) of the
samples. The primers were as follows: TCONS_00028585, forward:
5′-CGCAGAAGCACAGGGTTAAT-3′, reverse, 5′-GCTGTAGCTTGCTGGCTCTT-3′;
ENST00000439796, forward, 5′-TGAAAAACACCGCAGTCAAA-3′, reverse,
5′-AGGCCCTAAAGAAGGCCATA-3′; TCONS_l2_00009351, forward,
5′-ATGAAGCTGGGCAAATCAGT-3′, reverse, 5′-AGATGGCACAAATGCTCTCC-3′;
ENST00000448134, forward 5′-TTGGTGGACGGAACAGTGTA-3′, reverse,
5′-GAGTTAGCCAGTGGGGTGAA-3′; ENST00000607876, forward,
5′-CCCTCACACCTCTGTGTCCT-3′, reverse, 5′-GTGACACAGCCACAGTCCAC-3′;
and ACTB, forward, 5′-GTGGCCGAGGACTTTGATTG-3′ and reverse,
5′-CCTGTAACAACGCATCTCATATT-3′.
The thermal cycling conditions were set to 40 cycles
at 95°C for 10 sec and 60°C for 60 sec, following predegeneration
for 10 min at 95°C. Data were analyzed using the 2−∆∆Cq
method (21,22).
Functional group analysis
The cis target genes were analyzed through the gene
annotations provided by the University of California (Oakland, CA,
USA) (23), and trans target genes
were predicted using BLAT (24).
GO analysis, which contained three domains, namely the ‘biological
process’, ‘cellular component’ and ‘molecular function’, was
performed to explore the functions of differentially expressed
coding genes with the Database for Annotation, Visualization and
Integrated Discovery (http://david.abcc.ncifcrf.gov) (25). Pathway analysis was also adopted to
explore potential pathways of differentially expressed coding
genes, according to KEGG pathways (https://www.genome.jp/kegg/). The criteria were set as
follows: Fold-change of ≥2 or ≤-2, and P-value of <0.05 between
the DCM and control groups.
Analysis of the lncRNA-mRNA
co-expression network
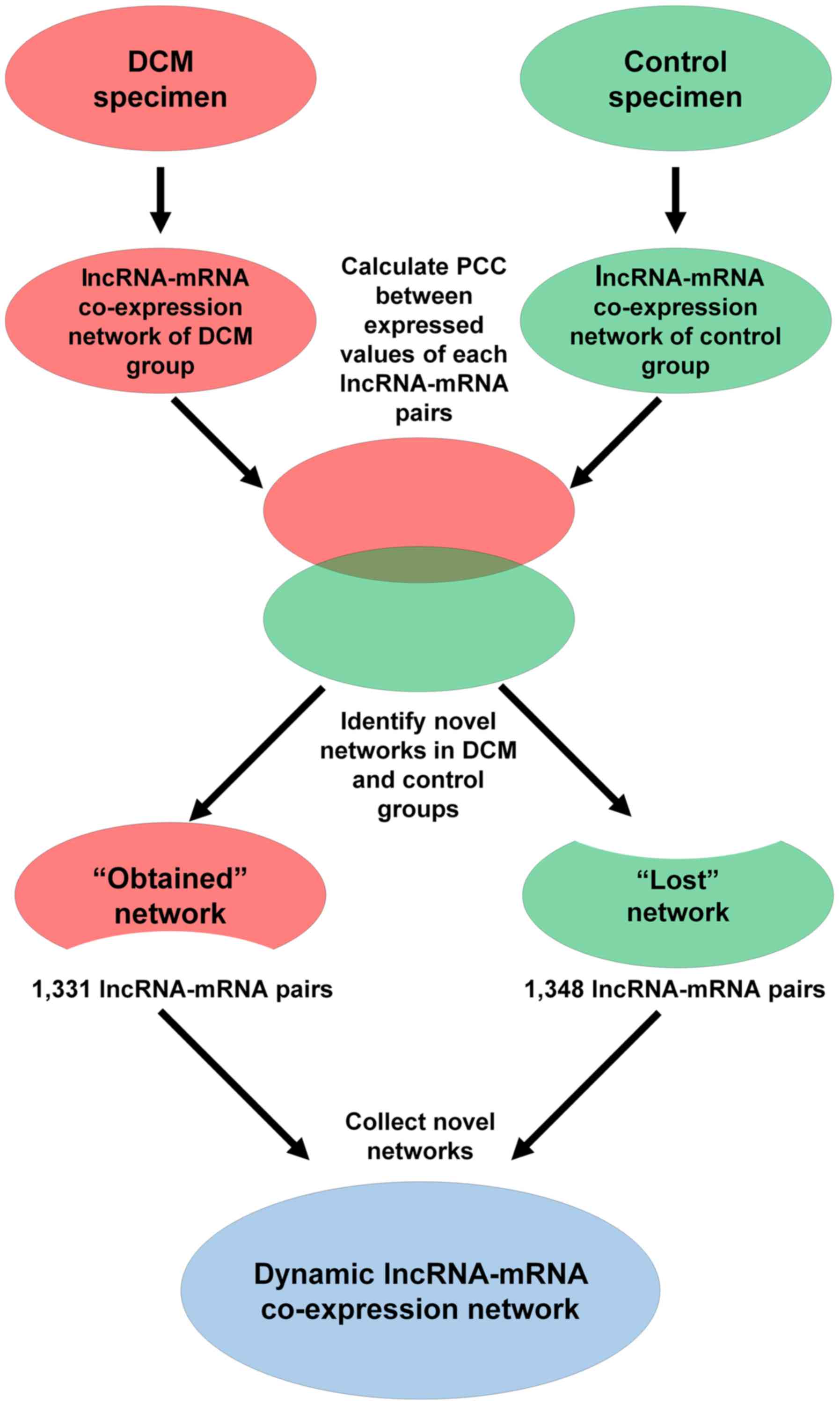
Pearson's correlation coefficient (PCC) was
calculated for each of the lncRNA-mRNA pairs across the DCM and
control groups. Co-expressed lncRNA-mRNA pairs with PCC>0.99 and
P<0.01 were selected. In total, two lncRNA-mRNA co-expression
networks were initially constructed based on co-expressed
lncRNA-mRNA pairs in the DCM and control groups. The ‘lost’ network
presents the lncRNA-mRNA co-expression pairs, which only appeared
in the control group, and the ‘obtained’ network presents the
co-expression pairs, which only appeared in the DCM group. Finally,
the dynamic lncRNA-mRNA co-expression network was constructed by
combining the ‘lost’ and ‘obtained’ networks. Subsequently, key
lncRNA nodes were identified in the combined network. According to
the mRNAs paired with key lncRNAs, GO analysis was performed to
separately identify the significantly enriched biological processes
in the DCM and the normal co-expression networks. P<0.05 was
considered to indicate a statistically significant difference.
