Introduction
Breast cancer (BC) is the most commonly diagnosed
cancer and the leading cause of death in women worldwide,
accounting for ~2.1 million new cases and 626,679 deaths (1). It is a heterogeneous disease with
different histopathological features and therefore has different
prognostic outcomes. Among those, 70–80% of patients with BC are
diagnosed with estrogen receptor (ER)-positive BC (ER+
BC) (2,3).
Metastasis is common in late-stage BC, which is a
complicated and multistep process that involves alterations in
multiple pathway stages and the accumulation of genetic mutations.
During metastasis, cancer cells leave the primary tumor, invade the
circulatory and lymphatic system, enter distant organs and form
metastases (4). In this process,
tumor cells undergo multiple changes that drive their spread,
invasion, migration and homing, and eventually cause them to form
colonies (5,6). The majority of solid human tumors are
of epithelial cell origin, which undergo a complicated process to
initiate metastasis and epithelial-mesenchymal transition (EMT)
(5–7). During EMT, epithelial cells lose
their polarity and adhesion capacity, and become mesenchymal-like
cells, which allows them to have migratory and invasive abilities
(5–7). Increasing evidence has demonstrated
that the TGF-β/SMADs, IL-6/STAT3 and PI3K/AKT signaling pathways
promote EMT during tumor metastasis (8–10).
Endocrine therapies using selective ER modulators,
antiestrogens and aromatase inhibitors are the primary treatment
strategy for ER+ BC (11–13).
Although there are a number of drugs used to treat ER+
BC, these drugs are often associated with drug resistance and
severe side effects (14,15). Therefore, developing more effective
medications with fewer side-effects to treat ER+ BC is
urgently required.
Narasin, a carboxylic polyether ionophore isolated
from Streptomyces albus, has widely been applied in
agriculture as an antibiotic to treat coccidiosis in poultry
(16). It is a derivative of
salinomycin, which has been confirmed to be a specific inhibitor of
cancer stem cells (17,18). Previous studies found that narasin
induces apoptosis, inhibits the proliferation of human hepatoma
HepG2 cell, deactivates NF-κB signaling and overcomes tumor
necrosis factor-related apoptosis-inducing ligand resistance in
glioma cells via endoplasmic reticulum stress (19–21).
Our previous study confirmed that salinomycin has active
antitumorigenic activities in human gastric cancer by suppressing
vascular endothelial growth factor receptor 2-mediated angiogenesis
(22). However, the role of
narasin in cancer treatment and the related molecular mechanisms
have not yet been fully elucidated. The present study reported that
narasin inhibited proliferation, migration and invasion of human
metastatic ER+ BC cells by inactivating the TGF-β/SMAD3-
and IL-6/STAT3-mediated EMT signaling pathways in vitro and
in vivo. These findings support the possibility of
repurposing narasin in the treatment of ER+ BC.
Materials and methods
Chemicals and reagents
Narasin was purchased from Sigma-Aldrich (Merck
KGaA), dissolved in 100% DMSO to generate a 10 mM solution and
stored at −80°C in small aliquots. Recombinant human TGF-β and IL-6
were purchased from PeproTech, Inc. Antibodies against
phosphorylated (p)-STAT3 (Tyr705; cat. no. 9145; 1:1,000), STAT3
(cat. no. 9139; 1:1,000), p-AKT (Ser473; cat. no. 4060; 1:1,000),
AKT (cat. no. 4691; 1:1,000), p-SMAD2 (Ser465/467)/SMAD3
(Ser423/425; cat. no. 8828; 1:1,000), SMAD3 (cat. no. 9523;
1:1,000), β-catenin (cat. no. ARG52651; 1:800; Arigo
Biolaboratories Corp.), ZEB1 (cat. no. ab203829; 1:500; Abcam) and
β-actin (cat. no. M1210-2; 1:10,000; Hangzhou HuaAn Biotechnology
Co., Ltd.), as well as an EMT Antibody Sampler kit (cat. no. 9782;
1:1,000) were purchased from Cell Signalling Technology, Inc. All
other reagents were acquired from Sigma-Aldrich (Merck KGaA).
Cell culture
Human ER+ BC cell lines, MCF-7 and T47D,
and human triple-negative BC cell line, MDA-MB-231, were purchased
from the American Type Culture Collection. All cells were cultured
in DMEM or RPMI-1640 medium supplemented with 10% fetal bovine
serum, and the medium was replaced every 48 h. Cells were
maintained in a humidified 37°C incubator with 5%
CO2.
Cell viability assay
Human BC cells were incubated in a 96-well plate at
a density of 4.5×104 to 5.5×104 cells per
well and treated with narasin at a final concentration of 0, 1,
2.5, 5, 10 µM for 72 h at 37°C. Cell viability was determined using
an MTS kit (CellTiter 96 Aqueous One Solution Reagent; Promega
Corporation), as previously described (23,24).
MTS solution (20 µl) was added to each well, the plates were
incubated for 2 h, and the absorbance was measured at 490 nm with a
microplate reader (Bio-Rad Laboratories, Inc.). All experiments
were repeated three times.
Wound healing assay
The wound healing assay was performed as previously
described (23,24). Briefly, human BC cell lines were
incubated in 6-well plates and allowed to grow to 80% confluence.
Cells were then washed three times with the medium, scratched with
a 200 µl pipette tip, and incubated in the growth medium with 10%
FBS and different concentrations of narasin. Images of cells were
taken at 24 h at ×40 magnification with an inverted microscope
(TE2000; Nikon Corporation). The migrated cells were counted in
three randomly selected fields. Cells in the growth medium without
narasin treatment served as the vehicle control. The inhibition
percentage was expressed as a percentage of vehicle control (100%).
The assay was repeated three times.
Transwell chamber invasion assay
The cell invasion assay was performed using
Matrigel-coated Boyden inserts (8 µm; BD Biosciences), as
previously described (23,24). Cells in the logarithmic growth
phase were digested using trypsin. Subsequently, 150 µl serum-free
culture medium containing 1×106 cells was plated into
the upper chamber, and 600 µl culture medium containing different
doses of narasin was plated into the lower chamber. The assays were
performed for 12 h. Invaded cells were fixed with 4%
paraformaldehyde at 4°C for 20 min, stained with 0.5% crystal
violet for 10 min at room temperature, and counted under a light
microscope in 4–7 randomly selected fields of view. Three
independent experiments were performed in triplicate.
RNA isolation, microarray,
bioinformatics analysis and reverse transcription
(RT)-semi-quantitative PCR
ER+ BC MCF-7 cells (2×106)
were seeded in 100-mm tissue culture dishes with or without narasin
treatment (0.05 µM) for 24 h. TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to isolate
RNA. The mRNA microarray analysis was carried out with the
Affymetrix GeneChip® PrimeView™ Human Gene Expression
Array (Affymetrix; Thermo Fisher Scientific, Inc.), according to
the manufacturer's instructions by the Gene Tech Company Limited
(Shanghai, China). Data were analyzed in The Database for
Annotation, Visualization and Integrated Discovery (DAVID) pathway
enrichment analysis for Kyoto Encyclopedia of Genes and Genomes
(KEGG) enrichment analysis (25–27)
(https://david.ncifcrf.gov/), Search Tool
for the Retrieval of Interacting Genes (STRING; http://string-db.org/) and the heatmap package in R
(28). The differentially
expressed genes (DEGs) were determined based on a false discovery
rate threshold of 0.05.
