Glucose plays an important role in metabolism. It is
not only the source of energy, but also the substrate of cell
composition biosynthesis (1). The
metabolic dysregulation of glucose homeostasis is the main
consequence of the development of diabetes and the major cause of
diabetic morbidity and mortality. There are two common types of
diabetes: Type 1 and type 2. Type 1 diabetes mellitus (T1DM) is
characterized by the insufficient secretion of insulin and the
excessive release of glucagon, which promotes hepatic lipolysis and
ketogenesis (2) and counteracts
hepatic anabolism. Type 2 diabetes mellitus (T2DM) is the more
common type of DM and is characterized by undetectable levels of
insulin, increased liver fat content, impaired insulin clearance
and hepatic insulin resistance (3).
DM is associated with various liver abnormalities,
including non-alcoholic fatty liver disease (NAFLD) (3) and excessive hepatic glycogenosis
(4). The liver serves a unique
role in glucose metabolism and is crucial for systemic glucose
homeostasis (5); it contributes to
the management of an enteral glucose load by inhibiting its own
glucose output, which aids the disposal of exogenous glucose by
extrahepatic tissues, such as adipose and skeletal muscle (6). The dysregulation of liver signaling
and metabolism predisposes individuals to NAFLD and/or T2DM, thus
certain liver-derived biomarkers (fetuin-A, alpha-hydroxybutyrate
and C-reactive protein) can be used for the diagnosis and prognosis
of DM and DM-associated complications (7). Therefore, the liver is also an
important target organ that regulates glucose homeostasis and can
be targeted by the administration of specific diabetic drugs
(8).
In the present review, the physiology and molecular
pathways of liver glucose homeostasis were investigated, as well as
the glucose metabolic disorders noted in DM. Hepatic lipogenesis
was also investigated since lipid-induced hepatic insulin
resistance is one of the main pathophysiological processes of
hepatic glucose metabolism disorder in T2DM.
During glucose homeostasis, the liver serves an
important role in carbohydrate synthesis, storage and
redistribution (9). The liver
performs opposite functions during hyperglycemic (glucose uptake
and glycogen synthesis) and hypoglycemic states (glycogenolysis and
gluconeogenesis), thus the physiological regulation of hepatic
glucose production is a complex process (6). Patients with T2DM and T1DM
demonstrate increased hepatic glucose production (HGP), of which
multiple extrahepatic mechanisms contribute to the physiological
regulation of HGP (10). However,
DM is a bi-hormonal disease and is not simply the result of insulin
deficiency (11,12). The pancreatic endocrine cell
hormones, glucagon (13) and
insulin (14), both serve central
roles in the regulation of both glucose and lipid metabolism.
Insulin inhibits the secretion of glucagon and promotes the storage
of lipids and carbohydrates, whereas glucagon facilitates
gluconeogenesis and glucose efflux from the liver (15). Previous studies have reported that
every type of diabetes is associated with hyperglucagonemia, the
suppression of which can reduce hyperglycemia (10,16).
For example, insulin exerts a strong regulatory effect on the
secretion of glucagon and causes the suppression of secretion of
glucagon from pancreatic α-cells; it has been reported that the
absence of this paracrine regulatory mode contributes to the
development of hyperglucagonemia and the increase of HGP in DM
(11,17). The main processes that contribute
to glucose homeostasis, include glycogenolysis, glycogen synthesis,
glycolysis and gluconeogenesis, all of which are regulated by
independent mechanisms (6,9). The dysregulation of these processes
is discussed in detail in the following section.
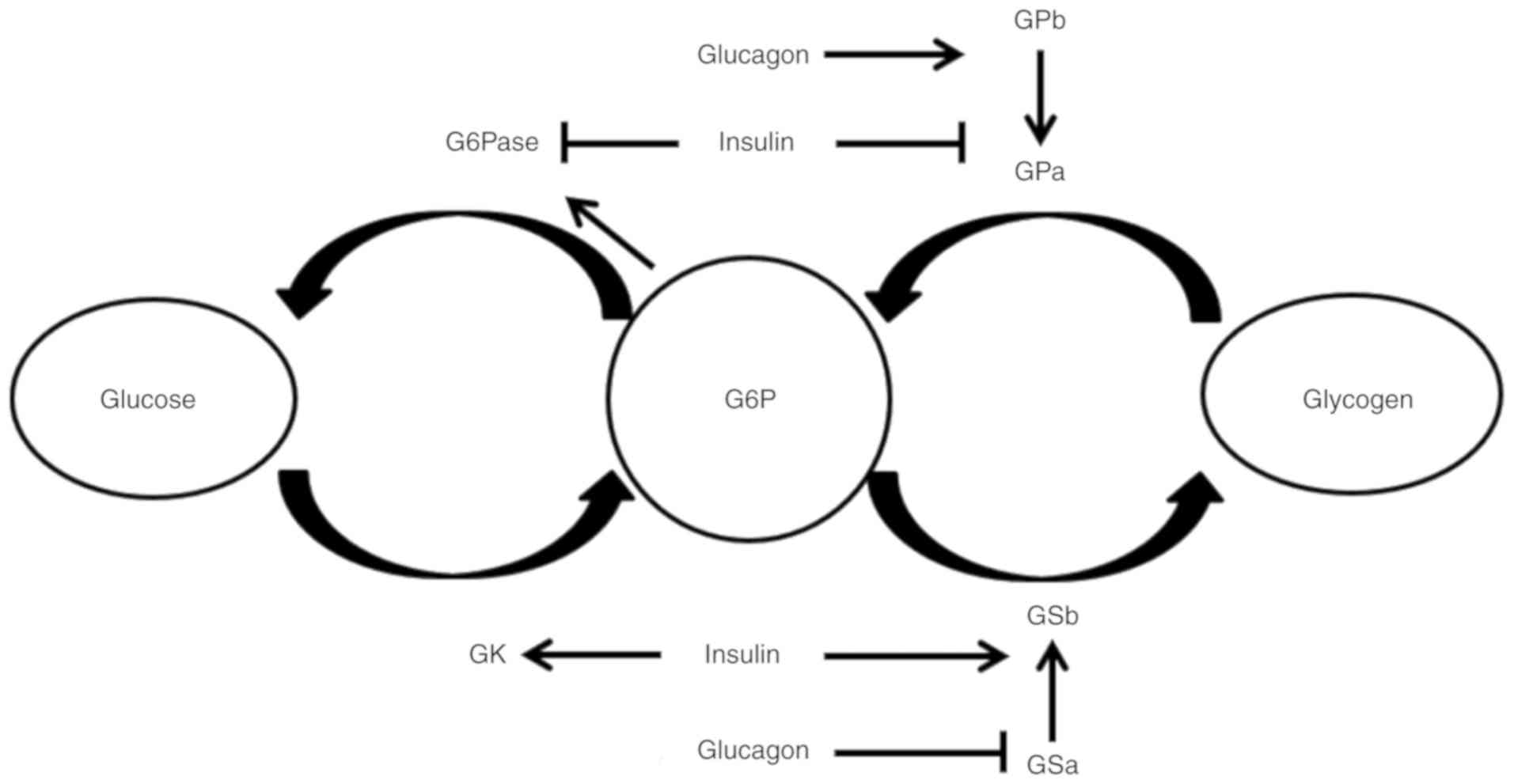
In hepatocytes, glucose is converted into G6P by
glucokinase (GK; the gatekeeper for glucose metabolism in
hepatocytes), and G6P is subsequently trapped in hepatocytes
(8). The affinity of GK (also
known as hexokinase IV) for glucose is low (S0.5; half-saturating
concentration, ~5 mmol/l) and the reaction rate exhibits a
sigmoidal dependence on the intracellular glucose concentration
(18). When the blood glucose
levels are <5 mmol/l (90 mg/dl), GK will not stimulate the
production of large amounts of G6P and subsequent steps are blocked
to ensure that hepatic glycogen synthesis is only highly active
when blood glucose levels are high (19). G6P is subsequently converted to
excess glycogen when insulin levels are high enough to activate
glycogen synthase (GSb) and inactivate glycogen phosphorylase (GPa)
(Fig. 1). In a diabetic state, it
was demonstrated that the glucose uptake in hepatocytes from
diabetic mice was significantly lower compared with the control
animals, which may be a result of the repressed GK synthesis in
response to a decreased insulin: Glucagon ratio (20). Glucagon has been demonstrated to
modify hepatic glucose uptake; for example, a previous animal study
indicated that under hyperglycemic and hyperinsulinemic conditions,
the physiological changes noted in arterial blood glucagon
dramatically changed the net hepatic glucose balance (21), whereas another study revealed that
elevated blood glucagon levels impaired the ability of the liver to
absorb and store glucose (22).