Results
Overview of lncRNAs between patients
with DCM and controls determined by RNA sequencing
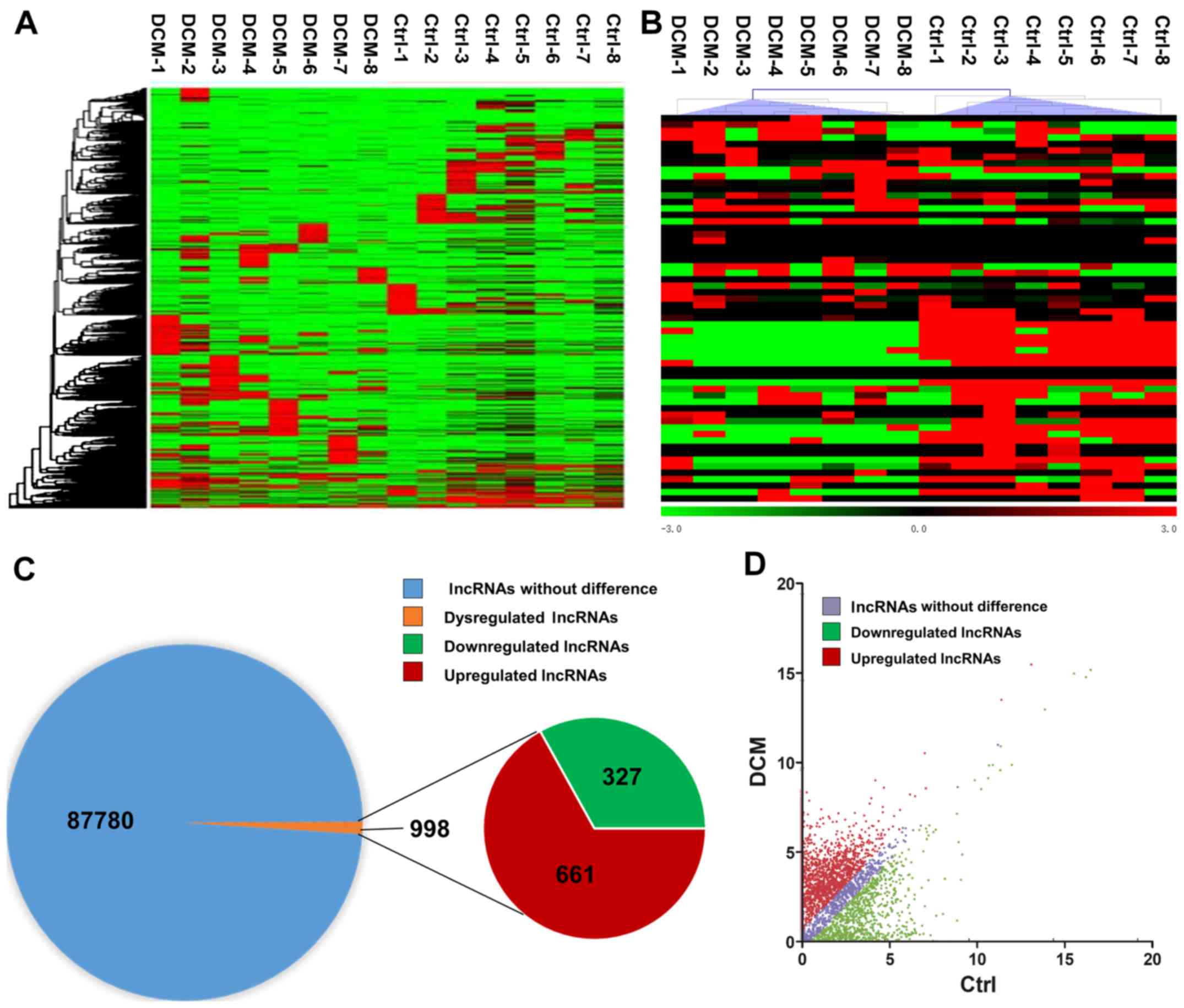
RNA sequencing detected a total of 88,768 lncRNAs in
the serum of DCM and control subjects. In order to assess the
differentially expressed lncRNAs, the criteria were set as
fold-change of ≥2 or ≤-2, and P-value of <0.05 between the DCM
and control groups. A hierarchical clustering approach was adopted
to verify the consistency in the specimens, according to
dysregulated lncRNAs (Fig. 1A). A
heat map of partially dysregulated lncRNAs is presented in Fig. 1B. Compared with the control group,
998 dysregulated lncRNAs were identified in patients with DCM,
including 661 unregulated lncRNAs and 327 downregulated lncRNAs
(Fig. 1C). In addition, a scatter
plot was implemented to identify differentially expressed lncRNAs
between DCM and control subjects (Fig.
1D).
Distribution profiles of
differentially expressed lncRNAs
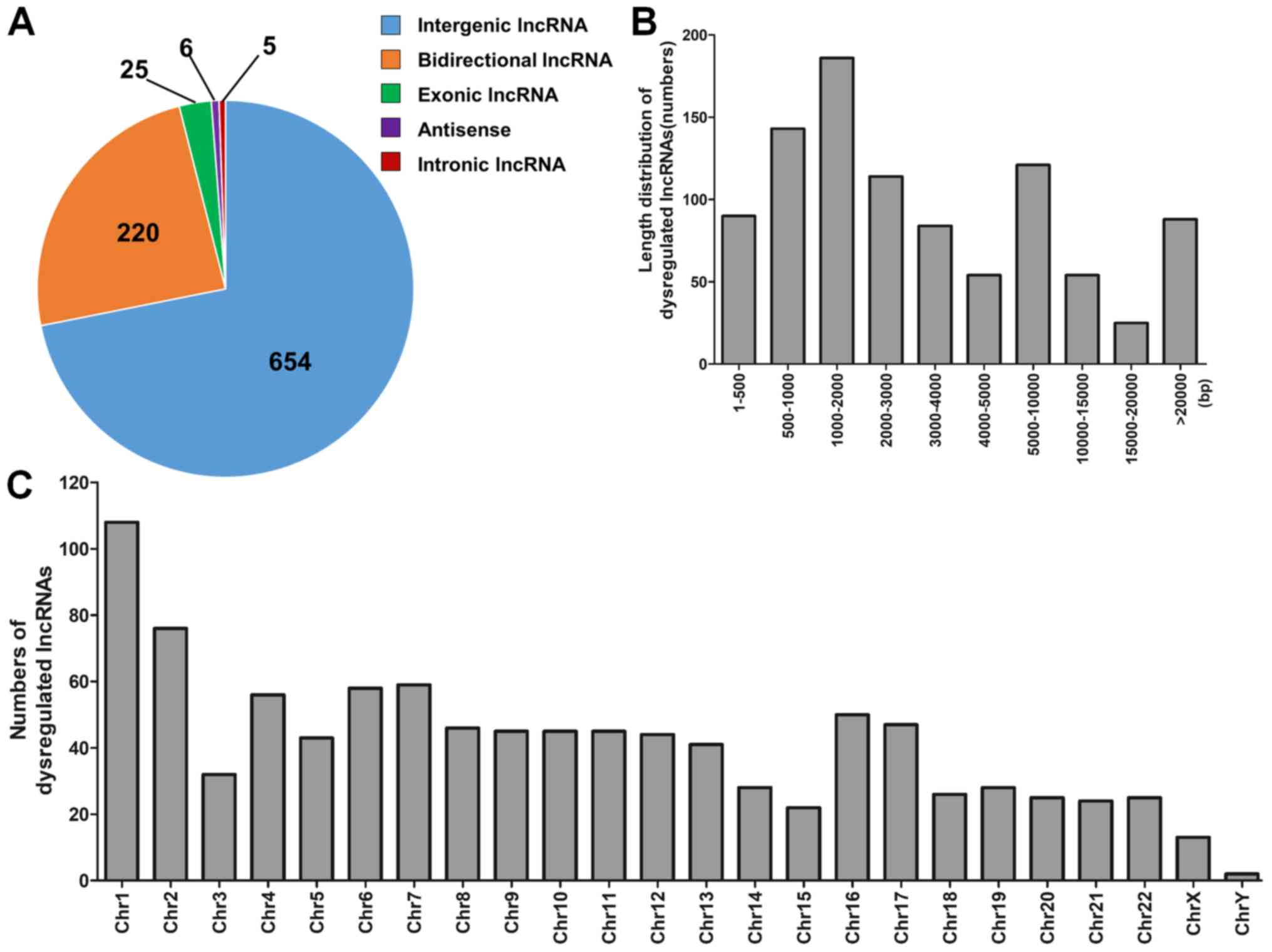
The dysregulated lncRNAs were subsequently
classified in five categories based on their genomic locations,
which included intergenic (n=654), bidirectional (n=220), exonic
(n=25), antisense (n=6) and intronic (n=5) lncRNAs (Fig. 2A). In terms of length distribution,
dysregulated lncRNAs ranged mainly between 500 and 10,000 base
pairs (bp; Fig. 2B). These
differentially expressed lncRNAs were scattered throughout the
human chromosomes (Fig. 2C).