Total RNA was reverse transcribed to cDNA using the
RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.). The following temperature protocol was used for
reverse transcription: 42°C for 60 min, 70°C for 5 min and 4°C
maintenance. PCR was performed using the SuperScript III Platinum
One-Step Quantitative RT-PCR System (Thermo Fisher Scientific,
Inc.). The following thermocycling conditions were used for qPCR:
94°C for 3 min; followed by 35 cycles of 94°C for 30 sec, annealing
temperature for 30 sec, 72°C for 35 sec; 72°C for 10 min; and
maintained at 4°C. The DNA products were run on a 2% agarose gel
containing ethidium bromide. DNA bands were visualized using an
ultraviolet imager and a Gel Imaging Analysis System (Bio-Rad
Laboratories, Inc.). GAPDH was used as the loading control. The
sequences of the primers used for qPCR were as follows: E-cadherin
forward, 5′-ACAGGATGGCTGAAGGTGAC-3′ and reverse,
3′-TCAGGATCTTGGCTGAGGAT-5′; N-cadherin forward,
5′-GGACCGAGAATCACCAAATG-3′ and reverse, 3′-CCATTAAGCCGAGTGATGGT-5′;
vimentin forward, 5′-TCGCCAACTACATCGACAAG-3′ and reverse,
3′-GACGCATTGTCAACATCCTG-5′; and GAPDH forward,
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse,
3′-TGTAGACCATGTAGTTGAGGTCA-5′. mRNA expression levels were
quantified using GeneSpan (version 7.04.02; SynGene).
Molecular docking analysis
Molecular docking analysis was performed to analyze
the binding ability of narasin to the TGF-β receptor using Autodock
4.2 (The Scripps Research Institute), as previously described
(23,24). TGF-β receptor (PDB ID, 3TZM) was
obtained from RCSB Protein Data Bank (http://www.rcsb.org/) and processed with UCSF Chimera
software (version 1.13.1; National Institutes of Health), which
included removal of water molecules, ions and ligands. The
molecular structure of hydrophilic narasin was obtained from NCBI
(https://www.ncbi.nlm.nih.gov/) without
the molecular optimization process.
Western blotting analysis
Western blotting was performed as previously
described (23,24). Briefly, cells were harvested and
lysed with RIPA buffer (4.25 g NaCl, 3.03 g Tris, 5 g SDS, 5 ml
NP-20, 2.5 g 10% sodium deoxycholate, pH 8.0). Protein
concentration was determined using a BCA assay kit (Sigma-Aldrich;
Merck KGaA). Samples containing 30–50 µg total proteins were
separated via SDS-PAGE on 8–12% gels, and subsequently transferred
to PVDF membranes by electroblotting (Bio-Rad Laboratories, Inc.).
Membranes were then blocked with 5% BSA (Sangon Biotech Co., Ltd.)
for 2 h, and then incubated overnight at 4°C with the following
primary antibodies: p-STAT3, STAT3, p-AKT, AKT, p-SMAD2/SMAD3 and
an EMT antibody kit. The next day, the membranes were incubated
with the appropriate anti-rabbit (1:2,500; cat. no. A21010; Abbkine
Scientific Co., Ltd.), anti-mouse (1:2,500; cat. no. A21020;
Abbkine Scientific Co., Ltd.) secondary antibodies for 1 h at room
temperature. After several washes, specifically bound antibodies
were visualized using an enhanced chemiluminescence system (Bio-Rad
Laboratories, Inc.). Protein expression levels were semi-quantified
using Image Lab software (version 5.2.1; Bio-Rad Laboratories,
Inc.).
Immunofluorescence (IF) assay
For the IF assay, cultured cells (2×104)
were treated with the indicated concentrations of narasin for 24 h,
followed by stimulation with 100 ng/ml IL-6 or 5 ng/ml TGF-β for 2
h at 37°C. Cells were fixed with 4% paraformaldehyde at 0°C for 20
min, permeabilized with 0.1% Triton X-100, and blocked with 1% BSA
in PBS at room temperature for 1 h. The cells were washed 3 times
with PBS after each step, followed by incubation with primary
antibodies (1:1,000) overnight at 4°C. After thorough washing, the
cells were stained with FITC-conjugated goat anti-mouse (1:1,000;
cat. no. A21020; Abbkine Scientific Co., Ltd.) or anti-rabbit IgG
(1:1,000; cat. no. A22120; Abbkine Scientific Co., Ltd.). Cells
were then washed and incubated with DAPI (Beijing Solarbio Science
& Technology Co., Ltd.) for 5–10 min at room temperature. After
the final wash, the samples were mounted with Antifade solution
(Beijing Solarbio Science & Technology Co., Ltd.) and
visualized under a fluorescence microscope (DM2000; Leica
Microsystems GmbH; magnification, ×20).
Xenograft and metastasis assays
A total of 46 four-week-old female BALB/c athymic
nude mice (weight, 20 g) were obtained from the Experimental Animal
Center of Ningxia Medical University. All mouse studies were
performed according to animal protocols approved by the
Institutional Animal Care and Use Committee of Ningxia Medical
University (Yinchuan, China). To measure metastasis, MCF-7-Luc
cells (1×105; Shanghai Key Regulatory Institute of
Biomedical Sciences) were injected into the left ventricle of
female BALB/c athymic nude mice (5-7 per group). Mice were treated
with narasin [0.5 mg/kg (n=6) or 1.5 mg/kg (n=5); i.p.] or DMSO
(control group; n=7) every 2 days for 50 consecutive days. The IVIS
detection system (Caliper Life Sciences) was used to detect tumor
growth and metastasis.
For the xenograft assays, MCF-7 cells
(1×107 cells per mouse; resuspended in compete medium)
were subcutaneously implanted into the axilla of the mice. When
tumors reached ~100 mm3, the mice were randomly divided
into three groups (n=9) and injected with narasin (0.5 or 1.5
mg/kg) or DMSO (control group) every 2 days for 2 consecutive weeks
(14 days). After 2 weeks of treatment, the maximal tumor volume was
1,400 mm3. Then, the mice were euthanized, and tumor
tissues were harvested, weighed and photographed. Mice were first
anesthetized with 2% pentobarbital 0.2 ml (20 mg/kg/per mouse) by
intraperitoneal injection, and then euthanized by cervical
dislocation. Cessation of breathing and heartbeat was used to
confirm death. There were no accidental deaths during this
experiment.
All mice were kept in specific-pathogen-free animal
laboratories at Ningxia Medical University, air purification degree
was at least 10,000 grade according to the standard requirements,
ammonia concentration ≤14 mg/m3, temperature was
18–29°C, daily temperature difference was ≤3°C, relative humidity
was 40–70%, air flow speed ≤0.18 m/sec, and room air pressure
gradient 20–50 Pa. There was a total of 6 mice in each cage, they
were fed and given water regularly every day, and the cage was
cleaned every 3 days.
Histology and
immunohistochemistry
Tumor tissues were removed, fixed with 10%
formaldehyde at room temperature overnight, and embedded in
paraffin (0.4–0.5 mm-thick sections) for immunohistochemical
staining analysis to detect the expression of CD31. Sections were
blocked with 30% sheep serum (JinJiang ZhongYi JinQiao Machinery
Co., Ltd.) at room temperature for 1 h. Subsequently, sections were
incubated with an anti-CD31 primary antibody (1:500; cat. no.
ab134168; Abcam) at 4°C overnight. Following primary incubation,
sections were incubated with a HRP-conjugated secondary antibody
(1:400; cat. no. A21020; Abbkine Scientific Co., Ltd.) for 1 h at
room temperature. Images were taken using a Leica DM4000 B photo
microscope (magnification, ×400; Leica Microsystems GmbH).
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software (version 6.0; GraphPad Software, Inc.). Comparisons
among groups were analyzed using one-way ANOVA followed by a
Dunnett's test. Data are presented as the mean ± SD. All
experiments were repeated three times. P<0.05 was considered to
indicate a statistically significant difference.
