Introduction
Eukaryotic cells have two major intracellular
degradation pathways to maintain cellular homeostasis: The
ubiquitin proteasome system (UPS) and autophagy (1). The UPS is a highly selective pathway
for the degradation of misfolded, short-lived and damaged proteins.
Moreover, the UPS pathway recruits the proteasome to selectively
recognize and degrade ubiquitinated proteins involved in a variety
of cellular functions, including cell survival and death (2,3). In
contrast to the UPS, autophagy is the preferred process via which
long-lived proteins and damaged cytoplasmic constituents are
degraded via the autophagosome-lysosome pathway, and it is involved
in cellular homeostasis and stress responses (4,5). The
UPS and autophagy were initially considered to function
independently of one another. However, accumulating evidence has
revealed crosstalk between the two degradation pathways (6,7).
Previous studies investigating this crosstalk have focused on the
compensatory and complementary relationship between autophagy and
the UPS (7–9). It has been shown that UPS dysfunction
can activate autophagy in vitro in cell culture and in
vivo in animal models via the direct or indirect modulation of
proteins associated with cell survival and death (10–12).
The activation of the proteasomal degradation pathway is usually
inversely correlated with autophagic degradation (7,13).
Several mediators, such as P53, p62 and ubiquitin, have been
predicted to link the UPS and autophagy (7,14).
However, the key mediators that induce disease mechanisms remain
largely unknown.
The UPS involves the cooperative action of three
enzymes: Ubiquitin-activating enzyme (E1), ubiquitin-transferring
enzyme (E2) and ubiquitin ligase (E3) (15). E3 ligases are major participants in
the UPS system, as they are the final executioners of ubiquitin
tagging (13). However, little is
known regarding the E3 ligases responsible for the ubiquitination
of substrates degraded by selective autophagy. Among the various E3
proteins, a major ~500 kDa protein known as HECT, UBA and WWE
domain containing E3 ubiquitin protein ligase 1 (Huwe1), encodes a
E5-APC terminus (HECT) domain ubiquitin ligase and has several
disparate substrates, such as P53, MCL1 apoptosis regulator Bcl2
family member, cell division cycle 6, histone deacetylase 2 and
N-myc (11,12). Paradoxically, two seemingly
divergent effects of Huwe1, involving the increased survival and
increased apoptosis, have been observed in different models, such
as neurogenesis and brain cancer, depending on the cell type and
context (16–18). Furthermore, Huwe1 may serve
critical roles in nervous system plasticity, regeneration and
disease (19). UPS dysfunction is
involved in brain ischemia/reperfusion (I/R), but exerts complex
and unambiguous direct and indirect effects in the context of
cerebral I/R due to of its pivotal role in several intracellular
pathways, such as the NF-κB, reactive oxygen species and JNK
signaling pathway (20–23).
Autophagy is vital to cellular homeostasis in the
brain and is increased after cerebral ischemia (24). The role of heightened autophagy in
ischemic neurons is a double-edged sword (25–27).
Previous reports in experimental models have revealed the
beneficial effects of promoting autophagy during ischemia (24,27,28)
Inversely, other studies have shown that autophagy has a
detrimental role in brain ischemia, and that autophagy inhibitors
can attenuate neuronal damage under cerebral ischemic conditions
(29–31). Moreover, previous studies have
suggested that autophagy is beneficial during ischemia, but harmful
during reperfusion (26,32,33).
However, the precise biological function and underlying mechanism
of Huwe1 in autophagy in the context of brain ischemia is yet to be
elucidated. Thus, the present study aimed to investigate the
effects and of Huwe1 on autophagy and its molecular regulatory
properties during oxygen-glucose deprivation and reperfusion
(OGD/R). An in vitro rat cortical neuron model of OGD/R was
established to evaluate the effects of Huwe1 and its downstream
signaling cascades on autophagy.
Materials and methods
Primary cortical neuron culture
Primary cortical neurons were obtained from 45
female Sprague-Dawley rats (weight, 180–200 g; Chengdu Dashuo
Laboratory Animal Co., Ltd.) at embryonic day 17. The rats were
raised in quiet animal housing for 1 week, without exposure to
strong light, at a temperature of 23°C with 55% humidity, 12-h
day/night cycles and free access to food and water. Cerebral cortex
neurons were separated from rat brains using a standardized
protocol, as described previously (34,35).
The cerebral cortex was dissected from the brain and treated with
0.125% trypsin solution (Sigma-Aldrich; Merck KGaA; cat. no. T4549)
and 0.05% DNase I (Sigma-Aldrich; Merck KGaA; cat. no. DN25) at
37°C for 20 min.
Neurons were suspended in DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.; cat. no. 10099-141) and filtered through a cell
strainer (70 µm; BD Falcon; BD Biosciences; cat. no. 352350). The
dissociated cortical cells were then plated at a density of
1.5×106 cells/well in 6-well plates precoated with 0.1
mg/ml poly-D-lysine (Sigma-Aldrich; Merck KGaA; cat. no. P0899)
overnight at 37°C. The cells were maintained in neurobasal medium
(Gibco; Thermo Fisher Scientific, Inc.; cat. no. 12348-017)
supplemented with 2% B27 (Gibco; Thermo Fisher Scientific, Inc.;
cat. no. 17504-044), 100 U/ml penicillin/streptomycin and 25 mM
GlutaMax (Gibco; Thermo Fisher Scientific, Inc.; cat. no.
35050-061). The neuronal cell cultures were maintained in an
incubator (5% CO2/95% air) at 37°C for 7–12 days in
vitro (DIV 7–12).
All protocols were in accordance with the Care and
Use of Laboratory Animals and the China Council on Animal Care and
the National Institutes of Health guide for the Care and Use of
Laboratory Animals (NIH Publication No. 80-23, revised 1985)
(36), and were approved by the
Animal Ethics Committee of Sichuan University (approval no.
2018013).
OGD/R
OGD/R was induced in primary cortical neurons as
described previously (37,38). At DIV 7, primary cortical neurons
were maintained in glucose-free DMEM (Gibco; Thermo Fisher
Scientific, Inc.; cat. no. 11966-025) in an atmosphere of 5%
CO2 and 0.3% O2 at 37°C for 3 h to mimic
ischemic insult (OGD). After 3 h, OGD was terminated by replacing
the glucose-free DMEM with complete neurobasal medium and
incubating the cultures under normoxic conditions for 24, 48 or 72
h to mimic reperfusion. Cells in the control group were treated
identically, except that these cells were not exposed to OGD/R. All
experiments were repeated three times independently.
Lentiviral vector construction and
lentiviral infection
A lentiviral vector for silencing Huwe1 (pGIPZ
system) was purchased from Open Biosystems, Inc. A third-generation
pseudotype lentivirus encoding green fluorescent protein
(GFP)-expressing Huwe1 short hairpin (sh)RNA (GFP-Huwe1 lentivirus)
was generated as previously described (26,27).
The probe sequence of the shRNA targeting the coding sequence of
Huwe1 (GenBank NC_005120.4; 5′-TCTAGTAGCCAAATTGGAG-3′) was produced
with a three-plasmid system via the transient transfection of 293FT
cells (Invitrogen; Thermo Fisher Scientific, Inc.). The pCMVdr-8.91
and pMD2G plasmids (Invitrogen; Thermo Fisher Scientific, Inc.)
were used as package systems. For lentivirus production, 293FT
cells plated at 70% confluency were cotransfected with
Lipofectamine® 3,000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). The medium was collected 48 and 72
h after transfection, filtered through a 0.45-µm pore size filter
and concentrated using ultracentrifugation at 12,000 × g for 2.5 h
at 4°C. The resulting pellets were resuspended in 150 µl PBS (pH
7.2), pooled and stored at −80°C. The efficiency of infection, as
measured by GFP fluorescence, was >90% for the GFP-Huwe1
lentivirus. Expression of knockdown in infected 293FT cells was
measured for all lentiviruses via reverse
transcription-quantitative PCR (RT-qPCR) using SYBR Green as
described previously (39,40).
The knockdown efficiency of the Huwe1 shRNA
lentivirus was assessed in primary cortical neurons. Primary cell
cultures were infected on the indicated DIV 3 by adding
concentrated lentivirus and 5 µg/ml polybrene (cat. no. AL-118;
Sigma-Aldrich; Merck KGaA,) to the growth medium at a multiplicity
of infection of 0.5. On DIV 3, 3/4 of the culture medium was
removed from the culture wells, and 100 µl Huwe1 shRNA lentivirus,
500 µl fresh normal culture medium and 5 µg/ml polybrene were added
to each well of the 6-well plates. The cells were incubated for 24
h at 37°C, the medium was changed and then the cells were
continuously incubated at 37°C for 4 days before being subjected to
OGD/R. The transduction efficiency was assessed using RT-qPCR and
western blot analysis of Huwe1. The scramble-GFP lentivirus was
used as a control in each experiment and compared with the
GFP-Huwe1 lentivirus.
Drug treatment
The autophagy promoter rapamycin (cat. no. V900930),
the autophagy inhibitor wortmannin (cat. no. W1628) and a JNK
inhibitor (SP600125; cat. no. S5567) were purchased from
Sigma-Aldrich (Merck KGaA) and dissolved in DMSO. Rapamycin (final
concentration of 100 nmol/l) or wortmannin (final concentration of
1 µM) was added to the medium at 37°C for 24 h at the onset of
reperfusion on DIV 7. On DIV 7, SP600125 (final concentration of 10
mM) was added to the medium 30 min before and throughout OGD/R at
37°C. The cells were harvested for subsequent experiments. All
cells were compared with the control group (treated with the same
amount of DMSO at 37°C for 24 h). The cells were harvested 24 h
after reperfusion.
TUNEL assay
Cell apoptosis was assessed using TUNEL staining.
TUNEL staining was performed using an in situ cell death
detection kit (Roche Diagnostics; cat. no. 11684817910) in
accordance with the manufacturer's instructions. Cells fixed on
coverslips with 4% paraformaldehyde for 30 min at room temperature
and then were treated with 0.1% Triton X-100 for 10 min at room
temperature. The cells were washed with PBS and incubated with 50
µl TUNEL reaction mixture at 37°C for 1 h. The cells were then
counterstained with DAPI (1:100) for 10 min at room temperature and
were visualized using an inverted fluorescence microscope (Nikon
Corporation). Cells were mounted with ProLong Gold antifade reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). In total, ten fields
at ×400 magnification were randomly selected to count apoptotic
cells and total cells. The apoptotic index (AI) was calculated as
follows: AI = (number of apoptotic cells/total number of cells
counted) ×100%.