RT-PCR validation of differentially
expressed lncRNAs
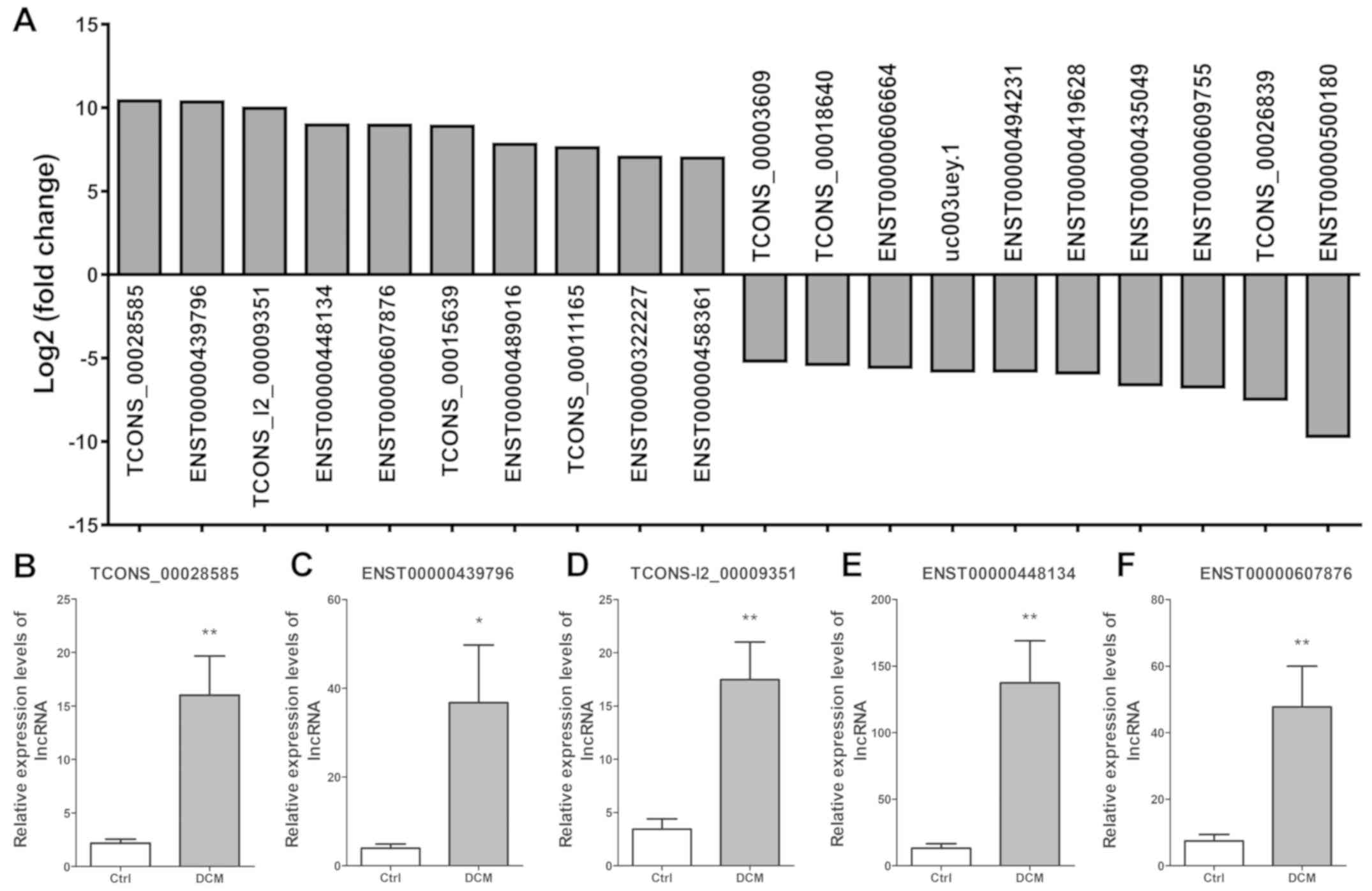
The top 10 upregulated lncRNAs presenting the
highest fold-change were TCONS_00028585, ENST00000439796,
TCONS_l2_00009351, ENST00000448134, ENST00000607876,
TCONS_00015639, ENST00000489016, TCONS_00011165, ENST00000322227
and ENST00000458361. The top 10 downregulated lncRNAs presenting
the highest fold-change were TCONS_00003609, TCONS_00018640,
ENST00000606664, uc003uey.1, ENST00000494231, ENST00000419628,
ENST00000435049, ENST00000609755, TCONS_00026839 and ENST00
000500180 (Fig. 3A). Next, five
dysregulated lncRNAs were selected to confirm the lncRNA
expression, including TCONS_00028585 (Fig. 3B), ENST00000439796 (Fig. 3C), TCONS_l2_00009351 (Fig. 3D), ENST00000448134 (Fig. 3E) and ENST00000607876 (Fig. 3F). The results verified that the
expression levels of these lncRNAs were significantly upregulated
in DCM patients as compared with those in control subjects.