Results
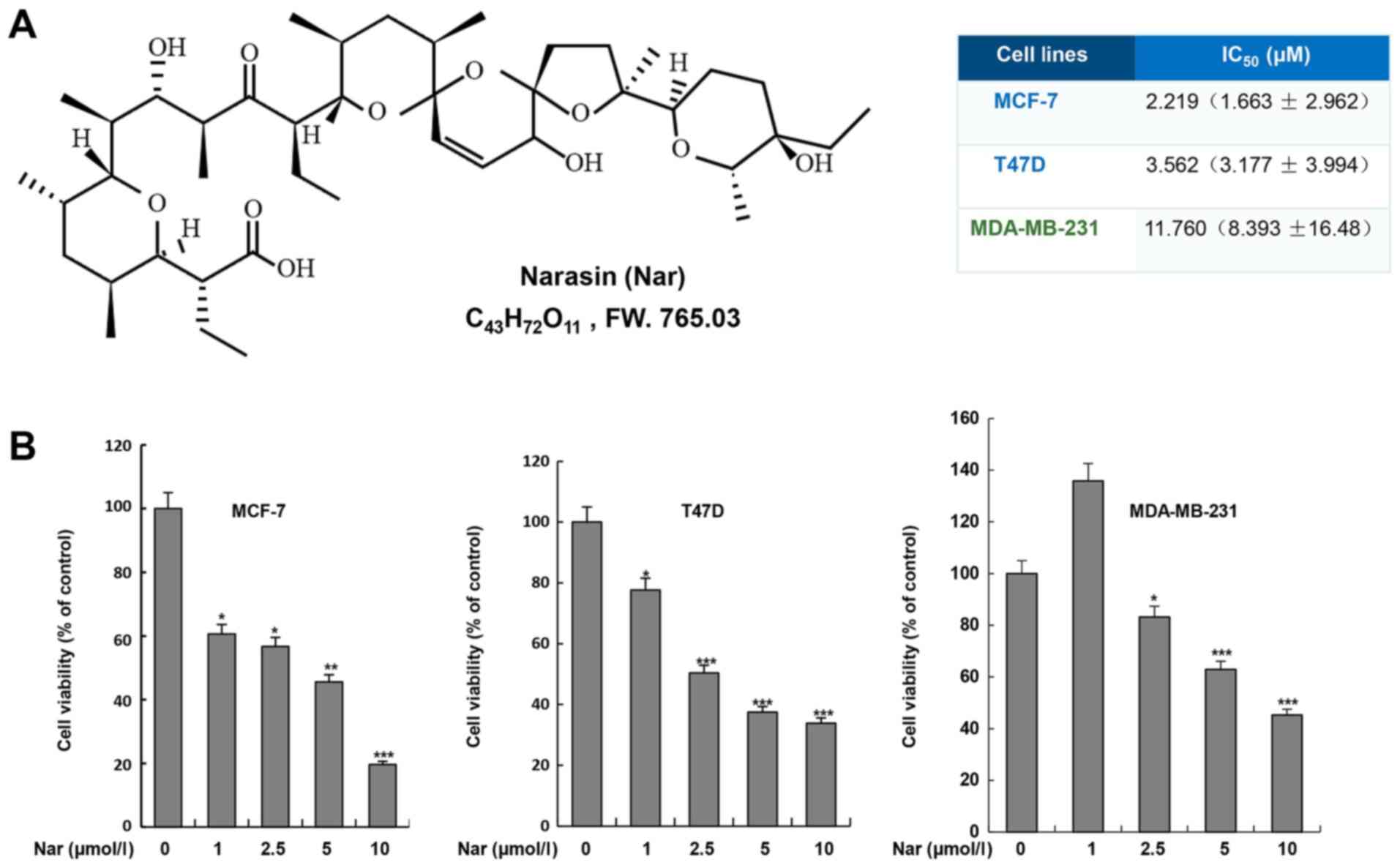
Narasin inhibits BC cell
viability
To determine the effect of narasin on BC cells, two
ER+ BC cell lines, MCF-7 and T47D, and one
triple-negative BC cell line, MDA-MB-231, were treated with narasin
at various concentrations for 72 h. Cell viability was assessed
using the MTS cell viability assay kit. As shown in Fig. 1B, narasin significantly inhibited
cell viability after treatment with different concentrations of
narasin (1–10 µM) for 72 h, with a half-maximal inhibitory
concentration (IC50) of 11.76 µM in MDA-MB-231 cells.
However, lower concentrations of narasin significantly reduced the
viability of MCF-7 and T47D cells, with IC50 values of
2.219 and 3.562 µM, respectively. Therefore, the data suggested
that narasin may have increased inhibitory effects on MCF-7 and
T47D ER+ BC cell lines compared with MDA-MB-231
triple-negative BC cell line.
Narasin inhibits cell migration and
invasion by suppressing EMT in ER+ BC cells
Cell migration and invasion are essential steps in
metastasis. Thus, the effect of narasin on the migration of BC
cells was investigated using a wound healing assay. Of note, it was
found that narasin concentrations ranging from 2.5 to 5 µM could
inhibit the migration of MDA-MB-231 cells. Whereas, narasin at
concentrations between 0.005 and 0.05 µM significantly inhibited
the migration of MCF-7 and T47D cells in a dose-dependent manner
(Fig. 2A). The Transwell invasion
assay further revealed that narasin dose-dependently suppressed the
invasive ability of BC cells in a similar manner (Fig. 2B). The concentrations of narasin
used in the migration and invasion experiments did not affect cell
viability (Fig. 2A and B). Thus,
this indicated that narasin can significantly inhibit the migration
and invasion of ER+ BC cells at lower concentrations due
to inhibition of cell migration rather than inhibition of cell
viability. Collectively, these findings suggested that narasin may
be a candidate drug for ER+ BC treatment.