Elevated blood-glucose in the postprandial state was found to
activate GK activity and subsequently increase hepatic G6P
production (23) (Fig. 1). In T2DM (24), impaired liver glucose uptake has
been reported, which subsequently lead to postprandial
hyperglycemia following the discovery that faulty hepatic GK
activation could lead to impaired glucose uptake in T2DM (25). A more recent study demonstrated
that the nicotinamide adenine dinucleotide (NAD)-dependent protein
deacetylase sirtuin-2 (Sirt2) promoted hepatic glucose uptake
through deacetylating the GK regulatory protein. Similary, in high
fat diet-fed obese diabetic mice, the overexpression of hepatic
Sirt2 increased the glucose uptake in the liver, attenuating
impaired glucose tolerance (26).
Overall, the imbalance between hepatic glucose
release and glucose uptake is an important factor in the
development of DM; therefore, the drugs used to treat of DM should
be designed to increase glucose uptake by activation of GK.
Unfortunately, the majority of the drugs that target GK activation
for the treatment of T2DM have not been clinically successful due
to side effects, such as hypoglycemia, steatohepatitis and loss of
efficacy over time (8). However,
TTP399, a hepato-elective GK activator, was assessed in a recently
reported double-blind, 6-month study and it was found that TTP399
did not cause hypoglycemia and exhibited limited or no detrimental
effect on plasma lipids or liver enzymes (34). Moreover, it is a liver specific GK
activator without side effects of hypertension, highlighting the
importance of liver selectivity when targeting GK activity
(34).
Net glycogen deposition in the liver depends on the
coordinated inhibition of glycogenolytic molecules and the
stimulation of glycogen synthesis molecules, of which
glycogenolysis and glycogen synthesis are regulated by complex
mechanisms (35,36). Generally, a useful simplification
is that glucose is the main inhibitor of hepatic glycogenolysis and
insulin is the main activator of hepatic glycogen synthesis. This
has been demonstrated in healthy individuals, using 13C nuclear
magnetic resonance spectroscopy (13C-NMRS) measurements of GS and
glycogen phosphorylase flux in the liver (37).
The synthesis of glycogen occurs through a process
called glycogenesis, which requires ATP and uridine-5′-triphosphate
(23). Glycogen can be synthesized
directly from glucose (from glucose to G6P to UDP-glucose to
glycogen) or indirectly (from glucose to G6P to pyruvate to G6P to
UDP-glucose to glycogen) in the liver (23). These pathways have similar effects
on hepatic glycogen synthesis (38). Three regulated enzymes have been
identified that exert a high degree of control over glycogen
metabolism in the liver: GK (39),
GS and GP (40). The changes in GK
activity are achieved by regulating GK protein expression or the
dissociation of GK from the GK regulatory protein (41). The activities of GS and GP are
determined by phosphorylation and dephosphorylation, which serve
opposite actions on the activity levels of GS and GP enzymes,
leading to a large change in glycogen synthesis. After
phosphorylation, GP has activity to decompose glycogen, while after
dephosphorylation, GS has activity to promote glycogen synthesis
(Fig. 1) (23).
Under T1DM conditions, blood glucose levels are high
and exogenous administration of insulin is elevated, which leads to
increased hepatic glycogen production and excessive glycogen
storage in the liver (42).
Hepatic glycogenosis is a relatively benign disease and does not
progress easily to fibrosis (43);
glycogenosis is the hepatic response to the excess circulating
insulin and glucose load in patients with T1DM, whereas
non-alcoholic steatohepatitis is the more common diagnosis when
elevated insulin and glucose levels occur in adults with T2DM
(43). The possible pathogenesis
of hepatic glycogenosis is considered to occur following the
accumulation of hepatic glycogen in patients with unstable DM as a
result of the increased flux of glucose into the hepatocytes
(44).
Moreover, 13C-NMRS and variable infusion dual-tracer
methodologies were used to study patients with T2DM and
non-diabetic volunteer control subjects, which demonstrated that
postprandial glycogen synthesis was decreased in mildly overweight
patients with T2DM (47). This was
accompanied by the impaired inhibition of hepatic glucose
production. Patients with T2DM also exhibited a 35% reduction of
hepatic glycogen content (47).
Del Prato et al (48) also
reported that glycogen synthesis was reduced in patients with T2DM.
These results suggested that the reduced glycogen storage in the
liver may lead to postprandial hyperglycemia in patients with T2DM.
Furthermore, in mice, a previous study highlighted that T2DM
hepatic glycogen was present as large aggregates, which may have
aided the inhibition of the interconversion between glucose and
glycogen in T2DM. In the same study, it was also found that the
size of branched glycogen particles was correlated with the glucose
release rate (49).
In patients with DM, hepatic glycogen synthesis is
impaired; in T1DM, insufficient insulin is responsible for the
reduced glycogen synthesis, whereas T2DM is accompanied by reduced
hepatic glycogen synthesis due to the diminished insulin signaling
induced by lipid accumulation in various organs (50). GP inhibitors could thereby increase
glycogen synthesis and have been previously used to treat T2DM
(8,51).
Glycogenolysis is not a simple reversal of
glycogenesis but a separate pathway, of which two pathways of
glycogen breakdown have been identified. The first glycogen
breakdown pathway is the canonical glycogenolysis route, including
GP and glycogen debranching enzymes (52) (Fig.
1). The other pathway is associated with autophagy (52). Autophagy-dependent glycogen
decomposition produces non-phosphorylated glucose through lysosomal
1,4-α-glucosidase (52); however,
the mechanism by which hepatocytes sense the decline in blood
glucose levels and activate selective glycophagy remains poorly
understood (53). Hepatic-specific
autophagy serves a role in the regulation of blood glucose levels,
while insulin has a dominant role over glucagon in the control of
liver autophagy (54).