GO and KEGG pathway analyses of
dysregulated lncRNAs
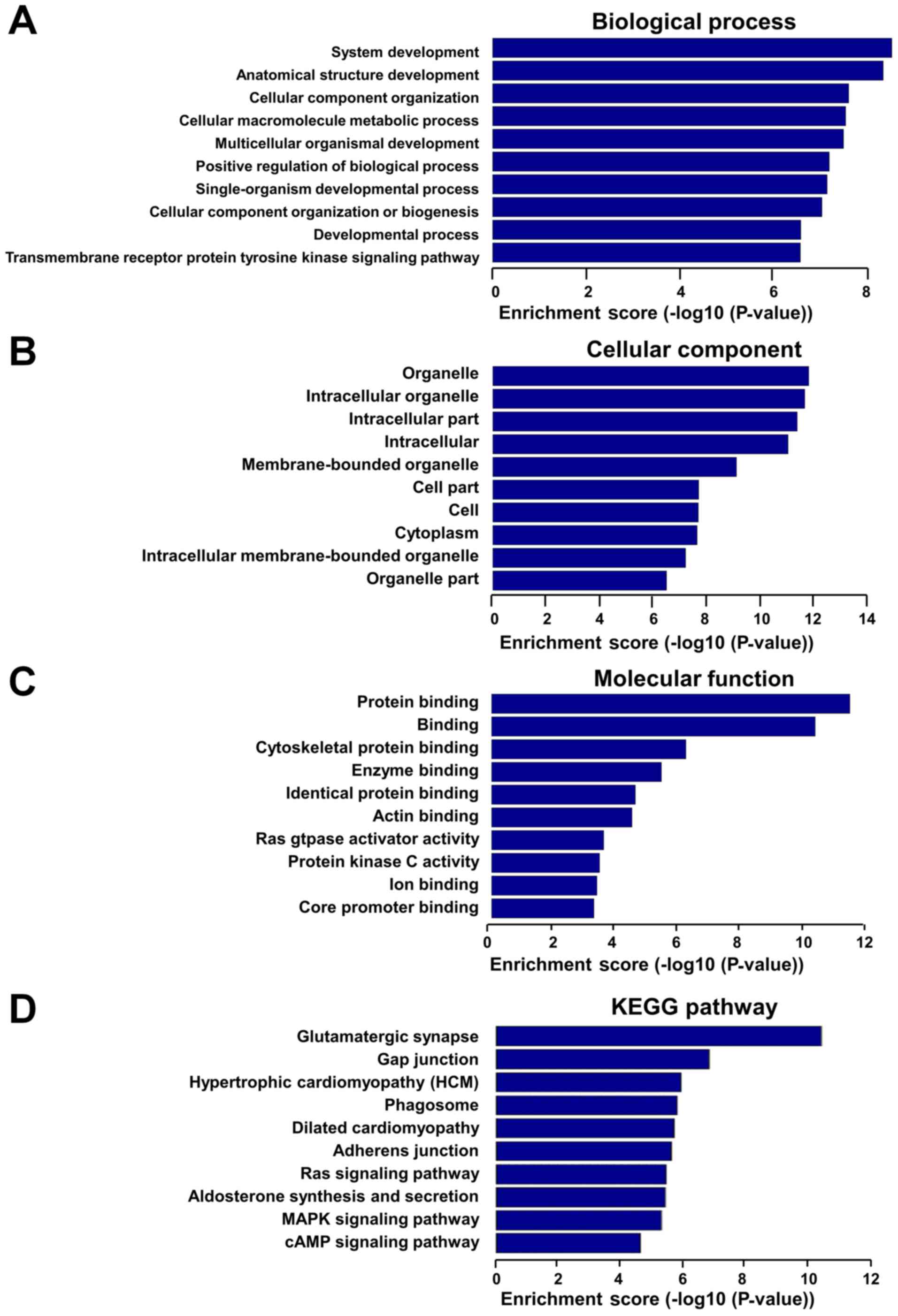
In order to assess the potential functions of these
dysregulated lncRNAs, GO and KEGG pathway analyses were performed
with the target coding genes of downregulated lncRNAs and
upregulated lncRNAs, separately. GO analysis was comprised of three
networks, including the ‘biological process’, ‘cellular component’
and ‘molecular function’. Through GO analysis of neighbor coding
genes, the downregulated lncRNAs were found to be mostly enriched
as follows: System development, anatomical structure development
and cellular component organization in the ‘biological process’
analysis (Fig. 4A); organelle,
intracellular organelle and intracellular part in the ‘cellular
component’ analysis (Fig. 4B); and
protein binding, cytoskeletal protein binding and enzyme binding in
the ‘molecular function’ analysis (Fig. 4C). KEGG pathway analysis revealed
that downregulated lncRNAs were mainly associated with gap
junction, hypertrophic cardiomyopathy, phagosomes and DCM pathways
(Fig. 4D). In addition,
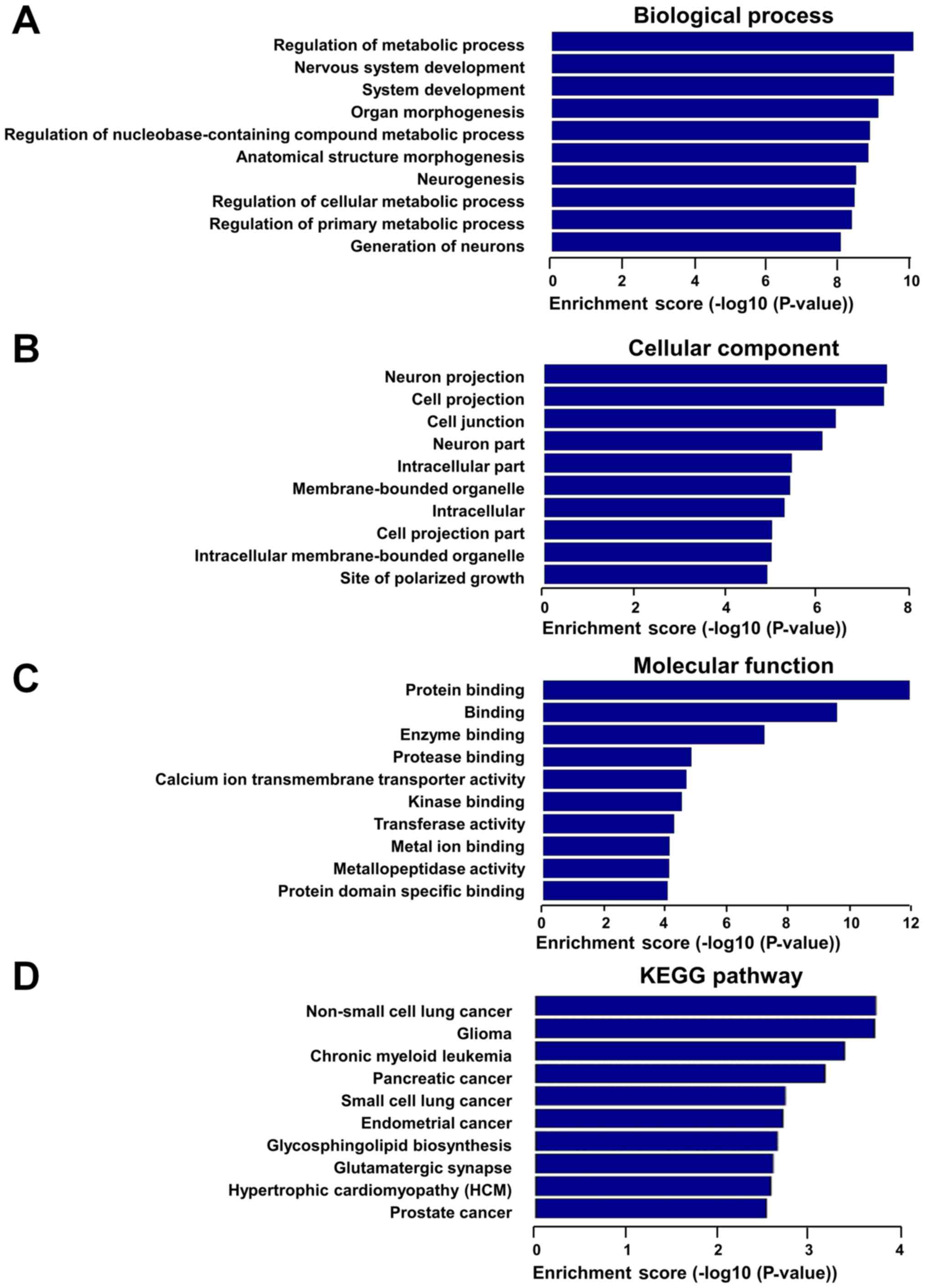
upregulated lncRNAs were mainly associated with the regulation of
metabolic process, organ morphogenesis and regulation of
nucleobase-containing compound metabolic process in the biological
process analysis (Fig. 5A), with
the cell projection, cell junction and intracellular part in the
‘cellular component’ analysis (Fig.