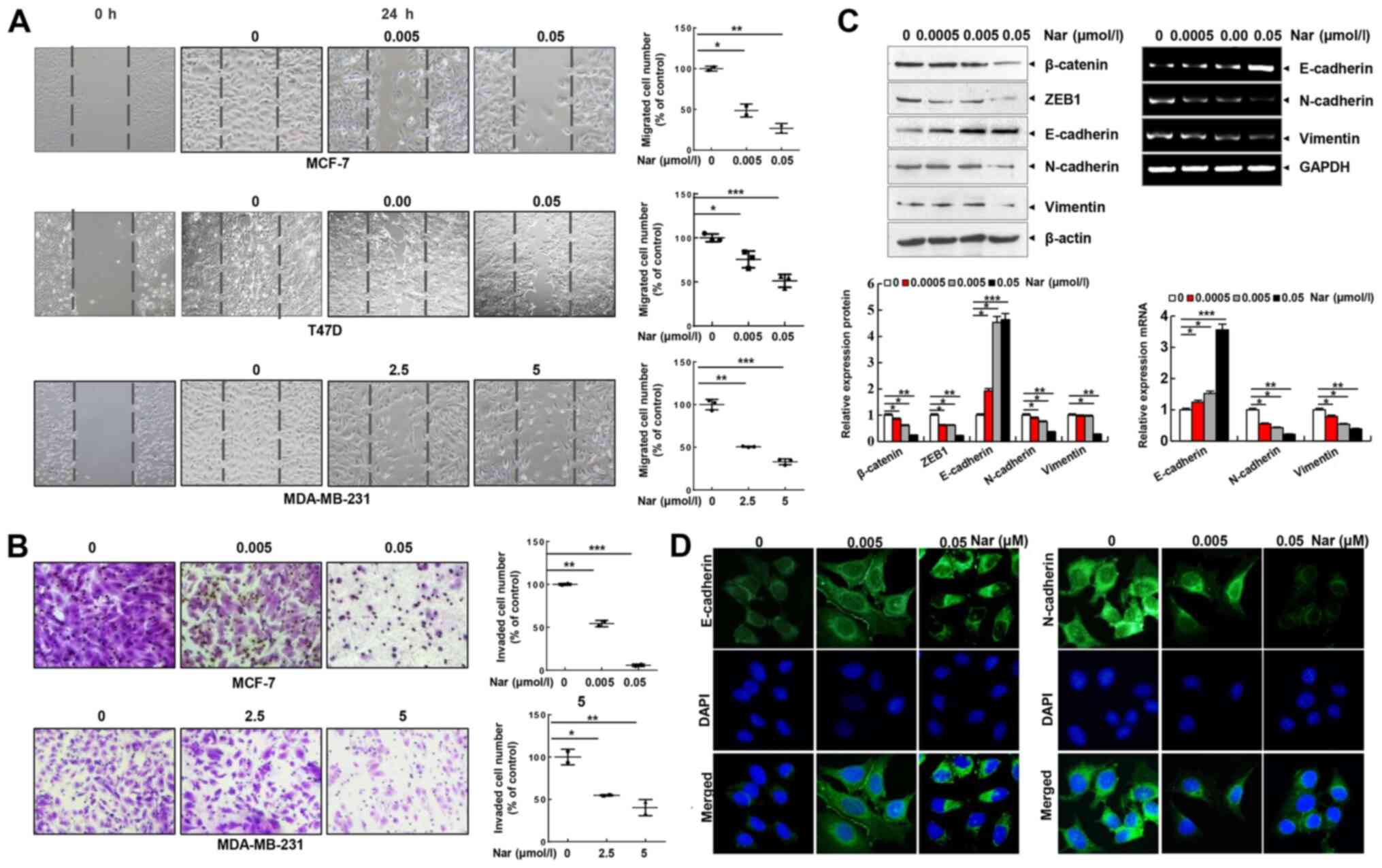 | Figure 2.Narasin inhibits BC cell migration
and invasion and reverses the EMT phenotype in vitro. (A)
Wound healing assay. BC cells were seeded into 6-well plates, a
wound was created after the cells reached 100% confluence, and then
different concentrations of narasin were added. Images were
acquired after 24 h. (B) Invasion assay. MDA-MB-231 and MCF-7 cells
were seeded into the upper chambers of Transwell inserts precoated
with Matrigel. After 24 h of incubation, images were obtained. (C)
Following treatment with narasin for 24 h, EMT-related markers were
probed by western blotting. MCF-7 cells were treated with narasin
for 24 h and then analyzed for EMT-related marker mRNA expression
by reverse transcription-semi-quantitative PCR. (D) MCF-7 cells
were treated with narasin for 24 h, and the levels of N-cadherin
(green) and E-cadherin (green) were detected by immunofluorescence.
Nuclei were stained with DAPI (blue). Scale bar, 40 µm. *P<0.05,
**P<0.01, ***P<0.001. BC, breast cancer; EMT,
epithelial-mesenchymal transition; ZEB1, and zinc finger
E-box-binding homeobox 1. |
EMT is considered to be the initial and critical
step towards the metastasis of cancer cells (29). To explore the potential regulatory
mechanisms of narasin in migration and invasion of ER+
BC cells, the expression of commonly known EMT-associated
biomarkers, including E-cadherin, N-cadherin and vimentin, were
examined by RT-semi-quantitative PCR and western blotting. At
concentrations between 0.005 and 0.05 µM, narasin downregulated the
expression of mesenchymal markers (N-cadherin and vimentin), and
upregulated the expression of epithelial cell marker E-cadherin in
MCF-7 cells (Fig. 2C). Moreover,
the EMT transcription factors β-catenin and zinc finger
E-box-binding homeobox 1 (ZEB1) were downregulated by narasin
compared with the control (Fig.
2C). Consistent with this result, an IF assay confirmed that
exposure to narasin reversed EMT, as indicated by the decreased
membrane-localized N-cadherin and increased E-cadherin (Figs. 2D and S1A and B). These results confirmed that
narasin had inhibitory effects on the migration and invasion of
ER+ BC cells by impairing the process of EMT.
Narasin inhibited TGF-β/SMAD3 and
IL-6/STAT3 signaling in ER+ BC cells
A number of reports have demonstrated that human BC
cell line MCF-7 is a more suitable model for studying pre-clinical
ER+ BC in vitro and in vivo (30–32).
Therefore, to identify the potential mechanisms of narasin-mediated
inhibition of ER+ BC metastasis and proliferation, the
ER+ breast cancer line, MCF-7, was chosen to investigate
the gene expression profiles involved in the inhibitory effects of
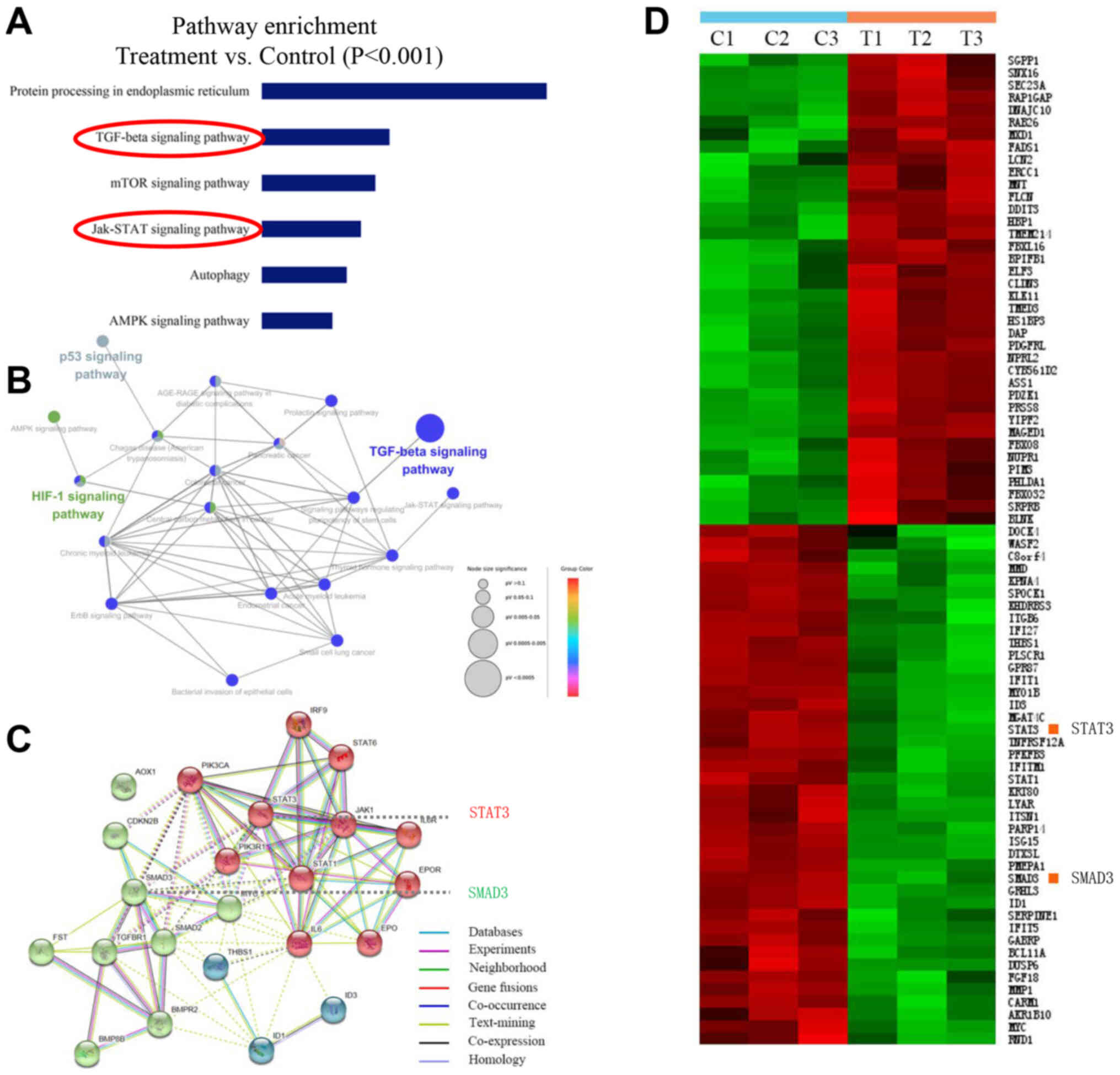
narasin by conducting a microarray analysis. Pathway enrichment
analysis, an analysis that maps genes to the KEGG pathways
(25–27), revealed the involvement of several
key signaling pathways, including ‘TGF-β signaling pathway’, ‘mTOR
signaling pathway’, ‘JAK-STAT signaling pathway’, ‘p53 signaling
pathway’ and ‘HIF-1 signaling pathway’, which play essential roles
in tumor progression and metastasis (Fig. 3A and B). Carcinogenic signaling
networks are usually represented as crosstalk between multiple
signaling pathways, rendering single protein measurements
ineffective in predicting complex cellular responses to drugs.