During insulin resistance, glucose inactivates the
stimulated form of GP (GPa) and thereby inhibits glycogenolysis
(23). During the period of energy
restriction in patients with T2DM, the decrease in fasting blood
glucose levels is largely due to decreased glycogenolysis, while
the change in gluconeogenesis is not significant (55). A previous study aimed to compare
the size distribution, degradation kinetics and branching structure
of normal and diabetic liver glycogen; it was observed that T2DM
hepatic glycogen existed as large, loosely bound aggregates that
were not present in the normal liver, which may have resulted in
the inflexibility of glucose and glycogen interconversion in T2DM
(49).
Impaired glycogenolysis is also noted in T1DM;
hepatic glycogen breakdown was significantly reduced in patients
with poorly controlled T1DM, as measured by 13C-MNRS (46). In another study, the contribution
of net hepatic glycogenolysis during insulin-induced hypoglycemia
was quantitatively analyzed using 13C-MNRS in 10 non-diabetic and 7
T1DM subjects (HbA1c 6.5±0.2%) and it was concluded that in
intensively treated T1DM subjects, hypoglycemia failed to stimulate
hepatic glycogen decomposition or activate endogenous glucose
production (56).
Gluconeogenesis, is essentially a reverse process of
the glycolytic pathway. In humans, gluconeogenesis occurs
predominantly in the liver (57)
and is regulated by a slower mechanism through changes in gene
expression (23) in response to
hormones, notably insulin and glucagon (36).
Increased glucose production is a consistent feature
of T2DM, which can be attributed to increased gluconeogenesis
rather than glycogenolysis rates (58,59).
The possible influencing factors include: i) Hepatic resistance to
the action of insulin, leading to the inappropriate inhibition of
hepatic glucose output (60); ii)
high levels of glucagon, leading to the overactivation of signaling
pathways, which are usually activated during fasting when glucose
supply is required (60); and iii)
the indirect regulation of gluconeogenesis by excess circulating
free fatty acids (FFAs) through the insulin receptor independent
pathway (61,62). A previous study using normal blood
glucose and hyperglycemic clamps provided evidence for deficiencies
in the rapid inhibition of hepatic glucose production in T2DM
(63). Non-invasive 13C-NMRS
studies have demonstrated that hepatic glycogen decomposition was
low and gluconeogenesis was increased in patients with T2DM
(58,64). In one study, specific small
molecules (SR-18292) were used to increase the acetylation of the
peroxisome proliferator-activated receptor gamma coactivator
1-alpha (PGC-1α). The study demonstrated that these molecules
inhibited the activity of PGC-1α-dependent gluconeogenesis, thereby
increasing insulin sensitivity and lowering blood glucose levels in
T2D mice (65).
T1DM rats were discovered to have ~90% lower insulin
and leptin levels, as well as 90% higher glucagon levels in the
plasma compared with control non-diabetic rats (66). Glucagon stimulates hepatic glucose
production by activating enzymes involved in gluconeogenesis and
glycogenolysis (60). In animal
experiments, the inhibition of glucagon action prevented the
metabolic disorders noted in T1DM mice (67). Another study indicated that
moderately controlled patients with T1DM exhibited increased
glucose production during both rest and exercise, which may account
for the increased gluconeogenesis rates (68).
Increased glucose production in the liver that
occurs due to enhanced gluconeogenesis is the major contributor to
the high blood glucose levels observed in DM (69). Thus, inhibiting gluconeogenesis may
provide the most direct route for decreasing glucose production in
the liver; for example, Metformin is the most commonly used therapy
for T2DM, since it decreases gluconeogenesis in the liver without
increasing insulin secretion (70). However, the underlying mechanism by
which metformin inhibits hepatic gluconeogenesis remains unknown.
The addition of metformin to insulin therapy in T1DM is still under
debate and previous findings of metformin in patients with T1DM
have resulted in conflicting results (71).
The pyruvate produced by glycolysis is an important
intermediate product in the conversion of carbohydrates to fatty
acids and cholesterol (73). The
hepatic abnormal regulation of pyruvate metabolism has been
reported in DM and NAFLD (74). In
insulin-resistant conditions, pyruvate is used for gluconeogenesis
and fatty acid synthesis rather than ATP generation, which is
promoted by the tricarboxylic acid (TCA) cycle, resulting in
hyperglycemia and hepatic steatosis (75,76).
The TCA cycle is affected by DM and the glycolytic activity is
weakened during the development of DM (77). The activation of the pyruvate
dehydrogenase complex promotes the conversion of pyruvate to
acetyl-CoA by oxidative decarboxylation (78). Go et al (76) reported that by inhibiting pyruvate
dehydrogenase kinase 2, the activity of the hepatic pyruvate
dehydrogenase complex was increased, ameliorating hepatic steatosis
and improving insulin sensitivity through regulating the TCA
cycle.
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is
an important enzyme used to catalyze the sixth step of glycolysis,
which is the step between the energy-requiring phase and the
energy-releasing phase (79).
GAPDH is a major target protein involved in oxidative stress
(80) and it is inactivated due to
the accumulation of reactive oxygen species (81). It is well known that oxidative
stress serves an important role in the incidence of diabetic
complications and hyperglycemia (82). Moreover, it leads to the excessive
production of mitochondrial superoxide, resulting in a 66% decrease
in GAPDH activity (83). The
imbalance of the nicotinamide adenine dinucleotide
(NADH)/NAD+ redox status in DM was revealed to inhibit
both GAPDH and dihydrolipoamide dehydrogenase in the pyruvate
dehydrogenase complex (84) and
the impairment of glycolysis led to the accumulation of
glyceraldehyde 3-phosphate (G3P), as reported in microvascular and
cardiovascular studies (85–87).
Therefore, all intermediate products prior to G3P (including G3P)
must be converted through the branch pathways of the glycolytic
pathway (82,87,88).
Moreover, in a previous study GAPDH inactivation resulted in a
shift in metabolic flux from glycolysis to the pentose phosphate
pathway (PPP) (89); however, to
the best of our knowledge, no direct evidence of decreased hepatic
GAPDH activity in DM has been previously reported.
In brief, hepatic glycolytic activity is weakened in
both T1DM and T2DM, which can enhance hepatic glucose utilization
for targeting hepatic glucose output. The alteration of
mitochondrial uncoupling can also enhance glucose utilization in
the liver by increasing fatty acid and glucose oxidation (90).
In the majority of organs, 80–90% of glucose
oxidation occurs via glycolysis and the remaining 10–20% occurs via
the PPP (91); the percentage of
glucose metabolized by PPP varies from 5 to 30% in different
tissues (92). In lipid- and
steroid-synthesizing tissues, such as the liver, lactating mammary
glands, white adipose tissue, adrenal glands and gonads, in
addition to the erythrocytes, the PPP produces the highest
proportion of glucose flux (92).
PPP is usually divided into an oxidative and a nonoxidative pathway
and the activity of the former is generally higher than that of the
latter (93). G6P dehydrogenase
(G6PD) is a rate limiting enzyme of the PPP and produces the
majority of nicotinamide adenine dinucleotide phosphate (NADPH) in
the cell, which is the most abundant reducing coenzyme in the cells
(94). NADPH is an important
cofactor involved in several enzymatic reactions in the cell, such
as nitric oxide production, fatty acid synthesis/oxidation and the
production of glutathione by glutathione reductase (95). In addition, NADPH is required to
remove excess hydrogen peroxide from the glutathione system
(94).
