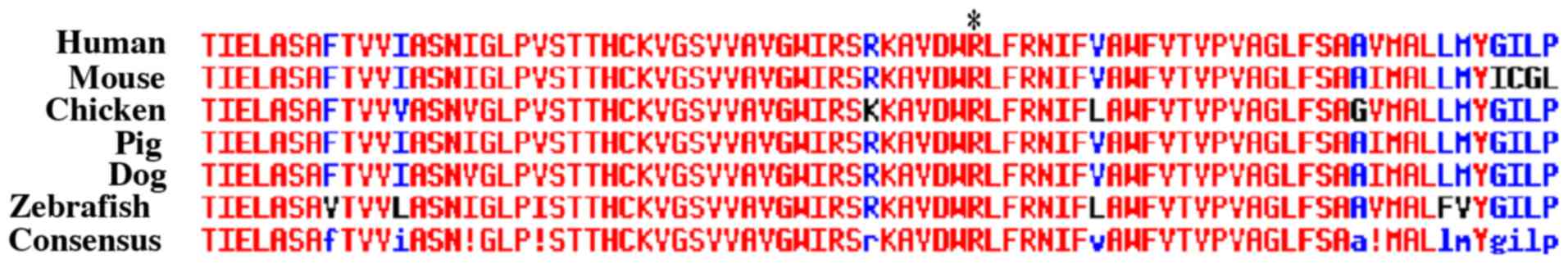Introduction
Hereditary multiple exostoses (HME) is an autosomal
dominant inheritance disease in children, with an estimated
incidence of 1 in 50,000 in the Western population in 1994
(1–3). While the clinical manifestation of
HME varies, it is characterised by cartilage-capped tumours (called
exostoses or osteochondromas) that occur on endochondral bone, and
the most commonly involved bones include the distal femur, proximal
tibia, fibula and humerus (1–3).
Currently, the detailed mechanism of HME remains
unclear. Numerous previous studies have reported that the
development of HME is associated with gene mutations in exostosin
(EXT)-1 and EXT-2 (4–6). The
EXT-1 gene is located on chromosome 8 (locus 8q24.1) whereas
the EXT-2 gene is located on chromosome 11 (locus 11p11-13)
(4–6). Previous studies have reported that
70–95% of patients with HME display mutations in the EXT-1
and EXT-2 genes (6).
EXT genes encode a protein product of the enzyme
glycosyltransferase, which serves an important role in the
synthesis of heparan sulfate proteoglycans (HSPGs). As an important
component of the extracellular matrix, HSPGs regulate the
proliferation and differentiation of chondrocytes (5). Synthesis defects in HSPGs due to
mutations in EXT-1 or EXT-2 cause the abnormal
proliferation and differentiation of chondrocytes and subsequently
lead to the development of HME.
Although the main causative genes for HME are
EXT-1 and EXT-2 (6,7),
there are numerous patients with HME without EXT-1 and
EXT-2 gene mutations (8,9).
Therefore, it is crucial to identify other candidate genes in
patients with HME. However, few genetic studies have focused on
patients with HME without EXT-1 and EXT-2 mutations
(10,11).
In the present study, whole-exome sequencing was
performed in a typical Chinese HME family without EXT-1 and
EXT-2 mutations. The aim of the present study was to
identify novel candidate genes for the development of HME.
Materials and methods
Patients
The patients recruited to the present study were
from a family in Guangdong Province, China between January 2016 and
September 2016. The proband (age, 4 years; sex, male) sought
medical advice at Guangzhou Women and Children's Medical Center due
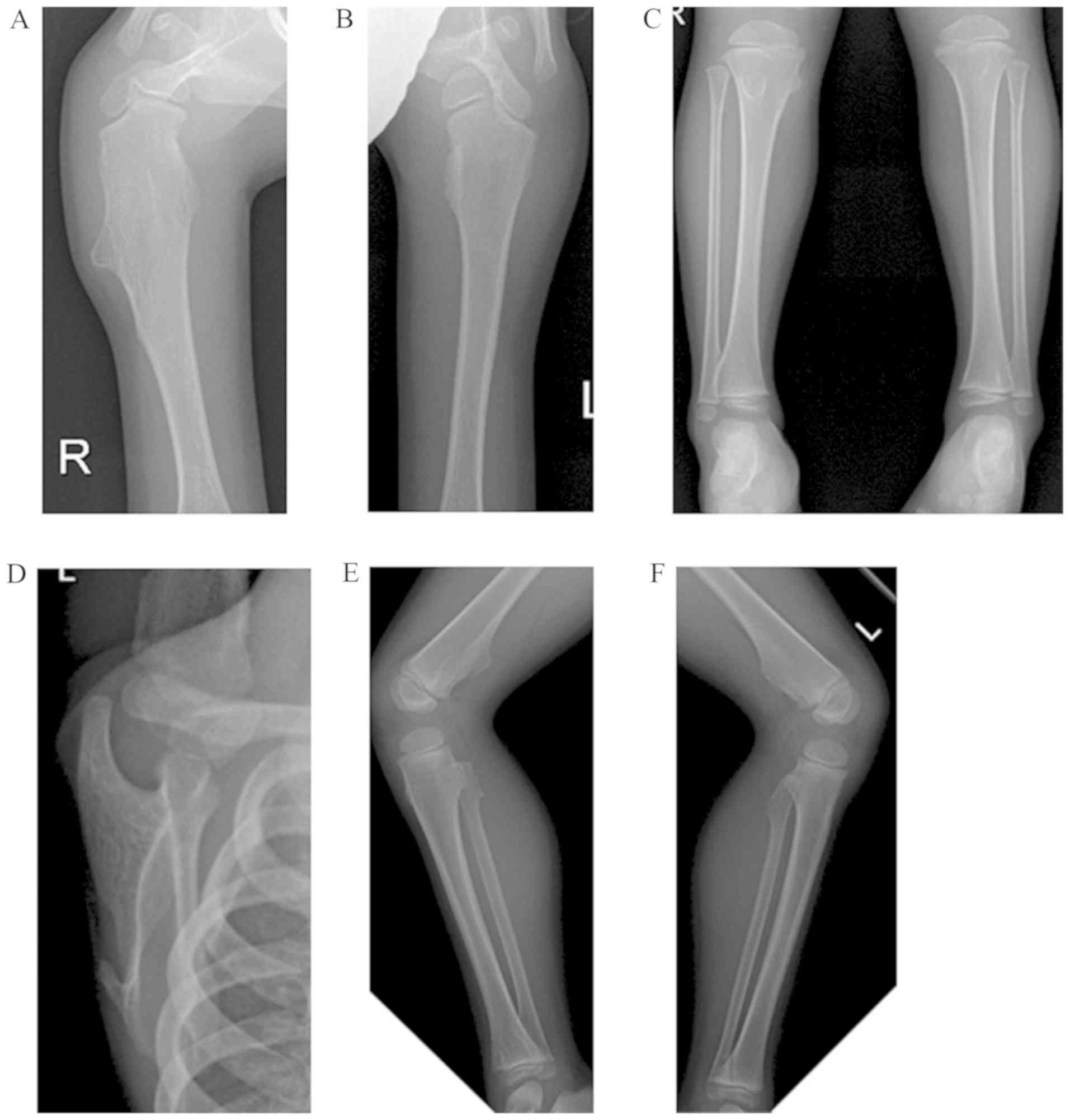
to multiple exostoses at the bilateral distal femur, proximal tibia
and fibula, distal tibia, proximal humerus and left scapula
(Fig. 1). The proband was
diagnosed with HME.