5B), and with protein binding, enzyme binding and calcium ion
transmembrane transporter activity in the ‘molecular function’
analysis (Fig. 5C). The
upregulated lncRNAs exhibited a strong association with
hypertrophic cardiomyopathy and various cancer pathways, according
to KEGG pathway analysis (Fig.
5D).
Overview of mRNAs between patients
with DCM and controls determined by RNA sequencing
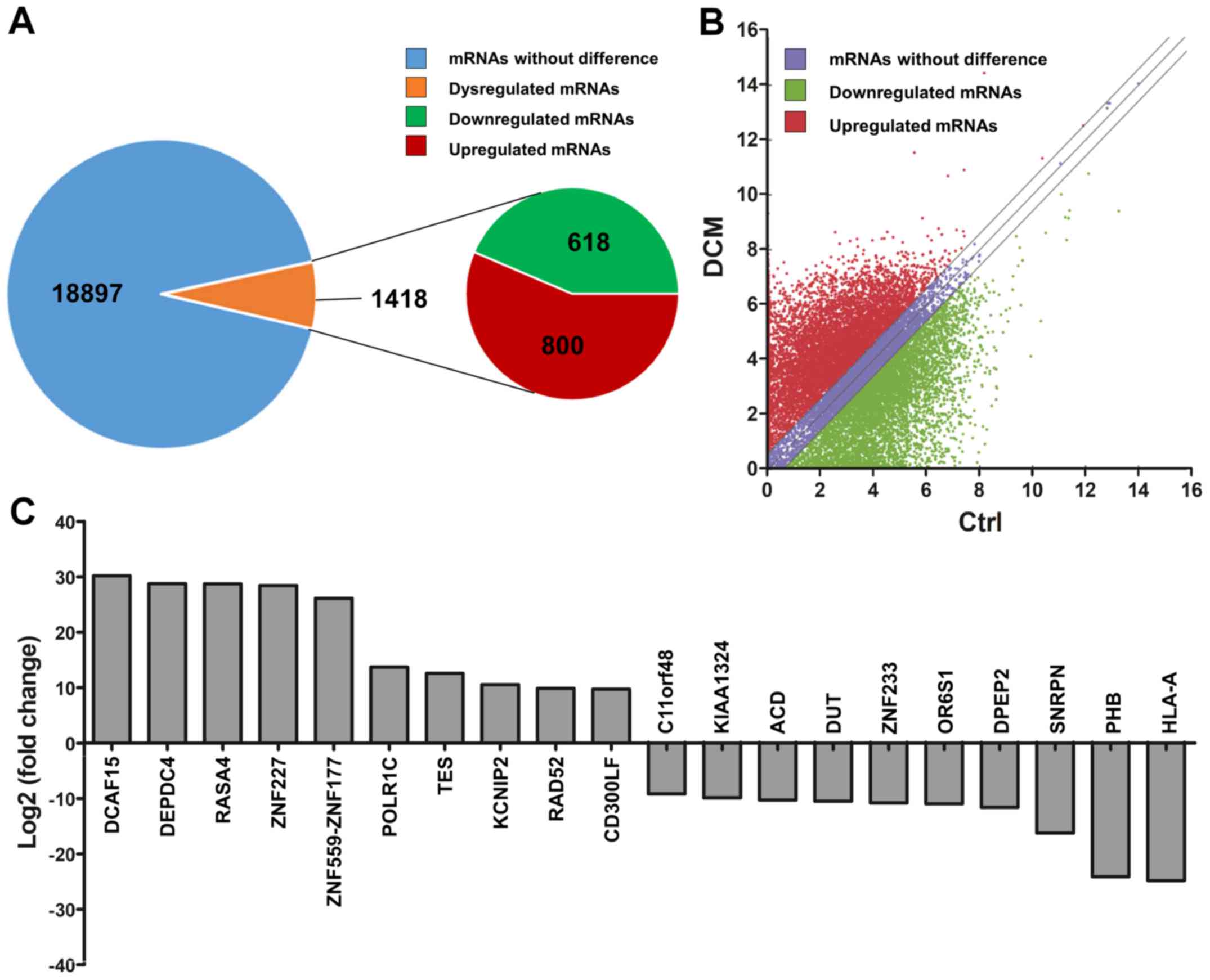
By RNA sequencing, a total of 1,418 differentially
expressed mRNAs (800 upregulated mRNAs and 618 downregulated mRNAs)
were identified among the 20,315 detected mRNAs between the DCM and
control groups (Fig. 6A),
according to the criteria of a fold-change of ≥2 or ≤-2, and
P<0.05. A scatter plot was implemented to identify the
differentially expressed mRNAs (Fig.
6B). The top 10 upregulated mRNAs were DCAF15, DEPDC4, RASA4,
ZNF227, ZNF559-ZNF177, POLR1C, TES, KCNIP2, RAD52 and CD300LF. The
top 10 downregulated mRNAs were HLA-A, PHB, SNRPN, DPEP2, OR6S1,
ZNF233, DUT, ACD, KIAA1324 and C11orf48 (Fig. 6C).
lncRNA-mRNA co-expression network
analysis
A dynamic lncRNA-mRNA co-expression network was
constructed based on the PCC to identify the correlation between
differentially expressed lncRNAs and mRNAs. Initially, the DCM and
control lncRNA-mRNA co-expression networks were constructed
individually. A total of 1,331 lncRNA-mRNA pairs were detected in
the DCM co-expression network (‘obtained’ network), while 1,348
lncRNA-mRNA pairs were identified in the control co-expression
network (‘lost’ network). Next, the two networks were combined into
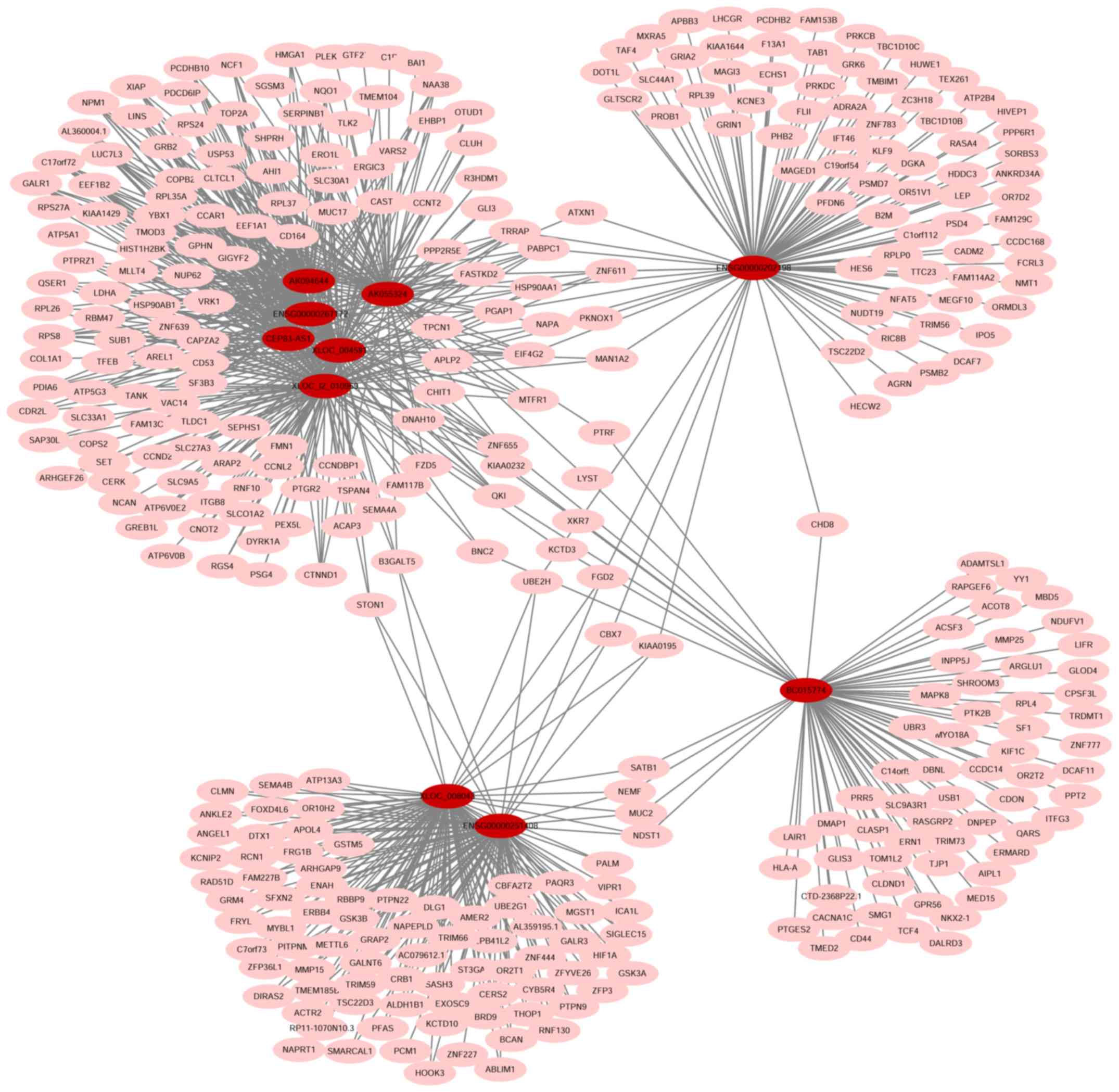
a total co-expression network containing 676 nodes (69 lncRNA nodes
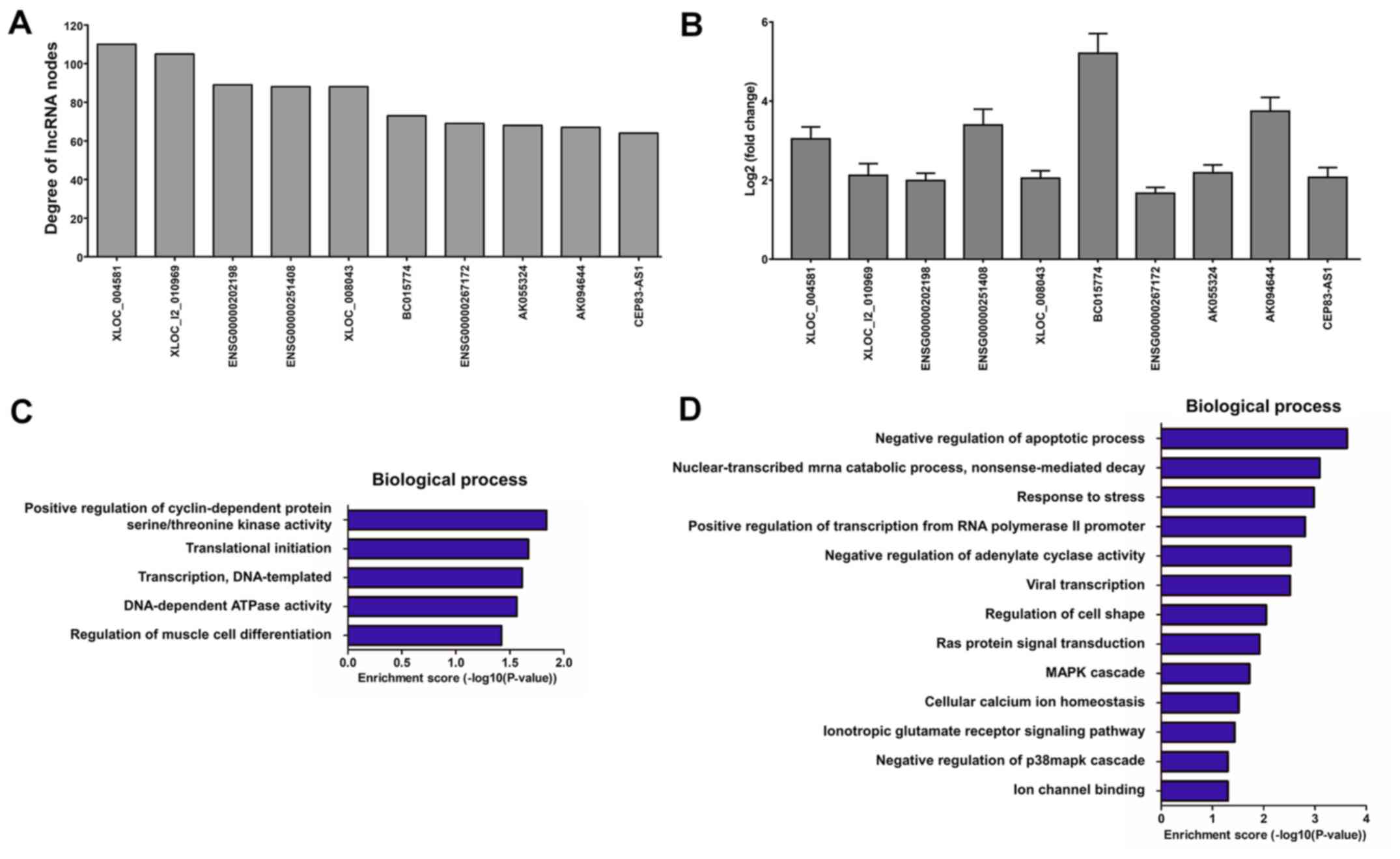
and 607 mRNA nodes) and 2,661 edges (Fig. 7). The top 10 lncRNA nodes (hub