To the best of our knowledge, the fate of the
glucose molecules that are metabolized by the PPP in the liver has
not been well studied. In DM, previous findings have demonstrated
that the activity of G6PD is inhibited and the content of NADPH is
reduced in the liver of T1DM rats (induced by STZ- and
alloxan-treatment) (96). Similar
findings have also been reported in the liver of patients with
chronic DM (97). In an animal
study of mild, moderate and severe hyperglycemia induced by STZ and
nicotinamide treatment, it was found that the levels of hepatocyte
glucose-6-phosphate dehydrogenase (G6PD) activity in mild
hyperglycemia remained similar to the normal values, whereas in
moderate and severely hyperglycemia, they were significantly
reduced (94). The
G6PD:NADPH/nicotine adenine dinucleotide phosphate
(NADP+) ratio and the glutathione levels exhibited a
negative correlation with the blood glucose concentration and a
positive direct correlation with insulin levels (94). In addition, in T2DM obese Zucker
rats, hepatic G6PD protein levels and activity were significantly
higher compared with those noted in lean rats (98,99).
Increased G6PD levels have also been noted in high-fat diet
STZ-treated-T2D rats (100).
Hepatic metabolic changes in patients with T2DM are
characterized via increased liver fat content, impaired insulin
clearance and hepatic insulin resistance (3,116).
Insulin promotes hepatic lipogenesis and in insulin resistance
states, this action is maintained, whereas its ability to reduce
hepatic gluconeogenesis is reported to be impaired (103,117). The production of glucose from
non-carbohydrate sources was increased in patients with NAFLD, a
phenomenon known as selective insulin resistance (103). This is one explanation for the
occurrence of NAFLD in T2DM, whereas another hypothesis is that
NAFLD may develop independently of insulin in the liver. Vatner
et al (118) reported that
in T2DM, the main source of hepatic lipid synthesis was the
esterification of preformed fatty acids, which was mainly dependent
on the transport of substrates and was largely independent of
hepatic insulin action. These findings demonstrated that plasma was
the main source of TG synthesis in T2DM albeit the role of insulin
in fatty acid esterification (if any) was not clear (119).
Increased liver fat has been discovered to be
associated with decreased insulin clearance and sensitivity in
patients with T2DM (116,120), whereas in T1DM, which is
characterized by decreased insulin secretion, the lipid synthesis
in patients with T1DM was reduced (11,114,121). T1DM is characterized by
insufficient insulin secretion and excessive glucagon production
(2) and the mechanism of NAFLD in
T1DM may be complicated; Regnell and Lernmark (122) proposed three hypotheses to
explain the occurrence of hepatic steatosis in patients with T1DM.
In the present review, two possible reasons are highlighted:
Initially, the establishment of insulin therapy was found to
promote lipogenesis (113) and
weight gain in patients with T1DM (123) and secondly, the increased
presence of hepatic lipids may be associated with hyperglycemia
that directly influence insulin and glucagon secretion (122). Abnormal autophagy may also have
an important role in aggravating lipid metabolic disorders
contributing to steatohepatitis in diabetes; Singh et al
(124) demonstrated that the
inhibition of autophagy in cultured hepatocytes and in mouse liver
tissues increased TG storage in lipid droplets.
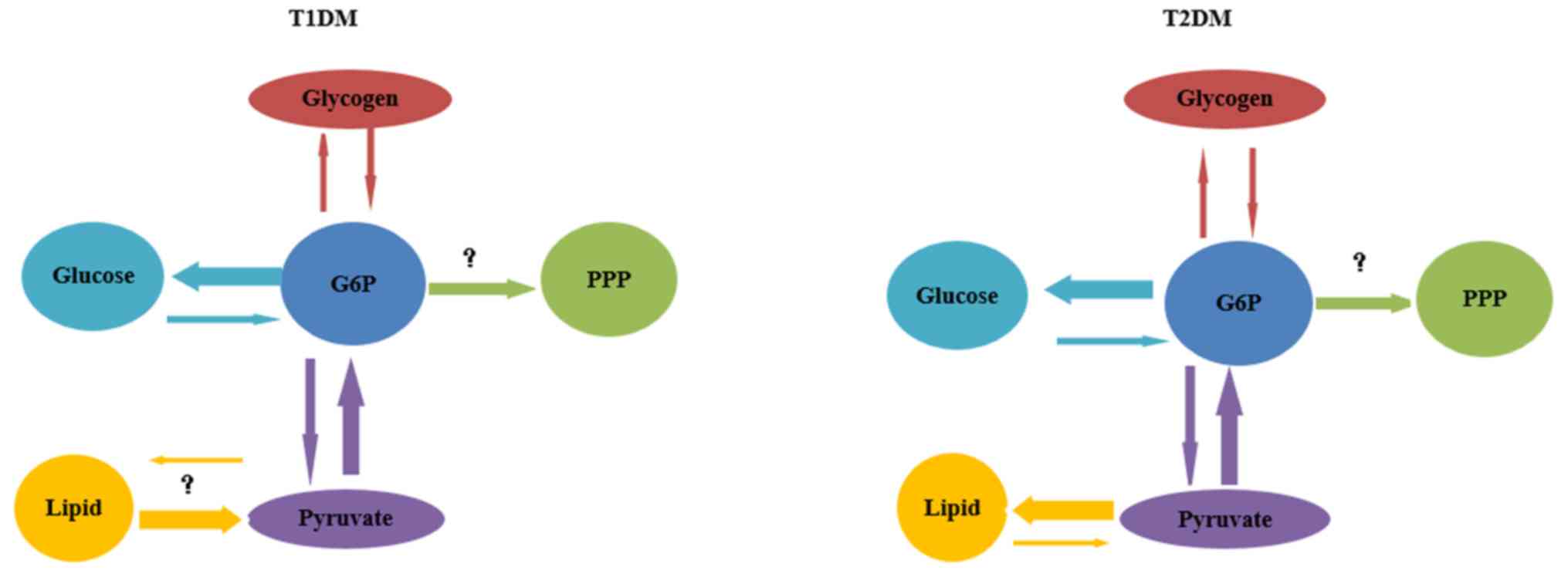
In brief, hyperglycemia in both T1DM and T2DM may be
explained by increased G/G6P cycling or by increased flux through
the gluconeogenesis pathway. Reduced flux of glycogenesis and
glycogenolysis may also explain the inefficiency of acute
regulation of hypoglycemia in fasting and of hyperglycemia
following a meal (Fig. 2).
In the current review, the findings suggested that
the dysregulation of hepatic glucose and lipid metabolism may be
the primary factor underlying the pathogenesis of T2DM and T1DM.
These two chronic diseases affect the body's ability to regulate
glucose and result in different hepatic pathological conditions
Overall, it is evident that the liver is crucial for systemic
glucose homeostasis and that the disorders of glucose metabolism
may contribute to the initiation, progression and exacerbation of
DM; however, there are still several unanswered questions and gaps
in our knowledge that must be addressed in the future.
Not applicable.