Additionally, the parents were evaluated according
to medical record reviews, physical examinations and patient
history to obtain detailed information about all family members. As
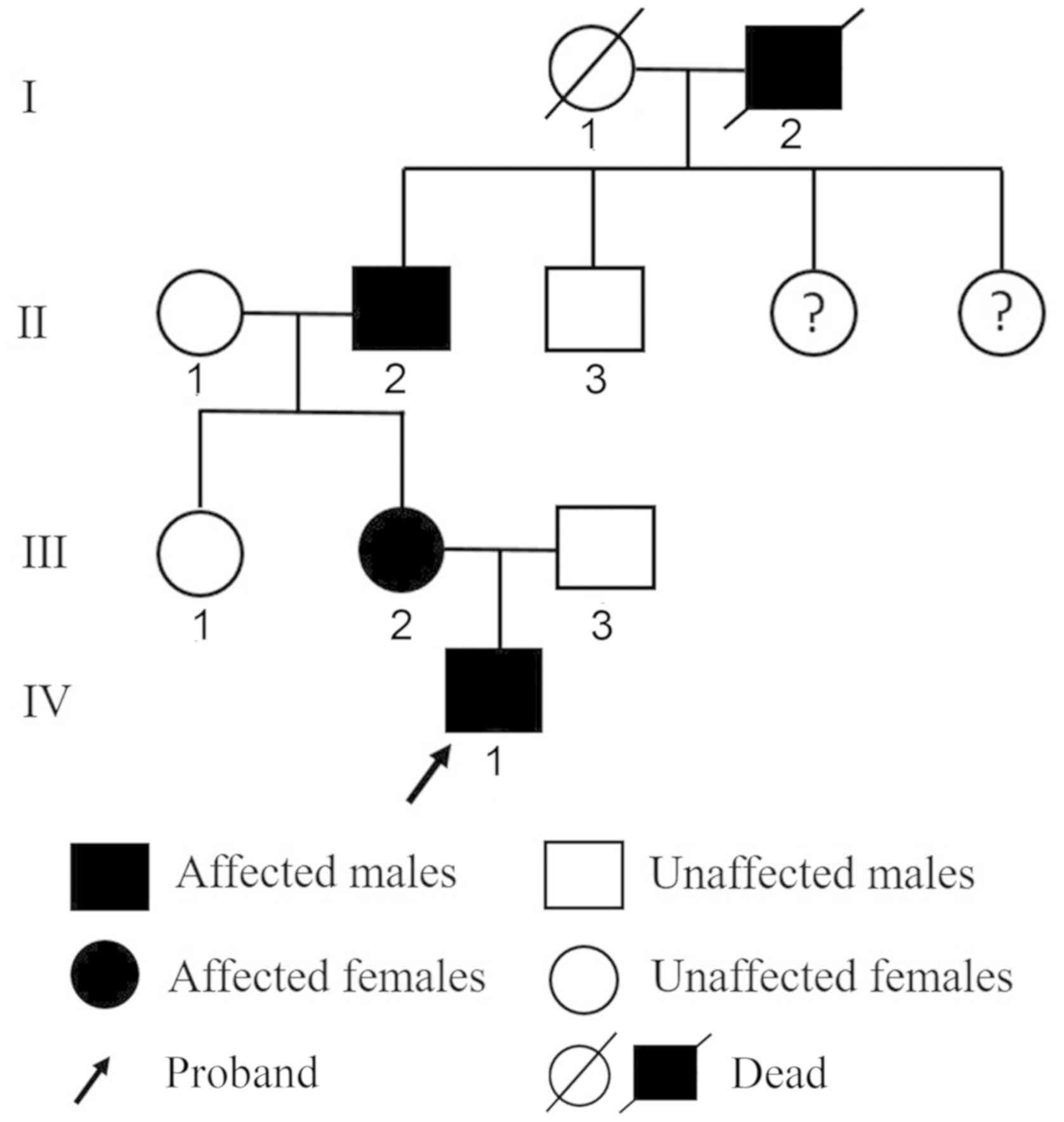
a result, the mother (33 years old) and grandfather (58 years old)
of the proband were also diagnosed with HME (Fig. 2). The great-grandfather of the
proband was also reported to have multiple exostoses, although
physical examination could not be performed to confirm this
information as the patient was deceased.
The present study protocol was approved by the Human
Ethics Committee of the Guangzhou Women and Children's Medical
Center and Guangzhou Medical University (approval no. 2017–320).
All patients or their guardians provided written informed consent.
The blood samples of all members in the family were obtained.
Library construction, whole-exome
sequencing and data analysis
Genomic DNA was extracted from peripheral blood
samples using the Genomic DNA Purification kit (Qiagen China Co.,
Ltd) and DNA concentrations were detected by a Qubit Fluorometer
(Thermo Fisher Scientific, Inc.). Sample integrity and purity were
detected by agarose gel electrophoresis. Exons of EXT-1 and
EXT-2 were sequenced by the Guangzhou Institute of
Pediatrics at the Guangzhou Women and Children's Medical Center via
Sanger sequencing to identify whether the family had mutations in
these established HME-associated causal genes (12); however, no mutations were found in
these genes. Thus, the results indicated that unknown mutations may
be responsible for the development of HME in this family.
Therefore, whole-exome sequencing was subsequently performed.
All the processes of whole-exome sequencing were
performed by the Beijing Genomics Institute. Briefly, extracted
genomic DNA was randomly fragmented to an average size of 250–300
bp on a Covaris s220 sonication machine (Covaris, Inc.). Fragmented
DNA was tested by agarose gel electrophoresis and purified using an
AxyPrep Mag PCR Clean kit (Axygen; Corning) according to the
manufacturer's protocol. The exonic DNA fragments were subjected to
end-repair, 3′ adenylation, adaptor ligation. Following adaptor
ligation, the DNA library was amplified according to a standard
Illumina protocols, and the PCR products were recovered using the
AxyPrep Mag PCR Clean kit. Then, the library was qualified by an
Agilent Technologies 2100 Bioanalyzer (Agilent Technologies, Inc.)
and an ABI StepOnePlus Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. The qualified libraries were sequenced on the
Illumina HiSeq Xten System (Beijing Genomics Institute) with 150 bp
end reads. Sequencing image data were then translated to primary
raw reads (paired-end reads) using Illumina Base Calling software
(CASAVA 1.8; Illumina, Inc). The human reference genome assembly
hg19 (GRCh37) was used as a reference sequence in this analysis
process.