nodes) with the highest degree were XLOC_004581, XLOC_l2_010969,
ENSG00000202198, ENSG00000251408, XLOC_008043, BC015774,
ENSG00000267172, AK055324, AK094644 and CEP83-AS1 (Fig. 8A).
Furthermore, the blood samples from another 20
patients with DCM and 20 healthy controls were collected to
identify the expression levels of the top 10 lncRNA nodes with the
highest degree using RT-PCR assay (Fig. 8B). According to the GO analysis,
the top 10 lncRNA-mRNA co-expression pairs with the highest degree
were mainly associated with positive regulation of cyclin-dependent
protein serine/threonine kinase activity, translational initiation,
DNA-templated transcription and regulation of muscle cell
differentiation in the DCM group (‘obtained’ network; Fig. 8C), and with negative regulation of
apoptotic process, nuclear-transcribed mRNA catabolic process and
response to stress in the control group (‘lost’ network; Fig. 8D). Finally, an lncRNA-mRNA
co-expression network was constructed in order to identify the
target mRNAs of the top 10 lncRNA nodes with the highest degree
(Fig. 9).
Discussion
In the present study, a genome-wide analysis of
dysregulated circulating lncRNAs between patients with DCM and
controls was performed by RNA sequencing. Subsequently, the study
attempted to elucidate the potential mechanism of DCM with
bioinformatics analysis. A total of 998 dysregulated lncRNAs and
1,418 differentially expressed mRNAs were identified between
patients with DCM and controls. Among these dysregulated lncRNAs,
the expression levels of several upregulated lncRNAs were verified
with RT-PCR; the expression levels of downregulated lncRNAs in DCM
should be conducted in the future. These lncRNAs were mainly
intergenic and bidirectional lncRNAs distributed among all human
chromosomes, with lengths mainly ranging between 500 and 10,000 bp.