Western blotting
Extracted total proteins were incubated with cold
RIPA lysis buffer (Beyotime Institute of Biotechnology). Protein
samples (20–60 µg) were separated on 10 or 15% SDS-PAGE gels and
blotted onto PVDF membranes (EMD Millipore). The membranes were
blocked with 5% non-fat milk at room temperature for 30 min.
Subsequently, the membranes were incubated with primary antibodies
at 4°C overnight. The primary antibodies were as follows: Rabbit
anti-Huwe1 (1:1,000; Lifespan Biosciences, Inc.; cat. no.
LS-B1359), rabbit anti-ubiquitin (1:1,000; Cell Signaling
Technology, Inc.; cat. no. 3933), rabbit anti-Beclin-1 (1:1,000;
Cell Signaling Technology, Inc.; cat. no. 3495), rabbit
anti-autophagy related (ATG)7 (1:10,000; Abcam; cat. no. ab133528),
rabbit anti-ATG3 (1:10,000; Abcam; cat. no. ab108282), rabbit
anti-ATG5 (1:1,000; Abcam; cat. no. ab109490), rabbit anti-p62
(1:1,000; Cell Signaling Technology, Inc.; cat. no. 16177), rabbit
anti-microtubule associated protein 1 light chain 3 α (LC3;
1:1,000; Cell Signaling Technology, Inc.; cat. no. 4108), rabbit
anti-cleaved caspase 3 (1:1,000; Cell Signaling Technology, Inc.;
cat. no. 9664P), rabbit anti-Bcl2 (1:500; ProteinTech Group, Inc.;
cat. no. 12789-1-AP), rabbit anti-Bax (1:500; ProteinTech Group,
Inc.; cat. no. 50599-2-Ig), rabbit anti-P53 (1:1,000; Abcam; cat.
no. ab183544) and mouse anti-tubulin (1:10,000; Zhengneng
Biotechnology; cat. no. 700608). After washing, the membrane was
incubated with horseradish peroxidase-conjugated secondary antibody
(1:10,000; Invitrogen; Thermo Fisher Scientific, Inc.; cat. no.
A24494) for 1 h at room temperature (RT). Immunoreactivity was
detected with an enhanced chemiluminescence detection reagent (EMD
Millipore) on film according to the manufacturer's instructions.
The optical densities of the bands were quantitatively analyzed
using ImageJ software (version 1.8.0; National Institutes of
Health). The protein levels were normalized to the expression of
tubulin.
Immunofluorescence
Cells were seeded (2×104) on coverslips,
which were placed in 24-well plates. After OGD/R treatment, the
cells were fixed in 4% paraformaldehyde for 10 min at room
temperature, washed three times with PBS (5 min each), treated with
0.2% Triton X-100 for 15 min, blocked with 3% BSA (Sigma-Aldrich;
Merck KGaA) at 4°C for 30 min and then incubated with primary
antibodies [rabbit anti-LC3B, 1:50, ProteinTech Group, Inc., cat.
no. 18725-1-AP; rabbit anti-growth associated protein 43 (Gap-43),
1:200, Cell Signaling Technology, Inc., cat. no. 8945; rabbit
anti-lysosomal associated membrane protein 2 (LAMP2), 1:50,
ProteinTech Group, Inc. cat. no. 10397-1-AP; and mouse
anti-neuron-specific class III β-tubulin (Tuj1), 1:1,250, Chemicon
International; Thermo Fisher Scientific, Inc., cat. no. MAB1637] at
4°C overnight. The coverslips were washed and incubated with
secondary antibodies (1:500) at room temperature for 2 h. The
secondary antibodies included Alexa 594-labeled goat F(ab′)2
anti-(mouse IgG) and Alexa 488-labeled goat F(ab′)2 anti-(rabbit
IgG), purchased from Invitrogen (Thermo Fisher Scientific, Inc.;
cat. nos. A-21208 and A-11016). The nuclei were stained with DAPI
(1:100) at room temperature for 10 min. The fluoresce images were
captured on a Nikon inverted fluorescence microscope
(magnification, ×40) and analyzed using Image Pro Plus software
(version 6.0; Media Cybernetics, Inc.) and Adobe Photoshop software
(version 16.0; Adobe Systems, Inc.).
RT-qPCR
SYBR Green-based RT-qPCR was performed to examine
changes in the mRNA expression of Huwe1. RNA was prepared from
primary rat cortical neurons treated as aforementioned. Total RNA
was isolated using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). Subsequently, total RNA was reverse
transcribed into cDNA using a reverse transcription kit (Takara
Biotechnology Co., Ltd.). The temperature protocol used for reverse
transcription was as follows: 25°C for 10 min, 42°C for 60 min and
95°C for 5 min. Subsequently, qPCR was performed using the ABI
Prism 7500 Sequence Detection system (Thermo Fisher Scientific,
Inc.). For RT-qPCR analysis, the reaction contained cDNA template
(500 ng), 1 µl forward and 1 µl reverse primers and 5 µl SYBR Green
I Master mix (Invitrogen; Thermo Fisher Scientific, Inc.). The
following thermocycling conditions were used for qPCR: 95°C for 60
sec; followed by 45 cycles of 95°C for 15 sec and 60°C for 60 sec.
GAPDH was used as an internal control. The following primers were
used for PCR: Huwe1 forward, 5′-CAAGTAGCCATCAGCAAGA-3′ and reverse,
5′-GTCCTCCAGTTCATTCTCAA-3′; and GAPDH forward,
5′-GCCAAAAGGGTCATCATCTC-3′ and reverse,
5′-GTAGAGGCAGGGATGATGTTC-3′. The mRNA levels were quantified using
the 2−∆∆Cq method and normalized to the internal
reference gene GAPDH (9).
Statistical analysis
Data are presented as the mean ± SEM. Statistical
analyses were performed using SPSS software (version 17.0; SPSS,
Inc.). Data from different time points were compared between groups
using unpaired Student's t-test (for comparisons between two
groups) or one-way ANOVA followed by the Tukey's test (for
comparisons of ≥3 groups). P<0.05 was considered to indicate a
statistically significant difference. Experiments were performed at
least three times.
Results
Silencing Huwe1 increases the
expression of autophagy-related proteins under OGD/R
conditions
OGD/R-induced neuronal injury mimics cerebral I/R
injury in vitro and results in neuronal insult (41). In our previous study, it was found
that primary cortical neuron autophagy increased at 24 h after 3 h
of OGD (42). To observe the
effect of the administration of treatments to interfere with
autophagy, reperfusion for 24 h was selected for further study.
Cortical neurons were treated with the Huwe1 shRNA lentivirus on
DIV 3 and then subjected to OGD for 3 h and reperfusion for 24 h on
DIV 7.
Huwe1 is a large (482 kDa) HECT ubiquitin ligase
(43). It was identified that the
mRNA and protein expression of Huwe1 was significantly increased at
24 h reperfusion after OGD 3 h compared with the control group
(Fig. 1A-C). Compared with
infection with the GFP-scramble lentivirus, infection with the
Huwe1 shRNA lentivirus significantly decreased the protein and mRNA
expression levels of Huwe1 at 24 h reperfusion after 3 h of OGD
(Fig. 1A-C). Moreover, we used
Huwe1 shRNA-treated cortex neuronal cells, and found that
shRNA-Huwe1also decreased the mRNA expression of Huwe1 24 h
post-transfection (Fig. S1).
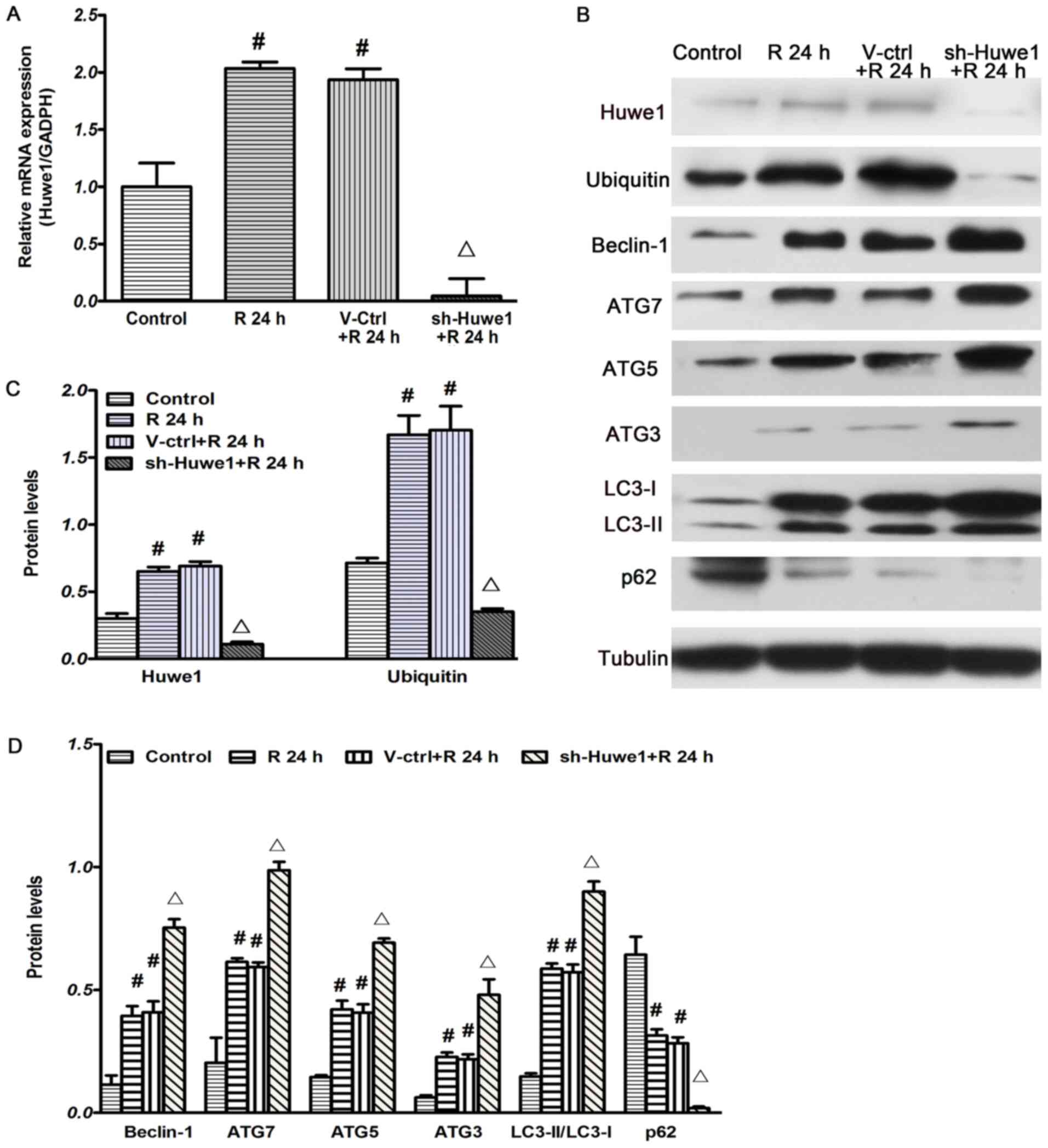 | Figure 1.Silencing Huwe1 induces the
expression of autophagy-related proteins under OGD/R conditions.