The detection of variants was performed using a
Genome Analysis Toolkit (GATK version 3.3.0; Broad Institute)
according to the manufacturer's protocol and all the variants were
saved as a variant call format (VCF) file. Tertiary analysis was
performed based on VCF files from the secondary analysis using
SnpEff software (version 4.1; snpeff.sourceforge.net/index.html),
including quality filtering, annotation, data aggregation and
functional prediction. According to the phenotype of the family,
there were two potential types of inheritance models, including
autosomal dominant inheritance and autosomal recessive inheritance.
The 1,000 Genomes Project and the Exome Aggregation Consortium
database were used to analyse the variants (13). A total of 3 databases [RefGene
(14), KnownGene (15) and Ensembl (16)] were used to annotate these
variations. All variants were filtered if the variant was annotated
as a nonsynonymous, frameshift or splicing mutation in one of these
databases. Finally, further harmful prediction was performed for
the mutations using common prediction tools [Sorting Intolerant
From Tolerant (SIFT; http://provean.jcvi.org), Polyphen2
(genetics.bwh.harvard.edu/pph2), Genomic Evolutionary Rate
Profiling (GERP++) (17),
Likelihood Ratio Test (LRT;
evomics.org/resources/likelihood-ratio-test) and MutationTaster
(www.mutationtaster.org)] and the
mutations that were predicted to be harmful by ≥3 of these tools
and that had no benign predictions were excluded.
Variant validations and selection of
the candidate gene
Candidate variants from whole-exome sequencing were
validated by Sanger sequencing. Subsequently, the function of the
validated gene was evaluated. Genes with a potential ability to
influence chondrocyte proliferation and differentiation were more
likely to be involved in the development of HME and were determined
to be the candidate genes for HME (3).
Alignment of the identified candidate
gene
If a gene was determined to be the pathologic
candidate gene for HME, alignment of the gene at the locus of
mutation among different species (human, mouse, chicken, pig, dog,
and zebrafish) was performed using MultAlin
(multalin.toulouse.inra.fr/multalin) to investigate whether the
mutation was located at a highly conserved region.
Magnetic resonance images (MRI)
The cranial MRI examination was performed on the
proband to detect pathological change of the brain. In particular,
brain calcification was diagnosed as a local lesion with low signal
on both T2-weighted imaging and T1 weighted imaging (18).
Positive rate of EXT-1 and EXT-2
mutations
PubMed (pubmed.ncbi.nlm.nih.gov) and Web of Science
(www.webofknowledge.com) databases were
searched to identify literature that reported the positive rate of
EXT-1 and EXT-2 mutations in different populations.
The rates were extracted and the mean value was calculated.
Results
Whole-exome sequencing
All samples from the family were successfully
sequenced and variants were detected with an average sequencing
depth on the target over 100 and 98% coverage of the target region.
The high-quality reads obtained from II-1, II-2, II-3, III-1,
III-2, III-3 and the proband (IV-1; Fig. 2) were 6,858, 5,668, 5,516, 5,917,
6,049, 5,989 and 5,560 Mb, respectively. VCF files for samples in
the family were imported together into SnpEff software for
posterior annotation and filtering. According to the phenotype of
the family, there were two potential types of inheritance models in
this family, including autosomal dominant inheritance and autosomal
recessive inheritance.
Under the autosomal dominant inheritance model, the
patients with HME had a heterozygous mutation and healthy controls
had no variants. A total of 3 databases, including refGene,
KnownGene and Ensembl, were used to annotate all the variants. As a
result, a total of 1,830 original variants were revealed to be
heterozygous mutations in the 3 patients with HME, which were not
present in the healthy controls (Data S1, autosomal_dominant.vcf).
Of these variants, 182 mutations were nonsynonymous mutations.
Mutations located at repeating regions of the genome were excluded,
resulting in 145 mutations. Furthermore, variants with mutation
frequencies >1% in the Asian population (The 1,000 Genomes
Project and the Exome Aggregation Consortium database) were
excluded and 28 mutations remained. Among these 28 variants, 2
variants did not result in amino acid polymorphisms and were
excluded. Further harmful prediction was performed for these 26
mutations using common prediction tools (SIFT, Polyphen2, GERP++,
LRT and MutationTaster; Table I)
and the mutations that were predicted to be harmful by ≥3 of these
tools and that had no benign predictions were excluded. This
resulted in a total of 2 mutations, c.C1849T in solute carrier
family 20 member 2 (SLC20A2) and c.G506A in leucine zipper and
EF-hand containing transmembrane protein 1 (LETM1). Both were
nonsynonymous single-nucleotide variant mutations and were selected
as the candidate variants.
 | Table I.Harmful prediction of the 26 gene
variations. |
Table I.
Harmful prediction of the 26 gene
variations.
|
|
|
|
|
| Harmful
prediction |
|---|
|
|
|
|
|
|
|
|---|
| Position | Gene | Transcript | Nucleotide
change | Amino acid
change | SIFT | Polyphen2 | LRT | GERP++ | Mutation
Taster |
|---|
| Chr1:
228475819 | OBSCN | NM_001098623 | c.C9869T | p.T3290M | T | D | N | N | N |
| Chr2:
219870747 | CFAP65 | NM_194302 | c.C4918T | p.H1640Y | T | B | N | N | D |
| Chr4: 1843162 | LETM1 | NM_012318 | c.G506A | p.R169H | D | D | D | D | D |
| Chr4: 7026948 | TBC1D14 | NM_001113363 | c.G1135A | p.A379T | T | B | D | D | D |
| Chr5: 68661455 | TAF9 | NM_001015892 | c.A110G | p.N37S | D | B | D | D | D |
| Chr5:168093596 | SLIT3 | NM_001271946 | c.C4456T | p.R1486C | T | D | N | D | D |
| Chr7:
143098436 | EPHA1 | NM_005232 | c.G413A | p.R138Q | T | D | D | D | D |
| Chr8: 42275431 | SLC20A2 | NM_001257180 | c.C1849T | p.R617C | D | D | D | D | D |
| Chr8: 67425859 | C8orf46 | NM_152765 | c.G427A | p.V143I | – | B | N | D | N |
| Chr9: 19347068 | DENND4C | NM_017925 | c.G4154C | p.S1385T | T | B | N | N | N |
| Chr9:
100693246 | HEMGN | NM_197978 | c.A431G | p.Q144R | T | B | N | N | N |
| Chr9:
116181419 | C9orf43 | NM_001278629 | c.A319G | p.I107V | T | B | N | N | N |
| Chr12:
88390214 | C12orf50 | NM_152589 | c.A418G | p.M140V | T | B | N | N | N |
| Chr12:
88505514 | CEP290 | NM_025114 | c.A2174C | p.E725A | T | D | D | D | D |
| Chr12:
95927927 | USP44 | NM_001042403 | c.G106A | p.E36K | T | D | D | D | D |
| Chr12:
126004130 | TMEM132B | NM_052907 | c.G1237A | p.V413I | T | B | N | N | D |
| Chr14:
32257075 | NUBPL | NM_001201574 | c.A54G | p.I18M | D | D | D | N | D |
| Chr14:
51446236 | TRIM9 | NM_015163 | c.A1939G | p.I647V | T | B | N | N | D |
| Chr15:
44695168 | CASC4 | NM_138423 | c.G1156A | p.V386I | T | D | D | D | D |
| Chr17: 8048267 | PER1 | NM_002616 | c.G2263A | p.A755T | T | B | N | D | D |
| Chr17:
10535909 | MYH3 | NM_002470 | c.C4840T | p.R1614W | D | D | – | N | D |
| Chr18:
21508694 | LAMA3 | NM_001127718 | c.C3406A | p.Q1136K | T | D | – | D | D |
| Chr18:
22805996 | ZNF521 | NM_001308225 | c.G1226A | p.R409H | T | D | D | D | D |
| Chr19:
18468286 | PGPEP1 | NM_001308366 | c.C298T | p.R100C | D | B | N | D | D |
| Chr19:
24309223 | ZNF254 | NM_001278678 | c.G166T | p.G56X | T | – | – | N | D |
| Chr21:
15884882 | SAMSN1 | NM_022136 | c.G292A | p.G98R | T | B | N | D | N |
Under the autosomal recessive inheritance model,
patients with HME had homozygous mutations, while healthy controls
had heterozygous mutations or no mutations. A total of 3 databases,
including refGene, KnownGene and Ensembl, were used to annotate all
variants. As a result, a total of 246 original variants were
revealed to be consistent with the recessive inheritance model
(Data S2, autosomal_recessive.vcf). Of these variants, 19 mutations
were nonsynonymous and were not located in repeating regions of the
genome. However, all 19 of these variants were excluded due to a
mutation frequency >1% in the Asian population (The 1,000
Genomes and the Exome Aggregation Consortium database) (13). Furthermore, harmful prediction
using common prediction tools was conducted and all variants were
excluded.