Bioinformatics analysis revealed that the dysregulated lncRNAs
exhibited a strong association with system development, organ
morphogenesis and metabolic regulation in the ‘biological process’
analysis. It was also observed that several potential pathways,
including the gap junction pathway, phagosome, and dilated and
hypertrophic cardiomyopathy pathways, may serve crucial roles in
the genesis and development of DCM. Through the biological analysis
of the top 10 lncRNA-mRNA pairs with the highest degree in the
dynamic lncRNA-mRNA co-expression network, it was observed that the
top 10 lncRNAs were mainly associated with DNA-templated
transcription, translational initiation and regulation of muscle
cell differentiation in the DCM group. However, these top 10
lncRNAs were associated with the negative regulation of apoptotic
process, nuclear-transcribed mRNA catabolic process and regulation
of response to stress in the control group.
Due to the severe outcome of patients with DCM, it
is imperative to uncover effective biomarkers and therapy targets
for the diagnosis and treatment of this disease. lncRNAs are
implicated in diverse biological processes in different diseases,
such as in cell-cycle control, differentiation, apoptosis,
chromatin remodeling, maintenance of the nuclear structure
integrity, and transcriptional and post-transcriptional processing
(26–29). Recent studies have demonstrated
that lncRNAs serve critical roles in cardiac development, and
participate in cardiac homeostasis and regeneration as epigenetic
regulators of cardiac gene expression (11,13,30).
Circulating lncRNAs have also been reported to be involved in the
pathophysiology of cardiovascular diseases. For instance, the
circulating lncRNA HOTAIR performed a protective role in acute
myocardial infarction via the negative regulation of miR-1
(31). Although a few pathogenic
lncRNAs have been reported in DCM (32–34),
the majority of these studies did not employ RNA sequencing tools
and bioinformatics analysis; therefore, more comprehensive
information remains to be elucidated. In the present study, the
genome-wide profile of lncRNAs was identified by comparing their
expression in the serum of patients with DCM and controls by RNA
sequencing. The present study identified the lncRNA variation in
DCM and provides a critical insight on the potential mechanisms
underlying this disease. In addition, the present results are
important for the identification of novel biomarkers of idiopathic
DCM.
Cardiac sarcomere proteins are vitally important in
maintaining a normal cardiac structure. Mutations of the genes
encoding these proteins, particularly the titin and myosin heavy
chain 7 genes, may result in the occurrence of DCM (35–38).
In the present study, it was observed that the titin gene was
regulated by several lncRNAs, including ENGS00000237298,
ENGS00000267784, ENGS00000442329, ENGS00000603415 and
ENGS00000603521. In addition, the myosin heavy chain 7 gene was
regulated by NR126491. These results suggested the potential
regulatory mechanisms of these sarcomere proteins, although
mutations in sarcomeric genes were not examined in the present
study. Furthermore, more potential genes were identified at the
genome-wide level, which may function as key regulators in the
development of DCM.
The lncRNA-mRNA co-expression network analysis was
performed by combining lncRNA-mRNA pairs identified separately in
the DCM and control groups. Biological processes were analyzed with
the target mRNAs of the top 10 lncRNA nodes with the highest degree
in the DCM and control groups. The function of the majority of
these lncRNAs remain unknown, apart from ENSG00000202198 (also
known as RN7SK), which was identified to be associated with
immunodeficiency syndrome (39).
Notably, these 10 lncRNAs participated in different biological
processes between the DCM and control groups. It is known that
lncRNAs are able to regulate multigene expression to participate in
biological processes (9,10). The findings of the present study
suggested that lncRNAs were also able to change their main target
genes, according to different disease circumstances. Modifying the
cardiac circumstances of patients with DCM may be an effective
therapy to relieve the progression of this disease.
In conclusion, the present study identified the
genome-wide profile of lncRNAs in DCM by RNA sequencing.
Bioinformatics analysis identified the potential functions of these
lncRNAs. The lncRNA-mRNA co-expression network analysis revealed
that the main target genes were different in DCM. Although the
study mainly focused on bioinformatics analysis without
experimental confirmation of the pathophysiology in DCM, the
findings offered a novel insight into the disease pathogenesis and
provided a theoretical basis for future studies of lncRNAs in
DCM.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81573095).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Gene Expression Omnibus database
(accession no. GSE101585; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE101585).
Authors' contributions
GH, WY and JL designed the study and conducted the
experiments. CY and YX contributed to the data acquisition and
collection. YW, JW and MC were responsible for analyzing and
interpreting the data. GH, WY and JL drafted the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures in the present study were approved by
the Ethics Committee of the Liaocheng People's Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Halliday BP, Baksi AJ, Gulati A, Ali A,
Newsome S, Izgi C, Arzanauskaite M, Lota A, Tayal U, Vassiliou VS,
et al: Outcome in dilated cardiomyopathy related to the extent,
location, and pattern of late gadolinium enhancement. JACC
Cardiovasc Imaging. Sep 6–2018.(Epub ahead of print). doi:
10.1016/j.jcmg.2018.07.015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Salerno M and Robinson AA: Risk
stratification in nonischemic dilated cardiomyopathy in the era of
personalized medicine: Can cardiac magnetic resonance with late
gadolinium imaging ‘enhance’ our strategy? JACC Cardiovasc Imaging.
11:1285–1287. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yancy CW, Jessup M, Bozkurt B, Butler J,
Casey DE Jr, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC,
Givertz MM, et al: 2017 ACC/AHA/HFSA Focused Update of the 2013
ACCF/AHA Guideline for the Management of Heart Failure: A Report of
the American College of Cardiology/American Heart Association Task
Force on Clinical Practice Guidelines and the Heart Failure Society
of America. Circulation. 136:e137–e161. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garfinkel AC, Seidman JG and Seidman CE:
Genetic pathogenesis of hypertrophic and dilated cardiomyopathy.
Heart Fail Clin. 14:139–146. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harakalova M and Asselbergs FW: Systems
analysis of dilated cardiomyopathy in the next generation
sequencing era. Wiley Interdiscip Rev Syst Biol Med. 10:e14192018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Merlo M, Cannatà A, Gobbo M, Stolfo D,
Elliott PM and Sinagra G: Evolving concepts in dilated
cardiomyopathy. Eur J Heart Fail. 20:228–239. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thum T and Condorelli G: Long noncoding
RNAs and microRNAs in cardiovascular pathophysiology. Circ Res.
116:751–762. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cabili MN, Trapnell C, Goff L, Koziol M,
Tazon-Vega B, Regev A and Rinn JL: Integrative annotation of human
large intergenic noncoding RNAs reveals global properties and
specific subclasses. Genes Dev. 25:1915–1927. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eades G, Zhang YS, Li QL, Xia JX, Yao Y
and Zhou Q: Long non-coding RNAs in stem cells and cancer. World J
Clin Oncol. 5:134–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Y, Li Y, Wang J and Lei P: Long
noncoding RNA ferritin heavy polypeptide 1 pseudogene 3 controls
glioma cell proliferation and apoptosis via regulation of the
microRNA-224-5p/tumor protein D52 axis. Mol Med Rep. 18:4239–4246.