Huwe1 shRNA lentivirus was infected into cortical neuronal cells on
DIV 3, and then the cells were subjected to OGD for 3 h and
reperfusion for 24 h on DIV 7. (A) Huwe1 shRNA lentivirus decreased
the mRNA expression of Huwe1 at 24 h after reperfusion, as
measuring via reverse transcription-quantitative PCR, with GAPDH as
an internal control. (B) Huwe1 shRNA decreased the protein
expression levels of (C) Huwe1 and ubiquitin at 24 h after
reperfusion, but significantly increased the expression of (D)
autophagy-related proteins (Beclin-1, ATG7, ATG5, ATG3, LC3 and
p62) compared with the V-ctrl group, as detected using western
blotting. Tubulin was used as an internal control for western
blotting. All experiments were repeated three times independently.
#P<0.05 vs. the control group; ∆P<0.05
vs. the V-ctrl group. DIV, days in vitro; V-ctrl, green
fluorescent protein-scramble control lentivirus-treated group; R,
reperfusion; OGD, Oxygen-glucose deprivation; sh, short hairpin
RNA; Huwe1, HECT, UBA and WWE domain containing E3 ubiquitin
protein ligase 1; ATG, autophagy related; LC3, microtubule
associated protein 1 light chain 3 α. |
Ubiquitin, an 8.5-kDa protein, is activated by E1
and then transferred to E2 before conjugation to its substrate
proteins by E3 ligase (10). The
results demonstrated that the expression of ubiquitin was
significantly increased at 24 h reperfusion after 3 h of OGD
compared with the control group (Fig.
1B and C). Moreover, compared with treatment with the
GFP-scramble lentivirus, infection with the Huwe1 shRNA lentivirus
significantly decreased the protein expression of ubiquitin at 24 h
reperfusion after 3 h OGD.
The influence of Huwe1 on autophagy under OGD/R
conditions was investigated by measuring the expression levels of
autophagy-related proteins during OGD/R. A number of
autophagy-related proteins have been reported to be involved in
autophagy (44), such as ATG5 and
LC3, which exists in two forms, LC3 I and its proteolytic
derivative LC3 II. Furthermore, the conversion of LC3 I (16 kDa) to
LC3 II (14 kDa) indicates ‘LC3 puncta processing’, and the
generation of LC3 II is used to detect autophagic activity
(45). The expression of
autophagy-related proteins (Beclin-1, ATG7, ATG3, ATG5 and
LC3-II/LC3-I) was significantly increased at 24 h reperfusion after
3 h of OGD compared with the control group. Compared with treatment
with the GFP-scramble control lentivirus, infection with Huwe1
shRNA significantly decreased the expression of p62, but
significantly increased the expression of ATG5 and the rate of
LC3-II/LC3-I at 24 h after reperfusion (Fig. 1B and D). In addition, other
autophagy-related proteins, such as Beclin-1, ATG7 and ATG3, were
examined. Significantly increased expression levels of Beclin-1,
ATG7 and ATG3 were identified at 24 h after reperfusion in cortical
neurons treated with Huwe1 shRNA compared with those infected with
the GFP-scramble control lentivirus (Fig. 1B and D). These findings indicated
an enhancement of neuronal autophagy after Huwe1 silencing under
OGD/R injury.
Immunofluorescence results for
LAMP2
Autophagy can be divided into three types:
Chaperone-mediated autophagy (CMA), microautophagy and
macroautophagy. Previous studies have focused on macroautophagy
under ischemic conditions, but there is evidence for the activation
of CMA after ischemia and hypoxia (46–48).
LAMP2 is involved in CMA (48,49).
Therefore, the present study examined the expression of LAMP2 in
cortical neurons using immunofluorescence, and LAMP2 was only
faintly detected in the cytosolic fraction of healthy control
cortical neurons (Fig. 2A).
OGD/R-treated cortical neurons exhibited a significantly increased
average optical value of LAMP2 at 24 h after reperfusion (Fig. 2B and E) compared with the control
cortical neurons. In addition, compared with treatment of
GFP-scramble control lentivirus (Fig.
2C), infection with the Huwe1 shRNA lentivirus significantly
increased the average optical value of LAMP2 at 24 h after OGD
(Fig. 2D and E). These results
suggested that silencing Huwe1 also increased the expression of
LAMP2 under OGD/R injury, as this protein is involved in CMA.
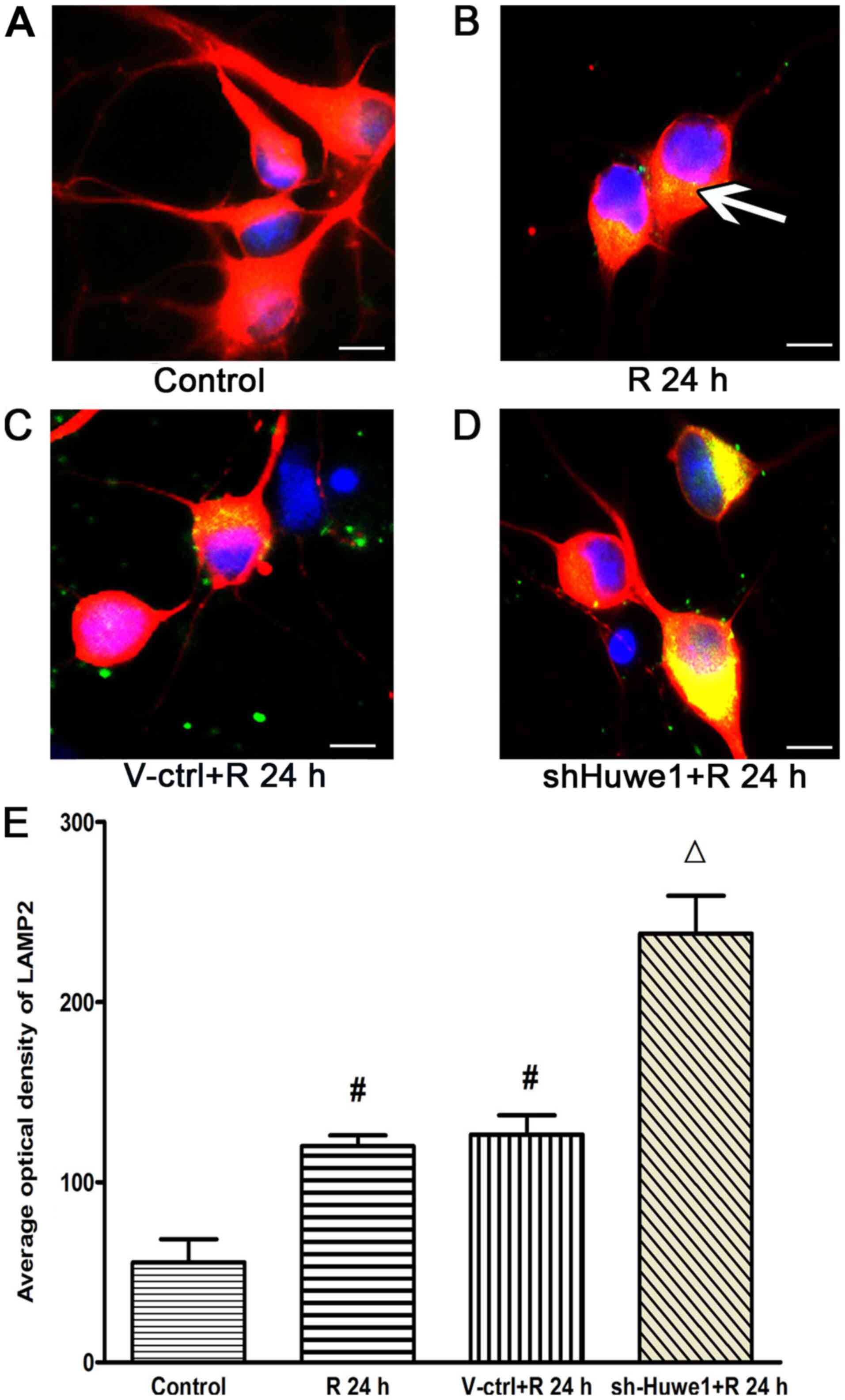 | Figure 2.Silencing Huwe1 affects the
expression of LAMP2, as determined via immunofluorescence. Primary
cortical neurons cultured on coverslips were infected with the
Huwe1 shRNA lentivirus and then exposed to OGD for 3 h and
reperfusion for 24 h. Neuronal cells were stained with LAMP2
(green) and Tuj1 (red) for a double label immunofluorescence assay.
DAPI (blue) was used to stain cell nuclei. LAMP2 immunofluorescence
was observed mainly around the cell cytoplasm in cortical neuronal
cells under (B) OGD/R conditions (indicated by white arrowheads),
compared with (A) normal control cells. Compared with cells (C)
treated with the GFP scramble lentivirus control, (D) infection
with Huwe1 shRNA markedly increased the expression of LAMP2 at 24
h. Scale bar, 10 µm. (E) Quantification of immunofluorescence
results. All experiments were repeated three times independently.
#P<0.05 vs. control group; ∆P<0.05 vs.
V-ctrl group. V-ctrl, green fluorescent protein-scramble control
lentivirus-treated group; R, reperfusion; OGD, Oxygen-glucose
deprivation; sh, short hairpin RNA; Huwe1, HECT, UBA and WWE domain
containing E3 ubiquitin protein ligase 1; LAMP, lysosomal
associated membrane protein 2. |
Silencing Huwe1 and inhibiting the JNK
pathway affects autophagy under OGD/R conditions
The potential mechanisms underlying the regulation
of Huwe1 in autophagy under OGD/R conditions were examined.
Healthy neurons in the control group developed
normal polarity, exhibiting a single axon and multiple dendrites,
and LC3B was at a basal level in normal neurons (Fig. 3A). Compared with these normal
cortical neurons, OGD/R-treated cortical neurons exhibited a
significantly increased average optical value of LC3B at 24 h after
reperfusion (Fig. 3B and M).