Variant validations by Sanger
sequencing
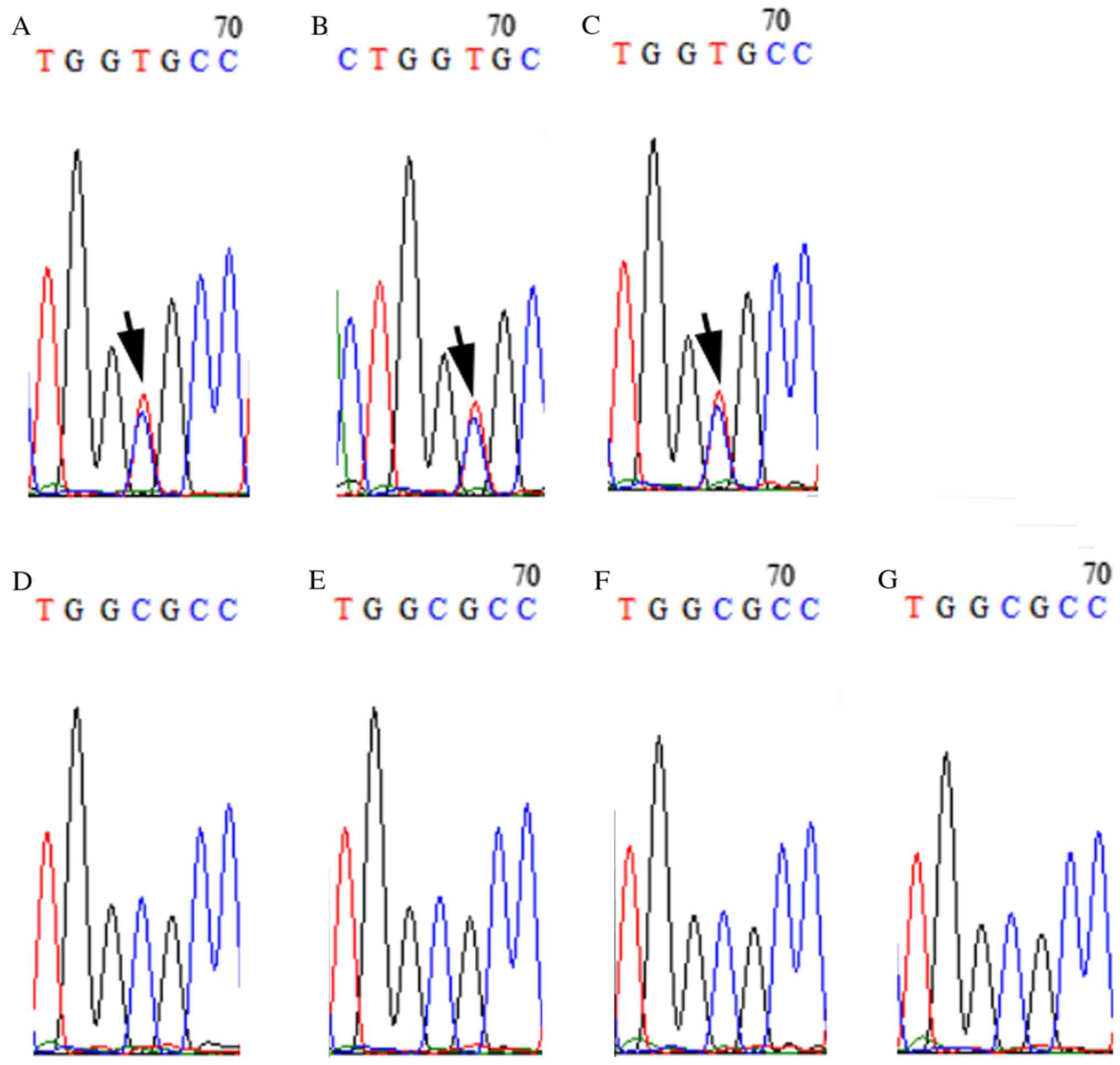
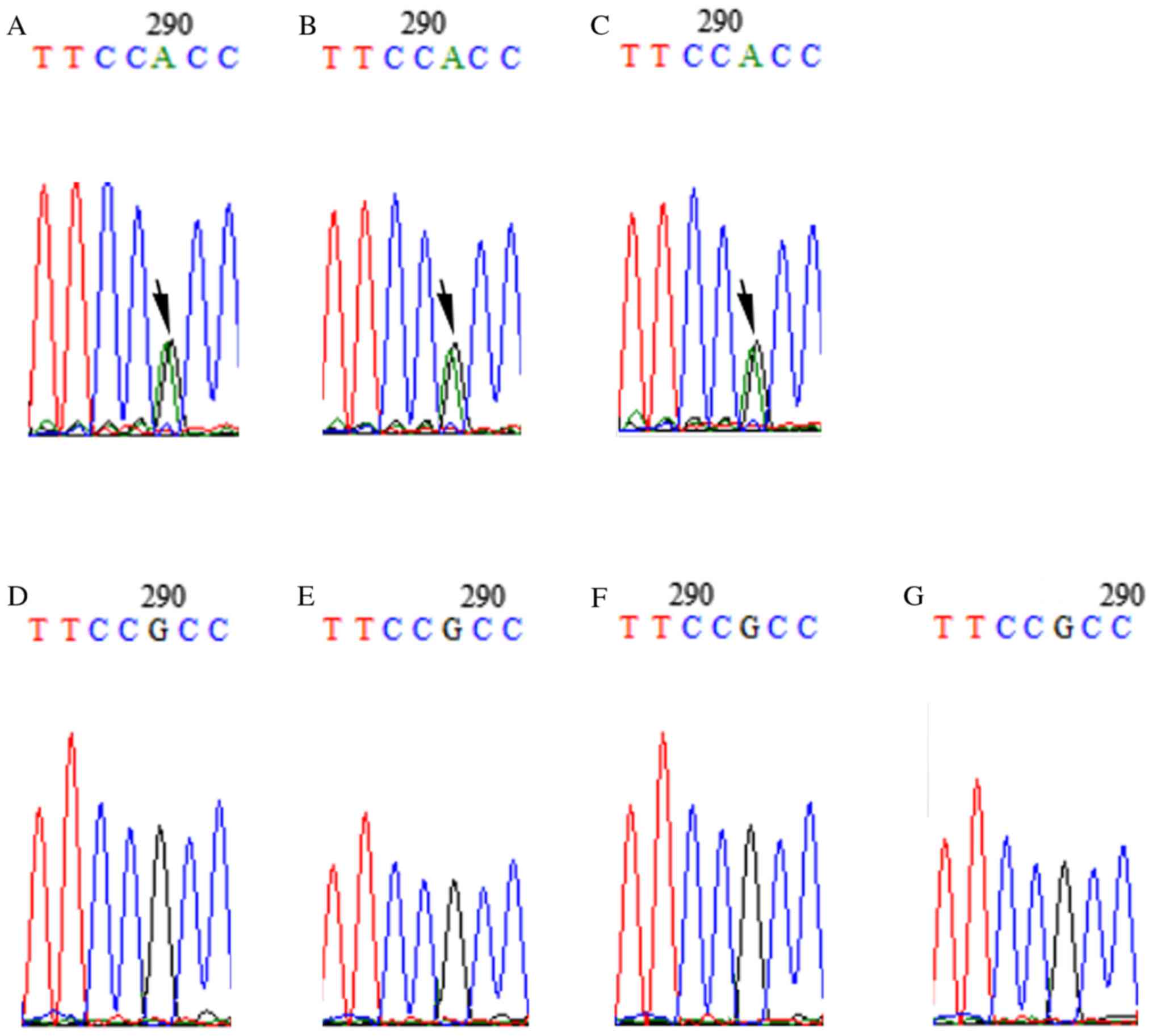
The selected candidate variants (c.C1849T in SLC20A2
and c.G506A in LETM1) were validated by Sanger sequencing. Sanger
sequencing results confirmed that these two mutations were present
in all patients with HME (II-2, III-2 and IV-1), while family
members who did not have HME did not have these mutations (Figs. 3 and 4). Of the two genes, SLC20A2 mutation may
induce metabolic disturbances of phosphates to regulate chondrocyte
proliferation and differentiation, and subsequently lead to the