Further enrichment analysis was performed to predict the underlying
interaction network based on the DEG profiles (33). The data showed that there were some
interactions between these signaling pathways (Fig. 3B and C). Then, the protein-protein
interaction networks were examined using data from the STRING
database. The results showed that the DEGs of signaling pathways
formed tightly connected networks (Fig. 3C), suggesting that these signaling
pathways work as a functional module at the protein level.
Essential genes that are involved in the function of TGF-β and
JAK/STAT3 signaling, including ‘SMAD3’ and ‘STAT3’, are highlighted
in Fig. 3C, and were significantly
downregulated by narasin treatment in the presence of 0.05 µM in
the heatmap (Fig. 3D). Further
pathway analysis of these DEGs revealed several pathways that were
affected by narasin inhibition. Among these pathways we chose to
focus on the top-ranked TGF-β/SMAD3 and IL-6/STAT3 signaling
pathways.
Effect of narasin on
TGF-β/SMAD3-mediated EMT in ER+ BC cells
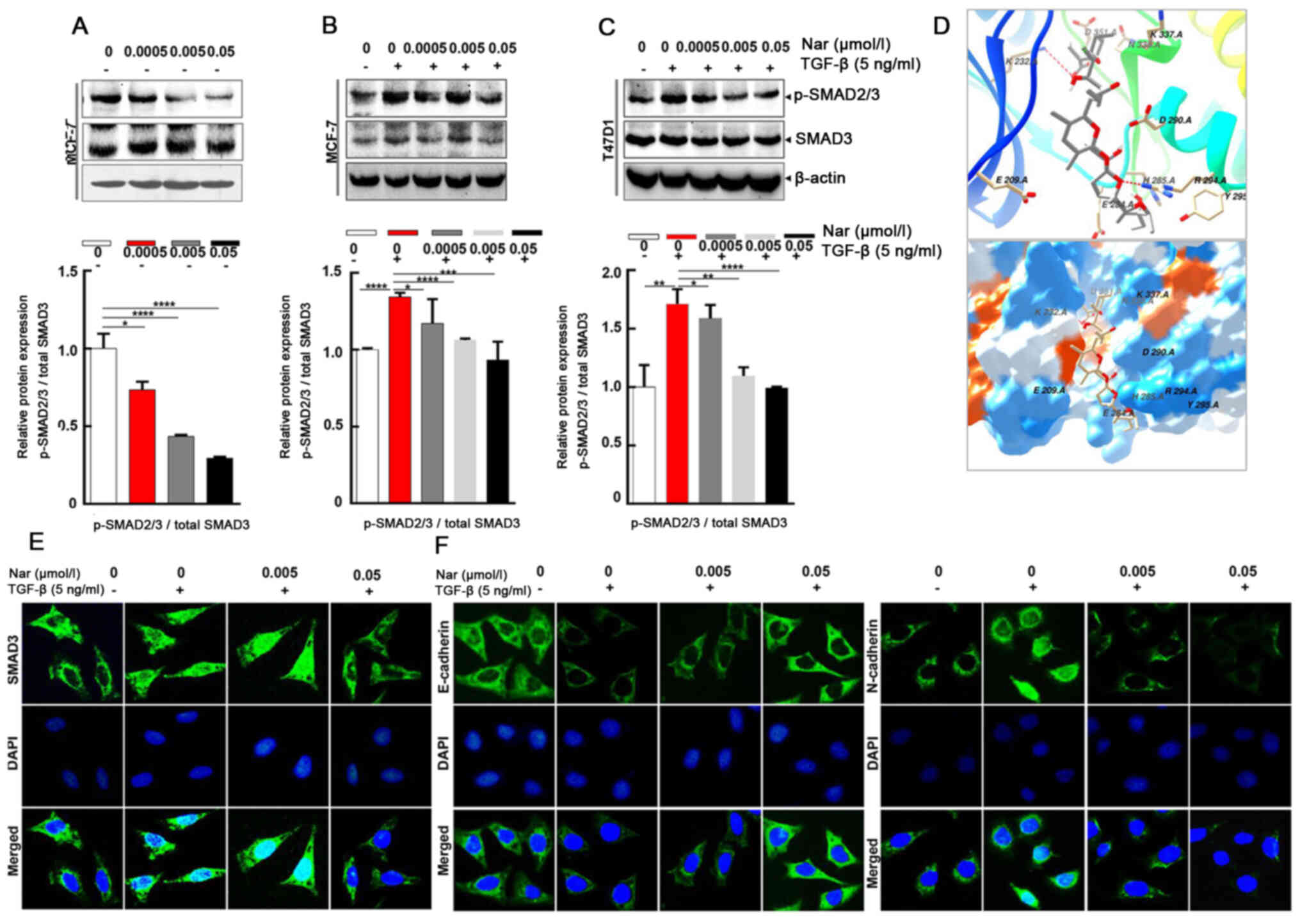
To further explore whether narasin regulated the
TGF-β/SMAD3 signaling pathway in ER+ BC cells (34), the effect of narasin on SMAD2/3
phosphorylation, which is a crucial mediator of TGF-β/SMAD3
signaling (35–39), was examined. Narasin at tested
concentrations (0.005 to 0.05 µmol/l) inhibited SMAD2/3
phosphorylation without TGF-β in a dose-dependent manner in
ER+ BC cell lines MCF-7 (Fig. 4A) and with or without TGF-β in a
dose-dependent manner in ER+ BC cell lines MCF-7 and
T47D (Fig. 4B-C). The results
showed that TGF-β triggered SMAD2/3 phosphorylation in MCF-7 and
T47D cells (Fig. 4B and C).
Treatment with narasin (0.005 to 0.05 µmol/l) notably inhibited
TGFβ-induced SMAD2/3 phosphorylation in a dose-dependent manner in
MCF-7 and T47D cells without affecting total SMAD2/3 expression
(Fig. 4B and C).
To explore the interaction between narasin and the
TGF-β receptor, a molecular docking experiment was performed using
Autodock. The optimized binding model of narasin with the TGF-β
receptor is shown in Fig. 4D. The
narasin molecule was bound to the ATP binding pocket of the TGF-β
receptor and was stabilized by hydrogen bonds and hydrophobic
interactions. Hydrogen bonds between the amino of Lys232 and the
hydroxyl of narasin, and guanine of Arg294 with oxygen of narasin,
were observed between the receptor and ligand, which can
significantly increase the binding ability. Moreover, the
hydrophobic interaction, formed by ALKYL 1 groups or aromatic ring
of Asp351, His285, Asp290, Tyr295, stabilized the binding complex
in aqueous solution. Hydrophilic interactions between the protein
and ligand, which included Lys337, Arg338, Glu284 and Glu209,
impacted the molecular chemical conformation of the ligand. These
three types of molecular interactions promoted the biological
function of narasin to inhibit tumor cell proliferation and
metastasis.