Silencing Huwe1 significantly increased the expression of LC3B at
24 h after OGD/R, compared with the scramble control (Fig. 3C, D and M). The cell body and
proximal end of neurites became shortened and degraded at 24 h
after OGD in the OGD/R + shRNA-Huwe1 group (Fig. 3D).
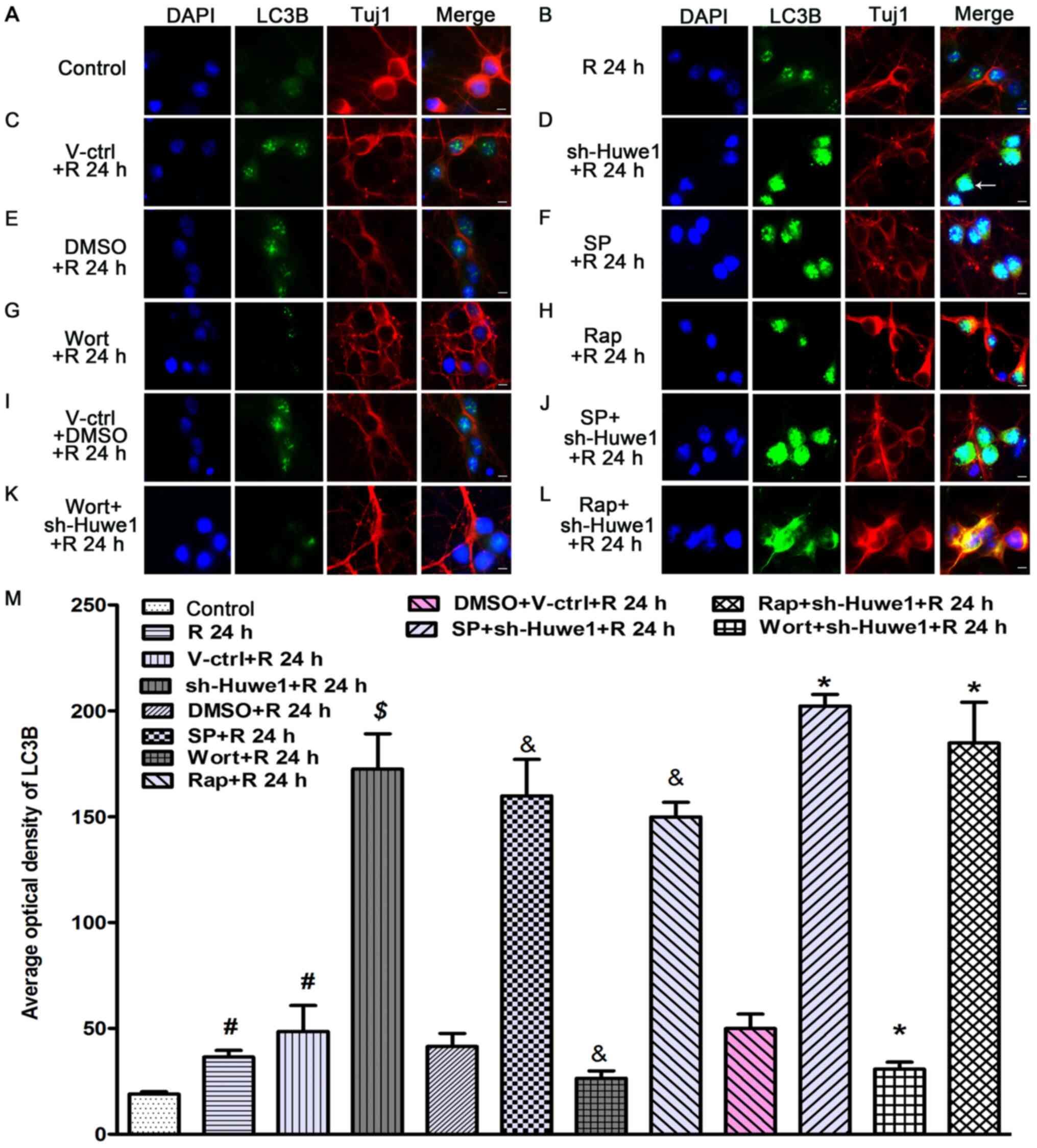 | Figure 3.Immunofluorescence staining results
for Tuj1 and LC3B. Cultured cortical neurons were infected with
Huwe1 shRNA with or without an autophagy inhibitor, an autophagy
inducer or a JNK pathway inhibitor, and then subjected to OGD/R for
24 h. Neuronal cells were stained with LC3B (green) and Tuj1 (red)
for a double label immunofluorescence assay. DAPI (blue) was used
to stain the cell nuclei. (A) Healthy, control neurons had normal
polarity, with most of the cells having a single axon and multiple
dendrites. LC3B was mainly localized to the cell nucleus in (B)
cortical neuronal cells under OGD/R conditions compared with
healthy control neurons. The cell bodies and proximal end of
neurites became shortened and degraded at 24 h after OGD. (C)
Compared with the control lentivirus, (D) treatment with Huwe1
shRNA increased the expression of LC3B. (E) Compared with DMSO, (F)
the JNK inhibitor or (H) an autophagy inducer increased the
expression of LC3B, as well as shortened neuronal axons at 24 h
after OGD. (I) Compared with cotreatment with the control
lentivirus and DMSO, (J) cotreatment with Huwe1 shRNA and a JNK
inhibitor also increased the level of LC3B at 24 h. Compared with
(E) DMSO, (G) Wort decreased the expression of LC3B and slightly
reduced neurite degradation at 24 h after OGD. Relatively intact
structure and polarity of some neurons was observed at 24 h upon
(K) cotreatment with Huwe1 shRNA and Wort. (L) Cotreatment with
Huwe1 shRNA and autophagy inducer also increased the level of LC3B
at 24 h. Scale bar, 10 µm (magnification, ×60). (M) Quantitation of
the staining results. #P<0.05 vs. control group;
$P<0.05 vs. V-ctrl group; &P<0.05
vs. DMSO group; *P<0.05 vs. the DMS + V-ctrl group. Wort,
wortmannin; V-ctrl, green fluorescent protein-scramble control
lentivirus-treated group; R, reperfusion; OGD, Oxygen-glucose
deprivation; sh, short hairpin RNA; Huwe1, HECT, UBA and WWE domain
containing E3 ubiquitin protein ligase 1; LC3, microtubule
associated protein 1 light chain 3 α; Tuj1, neuron-specific class
III β-tubulin; SP, SP600125; Rap, Rapamycin. |
The effects of the administration of treatments to
interfere with autophagy on the potential mechanisms underlying the
regulation of Huwe1 in autophagy were also evaluated. Compared with
DMSO (Fig. 3E), SP600125
significantly increased the expression of LC3B under OGD/R
conditions at 24 h after reperfusion (Fig. 3F and M). Compared with cotreatment
with DMSO and the scramble control lentivirus (Fig. 3I), cotreatment with SP600125 and
Huwe1 shRNA also significantly increased the level of LC3B at 24 h
and caused neuronal cells to lose polarity (Fig. 3J and M). Rapamycin significantly
enhanced the expression of LC3B at 24 h after reperfusion compared
with DMSO (Fig. 3H and M). It was
also found that, compared with cotreatment with DMSO and the
scramble control lentivirus, cotreatment with rapamycin and Huwe1
shRNA significantly enhanced the expression of LC3B at 24 h
(Fig. 3L and M), as well as caused
neuronal cells to lose polarity. The autophagy inhibitor wortmannin
decreased the average optical value of LC3B at 24 h after 3 h of
OGD compared with DMSO treatment (Fig.
3G). Moreover, compared with cotreatment with DMSO and the
scramble control lentivirus, cotreatment with wortmannin and Huwe1
shRNA inhibited the expression of LC3B at 24 h, and maintained the
relatively intact structure and polarity of some neurons after OGD
(Fig. 3K). Thus, a decrease in
activated JNK may be involved in the effect of Huwe1 on autophagy
under OGD/R injury.
Autophagy modulates hypoxia-induced
apoptosis under OGD/R conditions
Autophagy triggers either apoptosis or necrotic cell
death (50), and LC3, as a key
molecule involved in autophagy, is required for autophagic cell
death (4,24). The present study administered the
autophagy promoter, rapamycin, and the autophagy inhibitor,
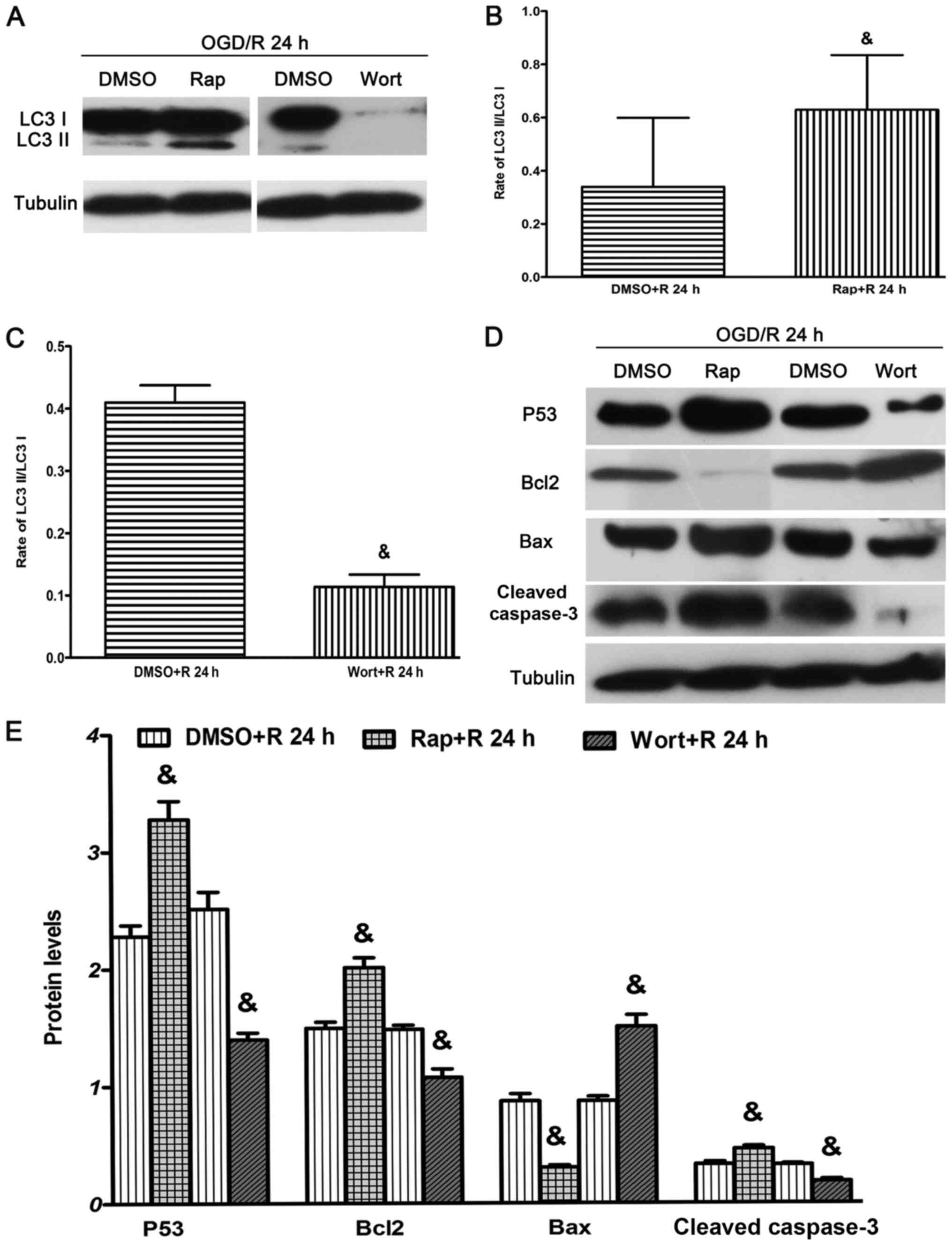
wortmannin, under OGD/R conditions. It was demonstrated that
rapamycin significantly increased the ratio of LC3-II/LC3-I at 24 h
after reperfusion (Fig. 4A and C).