The present work was supported in part by grants
from the Basic Scientific Research Projects of Wenzhou Municipal
Science and Technology Bureau (CN) (grant no. Y20180253 to SZJ),
NIEHS (T32-ES011564 to JY) and China-U.S. University of Louisville
Paediatric Research Exchange Training Program. Personnel expenses
and partial research-related expenses for Dr Saizhi Jiang and Dr
Kai Wang were provided by the First Affiliated Hospital of the
Wenzhou Medical University through a collaborative research
agreement between the University of Louisville and the First
Affiliated Hospital of the Wenzhou Medical University, Wenzhou,
China.
Data sharing is not applicable to this article, as
no data sets were generated or analyzed during the current
study.
SJ, LC, JLY, KW and YQ conceived and designed the
present study. All authors read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Towle HC: Glucose as a regulator of
eukaryotic gene transcription. Trends Endocrinol Metab. 16:489–494.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Habegger KM, Heppner KM, Geary N, Bartness
TJ, DiMarchi R and Tschop MH: The metabolic actions of glucagon
revisited. Nat Rev Endocrinol. 6:689–697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhatt HB and Smith RJ: Fatty liver disease
in diabetes mellitus. Hepatobiliary Surg Nutr. 4:101–108.
2015.PubMed/NCBI
|
|
4
|
Sumida Y and Yoneda M: Glycogen
hepatopathy: An under-recognized hepatic complication of
uncontrolled type 1 diabetes mellitus. Intern Med. 57:1063–1064.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moore MC, Coate KC, Winnick JJ, An Z and
Cherrington AD: Regulation of hepatic glucose uptake and storage in
vivo. Adv Nutr. 3:286–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Petersen MC, Vatner DF and Shulman GI:
Regulation of hepatic glucose metabolism in health and disease. Nat
Rev Endocrinol. 13:572–587. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dorcely B, Katz K, Jagannathan R, Chiang
SS, Oluwadare B, Goldberg IJ and Bergman M: Novel biomarkers for
prediabetes, diabetes, and associated complications. Diabetes Metab
Syndr Obes. 10:345–361. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rines AK, Sharabi K, Tavares CD and
Puigserver P: Targeting hepatic glucose metabolism in the treatment
of type 2 diabetes. Nat Rev Drug Discov. 15:786–804. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rui L: Energy metabolism in the liver.
Compr Physiol. 4:177–197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Girard J: Glucagon, a key factor in the
pathophysiology of type 2 diabetes. Biochimie. 143:33–36. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mittendorfer B and Klein S: Absence of
leptin triggers type 1 diabetes. Nat Med. 20:705–706. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Unger RH and Orci L: Paracrinology of
islets and the paracrinopathy of diabetes. Proc Natl Acad Sci USA.
107:16009–16012. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wewer Albrechtsen NJ, Pedersen J,
Galsgaard KD, Winther-Sørensen M, Suppli MP, Janah L, Gromada J,
Vilstrup H, Knop FK and Holst JJ: The liver-α cell axis and type 2
diabetes. Endocr Rev. 40:1353–1366. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bergman RN and Iyer MS: Indirect
Regulation of endogenous glucose production by insulin: The single
gateway hypothesis revisited. Diabetes. 66:1742–1747. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pearson MJ, Unger RH and Holland WL:
Clinical trials, triumphs, and tribulations of glucagon receptor
antagonists. Diabetes Care. 39:1075–1077. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Basco D, Zhang Q, Salehi A, Tarasov A,
Dolci W, Herrera P, Spiliotis I, Berney X, Tarussio D, Rorsman P
and Thorens B: α-cell glucokinase suppresses glucose-regulated
glucagon secretion. Nat Commun. 9:5462018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Quesada I, Tuduri E, Ripoll C and Nadal A:
Physiology of the pancreatic alpha-cell and glucagon secretion:
Role in glucose homeostasis and diabetes. J Endocrinol. 199:5–19.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu S, Ammirati MJ, Song X, Knafels JD,
Zhang J, Greasley SE, Pfefferkorn JA and Qiu X: Insights into
mechanism of glucokinase activation: Observation of multiple
distinct protein conformations. J Biol Chem. 287:13598–13610. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Giordano S, Martocchia A, Toussan L,
Stefanelli M, Pastore F, Devito A, Risicato MG, Ruco L and Falaschi
P: Diagnosis of hepatic glycogenosis in poorly controlled type 1
diabetes mellitus. World J Diabetes. 5:882–888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Barzilai N and Rossetti L: Role of
glucokinase and glucose-6-phosphatase in the acute and chronic
regulation of hepatic glucose fluxes by insulin. J Biol Chem.
268:25019–25025. 1993.PubMed/NCBI
|
|
21
|
Holste LC, Connolly CC, Moore MC, Neal DW
and Cherrington AD: Physiological changes in circulating glucagon
alter hepatic glucose disposition during portal glucose delivery.
Am J Physiol. 273:E488–E496. 1997.PubMed/NCBI
|
|
22
|
Ramnanan CJ, Edgerton DS, Kraft G and
Cherrington AD: Physiologic action of glucagon on liver glucose
metabolism. Diabetes Obes Metab. 13 (Suppl 1):S118–S125. 2011.
View Article : Google Scholar
|
|
23
|
Agius L: Glucokinase and molecular aspects
of liver glycogen metabolism. Biochem J. 414:1–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iozzo P, Hallsten K, Oikonen V, Virtanen
KA, Kemppainen J, Solin O, Ferrannini E, Knuuti J and Nuutila P:
Insulin-mediated hepatic glucose uptake is impaired in type 2
diabetes: Evidence for a relationship with glycemic control. J Clin
Endocrinol Metab. 88:2055–2060. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Coate KC, Kraft G, Shiota M, Smith MS,
Farmer B, Neal DW, Williams P, Cherrington AD and Moore MC: Chronic
overeating impairs hepatic glucose uptake and disposition. Am J
Physiol Endocrinol Metab. 308:E860–E867. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Watanabe H, Inaba Y, Kimura K, Matsumoto
M, Kaneko S, Kasuga M and Inoue H: Sirt2 facilitates hepatic
glucose uptake by deacetylating glucokinase regulatory protein. Nat
Commun. 9:302018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van Dijk TH, van der Sluijs FH, Wiegman
CH, Baller JF, Gustafson LA, Burger HJ, Herling AW, Kuipers F,
Meijer AJ and Reijngoud DJ: Acute inhibition of hepatic
glucose-6-phosphatase does not affect gluconeogenesis but directs
gluconeogenic flux toward glycogen in fasted rats. A
pharmacological study with the chlorogenic acid derivative S4048. J
Biol Chem. 276:25727–25735. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Foufelle F and Ferré P: New perspectives
in the regulation of hepatic glycolytic and lipogenic genes by
insulin and glucose: A role for the transcription factor sterol
regulatory element binding protein-1c. Biochem J. 366:377–391.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Clore JN, Stillman J and Sugerman H:
Glucose-6-phosphatase flux in vitro is increased in type 2
diabetes. Diabetes. 49:969–974. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bandsma RH, Grefhorst A, van Dijk TH, van
der Sluijs FH, Hammer A, Reijngoud DJ and Kuipers F: Enhanced
glucose cycling and suppressed de novo synthesis of
glucose-6-phosphate result in a net unchanged hepatic glucose
output in ob/ob mice. Diabetologia. 47:2022–2031. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rooney DP, Neely RD, Beatty O, Bell NP,
Sheridan B, Atkinson AB, Trimble ER and Bell PM: Contribution of
glucose/glucose 6-phosphate cycle activity to insulin resistance in
type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia.