The SMAD2/3 complex translocates into the nucleus to
regulate the expression of target genes with other DNA-binding
transcription factors (40,41).
Therefore, IF staining was used to verify the effect of narasin on
SMAD3 nuclear translocation in ER+ BC cells. The results
showed that narasin suppressed TGF-β-induced SMAD3 nuclear
translocation in a concentration-dependent manner, with the
effective inhibition concentration at 0.005 to 0.05 µmol/l
(Figs. 4E and S1C). To examine whether TGF-β-induced
EMT was suppressed by narasin, the TGF-β-induced expression of
E-cadherin and N-cadherin was examined by IF staining in MCF-7
cells.
Of note, MCF-7 cells treated with 5 ng/ml TGF-β led
to a significant reduction in epithelial phenotype marker
E-cadherin and an increase in the mesenchymal phenotype marker
N-cadherin (Figs. 4F and S1D and E). However, cells exposed to
narasin (0.005 to 0.05 µmol/l) showed a dose-dependent increase in
the expression of E-cadherin and a decrease in the expression of
N-cadherin compared with the TGF-β only group (Fig. 4F). Collectively these results
indicated that narasin could inhibit the phosphorylation of SMAD3,
block the binding of the TGF-β receptor with its ligand, and thus
inactivate the downstream SMAD2/3-mediated EMT signaling
pathway.
Effect of narasin on IL-6/STAT3
signaling-mediated EMT in ER+ BC cells
Aside from the TGF-β/SMAD signaling-mediated EMT
process, STAT3 may also directly mediate EMT in cancer metastasis
(9,42). IL-6 is a direct upstream mediator
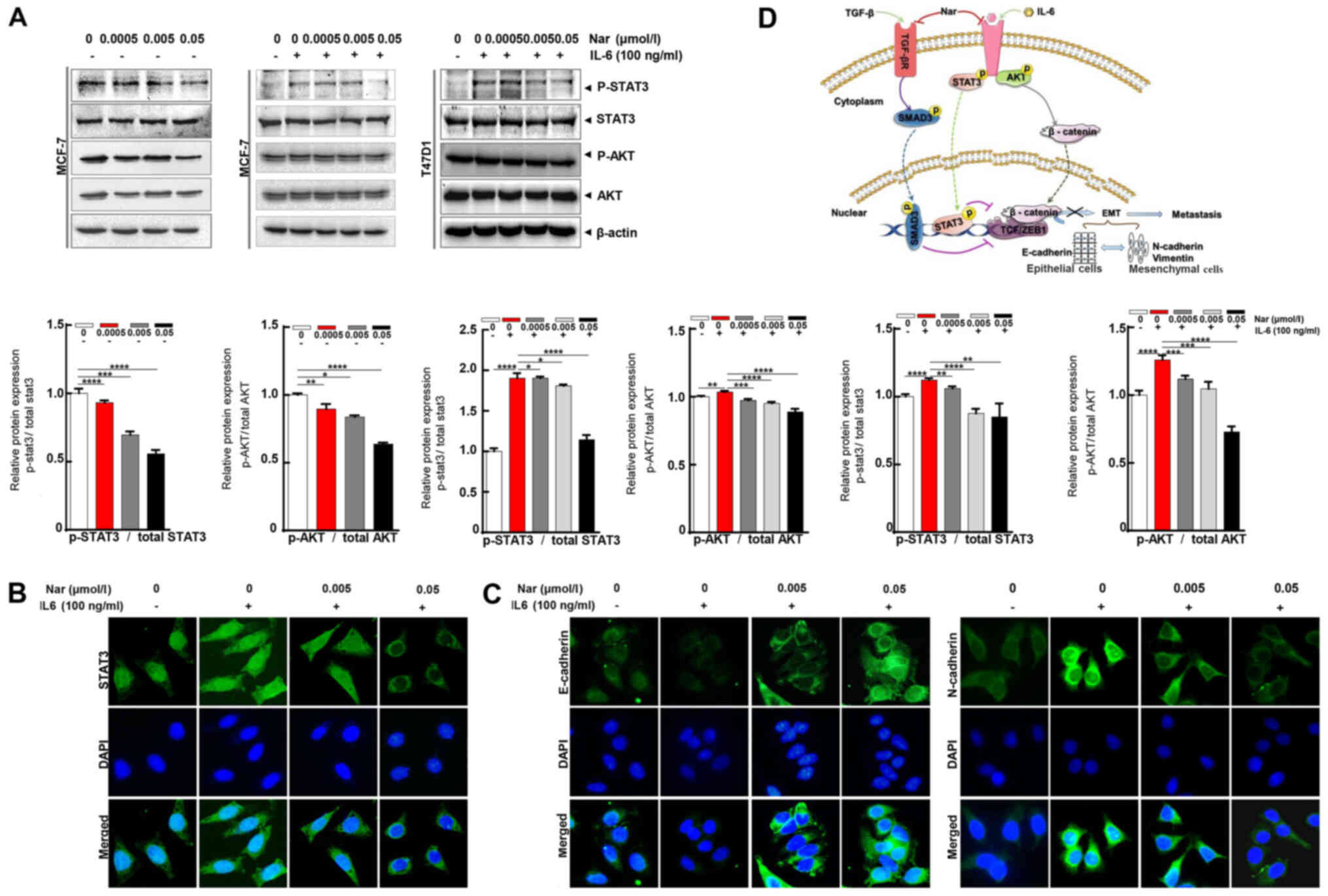
of p-STAT3. In the present study, the effect of narasin on
IL-6-induced STAT3 phosphorylation was investigated. The results
showed that 100 ng/ml IL-6 notably upregulated STAT3
phosphorylation in ER+ BC cells, and that 0.05 µmol/l
narasin effectively inhibited STAT3 phosphorylation in MCF-7 and
T47D cells without affecting total STAT3 expression (Fig. 5A). As shown in Fig. 5A, with or without IL-6, narasin
(0.005 to 0.05 µmol/l) also inhibited AKT phosphorylation and did
not affect total AKT expression. Constitutive or inducible
phosphorylation at Tyr705 of STAT3 is vital for promoting the
biological functions of STAT3, including dimerization and
cytoplasmic-to-nuclear translocation of STAT3. It was found that
narasin inhibited IL-6-induced STAT3 nuclear accumulation (Figs. 5B and S1F), with the maximum inhibition
observed at 0.005 to 0.05 µmol/l.
To further determine whether narasin suppressed
IL-6-induced EMT, the IL-6-induced expression and distribution of
E-cadherin and N-cadherin were examined by IF staining in MCF-7
cells. Treatment with IL-6 decreased the expression of E-cadherin
and increased the expression of N-cadherin (Figs. 5C and S1G and H). In contrast, cells exposed to
narasin at concentrations of 0.005 to 0.05 µmol/l showed a
dose-dependent increase in E-cadherin expression and a decrease in
N-cadherin expression. These data indicated that narasin could
inhibit EMT via TGF-β/SMAD3 and IL-6/STAT3 signaling, which
suppresses the migratory and invasive abilities of ER+
BC cells.