development of HME.
Alignment of the identified candidate
gene
SLC20A2 was identified as the pathologic candidate
gene. SLC20A2 alignment at the locus of mutation among different
species was performed. The mutation locus of SLC20A2 located at a
highly conserved region (Fig. 5).
Mutations in c.C1849T in SLC20A2 led to a change in an amino acid
(p.R617C).
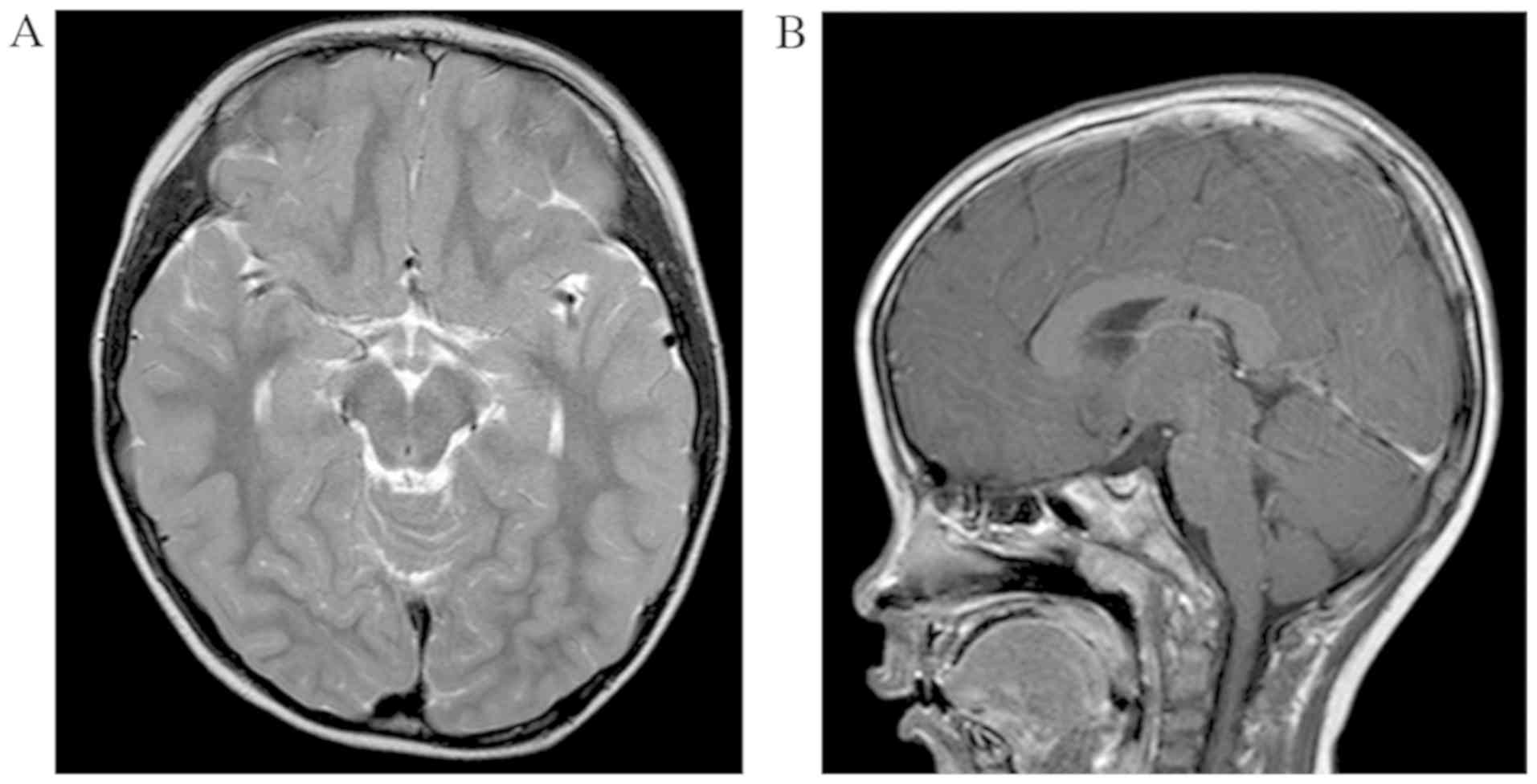
Results of cranial MRI
Cranial MRI indicated the proband did not exhibit
any brain pathologic changes, such brain calcification, tumor or
hydrocephalus (Fig. 6).