However, wortmannin significantly decreased the ratio of
LC3-II/LC3-I at 24 h after reperfusion (Fig. 4A and D).
Apoptosis is involved in DNA damage and involves
various regulators of the cell cycle, including P53, Bcl2 and Bax,
and the protein downstream cleaved caspase 3. Bcl2 is an
anti-apoptotic protein, while Bax is a pro-apoptotic protein that
belongs to the Bcl2 family of evolutionarily related proteins
(51–54). The results suggested that rapamycin
significantly increased the expression levels of P53, Bcl2 and
cleaved caspase 3, but significantly decreased the expression of
Bax at 24 h after reperfusion (Fig. 4B
and E) compared with treatment with DMSO. Moreover, wortmannin
significantly reduced the expression levels of P53, Bcl2 and
cleaved caspase 3, but increased the expression of Bax at 24 h
after reperfusion (Fig. 4B and E)
compared with treatment with DMSO.
Subsequently, neurons were prepared to detect
apoptosis using TUNEL staining at 24 h after OGD, and the results
were quantified (Fig. 5J). An
increased level of autophagy is harmful to neuronal cells in the
context of OGD/R (54). OGD
induced neuron cell apoptosis at 24 h compared with the control
group (Fig. 5A). Compared with the
scramble control lentivirus (Fig.
5B; AI, 41.29±1.82%), Huwe1 shRNA significantly decreased the
number of TUNEL-positive cells (Fig.
5C; AI, 23.01±2.03%). Compared with treatment with DMSO
(Fig. 5D; AI, 40.66±4.17%),
treatment with wortmannin significantly decreased neuronal
apoptosis at 24 h after OGD/R (Fig.
5E; AI, 21.45±8.97%). Moreover, compared with cotreatment with
DMSO and the scramble control lentivirus, infection with Huwe1
shRNA and treated with wortmannin significantly decreased the
number of TUNEL-positive cells (Fig.
5H; AI, 23.20±2.96%). Compared with treatment with DMSO (AI,
40.66±4.17%), treatment with SP600125 (Fig. 5F; AI, 21.34±8.05) also
significantly decreased the number of TUNEL-positive cells at 24 h
after OGD. The results demonstrate that, compared with cotreatment
with DMSO and the scramble control lentivirus (Fig. 5G; AI, 42.05±10.07%), cotreatment
with Huwe1 shRNA and SP600125 (Fig.
5I; AI, 17.15±8.25%) significantly decreased the number of
TUNEL-positive cells.
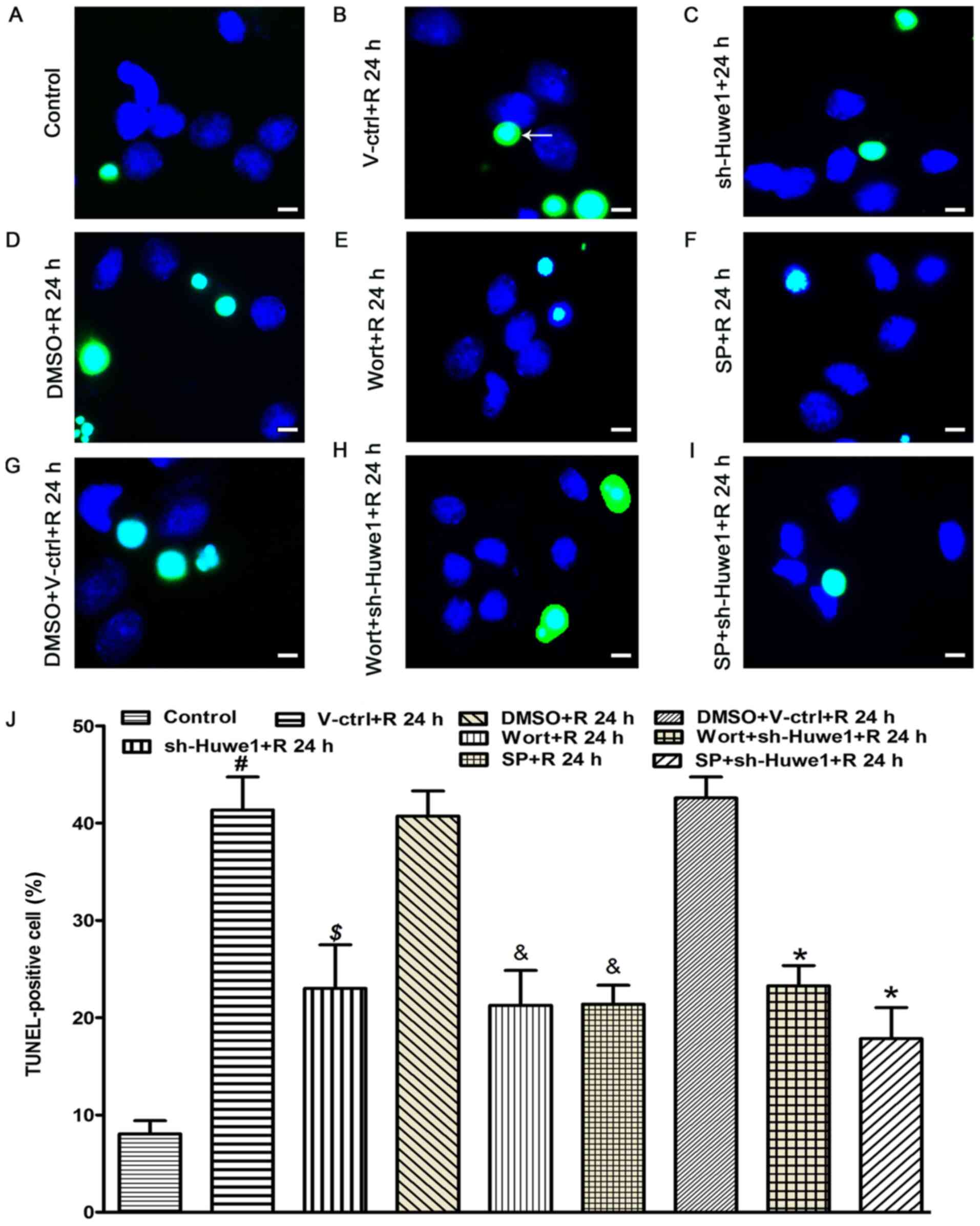 | Figure 5.Apoptosis of neurons, as determined
via TUNEL assay. Primary cortex neuronal cells were exposed to OGD
for 3 h and reperfusion for 24 h. Neuronal cells were treated with
the autophagy inhibitor Wort at the onset of reperfusion or a JNK
inhibitor (SP). Apoptotic neurons were stained with TUNEL (green)
for the fluorescence assay. DAPI (blue) was used to stain cell
nuclei. Compared with (A) healthy control cortical neurons,
TUNEL-positive cells (indicated by white arrowheads) were detected
in (B) cortical neurons at 24 h after OGD. (C) Treatment with Huwe1
shRNA decreased neuronal apoptosis at 24 h after OGD. Compared with
(D) DMSO treated cells, (E) Wort and a (F) JNK inhibitor reduced
cellular apoptosis at 24 h after OGD. (G) Compared with the DMSO +
V-ctrl group, (H) cotreatment with Huwe1 shRNA and Wortmannin also
reduced cellular apoptosis at 24 h. (I) Cotreatment with Huwe1
shRNA and JNK inhibitor decreased neuronal apoptosis at 24 h. Scale
bar, 10 µm. (J) Quantification of TUNEL assay results. All
experiments were repeated three times independently.
#P<0.05 vs. control group; $P<0.05 vs.
V-ctrl group; &P<0.05 vs. DMSO group; *P<0.05
vs. DMSO + V-ctrl group. R, reperfusion; OGD, Oxygen-glucose
deprivation; Wort, wortmannin; SP, SP600125; V-ctrl, green
fluorescent protein-scramble control lentivirus-treated group; sh,
short hairpin RNA; Huwe1, HECT, UBA and WWE domain containing E3
ubiquitin protein ligase 1. |
Immunofluorescence results for
Gap-43
Gap-43 is abundantly synthesized, especially in
growth cones that are associated with axonal growth, and serves a
critical role in neuronal differentiation, plasticity and
regeneration (55). It was
identified that OGD/R increased the average optical value of Gap-43
at 24 h reperfusion after 3 h of OGD compared with normal cortical
neurons (Fig. 6B). Huwe1 silencing
(Fig. 6D) and the autophagy
inhibitor wortmannin (Fig. 6F)
also increased the average optical value of Gap-43 at 24 h
reperfusion after 3 h of OGD. However, compared with cotreatment
with DMSO and the scramble control lentivirus (Fig. 6G), cotreatment with wortmannin and
Huwe1 shRNA did not increase the expression of Gap-43 at 24 h after
OGD (Fig. 6H).