Narasin attenuates ER+ BC
metastasis
As commonly known, the lungs, brain and bones are
the primary sites for BC metastasis (43), although nearly any tissue in the
body can be metastasized by BC. In the present study, the widely
used mouse left ventricle injection tumor metastasis model with
MCF-7-Luc cells was used to investigate the potential inhibitory
effects of narasin against BC metastasis. Following cancer cell
injection, mice were treated with narasin for the duration of the
experiment, and then a bioluminescence imaging assay was used to
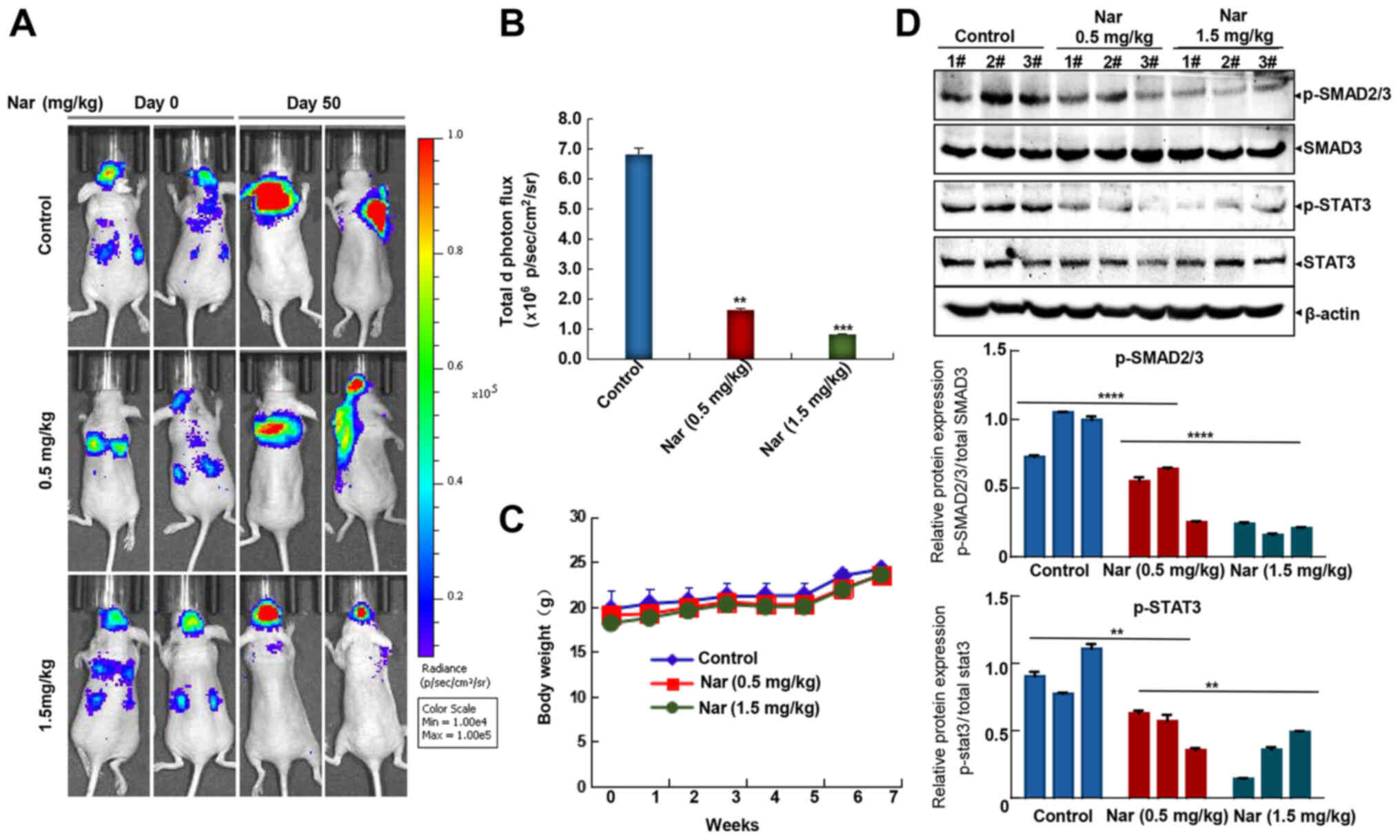
examine the effects of this treatment. As shown in Fig. 6A and B, intraperitoneal injection
of narasin at doses of 0.5 and 1.5 mg/kg reduced brain and lung
metastases, achieving ~76.2 and 87.5% inhibition compared with the
untreated control group on day 50. Besides, no significant
differences in body weight were detected among the three groups
during treatment periods (Fig.
6C), which may indicate that the toxicity of narasin was low at
the effective dose used to inhibit tumor growth. Besides, Fig. 6D shows that narasin effectively
suppressed the phosphorylation of both p-SMAD2/3 and p-STAT3,
without altering the total protein expression in the treatment
groups as compared with the controls.
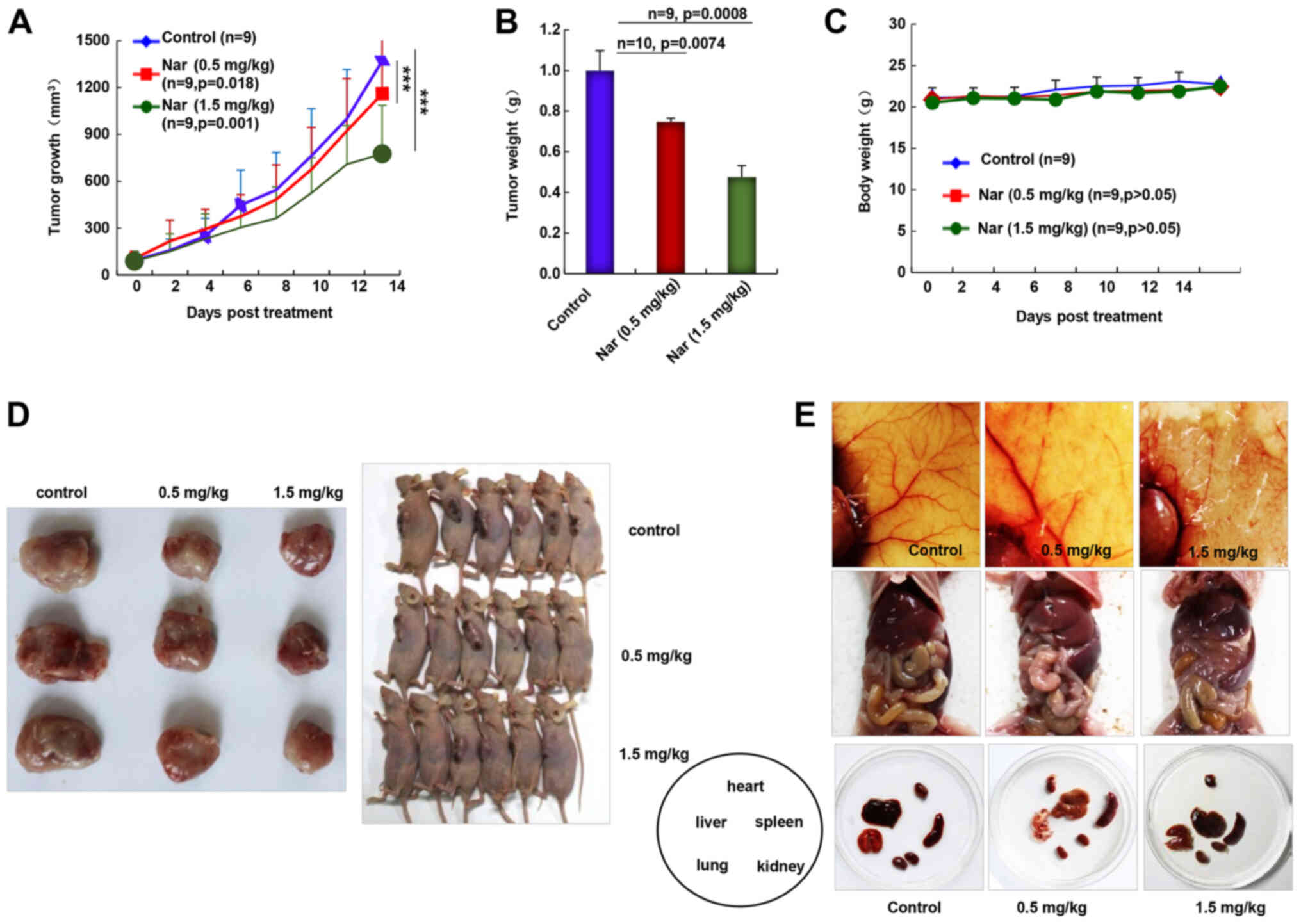
Narasin inhibits tumor growth and
angiogenesis in ER+ breast cancer
As the inhibition of primary BC tumor metastasis was
observed following narasin treatment, the in vivo antitumor
activities of narasin were investigated using the nude mouse
subcutaneous xenograft model with MCF-7 cells. Intraperitoneal
administration of narasin (0.5 and 1.5 mg/kg, 14 days)
significantly decreased tumor volume (Fig. 7A) and tumor weight (Fig. 7B) without noticeable weight changes
(Fig. 7C). The percentage of tumor
growth inhibition of narasin was 14.9 and 40.1% at 0.5 and 1.5
mg/kg, respectively. When pulling the skin of each mouse back to
expose an entire tumor, it was found that narasin-mediated
suppression of tumor growth was associated with angiogenesis
inhibition, as shown in the representative image from each group
(Fig. 7D and E). The number of
CD31+ endothelial cells was notably decreased in the
high dose treatment group (Fig.