36:106–112. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Henly DC, Phillips JW and Berry MN:
Suppression of glycolysis is associated with an increase in glucose
cycling in hepatocytes from diabetic rats. J Biol Chem.
271:11268–11271. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Torres TP, Catlin RL, Chan R, Fujimoto Y,
Sasaki N, Printz RL, Newgard CB and Shiota M: Restoration of
hepatic glucokinase expression corrects hepatic glucose flux and
normalizes plasma glucose in zucker diabetic fatty rats. Diabetes.
58:78–86. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vella A, Freeman JLR, Dunn I, Keller K,
Buse JB and Valcarce C: Targeting hepatic glucokinase to treat
diabetes with TTP399, a hepatoselective glucokinase activator. Sci
Transl Med. 11:eaau34412019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ferrer JC, Favre C, Gomis RR,
Fernández-Novell JM, García-Rocha M, de la Iglesia N, Cid E and
Guinovart JJ: Control of glycogen deposition. FEBS Lett.
546:127–132. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin HV and Accili D: Hormonal regulation
of hepatic glucose production in health and disease. Cell Metab.
14:9–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Petersen KF, Laurent D, Rothman DL, Cline
GW and Shulman GI: Mechanism by which glucose and insulin inhibit
net hepatic glycogenolysis in humans. J Clin Invest. 101:1203–1209.
1998. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Soares AF, Viega FJ, Carvalho RA and Jones
JG: Quantifying hepatic glycogen synthesis by direct and indirect
pathways in rats under normal ad libitum feeding conditions. Magn
Reson Med. 61:1–5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Agius L, Peak M, Newgard CB, Gomez-Foix AM
and Guinovart JJ: Evidence for a role of glucose-induced
translocation of glucokinase in the control of hepatic glycogen
synthesis. J Biol Chem. 271:30479–30486. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Aiston S, Hampson L, Gómez-Foix AM,
Guinovart JJ and Agius L: Hepatic glycogen synthesis is highly
sensitive to phosphorylase activity: Evidence from metabolic
control analysis. J Biol Chem. 276:23858–23866. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matschinsky FM and Magnuson MA:
Glucokinase and Glycemic Diseases: From Basics to Novel
Therapeutics. Karger; Basel: pp. 1–9. 2004
|
|
42
|
Satyarengga M, Zubatov Y, Frances S,
Narayanswami G and Galindo RJ: Glycogenic hepatopathy: A
complication of uncontrolled diabetes. AACE Clin Case Rep.
3:e255–e259. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chatila R and West AB: Hepatomegaly and
abnormal liver tests due to glycogenosis in adults with diabetes.
Medicine (Baltimore). 75:327–333. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Julián MT, Alonso N, Ojanguren I, Pizarro
E, Ballestar E and Puig-Domingo M: Hepatic glycogenosis: An
underdiagnosed complication of diabetes mellitus? World J Diabetes.
6:321–325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hwang JH, Perseghin G, Rothman DL, Cline
GW, Magnusson I, Petersen KF and Shulman GI: Impaired net hepatic
glycogen synthesis in insulin-dependent diabetic subjects during
mixed meal ingestion. A 13C nuclear magnetic resonance spectroscopy
study. J Clin Invest. 95:783–787. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bischof MG, Krssak M, Krebs M, Bernroider
E, Stingl H, Waldhäusl W and Roden M: Effects of short-term
improvement of insulin treatment and glycemia on hepatic glycogen
metabolism in type 1 diabetes. Diabetes. 50:392–398. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Krssak M, Brehm A, Bernroider E, Anderwald
C, Nowotny P, Dalla Man C, Cobelli C, Cline GW, Shulman GI,
Waldhäusl W and Roden M: Alterations in postprandial hepatic
glycogen metabolism in type 2 diabetes. Diabetes. 53:3048–3056.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Del Prato S, Bonadonna RC, Bonora E, Gulli
G, Solini A, Shank M and DeFronzo RA: Characterization of cellular
defects of insulin action in type 2 (non-insulin-dependent)
diabetes mellitus. J Clin Invest. 91:484–494. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Besford QA, Zeng XY, Ye JM and Gray-Weale
A: Liver glycogen in type 2 diabetic mice is randomly branched as
enlarged aggregates with blunted glucose release. Glycoconj J.
33:41–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Samuel VT and Shulman GI: The pathogenesis
of insulin resistance: Integrating signaling pathways and substrate
flux. J Clin Invest. 126:12–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Henke BR and Sparks SM: Glycogen
phosphorylase inhibitors. Mini Rev Med Chem. 6:845–857. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ha J, Guan KL and Kim J: AMPK and
autophagy in glucose/glycogen metabolism. Mol Aspects Med.
46:46–62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Madrigal-Matute J and Cuervo AM:
Regulation of liver metabolism by autophagy. Gastroenterology.
150:328–339. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ezaki J, Matsumoto N, Takeda-Ezaki M,
Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K,
Yoshida M, et al: Liver autophagy contributes to the maintenance of
blood glucose and amino acid levels. Autophagy. 7:727–736. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Christiansen MP, Linfoot PA, Neese RA and
Hellerstein MK: Effect of dietary energy restriction on glucose
production and substrate utilization in type 2 diabetes. Diabetes.
49:1691–1699. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kishore P, Gabriely I, Cui MH, Di Vito J,
Gajavelli S, Hwang JH and Shamoon H: Role of hepatic glycogen
breakdown in defective counterregulation of hypoglycemia in
intensively treated type 1 diabetes. Diabetes. 55:659–666. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ekberg K, Landau BR, Wajngot A,
Chandramouli V, Efendic S, Brunengraber H and Wahren J:
Contributions by kidney and liver to glucose production in the
postabsorptive state and after 60 h of fasting. Diabetes.
48:292–298. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hundal RS, Krssak M, Dufour S, Laurent D,
Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF,
Landau BR and Shulman GI: Mechanism by which metformin reduces
glucose production in type 2 diabetes. Diabetes. 49:2063–2069.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rizza RA: Pathogenesis of fasting and
postprandial hyperglycemia in type 2 diabetes: Implications for
therapy. Diabetes. 59:2697–2707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sharabi K, Tavares CD, Rines AK and
Puigserver P: Molecular pathophysiology of hepatic glucose
production. Mol Aspects Med. 46:21–33. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kehlenbrink S, Koppaka S, Martin M,
Relwani R, Cui MH, Hwang JH, Li Y, Basu R, Hawkins M and Kishore P:
Elevated NEFA levels impair glucose effectiveness by increasing net
hepatic glycogenolysis. Diabetologia. 55:3021–3028. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu
W, Li C, Chen H, Monks BR, Chen J, Rabinowitz JD and Birnbaum MJ:
Direct Hepatocyte insulin signaling is required for lipogenesis but
is dispensable for the suppression of glucose production. Cell
Metab. 23:1154–1166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hawkins M, Gabriely I, Wozniak R, Reddy K,
Rossetti L and Shamoon H: Glycemic control determines hepatic and
peripheral glucose effectiveness in type 2 diabetic subjects.