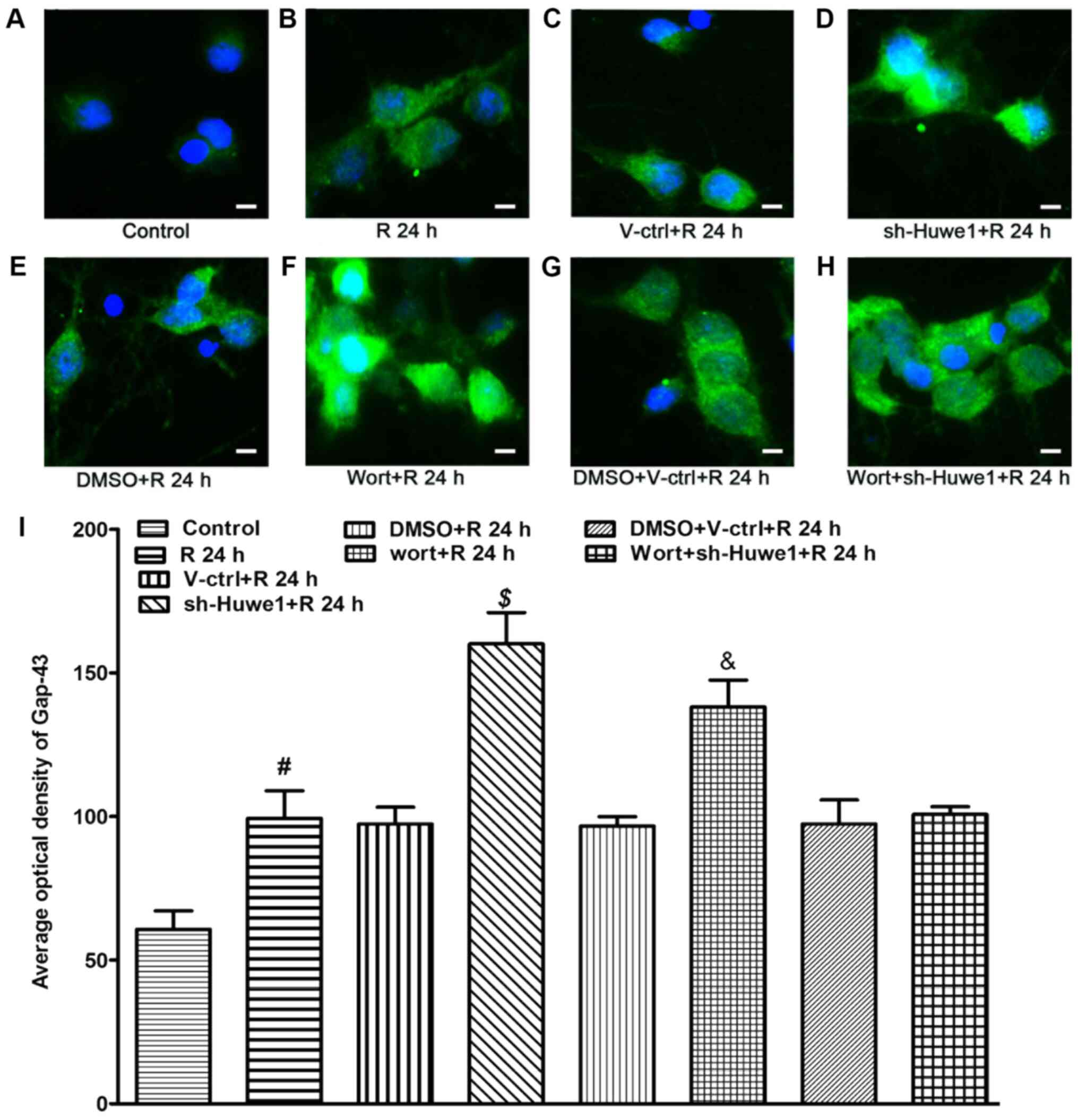 | Figure 6.Immunofluorescence staining results
for Gap-43. Neuronal cells were treated with Huwe1 shRNA with or
without the autophagy inhibitor Wort. Neuronal cells were stained
with Gap-43 (green), and DAPI (blue) was used to stain cell nuclei.
(A) Compared with normal control cells, (B) the expression of
Gap-43 at 24 h after OGD was increased. (C) Compared with the
control lentivirus, (D) treatment with Huwe1 shRNA also increased
the level of Gap-43 at 24 h. (E) Compared with DMSO, (F) an
autophagy inhibitor (Wort) increased the expression of Gap-43 at 24
h. (G) Compared with cotreatment with DMSO and the green
fluorescent protein scramble lentivirus, (H) cotreatment with Huwe1
shRNA and wortmannin induced the relative expression of Gap-43 at
24 h. Scale bar, 10 µm. (I) Quantitation of the staining results.
All experiments were repeated three times independently.
#P<0.05 vs. control group; $P<0.05 vs.
V-ctrl group; &P<0.05 vs. DMSO group. R,
reperfusion; OGD, Oxygen-glucose deprivation; Wort, wortmannin;
V-ctrl, green fluorescent protein-scramble control
lentivirus-treated group; sh, short hairpin RNA; Huwe1, HECT, UBA
and WWE domain containing E3 ubiquitin protein ligase 1; Gap-43,
growth associated protein 43. |
Discussion
The UPS and autophagy were previously suggested to
be networks with separate components, mechanisms and substrate
selectivity (13). Recently,
however, studies have reported that crosstalk between autophagy and
the UPS occurs at several molecular and functional sites shared
between these pathway, as either regulators or substrates (7,12,56).
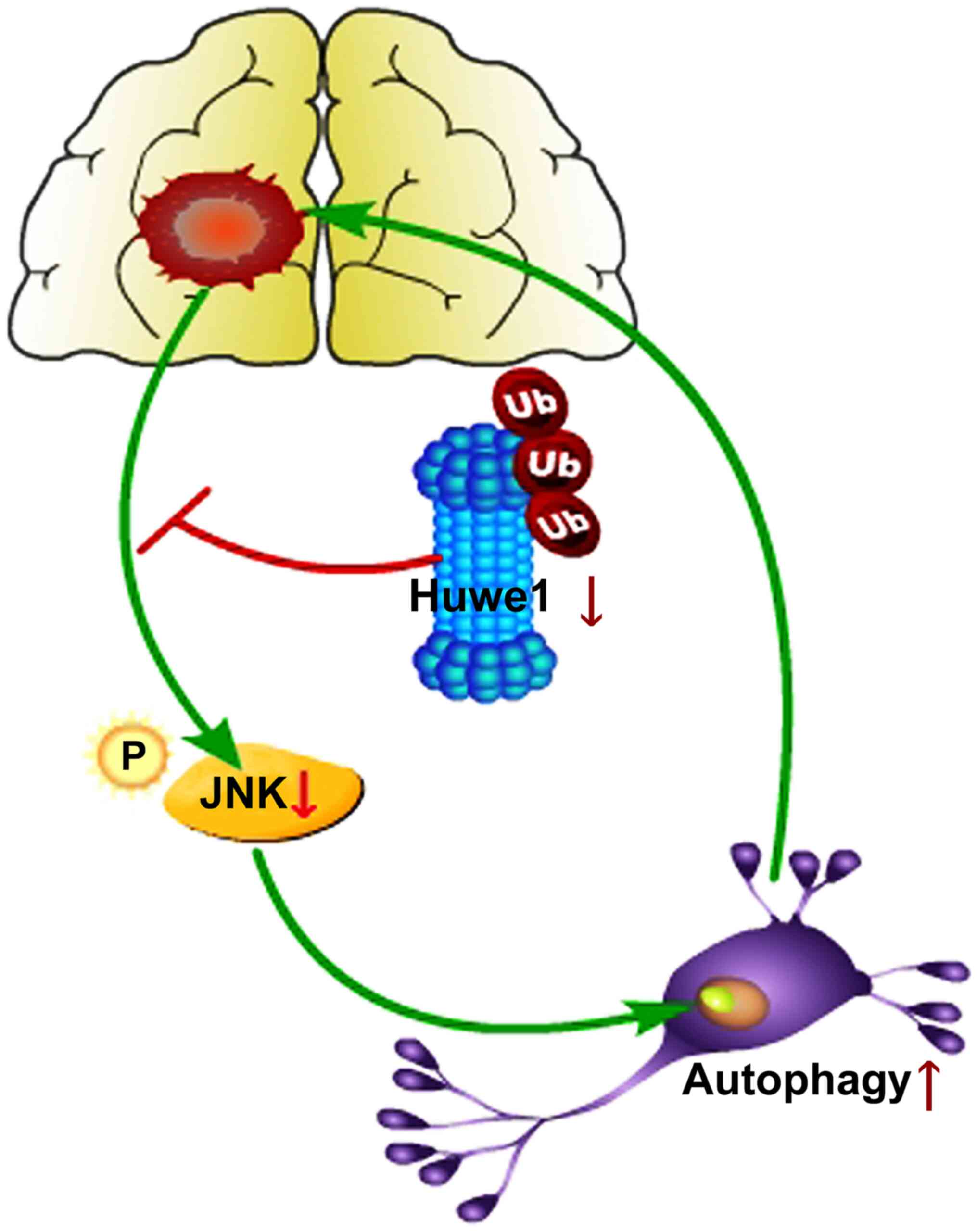
In the present study, it was demonstrated that: i) Silencing Huwe1
induced neuronal autophagy at 24 h after OGD/R; ii) silencing Huwe1
increased neuronal autophagy via the JNK pathway under OGD/R
conditions; and iii) silencing Huwe1 and inhibiting autophagy
affected apoptosis at 24 h after OGD/R. Based on these findings,
the present study proposed a model that involves a role for JNK in
the regulation of autophagy when there is dysfunction in Huwe1
(Fig. 7).
The UPS regulates numerous cellular processes and
serves a key role in various stress conditions, such as ischemia
(17). Autophagy can act as a
compensatory mechanism upon the impairment of proteasomal
degradation (7,57,58).
The UPS comprises the E1, E2 and E3 enzyme cascades. A previous
study revealed that E3 ligases, including the EI24-mediated
degradation of RING-domain E3 ligases, serves a critical role at
the interface between the UPS and autophagy by mediating the
proteasomal degradation of a subset of substrates (13). Another study observed that Huwe1
encodes a HECT domain ubiquitin ligase and is important for
neurogenesis in the cerebral cortex (19). The present results suggested that
silencing Huwe1 increased autophagy under OGD/R conditions.