Reported positive rate of EXT-1 and
EXT-2 mutations
A total of 14 papers from PubMed and Web of Science
were extracted that reported the positive rate of EXT-1 and
EXT-2 mutations in populations (Table II) (7–9,19–29).
The worldwide prevalence of EXT-1 and EXT-2 gene
mutations (1999-2018) was 47.2–95.7% in patients with HME. The
overall positive rate in patients with HME was 76.8%.
| Table II.Positive rates of exostosin (EXT)-1
and EXT-2 genes in patients with hereditary multiple exostoses as
reported by various studies. |
Table II.
Positive rates of exostosin (EXT)-1
and EXT-2 genes in patients with hereditary multiple exostoses as
reported by various studies.
| First author
(refs.) | Year | Journal | Country | Positive rate
(%) |
|---|
| Xu (19) | 1999 | Hum Genet | China | 47.2 (17/36) |
| Francannet
(20) | 2001 | J Med Genet | France | 85.7 (36/42) |
| Wuyts (21) | 2005 | Clin Genet | Belgium | 74 (37/50) |
| Vink (22) | 2005 | Eur J Hum
Genet | Netherlands | 63 (22/35) |
| Lonie (23) | 2006 | Hum Mutat | United Kingdom | 78 (56/72) |
| Kojima (24) | 2008 | Genet Test | Japan | 54 (23/43) |
| Jennes (25) | 2008 | J Mol Diagn | Belgium | 68.3 (43/68) |
| Heinritz (7) | 2009 | Ann Hum Genet | Germany | 95.7 (22/23) |
| Kang (26) | 2012 | Gene | China | 90 (9/10) |
| Sarrión (27) | 2013 | Sci Rep | Span | 95 (37/39) |
| Jamsheer (28) | 2014 | J Appl Genet | Poland | 84.9 (28/33) |
| Ishimaru (8) | 2016 | BMC Genet | Japan | 66.2 (47/71) |
| Santos (9) | 2018 | Mol Genet Genomic
Med | Brazil | 83 (95/114) |
| Li (29) | 2018 | Medicine
(Baltimore) | China | 93 (68/73) |
| Total | – | – | – | 76.8 (562/732) |
Discussion
Mutations in the EXT-1 and EXT-2 genes
are considered to be the primary cause of HME (5,7).
Numerous studies have identified various mutation sites in
EXT-1 and EXT-2 (30–32).
The reported worldwide prevalence of EXT-1 and EXT-2
gene mutations (1999–2018) is 47.2–95.7% in patients with HME
(8–10,19–29).
However, 23.2% of living patients with HME have no EXT-1 or
EXT-2 mutations. Presently, few studies have focused on
patients with HME without EXT-1 and EXT-2 mutations
(11,12). Therefore, it is crucial to identify
novel candidate genes in patients with HME.
The present study revealed a novel deleterious
mutation, c.C1849T in SLC20A2 using SIFT, PolyPhe2, LRT, GERP++ and
MutationTaster database analyses. As a member of the SLC20 family
of solute carriers (33,34), the SLC20A2 protein is comprised of
652 amino acids with a molecular mass of 7,0392 Da (33,34).
The SLC20A2 protein, also known as Pit-2, is a type III
sodium-phosphate cotransporter that mediates the transmembrane
movement of sodium and phosphate (34–36).
SLC20A2 is expressed on the basolateral membranes of polarized
epithelial cells and is involved in cellular phosphate homeostasis
(33). It is widely expressed in
numerous tissues and organs, including the kidneys and intestines
(33). Furthermore, previous
studies have reported that SLC20A2 is also expressed by
chondrocytes (35–37). In the present study, mutations in
c.C1849T in SLC20A2 led to a change in an amino acid (p.R617C).
Alignments amongst species indicated that this mutation was located
at a highly conserved amino acid sequence at which mutations have
increased probability to induce structural changes and SLC20
dysfunction, subsequently leading to a disturbance in the uptake of
inorganic phosphorus in cells (38). Mutations in SLC20A2 have been
increasingly reported in primary familial brain calcification cases
(39,40) and, to the best of our knowledge,
the present study is the first to report SLC20A2 mutation in
patients with HME. Sekine et al (38) reported that SLC20A2 variants
significantly decreased inorganic phosphate transport activity in
endothelial cells induced from induced pluripotent stem cells
(iPS-Cs) derived from patients with idiopathic basal ganglia
calcification patients compared with control iPS-ECs. Additionally,
SLC20A2 is a physiological regulator of tissue mineralization and
serves an important role in the determination of bone quality and
strength (41). However, cranial
magnetic resonance images indicated the proband in the present
study did not exhibit brain calcification.
SLC20A2 mutation-induced metabolic disturbances of
phosphates may be involved in the development of HME (38). Previous studies have demonstrated
that phosphate serves an important role in regulating the
proliferation and differentiation of chondrocytes (42,43).
Zalutskaya et al (42)
utilized a mouse metatarsal culture model to evaluate the effect of
various concentrations of phosphate on endochondral bone formation.
The results demonstrated that a phosphate-high concentration (7 nM)
significantly decreased the proliferation of chondrocytes and
promoted the terminal differentiation of hypertrophic chondrocytes.
However, Liu et al (44)
revealed that low phosphate concentrations inhibited the
differentiation of chondrocytes through the parathyroid
hormone-related peptide signaling pathway in a mouse metatarsal
culture model. Furthermore, Magne et al (45) reported similar results. Previous
studies have demonstrated that HME is characterized by the abnormal
proliferation and differentiation of chondrocytes (46,47).
The present study hypothesized that SLC20A2 mutation-induced
metabolic disorder of phosphates in chondrocytes may lead to their
abnormal proliferation and differentiation and subsequently result
in the development of HME.
Additionally, mutations in c.G506A in LETM1 were
predicted to be harmful using common prediction tools. LETM1
encodes a protein that is localized to the inner mitochondrial
membrane (48). Studies have
reported that mutations in this gene cause Wolf-Hirschhorn syndrome
(49,50). To the best of our knowledge, no
studies concerning the effect of the LETM1 gene on cartilages and
bones have been conducted. Thus, further studies are required to
evaluate the effect of mutations in c.G506A in LETM1 on the
proliferation and differentiation of chondrocytes.
There were limitations in the present study. In the
present study, whole-exome sequencing was performed and identified
a novel mutation of SLC20A2. Although mutations in SLC20A2 were
predicted to lead to disturbance of uptake of inorganic phosphorus,
the serum phosphorus levels of the patients was not assessed. Thus,
further research is required to investigate the effect of mutation
in c.C1849T in SLC20A2 on the expression and function of
SLC20A2.