Diabetes. 51:2179–2189. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Magnusson I, Rothman DL, Katz LD, Shulman
RG and Shulman GI: Increased rate of gluconeogenesis in type II
diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin
Invest. 90:1323–1327. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sharabi K, Lin H, Tavares CDJ, Dominy JE,
Camporez JP, Perry RJ, Schilling R, Rines AK, Lee J, Hickey M, et
al: Selective chemical inhibition of PGC-1α gluconeogenic activity
ameliorates type 2 diabetes. Cell. 169:148–160.e15. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Perry RJ, Zhang XM, Zhang D, Kumashiro N,
Camporez JP, Cline GW, Rothman DL and Shulman GI: Leptin reverses
diabetes by suppression of the hypothalamic-pituitary-adrenal axis.
Nat Med. 20:759–763. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lee Y, Wang MY, Du XQ, Charron MJ and
Unger RH: Glucagon receptor knockout prevents insulin-deficient
type 1 diabetes in mice. Diabetes. 60:391–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Petersen KF, Price TB and Bergeron R:
Regulation of net hepatic glycogenolysis and gluconeogenesis during
exercise: Impact of type 1 diabetes. J Clin Endocrinol Metab.
89:4656–4664. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hatting M, Tavares CDJ, Sharabi K, Rines
AK and Puigserver P: Insulin regulation of gluconeogenesis. Ann NY
Acad Sci. 1411:21–35. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Madiraju AK, Erion DM, Rahimi Y, Zhang XM,
Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald
MJ, et al: Metformin suppresses gluconeogenesis by inhibiting
mitochondrial glycerophosphate dehydrogenase. Nature. 510:542–546.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sabet S, Condren ME, Boston AF, Doak LC
and Chalmers LJ: Evolving pharmacotherapeutic strategies for type 1
diabetes mellitus. J Pediatr Pharmacol Ther. 23:351–361.
2018.PubMed/NCBI
|
|
72
|
Abdulrazaq NB, Cho MM, Win NN, Zaman R and
Rahman MT: Beneficial effects of ginger (Zingiber
officinale) on carbohydrate metabolism in
streptozotocin-induced diabetic rats. Br J Nutr. 108:1194–1201.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Gray LR, Tompkins SC and Taylor EB:
Regulation of pyruvate metabolism and human disease. Cell Mol Life
Sci. 71:2577–2604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cotter DG, Ercal B, Huang X, Leid JM,
d'Avignon DA, Graham MJ, Dietzen DJ, Brunt EM, Patti GJ and
Crawford PA: Ketogenesis prevents diet-induced fatty liver injury
and hyperglycemia. J Clin Invest. 124:5175–5190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kumashiro N, Beddow SA, Vatner DF,
Majumdar SK, Cantley JL, Guebre-Egziabher F, Fat I, Guigni B,
Jurczak MJ, Birkenfeld AL, et al: Targeting pyruvate carboxylase
reduces gluconeogenesis and adiposity and improves insulin
resistance. Diabetes. 62:2183–2194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Go Y, Jeong JY, Jeoung NH, Jeon JH, Park
BY, Kang HJ, Ha CM, Choi YK, Lee SJ, Ham HJ, et al: Inhibition of
pyruvate dehydrogenase kinase 2 protects against hepatic steatosis
through modulation of tricarboxylic acid cycle anaplerosis and
ketogenesis. Diabetes. 65:2876–2887. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Chen M, Zheng H, Xu M, Zhao L, Zhang Q,
Song J, Zhao Z, Lu S, Weng Q, Wu X, et al: Changes in hepatic
metabolic profile during the evolution of STZ-induced diabetic rats
via an 1H NMR-based metabonomic investigation. Biosci
Rep. Apr 23–2019.(Epub ahead of print). doi:
10.1042/BSR20181379.
|
|
78
|
Sugden MC and Holness MJ: Recent advances
in mechanisms regulating glucose oxidation at the level of the
pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol
Metab. 284:E855–E862. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Seidler NW: GAPDH and intermediary
metabolism. Adv Exp Med Biol. 985:37–59. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hwang NR, Yim SH, Kim YM, Jeong J, Song
EJ, Lee Y, Lee JH, Choi S and Lee KJ: Oxidative modifications of
glyceraldehyde-3-phosphate dehydrogenase play a key role in its
multiple cellular functions. Biochem J. 423:253–264. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yun J, Mullarky E, Lu C, Bosch KN,
Kavalier A, Rivera K, Roper J, Chio II, Giannopoulou EG, Rago C, et
al: Vitamin C selectively kills KRAS and BRAF mutant colorectal
cancer cells by targeting GAPDH. Science. 350:1391–1396. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Du XL, Edelstein D, Rossetti L, Fantus IG,
Goldberg H, Ziyadeh F, Wu J and Brownlee M: Hyperglycemia-induced
mitochondrial superoxide overproduction activates the hexosamine
pathway and induces plasminogen activator inhibitor-1 expression by
increasing Sp1 glycosylation. Proc Natl Acad Sci USA.
97:12222–12226. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wu J, Jin Z, Zheng H and Yan LJ: Sources
and implications of NADH/NAD(+) redox imbalance in diabetes and its
complications. Diabetes Metab Syndr Obes. 9:145–153.
2016.PubMed/NCBI
|
|
85
|
Funk SD, Yurdagul A Jr and Orr AW:
Hyperglycemia and endothelial dysfunction in atherosclerosis:
Lessons from type 1 diabetes. Int J Vasc Med.
2012:5696542012.PubMed/NCBI
|
|
86
|
Rask-Madsen C and King GL: Vascular
complications of diabetes: Mechanisms of injury and protective
factors. Cell Metab. 17:20–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yan LJ: Pathogenesis of chronic
hyperglycemia: From reductive stress to oxidative stress. J
Diabetes Res. 2014:1379192014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Brownlee M: The pathobiology of diabetic
complications: A unifying mechanism. Diabetes. 54:1615–1625. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ralser M, Wamelink MM, Kowald A, Gerisch
B, Heeren G, Struys EA, Klipp E, Jakobs C, Breitenbach M, Lehrach H
and Krobitsch S: Dynamic rerouting of the carbohydrate flux is key
to counteracting oxidative stress. J Biol. 6:102007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Perry RJ, Kim T, Zhang XM, Lee HY, Pesta
D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, et al:
Reversal of hypertriglyceridemia, fatty liver disease, and insulin
resistance by a liver-targeted mitochondrial uncoupler. Cell Metab.
18:740–748. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wamelink MM, Struys EA and Jakobs C: The
biochemistry, metabolism and inherited defects of the pentose
phosphate pathway: A review. J Inherit Metab Dis. 31:703–717. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Riganti C, Gazzano E, Polimeni M, Aldieri
E and Ghigo D: The pentose phosphate pathway: An antioxidant
defense and a crossroad in tumor cell fate. Free Radic Biol Med.
53:421–436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Cabezas H, Raposo RR and Meléndez-Hevia E:
Activity and metabolic roles of the pentose phosphate cycle in
several rat tissues. Mol Cell Biochem. 201:57–63. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Díaz-Flores M, Ibáñez-Hernández MA, Galván
RE, Gutiérrez M, Durán-Reyes G, Medina-Navarro R, Pascoe-Lira D,
Ortega-Camarillo C, Vilar-Rojas C, Cruz M and Baiza-Gutman LA:
Glucose-6-phosphate dehydrogenase activity and
NADPH/NADP+ ratio in liver and pancreas are dependent on
the severity of hyperglycemia in rat. Life Sci. 78:2601–2607. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Spaans SK, Weusthuis RA, van der Oost J
and Kengen SW: NADPH-generating systems in bacteria and archaea.