2018.PubMed/NCBI
|
|
11
|
Lorenzen JM, Martino F and Thum T:
Epigenetic modifications in cardiovascular disease. Basic Res
Cardiol. 107:2452012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ounzain S, Crippa S and Pedrazzini T:
Small and long non-coding RNAs in cardiac homeostasis and
regeneration. Biochim Biophys Acta. 1833:923–933. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calore M, De Windt LJ and Rampazzo A:
Genetics meets epigenetics: Genetic variants that modulate
noncoding RNA in cardiovascular diseases. J Mol Cell Cardiol.
89:27–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xie Z, Xia W and Hou M: Long intergenic
noncoding RNA-p21 mediates cardiac senescence via the Wnt/β-catenin
signaling pathway in doxorubicin-induced cardiotoxicity. Mol Med
Rep. 17:2695–2704. 2018.PubMed/NCBI
|
|
15
|
Yang KC, Yamada KA, Patel AY, Topkara VK,
George I, Cheema FH, Ewald GA, Mann DL and Nerbonne JM: Deep RNA
sequencing reveals dynamic regulation of myocardial noncoding RNAs
in failing human heart and remodeling with mechanical circulatory
support. Circulation. 129:1009–1021. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang ZP, Ding Y, Chen J, Wu G, Kataoka M,
Hu Y, Yang JH, Liu J, Drakos SG, Selzman CH, et al: Long non-coding
RNAs link extracellular matrix gene expression to ischemic
cardiomyopathy. Cardiovasc Res. 112:543–554. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu R, Liao X, Li X, Wei H, Liang Q, Zhang
Z, Yin M, Zeng X, Liang Z and Hu C: Expression profiles of long
noncoding RNAs and mRNAs in post-cardiac arrest rat brains. Mol Med
Rep. 17:6413–6424. 2018.PubMed/NCBI
|
|
18
|
Taichman DB, McGoon MD, Harhay MO,
Archer-Chicko C, Sager JS, Murugappan M, Chakinali MM, Palevsky HI
and Gallop R: Wide variation in clinicians' assessment of New York
Heart Association/World Health Organization functional class in
patients with pulmonary arterial hypertension. Mayo Clin Proc.
84:586–592. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maron BJ, Towbin JA, Thiene G,
Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE and Young
JB; American Heart Association; Council on ClinicalCardiology,
Heart Failure and Transplantation Committee; Quality of Care and
Outcomes Research and Functional Genomics and Translational
BiologyInterdisciplinary Working Groups; Council on Epidemiology
and Prevention, : Contemporary definitions and classification of
the cardiomyopathies: An American Heart Association Scientific
Statement from the Council on Clinical Cardiology, Heart Failure
and Transplantation Committee; Quality of Care and Outcomes
Research and Functional Genomics and Translational Biology
Interdisciplinary Working Groups; and Council on Epidemiology and
Prevention. Circulation. 113:1807–1816. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
WRITING COMMITTEE MEMBERS, Yancy CW,
Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC,
Geraci SA, Horwich T, et al: 2013 ACCF/AHA guideline for the
management of heart failure: A report of the American College of
Cardiology Foundation/American Heart Association Task Force on
practice guidelines. Circulation. 128:e240–e327. 2013.PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang W, Han Y, Yang C, Chen Y, Zhao W, Su
X, Yang K and Jin W: MicroRNA-19b-1 reverses ischaemia-induced
heart failure by inhibiting cardiomyocyte apoptosis and targeting
Bcl2 l11/BIM. Heart Vessels. Jan 3–2019.(Epub ahead of print). doi:
10.1007/s00380-018-01336-3. View Article : Google Scholar
|
|
23
|
Haeussler M, Zweig AS, Tyner C, Speir ML,
Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN,
et al: The UCSC Genome Browser database: 2019 update. Nucleic Acids
Res. 47:D853–D858. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kent WJ: BLAT-the BLAST-like alignment
tool. Genome Res. 12:656–664. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fatica A and Bozzoni I: Long non-coding
RNAs: New players in cell differentiation and development. Nat Rev
Genet. 15:7–21. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang N, Chen J, Zhang H, Wang X, Yao H,
Peng Y and Zhang W: lncRNA OIP5-AS1 loss-induced microRNA-410
accumulation regulates cell proliferation and apoptosis by
targeting KLF10 via activating PTEN/PI3K/AKT pathway in multiple
myeloma. Cell Death Dis. 8:e29752017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cajigas I, Leib DE, Cochrane J, Luo H,
Swyter KR, Chen S, Clark BS, Thompson J, Yates JR III, Kingston RE
and Kohtz JD: Evf2 lncRNA/BRG1/DLX1 interactions reveal
RNA-dependent inhibition of chromatin remodeling. Development.
142:2641–2652. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang R, Shi Y, Chen L, Jiang Y, Mao C, Yan
B, Liu S, Shan B, Tao Y and Wang X: The ratio of FoxA1 to FoxA2 in
lung adenocarcinoma is regulated by lncRNA HOTAIR and chromatin
remodeling factor LSH. Sci Rep. 5:178262015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Devaux Y, Zangrando J, Schroen B, Creemers
EE, Pedrazzini T, Chang CP, Dorn GW II, Thum T and Heymans S;
Cardiolinc network, : Long noncoding RNAs in cardiac development
and ageing. Nat Rev Cardiol. 12:415–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao L, Liu Y, Guo S, Yao R, Wu L, Xiao L,
Wang Z, Liu Y and Zhang Y: Circulating long noncoding RNA HOTAIR is
an essential mediator of acute myocardial infarction. Cell Physiol
Biochem. 44:1497–1508. 2017.PubMed/NCBI
|
|
32
|
Zhang Y, Zhang M, Xu W, Chen J and Zhou X:
The long non-coding RNA H19 promotes cardiomyocyte apoptosis in
dilated cardiomyopathy. Oncotarget. 8:28588–28594. 2017.PubMed/NCBI
|
|
33
|
Wang K, Long B, Zhou LY, Liu F, Zhou QY,
Liu CY, Fan YY and Li PF: CARL lncRNA inhibits anoxia-induced
mitochondrial fission and apoptosis in cardiomyocytes by impairing
miR-539-dependent PHB2 downregulation. Nat Commun. 5:35962014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ounzain S, Micheletti R, Beckmann T,
Schroen B, Alexanian M, Pezzuto I, Crippa S, Nemir M, Sarre A,
Johnson R, et al: Genome-wide profiling of the cardiac
transcriptome after myocardial infarction identifies novel
heart-specific long non-coding RNAs. Eur Heart J. 36:353–368a.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fatkin D, Seidman CE and Seidman JG:
Genetics and disease of ventricular muscle. Cold Spring Harb
Perspect Med. 4:a0210632014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Burke MA, Cook SA, Seidman JG and Seidman
CE: Clinical and mechanistic insights into the genetics of
cardiomyopathy. J Am Coll Cardiol. 68:2871–2886. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Herman DS, Lam L, Taylor MR, Wang L,
Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B,
Sparks E, et al: Truncations of titin causing dilated
cardiomyopathy. N Eng J Med. 366:619–628. 2012. View Article : Google Scholar
|
|
38
|
Muelas N, Hackman P, Luque H,
Garcés-Sánchez M, Azorín I, Suominen T, Sevilla T, Mayordomo F,
Gómez L, Martí P, et al: MYH7 gene tail mutation causing myopathic
profiles beyond Laing distal myopathy. Neurology. 75:732–741. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Eilebrecht S, Benecke BJ and Benecke AG:
Latent HIV-1 TAR regulates 7SK-responsive P-TEFb target genes and
targets cellular immune responses in the absence of Tat. Genomics
Proteomics Bioinformatics. 15:313–323. 2017. View Article : Google Scholar : PubMed/NCBI
|