In conclusion, the present study revealed two
mutations, c.C1849T in SLC20A2 and c.G506A in LETM1, in a Chinese
family with HME. Mutation in c.C1849T in SLC20A2 is more likely to
be involved in the development of HME. Considering that the present
study was an exploratory study of a family, additional
investigations may be required to verify the preliminary results of
the present study.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was sponsored by the National
Nature Science Foundation of China (grant no. 81702116).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YL, XL, HX designed the present study and
methodology, and supervised, reviewed and edited the manuscript. YL
wrote the original draft of the manuscript and acquired funding. HX
and YL provided resources. XL, MZ, FX, JL and ZY performed the
experiments. YL and MZ conducted data analysis. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The protocol of the present study was approved by
the Human Ethics Committee of the GuangZhou Women and Children's
Medical Center and GuangZhou Medical University (approval no.
2017-320). Written informed consent to participate was obtained
from all patients. In the case of children (<18 years), written
informed consent was obtained from the parents.
Patient consent for publication
Written informed consent for publishing data and
images was obtained from all patients. In the case of children
(<18 years), written informed consent for publishing data and
images was obtained from the parents.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SLC20A2
|
solute carrier family 20 member 2
|
|
HME
|
hereditary multiple exostoses
|
|
EXT-1
|
exostosin-1
|
|
EXT-2
|
exostosin-2
|
|
HSPG
|
heparan sulfate proteoglycan
|
|
VCF
|
variant call format
|
|
LETM1
|
leucine zipper and EF-hand containing
transmembrane protein
|
References
|
1
|
Beltrami G, Ristori G, Scoccianti G,
Tamburini A and Capanna R: Hereditary multiple exostoses: A review
of clinical appearance and metabolic pattern. Clin Cases Miner Bone
Metab. 13:110–118. 2016.PubMed/NCBI
|
|
2
|
McFarlane J, Knight T, Sinha A, Cole T,
Kiely N and Freeman R: Exostoses, enchondromatosis and
metachondromatosis; diagnosis and management. Acta Orthop Belg.
82:102–105. 2016.PubMed/NCBI
|
|
3
|
Pacifici M: Hereditary multiple exostoses:
New insights into pathogenesis, clinical complications, and
potential treatments. Curr Osteoporos Rep. 15:142–152. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang C, Zhang R, Lin H and Wang H:
Insights into the molecular regulatory network of pathomechanisms
in osteochondroma. J Cell Biochem. 120:16362–16369. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jochmann K, Bachvarova V and Vortkamp A:
Heparan sulfate as a regulator of endochondral ossification and
osteochondroma development. Matrix Biol. 34:55–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jennes I, Pedrini E, Zuntini M, Mordenti
M, Balkassmi S, Asteggiano CG, Casey B, Bakker B, Sangiorgi L and
Wuyts W: Multiple osteochondromas: Mutation update and description
of the multiple osteochondromas mutation database (MOdb). Hum
Mutat. 30:1620–1627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heinritz W, Hüffmeier U, Strenge S,
Miterski B, Zweier C, Leinung S, Bohring A, Mitulla B, Peters U and
Froster UG: New mutations of EXT1 and EXT2 genes in German patients
with Multiple Osteochondromas. Ann Hum Genet. 73:283–291. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ishimaru D, Gotoh M, Takayama S, Kosaki R,
Matsumoto Y, Narimatsu H, Sato T, Kimata K, Akiyama H, Shimizu K
and Matsumoto K: Large-scale mutational analysis in the EXT1 and
EXT2 genes for Japanese patients with multiple osteochondromas. BMC
Genet. 17:522016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santos SCL, Rizzo I, Takata RI,
Speck-Martins CE, Brum JM and Sollaci C: Analysis of mutations in
EXT1 and EXT2 in Brazilian patients with multiple osteochondromas.
Mol Genet Genomic Med. 6:382–392. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Panagopoulos I, Bjerkehagen B, Gorunova L,
Taksdal I and Heim S: Rearrangement of chromosome bands 12q14~15
causing HMGA2-SOX5 gene fusion and HMGA2 expression in
extraskeletal osteochondroma. Oncol Rep. 34:577–584. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Hul W, Wuyts W, Hendrickx J, Speleman
F, Wauters J, De Boulle K, Van Roy N, Bossuyt P and Willems PJ:
Identification of a third EXT-like gene (EXTL3) belonging to the
EXT gene family. Genomics. 47:230–237. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sanger F, Nicklen S and Coulson AR: DNA
sequencing with chain-terminating inhibitors. Proc Natl Acad Sci
USA. 74:5463–5467. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the american college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pruitt KD, Tatusova T and Maglott DR: NCBI
reference sequences (RefSeq): A curated non-redundant sequence
database of genomes, transcripts and proteins. Nucleic Acids Res 35
(Database Issue). D61–D65. 2007. View Article : Google Scholar
|
|
15
|
Hsu F, Kent WJ, Clawson H, Kuhn RM,
Diekhans M and Haussler D: The UCSC known genes. Bioinformatics.
22:1036–1046. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Flicek P, Amode MR, Barrell D, Beal K,
Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G,
Fitzgerald S, et al: Ensembl 2014. Nucleic Acids Res 42 (Database
Issue). D749–D755. 2014. View Article : Google Scholar
|
|
17
|
Cooper GM, Stone EA, Asimenos G; NISC
Comparative Sequencing Program, ; Green ED, Batzoglou S and Sidow
A: Distribution and intensity of constraint in mammalian genomic
sequence. Genome Res. 15:901–913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Avrahami E, Cohn DF, Feibel M and Tadmor
R: MRI demonstration and CT correlation of the brain in patients
with idiopathic intracerebral calcification. J Neurol. 241:381–384.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu L, Xia J, Jiang H, Zhou J, Li H, Wang
D, Pan Q, Long Z, Fan C and Deng HX: Mutation analysis of
hereditary multiple exostoses in the Chinese. Hum Genet. 105:45–50.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Francannet C, Cohen-Tanugi A, Le Merrer M,
Munnich A, Bonaventure J and Legeai-Mallet L: Genotype-phenotype
correlation in hereditary multiple exostoses. J Med Genet.
38:430–434. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wuyts W, Radersma R, Storm K and Vits L:
An optimized DHPLC protocol for molecular testing of the EXT1 and
EXT2 genes in hereditary multiple osteochondromas. Clin Genet.
68:542–547. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vink GR, White SJ, Gabelic S, Hogendoorn
PC, Breuning MH and Bakker E: Mutation screening of EXT1 and EXT2
by direct sequence analysis and MLPA in patients with multiple
osteochondromas: Splice site mutations and exonic deletions account
for more than half of the mutations. Eur J Hum Genet. 13:470–474.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lonie L, Porter DE, Fraser M, Cole T, Wise
C, Yates L, Wakeling E, Blair E, Morava E, Monaco AP and Ragoussis
J: Determination of the mutation spectrum of the EXT1/EXT2 genes in
British Caucasian patients with multiple osteochondromas, and
exclusion of six candidate genes in EXT negative cases. Hum Mutat.