Autophagy, closely resembling the UPS, is achieved
via the combined action of several ATGs (59). Moreover, Guo et al (59) demonstrated that when the UPS is
impaired, autophagy is activated. In the current study, treatment
with Huwe1 shRNA increased ATG7, ATG5 and ATG3 expression levels
after reperfusion for 24 h. LC3-II is the cleaved form of LC3-I,
and an increased LC3-II/LC3-I ratio reflects active autophagy
(24,60). The ratio of LC3-II/LC3-I was also
increased by treatment with shRNA-Huwe1 under OGD/R conditions in
the present study, and thus the results suggested that silencing
the expression of Huwe1 induced autophagy under OGD/R conditions.
It was also identified that silencing Huwe1 increased the
expression of LAMP2 under OGD/R conditions. Therefore, the current
findings indicated that autophagy can act as a compensatory
mechanism upon the impairment of the E3 ligase Huwe1. However,
there is limited evidence regarding the precise mechanism(s) of
this crosstalk.
JNK is an important component of mitogen-activated
protein kinase (MAPK) cascades, which serves an important role in
transducing signals involved in autophagy (46,49,61–63).
Our previous study also revealed that OGD/R-treated neurons exhibit
decreased phosphorylation of JNK at 24 h compared with normal
neuronal cells (42). Treatment
with the Huwe1 shRNA lentivirus also decreases the ratio of
phosphorylated-JNK/JNK upon reperfusion for 24 h after 3 h of OGD,
while total JNK content is unchanged (50). Tuj1 is a somatodendritic marker of
dendrites and axons, while LC3B has been used to monitor autophagy
via measuring increases in punctate LC3 (61).
For example, previous studies of non-neuronal cells
have implicated JNK in the induction of autophagy (64–66).
In contrast to the pro-autophagic role of JNK in non-neuronal
cells, neuronal JNK suppresses autophagy (67). In the current study, it was
identified that the loss of neuronal JNK function caused the
activation of autophagy. Moreover, the present results suggested
that silencing Huwe1 decreased the phosphorylation of JNK under
OGD/R conditions. It was also demonstrated that cotreatment with a
JNK inhibitor and Huwe1 shRNA significantly increased the
expression levels of autophagy-related proteins. Collectively,
these findings indicated that increased autophagy in
Huwe1-deficient neurons may be mediated via the JNK pathway under
OGD/R conditions.
The current results demonstrated that the induction
of autophagy was accompanied by the compensatory silencing of Huwe1
under OGD/R conditions. Whether increased autophagic activity
serves a protective or harmful role is controversial. Autophagy is
a double-edged sword in ischemia (24,25,68–70).
Previous studies have reported that excessive autophagy in cerebral
ischemia contributes to neuronal death, including autophagic cell
death. Furthermore, the inhibition of excessive autophagy can
attenuate cerebral ischemia-associated neuronal damage (71–73).
Sciarretta et al (27)
revealed that autophagy was beneficial during ischemia but harmful
during reperfusion. In addition, a number of studies have reported
that autophagy can serve as a trigger of apoptosis or necrosis, as
well as act as an independent mechanism of cell death (24,53,54,74).
Silencing Huwe1 or inhibiting autophagy favors cell
survival under OGD/R conditions. However, silencing Huwe1 also
induces autophagy. Under OGD/R conditions, neuronal recruitment in
autophagy is accompanied by Huwe1 degradation in response to I/R
injury (17,42,71,75).
Delgado et al (52) showed
that a three-part signaling system involving autophagy, the UPS and
apoptosis balances cell death vs. survival decisions. There are
three main axes of the three-part signaling system
(autophagy-apoptosis, UPS-apoptosis and autophagy-UPS), and
crosstalk in the system is bidirectional (52). However, whether there is a
three-part signaling system or feedback mechanism between Huwe1,
autophagy and apoptosis under OGD/R conditions requires further
investigation. Autophagy and apoptosis share several common
modulators and signaling pathways, and may antagonize or promote
one another in certain cases, such as brain injury and ischemia
(73). Previous studies have
suggested that JNK inhibitors effectively reduce I/R-induced cell
death (76). In a previous study,
it was identified that silencing Huwe1 decreased the level of p-JNK
under OGD/R conditions (50). A
JNK inhibitor increased the expression of autophagy-related
proteins but decreased the expression levels of apoptosis-related
proteins. p-JNK may be a key mediator of the antagonistic
interaction between apoptosis and autophagy in response to UPS
impairment in primary cortical neuronal cells (61). However, the role of p-JNK in the
three-part signaling system of autophagy, the UPS and apoptosis to
balance cell death vs. survival decisions in response to Huwe1
impairment under OGD/R conditions requires further
investigation.
Huwe1 is involved in the ubiquitination and
degradation of multiple proteins, including P53 (19). Proteasome inhibition in neurons has
been shown to induce autophagy via the metabolic stabilization of
the transcription factor P53 (9,61,74).
Recently, it has been revealed that apoptosis signaling exhibits
substantial molecular crosstalk with the UPS, as well as autophagy.
Whether there is a compensatory role for autophagy following the
impairment of Huwe1 via the polyubiquitination of P53, and if P53
is involved in the three-part signaling system of Huwe1, autophagy
and apoptosis under OGD/R conditions is yet to be elucidated. Based
on these aforementioned findings, further studies will examine the
levels of the autophagy-related proteins upon the co-administration
of small interfering RNA of P53 and Huwe1.
In conclusion, the present results suggested that
Huwe1 silencing was accompanied by a compensatory induction of
autophagy under OGD/R conditions. The JNK pathway may be a key
mediator of the interaction between Huwe1 and autophagy in response
to UPS impairment. However, in the context of brain ischemia, the
precise mechanisms underlying the interaction between autophagy and
Huwe1 require further clarification, and an additional
comprehensive study is necessary.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by The New Bud
Research Foundation of West China Second University Hospital of
China (grant no. kx100) and the Science and Technology Support
Projects in Sichuan Province (grant no. 2019YFS0411).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GQH, WZ and GLH conceived and designed the study.
GQH, YC and HJL performed the experiments. WZ and WMX performed the
data analysis. GHQ and GLH wrote the manuscript. WZ and GLH revised
the article. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Ethics
Committee of Sichuan University (approval no. 2018013).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
UPS
|
ubiquitin proteasome system
|
|
I/R
|
ischemia/reperfusion injury
|
|
OGD/R
|
Oxygen-glucose deprivation and
reperfusion
|
References
|
1
|
Dikic I: Proteasomal and autophagic
degradation systems. Annu Rev Biochem. 86:193–224. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ardley HC and Robinson PA: E3 ubiquitin
ligases. Essays Biochem. 41:15–30. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Finley D: Recognition and processing of
ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem.
78:477–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zaffagnini G and Martens S: Mechanisms of
selective autophagy. J Mol Biol. 428:1714–1724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martens S and Behrends C: Molecular
mechanisms of selective autophagy. J Mol Biol. 432:1–2. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schreiber A and Peter M: Substrate
recognition in selective autophagy and the ubiquitin-proteasome
system. Biochim Biophys Acta. 1843:163–181. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ji CH and Kwon YT: Crosstalk and interplay
between the ubiquitin-proteasome system and autophagy. Mol Cells.
40:441–449. 2017.PubMed/NCBI
|
|
8
|
Bao X, Ren T, Huang Y, Ren C, Yang K,
Zhang H and Guo W: Bortezomib induces apoptosis and suppresses cell
growth and metastasis by inactivation of Stat3 signaling in
chondrosarcoma. Int J Oncol. 50:477–486. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lagunas-Martinez A, Garcia-Villa E,
Arellano-Gaytán M, Contreras-Ochoa CO, Dimas-González J,
López-Arellano ME, Madrid-Marina V and Gariglio P: MG132 plus
apoptosis antigen-1 (APO-1) antibody cooperate to restore p53
activity inducing autophagy and p53-dependent apoptosis in HPV16
E6-expressing keratinocytes. Apoptosis. 22:27–40. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kwon YT and Ciechanover A: The ubiquitin
code in the ubiquitin-proteasome system and autophagy. Trends
Biochem Sci. 42:873–886. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nedelsky NB, Todd PK and Taylor JP:
Autophagy and the ubiquitin-proteasome system: Collaborators in
neuroprotection. Biochim Biophys Acta. 1782:691–699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan T, Huang Z, Chen L, Wang W, Zhang B,
Xu Y, Pan S, Mao Z, Hu H and Geng Q: Associations between
autophagy, the ubiquitin-proteasome system and endoplasmic
reticulum stress in hypoxia-deoxygenation or ischemia-reperfusion.
Eur J Pharmacol. 791:157–167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nam T, Han JH, Devkota S and Lee HW:
Emerging paradigm of crosstalk between autophagy and the
ubiquitin-proteasome system. Mol Cells. 40:897–905. 2017.PubMed/NCBI
|
|
14
|
Zaffagnini G, Savova A, Danieli A, Romanov
J, Tremel S, Ebner M, Peterbauer T, Sztacho M, Trapannone R,
Tarafder AK, et al: p62 filaments capture and present ubiquitinated
cargos for autophagy. EMBO J. 37:e983082018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
French ME, Klosowiak JL, Aslanian A, Reed
SI, Yates JR III and Hunter T: Mechanism of ubiquitin chain
synthesis employed by a HECT domain ubiquitin ligase. J Biol Chem.
292:10398–10413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao X, D'Arca D, Lim WK, Brahmachary M,
Carro MS, Ludwig T, Cardo CC, Guillemot F, Aldape K, Califano A, et
al: The N-Myc-DLL3 cascade is suppressed by the ubiquitin ligase
Huwe1 to inhibit proliferation and promote neurogenesis in the
developing brain. Dev Cell. 17:210–221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao X, Heng JI, Guardavaccaro D, Jiang R,
Pagano M, Guillemot F, Iavarone A and Lasorella A: The HECT-domain
ubiquitin ligase Huwe1 controls neural differentiation and
proliferation by destabilizing the N-Myc oncoprotein. Nat Cell
Biol. 10:643–653. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
The MULE/HUWE1 E3 ubiquitin ligase is a
tumor suppressor. Cancer Discov. 3:OF322013.PubMed/NCBI
|
|
19
|
Zhou J, Liu Q, Mao M and Tong Y: Huwe1 as
a therapeutic target for neural injury. Genet Mol Res.
13:4320–4325. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y, Luo Y, Luo T, Lu B, Wang C, Zhang Y,
Piao M, Feng C and Ge P: Trehalose inhibits protein aggregation
caused by transient ischemic insults through preservation of
proteasome activity, not via induction of autophagy. Mol Neurobiol.
54:6857–6869. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Min JW, Lü L, Freeling JL, Martin DS and
Wang H: USP14 inhibitor attenuates cerebral
ischemia/reperfusion-induced neuronal injury in mice. J Neurochem.