Front Microbiol. 6:7422015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Aragno M, Tamagno E, Gatto V, Brignardello
E, Parola S, Danni O and Boccuzzi G: Dehydroepiandrosterone
protects tissues of streptozotocin-treated rats against oxidative
stress. Free Radic Biol Med. 26:1467–1474. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Cédola N, Cabarrou A, Auciello N, Doria I,
Ponce de León H and Baylon N: The liver in human diabetes.
Concentration of some induced enzymes. Acta Diabetol Lat.
12:263–271. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gupte RS, Floyd BC, Kozicky M, George S,
Ungvari ZI, Neito V, Wolin MS and Gupte SA: Synergistic activation
of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src
kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat
liver. Free Radic Biol Med. 47:219–228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Shepherd A and Cleary MP: Metabolic
alterations after dehydroepiandrosterone treatment in Zucker rats.
Am J Physiol. 246:E123–E128. 1984.PubMed/NCBI
|
|
100
|
Dong K, Ni H, Wu M, Tang Z, Halim M and
Shi D: ROS-mediated glucose metabolic reprogram induces insulin
resistance in type 2 diabetes. Biochem Biophys Res Commun.
476:204–211. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kolderup A and Svihus B: Fructose
metabolism and relation to atherosclerosis, type 2 diabetes, and
obesity. J Nutr Metab. 2015:8230812015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lambert JE, Ramos-Roman MA, Browning JD
and Parks EJ: Increased de novo lipogenesis is a distinct
characteristic of individuals with nonalcoholic fatty liver
disease. Gastroenterology. 146:726–735. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Softic S, Cohen DE and Kahn CR: Role of
dietary fructose and hepatic de novo lipogenesis in fatty liver
disease. Dig Dis Sci. 61:1282–1293. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Barros BSV, Santos DC, Pizarro MH, del
Melo LGN and Gomes MB: Type 1 diabetes and non-alcoholic fatty
liver disease: When should we be concerned? A nationwide study in
Brazil. Nutrients. 9:E8782017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Calzadilla Bertot L and Adams LA: The
natural course of non-alcoholic fatty liver disease. Int J Mol Sci.
17:E7742016. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Targher G, Bertolini L, Padovani R,
Rodella S, Zoppini G, Pichiri I, Sorgato C, Zenari L and Bonora E:
Prevalence of non-alcoholic fatty liver disease and its association
with cardiovascular disease in patients with type 1 diabetes. J
Hepatol. 53:713–718. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kummer S, Klee D, Kircheis G, Friedt M,
Schaper J, Häussinger D, Mayatepek E and Meissner T: Screening for
non-alcoholic fatty liver disease in children and adolescents with
type 1 diabetes mellitus: A cross-sectional analysis. Eur J
Pediatr. 176:529–536. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Lăpădat AM, Jianu IR, Ungureanu BS,
Florescu LM, Gheonea DI, Sovaila S and Gheonea IA: Non-invasive
imaging techniques in assessing non-alcoholic fatty liver disease:
A current status of available methods. J Med Life. 10:19–26. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Guiu B, Petit JM, Loffroy R, Ben Salem D,
Aho S, Masson D, Hillon P, Krause D and Cercueil JP; Quantification
of liver fat content, : Comparison of triple-echo chemical shift
gradient-echo imaging and in vivo proton MR spectroscopy.
Radiology. 250:95–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Petit JM, Pedro L, Guiu B, Duvillard L,
Bouillet B, Jooste V, Habchi M, Crevisy E, Fourmont C, Buffier P,
et al: Type 1 diabetes is not associated with an increased
prevalence of hepatic steatosis. Diabet Med. 32:1648–1651. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Perseghin G, Lattuada G, De Cobelli F,
Esposito A, Costantino F, Canu T, Scifo P, De Taddeo F, Maffi P,
Secchi A, et al: Reduced intrahepatic fat content is associated
with increased whole-body lipid oxidation in patients with type 1
diabetes. Diabetologia. 48:2615–2621. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Cusi K, Sanyal AJ, Zhang S, Hartman ML,
Bue-Valleskey JM, Hoogwerf BJ and Haupt A: Non-alcoholic fatty
liver disease (NAFLD) prevalence and its metabolic associations in
patients with type 1 diabetes and type 2 diabetes. Diabetes Obes
Metab. 19:1630–1634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Regnell SE, Peterson P, Trinh L, Broberg
P, Leander P, Lernmark Å, Månsson S and Elding Larsson H: Magnetic
resonance imaging reveals altered distribution of hepatic fat in
children with type 1 diabetes compared to controls. Metabolism.
64:872–878. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Jiang S, Tang X, Wang K, Liang Y, Qian Y,
Lu C and Cai L: Hepatic functional and pathological changes of type
1 diabetic mice in growing and maturation time. J Cell Mol Med.
23:5794–5807. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Torbenson M, Chen YY, Brunt E, Cummings
OW, Gottfried M, Jakate S, Liu YC, Yeh MM and Ferrell L: Glycogenic
hepatopathy: An underrecognized hepatic complication of diabetes
mellitus. Am J Surg Pathol. 30:508–513. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Kotronen A, Juurinen L, Tiikkainen M,
Vehkavaara S and Yki-Järvinen H: Increased liver fat, impaired
insulin clearance, and hepatic and adipose tissue insulin
resistance in type 2 diabetes. Gastroenterology. 135:122–130. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Titchenell PM, Lazar MA and Birnbaum MJ:
Unraveling the regulation of hepatic metabolism by insulin. Trends
Endocrinol Metab. 28:497–505. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Vatner DF, Majumdar SK, Kumashiro N,
Petersen MC, Rahimi Y, Gattu AK, Bears M, Camporez JP, Cline GW,
Jurczak MJ, et al: Insulin-independent regulation of hepatic
triglyceride synthesis by fatty acids. Proc Natl Acad Sci USA.
112:1143–1148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Osório J: Diabetes: Hepatic lipogenesis
independent of insulin in type 2 diabetes mellitus-a paradox
clarified. Nat Rev Endocrinol. 11:1302015. View Article : Google Scholar
|
|
120
|
Alwahsh SM, Dwyer BJ, Forbes S, Thiel DH,
Lewis PJ and Ramadori G: Insulin production and resistance in
different models of diet-induced obesity and metabolic syndrome.
Int J Mol Sci. 18(pii): E2852017. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
American Diabetes Association: Diagnosis
and classification of diabetes mellitus. Diabetes Care. 35 (Suppl
1):S64–S71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Regnell SE and Lernmark Å: Hepatic
steatosis in type 1 diabetes. Rev Diabet Stud. 8:454–467. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Purnell JQ, Zinman B and Brunzell JD;
DCCT/EDIC Research Group, : The effect of excess weight gain with
intensive diabetes mellitus treatment on cardiovascular disease
risk factors and atherosclerosis in type 1 diabetes mellitus:
Results from the diabetes control and complications
trial/epidemiology of diabetes interventions and complications
study (DCCT/EDIC) study. Circulation. 127:180–187. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|