27:11602006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kojima H, Wada T, Seki H, Kubota T, Wakui
K and Fukushima Y: One third of Japanese patients with multiple
osteochondromas may have mutations in genes other than EXT1 or
EXT2. Genet Test. 12:557–561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jennes I, Entius MM, Van Hul E, Parra A,
Sangiorgi L and Wuyts W: Mutation screening of EXT1 and EXT2 by
denaturing high-performance liquid chromatography, direct
sequencing analysis, fluorescence in situ hybridization, and a new
multiplex ligation-dependent probe amplification probe set in
patients with multiple osteochondromas. J Mol Diagn. 10:85–92.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang Z, Peng F and Ling T: Mutation
screening of EXT genes in Chinese patients with multiple
osteochondromas. Gene. 506:298–300. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sarrión P, Sangorrin A, Urreizti R,
Delgado A, Artuch R, Martorell L, Armstrong J, Anton J, Torner F,
Vilaseca MA, et al: Mutations in the EXT1 and EXT2 genes in Spanish
patients with multiple osteochondromas. Sci Rep. 3:13462013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jamsheer A, Socha M, Sowińska-Seidler A,
Telega K, Trzeciak T and Latos-Bieleńska A: Mutational screening of
EXT1 and EXT2 genes in Polish patients with hereditary multiple
exostoses. J Appl Genet. 55:183–188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Y, Wang J, Tang J, Wang Z, Han B, Li N,
Yu T, Chen Y and Fu Q: Heterogeneous spectrum of EXT gene mutations
in Chinese patients with hereditary multiple osteochondromas.
Medicine (Baltimore). 97:e128552018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo X, Lin M, Yan W, Chen W and Hong G: A
novel splice mutation induces exon skipping of the EXT1 gene in
patients with hereditary multiple exostoses. Int J Oncol.
54:859–868. 2019.PubMed/NCBI
|
|
31
|
Yang A, Kim J, Jang JH, Lee C, Lee JE, Cho
SY and Jin DK: Identification of a novel mutation in EXT2 in a
fourth-generation Korean family with multiple osteochondromas and
overview of mutation spectrum. Ann Hum Genet. 83:160–170. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xia P, Xu H, Shi Q and Li D:
Identification of a novel frameshift mutation of the EXT2 gene in a
family with multiple osteochondroma. Oncol Lett. 11:105–110. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Collins JF, Bai L and Ghishan FK: The
SLC20 family of proteins: Dual functions as sodium-phosphate
cotransporters and viral receptors. Pflugers Arch. 447:647–652.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Forster IC, Hernando N, Biber J and Murer
H: Phosphate transporters of the SLC20 and SLC34 families. Mol
Aspects Med. 34:386–395. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Virkki LV, Biber J, Murer H and Forster
IC: Phosphate transporters: A tale of two solute carrier families.
Am J Physiol Renal Physiol. 293:F643–F654. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Solomon DH, Wilkins RJ, Meredith D and
Browning JA: Characterisation of inorganic phosphate transport in
bovine articular chondrocytes. Cell Physiol Biochem. 20:99–108.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Orfanidou T, Malizos KN, Varitimidis S and
Tsezou A: 1,25-Dihydroxyvitamin D(3) and extracellular inorganic
phosphate activate mitogen-activated protein kinase pathway through
fibroblast growth factor 23 contributing to hypertrophy and
mineralization in osteoarthritic chondrocytes. Exp Biol Med
(Maywood). 237:241–253. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sekine SI, Nishii K, Masaka T, Kurita H,
Inden M and Hozumi I: SLC20A2 variants cause dysfunctional
phosphate transport activity in endothelial cells induced from
Idiopathic Basal Ganglia Calcification patients-derived iPSCs.
Biochem Biophys Res Commun. 510:303–308. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kuroi Y, Akagawa H, Yoneyama T, Kikuchi A,
Maegawa T, Onda H and Kasuya H: Novel SLC20A2 mutation in a
Japanese pedigree with primary familial brain calcification. J
Neurol Sci. 399:183–185. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lemos RR, Ramos EM, Legati A, Nicolas G,
Jenkinson EM, Livingston JH, Crow YJ, Campion D, Coppola G and
Oliveira JR: Update and mutational analysis of SLC20A2: A major
cause of primary familial brain calcification. Hum Mutat.
36:489–495. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Beck-Cormier S, Lelliott CJ, Logan JG,
Lafont DT, Merametdjian L, Leitch VD, Butterfield NC, Protheroe HJ,
Croucher PI, Baldock PA, et al: Slc20a2, encoding the phosphate
transporter PiT2, is an important genetic determinant of bone
quality and strength. J Bone Miner Res. 34:1101–1114. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zalutskaya AA, Cox MK and Demay MB:
Phosphate regulates embryonic endochondral bone development. J Cell
Biochem. 108:668–674. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Khoshniat S, Bourgine A, Julien M, Weiss
P, Guicheux J and Beck L: The emergence of phosphate as a specific
signaling molecule in bone and other cell types in mammals. Cell
Mol Life Sci. 68:205–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu ES, Zalutskaya A, Chae BT, Zhu ED,
Gori F and Demay MB: Phosphate interacts with PTHrP to regulate
endochondral bone formation. Endocrinology. 155:3750–3756. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Magne D, Bluteau G, Faucheux C, Palmer G,
Vignes-Colombeix C, Pilet P, Rouillon T, Caverzasio J, Weiss P,
Daculsi G and Guicheux J: Phosphate is a specific signal for ATDC5
chondrocyte maturation and apoptosis-associated mineralization:
Possible implication of apoptosis in the regulation of endochondral
ossification. J Bone Miner Res. 18:1430–1442. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Milgram JW: The origins of osteochondromas
and enchondromas. A histopathologic study. Clin Orthop Relat Res.
174:264–284. 1983.
|
|
47
|
Benoist-Lasselin C, de Margerie E, Gibbs
L, Cormier S, Silve C, Nicolas G, LeMerrer M, Mallet JF, Munnich A,
Bonaventure J, et al: Defective chondrocyte proliferation and
differentiation in osteochondromas of MHE patients. Bone. 39:17–26.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Durigon R, Mitchell AL, Jones AW, Manole
A, Mennuni M, Hirst EM, Houlden H, Maragni G, Lattante S, Doronzio
PN, et al: LETM1 couples mitochondrial DNA metabolism and nutrient
preference. EMBO Mol Med. 10:e85502018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
McQuibban AG, Joza N, Megighian A,
Scorzeto M, Zanini D, Reipert S, Richter C, Schweyen RJ and
Nowikovsky K: A Drosophila mutant of LETM1, a candidate gene for
seizures in Wolf-Hirschhorn syndrome. Hum Mol Genet. 19:987–1000.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schlickum S, Moghekar A, Simpson JC,
Steglich C, O'Brien RJ, Winterpacht A and Endele SU: LETM1, a gene
deleted in Wolf-Hirschhorn syndrome, encodes an evolutionarily
conserved mitochondrial protein. Genomics. 83:254–261. 2004.
View Article : Google Scholar : PubMed/NCBI
|