140:826–833. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baptista MS, Duarte CB and Maciel P: Role
of the ubiquitin-proteasome system in nervous system function and
disease: Using C. elegans as a dissecting tool. Cell Mol Life Sci.
69:2691–2715. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang P, Shao BZ, Deng Z, Chen S, Yue Z and
Miao CY: Autophagy in ischemic stroke. Prog Neurobiol.
163-164:98–117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rami A and Kögel D: Apoptosis meets
autophagy-like cell death in the ischemic penumbra: Two sides of
the same coin? Autophagy. 4:422–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wolf MS, Bayir H, Kochanek PM and Clark
RSB: The role of autophagy in acute brain injury: A state of flux?
Neurobiol Dis. 122:9–15. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sciarretta S, Hariharan N, Monden Y,
Zablocki D and Sadoshima J: Is autophagy in response to ischemia
and reperfusion protective or detrimental for the heart? Pediatr
Cardiol. 32:275–281. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan W, Zhang H, Bai X, Lu Y, Dong H and
Xiong L: Autophagy activation is involved in neuroprotection
induced by hyperbaric oxygen preconditioning against focal cerebral
ischemia in rats. Brain Res. 1402:109–121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang WW, Huang BS, Han Y, Deng LH and Wu
LX: Sodium hydrosulfide attenuates cerebral ischemia/reperfusion
injury by suppressing overactivated autophagy in rats. FEBS Open
Bio. 7:1686–1695. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Su J, Zhang T, Wang K, Zhu T and Li X:
Autophagy activation contributes to the neuroprotection of remote
ischemic perconditioning against focal cerebral ischemia in rats.
Neurochem Res. 39:2068–2077. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qi Z, Dong W, Shi W, Wang R, Zhang C, Zhao
Y, Ji X, Liu KJ and Luo Y: Bcl-2 phosphorylation triggers autophagy
switch and reduces mitochondrial damage in limb remote ischemic
conditioned rats after ischemic stroke. Transl Stroke Res.
6:198–206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wei K, Wang P and Miao CY: A double-edged
sword with therapeutic potential: An updated role of autophagy in
ischemic cerebral injury. CNS Neurosci Ther. 18:879–886. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L,
Su L and Zhang Y: Inhibition of autophagy contributes to ischemic
postconditioning-induced neuroprotection against focal cerebral
ischemia in rats. PLoS One. 7:e460922012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu SY, Wu YM, Ji Z, Gao XY and Pan SY: A
modified technique for culturing primary fetal rat cortical
neurons. J Biomed Biotechnol. 2012:8039302012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fath T, Ke YD, Gunning P, Götz J and
Ittner LM: Primary support cultures of hippocampal and substantia
nigra neurons. Nat Protoc. 4:78–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Z, Chen Z, Wang J, Zhang M, Li Z, Wang
S, Dong B, Zhang C, Gao J and Shen L: Mouse avatar models of
esophageal squamous cell carcinoma proved the potential for
EGFR-TKI afatinib and uncovered Src family kinases involved in
acquired resistance. J Hematol Oncol. 11:1092018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gertz K, Kronenberg G, Kälin RE, Baldinger
T, Werner C, Balkaya M, Eom GD, Hellmann-Regen J, Kröber J, Miller
KR, et al: Essential role of interleukin-6 in post-stroke
angiogenesis. Brain. 135:1964–1980. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jones SM, Novak AE and Elliott JP: Primary
culture of cellular subtypes from postnatal mouse for in vitro
studies of oxygen glucose deprivation. J Neurosci Methods.
199:241–248. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ding B and Kilpatrick DL: Lentiviral
vector production, titration, and transduction of primary neurons.
Methods Mol Biol. 1018:119–131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Reich A, Spering C, Gertz K, Harms C,
Gerhardt E, Kronenberg G, Nave KA, Schwab M, Tauber SC, Drinkut A,
et al: Fas/CD95 regulatory protein Faim2 is neuroprotective after
transient brain ischemia. J Neurosci. 31:225–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nagai N, Yamamoto S, Tsuboi T, Ihara H,
Urano T, Takada Y, Terakawa S and Takada A: Tissue-type plasminogen
activator is involved in the process of neuronal death induced by
oxygen-glucose deprivation in culture. J Cereb Blood Flow Metab.
21:631–634. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
He G, Xu W, Tong L, Li S, Su S, Tan X and
Li C: Gadd45b prevents autophagy and apoptosis against rat cerebral
neuron oxygen-glucose deprivation/reperfusion injury. Apoptosis.
21:390–403. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhong Q, Gao W, Du F and Wang X:
Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the
polyubiquitination of Mcl-1 and regulates apoptosis. Cell.
121:1085–1095. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu F, Gu JH and Qin ZH: Neuronal autophagy
in cerebral ischemia. Neurosci Bull. 28:658–666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hale AN, Ledbetter DJ, Gawriluk TR and
Rucker EB III: Autophagy: Regulation and role in development.
Autophagy. 9:951–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li W, Zhu J, Dou J, She H, Tao K, Xu H,
Yang Q and Mao Z: Phosphorylation of LAMP2A by p38 MAPK couples ER
stress to chaperone-mediated autophagy. Nat Commun. 8:17632017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bejarano E and Cuervo AM:
Chaperone-mediated autophagy. Proc Am Thorac Soc. 7:29–39. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Penke B, Bogár F, Crul T, Sántha M, Tóth
ME and Vígh L: Heat shock proteins and autophagy pathways in
neuroprotection: From molecular bases to pharmacological
interventions. Int J Mol Sci. 19:3252018. View Article : Google Scholar
|
|
49
|
Zhang Q, Bian H, Guo L and Zhu H:
Pharmacologic preconditioning with berberine attenuating
ischemia-induced apoptosis and promoting autophagy in neuron. Am J
Transl Res. 8:1197–1207. 2016.PubMed/NCBI
|
|
50
|
Lalaoui N, Lindqvist LM, Sandow JJ and
Ekert PG: The molecular relationships between apoptosis, autophagy
and necroptosis. Semin Cell Dev Biol. 39:63–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Thorburn A: Apoptosis and autophagy:
Regulatory connections between two supposedly different processes.
Apoptosis. 13:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Delgado ME, Dyck L, Laussmann MA and Rehm
M: Modulation of apoptosis sensitivity through the interplay with
autophagic and proteasomal degradation pathways. Cell Death Dis.
5:e10112014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shi R, Weng J, Zhao L, Li XM, Gao TM and
Kong J: Excessive autophagy contributes to neuron death in cerebral
ischemia. CNS Neurosci Ther. 18:250–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Grasselli G and Strata P: Structural
plasticity of climbing fibers and the growth-associated protein
GAP-43. Front Neural Circuits. 7:252013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lamb CA, Yoshimori T and Tooze SA: The
autophagosome: Origins unknown, biogenesis complex. Nat Rev Mol
Cell Biol. 14:759–774. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen K, Cheng HH and Zhou RJ: Molecular
mechanisms and functions of autophagy and the ubiquitin-proteasome
pathway. Yi Chuan. 34:5–18. 2012.(In Chinese). View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hu Z, Yang B, Mo X and Xiao H: Mechanism
and regulation of autophagy and its role in neuronal diseases. Mol
Neurobiol. 52:1190–1209. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guo F, He XB, Li S and Le W: A central
role for phosphorylated p38α in linking proteasome
inhibition-induced apoptosis and autophagy. Mol Neurobiol.
54:7597–7609. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu C, Gao Y, Barrett J and Hu B:
Autophagy and protein aggregation after brain ischemia. J
Neurochem. 115:68–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Thornton C, Leaw B, Mallard C, Nair S,
Jinnai M and Hagberg H: Cell death in the developing brain after
hypoxia-ischemia. Front Cell Neurosci. 11:2482017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nozaki K, Nishimura M and Hashimoto N:
Mitogen-activated protein kinases and cerebral ischemia. Mol
Neurobiol. 23:1–19. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wu D and Cederbaum AI: Inhibition of
autophagy promotes CYP2E1-dependent toxicity in HepG2 cells via
elevated oxidative stress, mitochondria dysfunction and activation
of p38 and JNK MAPK. Redox Biol. 1:552–565. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Insulin-like growth factor-1 and TNF-alpha regulate autophagy
through c-jun N-terminal kinase and Akt pathways in human
atherosclerotic vascular smooth cells. Immunol Cell Biol.
84:448–454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Haberzettl P and Hill BG: Oxidized lipids
activate autophagy in a JNK-dependent manner by stimulating the
endoplasmic reticulum stress response. Redox Biol. 1:56–64. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Xu P, Das M, Reilly J and Davis RJ: JNK
regulates FoxO-dependent autophagy in neurons. Genes Dev.
25:310–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
He GQ, Xu WM, Liao HJ, Jiang C, Li CQ and
Zhang W: Silencing Huwe1 reduces apoptosis of cortical neurons
exposed to oxygen-glucose deprivation and reperfusion. Neural Regen
Res. 14:1977–1985. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wen YD, Sheng R, Zhang LS, Han R, Zhang X,
Zhang XD, Han F, Fukunaga K and Qin ZH: Neuronal injury in rat
model of permanent focal cerebral ischemia is associated with
activation of autophagic and lysosomal pathways. Autophagy.
4:762–769. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kost A, Kasprowska D, Labuzek K,
Wiaderkiewicz R and Gabryel B: Autophagy in brain ischemia. Postepy
Hig Med Dosw (Online). 65:524–533. 2011.(In Polish). View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Balduini W, Carloni S and Buonocore G:
Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal
Neonatal Med. 25 (Suppl 1):S30–S34. 2012. View Article : Google Scholar
|
|
72
|
Lu Q, Harris VA, Kumar S, Mansour HM and
Black SM: Autophagy in neonatal hypoxia ischemic brain is
associated with oxidative stress. Redox Biol. 6:516–523. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Qiao L, Fu J, Xue X, Shi Y, Yao L, Huang
W, Li J, Zhang D, Liu N, Tong X, et al: Neuronalinjury and roles of
apoptosis and autophagy in a neonatal rat model of
hypoxia-ischemia-induced periventricular leukomalacia. Mol Med Rep.
17:5940–5949. 2018.PubMed/NCBI
|
|
74
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
He GQ, Xu WM, Li JF, Li SS, Liu B, Tan XD
and Li CQ: Huwe1 interacts with Gadd45b under oxygen-glucose
deprivation and reperfusion injury in primary Rat cortical neuronal
cells. Mol Brain. 8:882015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Ischemia/reperfusion. Compr Physiol. 7:113–170. 2016.
View Article : Google Scholar : PubMed/NCBI
|