S2). Additionally, the skin color and organs of the nude mouse
were mostly normal, as shown in Fig.
7E, which may suggest that the mice could tolerate the
effective doses of narasin.
Discussion
The incidence and mortality of BC ranks first among
female malignancies; 70–80% of patients with BC are diagnosed as
ER+ (2). A number of
studies have reported that the primary cause of death in >90% of
patients with BC is due to BC metastasis (44,45).
Tamoxifen was the first FDA-approved drug for ER+ BC
(12). Subsequently, a large
number of drugs targeting ER+ BC have been discovered
and put into clinical use, such as everolimus and palbociclib.
However, the current compounds used to treat ER+ BC have
different degrees of side effects and drug resistance, thus
limiting their long-term use. There is an urgent need to identify
and develop a drug that is safe, economical and immune to drug
resistance to inhibit the metastasis of ER+ BC.
The present study reported a significant finding
that narasin, a widely used agricultural antibiotic approved by the
FDA, can act directly on tumor cells and inhibit various properties
associated with ER+ BC, including tumor cell
proliferation, migration and invasion, in vitro at lower
concentrations. The MCF-7 and T47D ER+ BC cell lines and
the MDA-MB-231 triple-negative BC cell line were chosen as the
in vitro models. The results showed that narasin had a
significant dose-dependent inhibitory effect on the cell viability
of BC cell lines, as aforementioned. The IC50 values of
narasin in the MCF-7, T47D and MDA-MB-231 cell lines were 2.219,
3.562 and 11.76 µM, respectively. When further comparing the
effects of narasin on BC cell migration and invasion, narasin at
the much lower concentration of 0.005 µmol/l was found to
significantly inhibit the migration and invasion of MCF-7 and T47D
ER+ BC cells; whereas, narasin inhibited MDA-MB-231
triple-negative BC cells at 2.5 µmol/l. The inhibitory effects on
cell migration and invasion continued to increase as the drug
concentration was increased. A comparison of those two results
suggested that narasin had a more pronounced inhibitory effect on
the migration and invasion of the MCF-7 and T47D ER+ BC
cell lines, and that it is a potential target for candidate drugs
to treat ER+ BC.
EMT plays a vital role in tumor progression,
invasion and metastasis, which is a common molecular mechanism of
tumor metastasis (7,46–49).
During the process of EMT, tumor cells lose their epithelial cell
characteristics and acquire mesenchymal cell characteristics, which
gives them invasive abilities (7).
Multiple signaling pathways, such as the TGF-β/SMAD, IL-6/STAT3 and
PI3K/AKT pathways, have been reported to promote EMT during tumor
progression (8,9,50).
However, the specific molecular mechanism by which narasin inhibits
ER+ BC cell migration and invasion has not yet been
revealed. In the present study, a microarray assay was used to
screen and identify potential signaling pathways that contribute to
the narasin-mediated inhibition on ER+ BC MCF-7
metastasis and proliferation. The microarray analysis revealed that
narasin could significantly block the TGF-β/SMAD3 and IL-6/STAT3
signaling pathways, and the downstream signaling molecules in
MCF-7. This result was confirmed by independent immunoblotting
analysis. It is of note that several key genes associated with
TGF-β, mTOR, JAK-STAT, p53 and HIF-1 signaling pathways were
chosen, such as AKT, mTOR, S6K, JAK2, STAT3, p53, HIF-1 and NF-κB.
Among these signaling molecules, no notable changes were observed
at the protein level of mTOR, S6K and HIF-1 in MCF-7 and T47D cell
lines, as measured via western blotting in our preliminary
experiments (data not shown). However, narasin markedly suppressed
the phosphorylation of TGF-β-induced SMAD2/3 and IL-6-induced STAT3
phosphorylation. Therefore, the TGF-β/SMAD3 and IL-6/STAT3
signaling pathways were selected for further study. It is hoped
that we can address these points in our future studies due to the
scope of the present study.
A number of studies have reported that TGF-β binds
to receptor 1 on the tumour cell membrane to activate the
downstream SMAD2 and SMAD3 proteins, while p-SMAD2/3 proteins bind
to cytoplasmic SMAD4 protein to form trimers. Following this, they
translocate to the nucleus and combine with DNA to play a role in
regulating the expression of related transcription factors
(51), which leads to a reduction
in the expression of E-cadherin and an increase in the expression
of N-cadherin and vimentin, thereby inducing EMT. The present
results showed that narasin significantly inhibited SMAD2/3
activation and translocation into the nucleus with or without TGF-β
treatment in ER+ BC cells. Using a cellular IF assay, it
was also found that narasin increased E-cadherin expression and
decreased N-cadherin expression in a dose-dependent manner upon
TGF-β stimulation.
Studies have demonstrated that IL-6 can increase the
growth and metastasis of ER+ BC by activating STAT3
phosphorylation and promoting EMT (9). In the present study, it was found
that narasin inhibited the IL-6-induced activation of STAT3, as
well as STAT3 translocation into the nucleus in MCF-7 and T47D BC
cells at the effective concentrations (0.005 to 0.05 µmol/l). Using
an IF assay, it was found that narasin dose-dependently increased
E-cadherin expression and inhibited N-cadherin expression induced
by IL-6. The aforementioned results indicated that narasin may
block EMT progression to suppress ER+ BC metastasis via
the TGF-β/SMAD3 and IL-6/STAT3 pathways. Recent studies revealed
that the IL-6/STAT3 signaling pathway was required for the
TGF-β-induced EMT process in lung cancer cells (52,53),
and STAT3 could selectively interact with SMAD3 to antagonize TGF-β
signalling (52,53). There may be crosstalk between the
IL-6/STAT3 and TGF-β/p-SMAD3 signaling pathway in certain
conditions (52,53), indicating that dual suppression of
the TGF-β/SMAD3 and IL-6/STAT3 pathways could inhibit the
progression of EMT more effectively.
Numerous chemotherapeutic drugs cannot penetrate the
blood-brain barrier, resulting in insufficient drug concentrations
at the brain lesion and therapeutic resistance to brain metastases.
In the present report, daily treatment with 0.5 and 1.5 mg/kg
narasin was effective in reducing brain and lung tumor metastasis,
as well as tumor growth in mouse tumor models, without inducing any
apparent toxicity. Collectively, these findings indicated that
narasin is a novel inhibitor of ER+ BC cells and may be
a promising anticancer drug candidate. However, further studies are
needed to substantiate this hypothesis.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Fen Wang
(Center for Translational Cancer Research, Institute of Biosciences
and Technology, Texas A&M, Texas, USA) for their help with the
English.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81560485,
816660500 and 81760525), the West China Top Class Discipline
Project in Basic Medical Sciences, Ningxia Medical University
(grant nos. NXYLXK2017B07 and NXYLXK2017A05); the Ningxia Municipal
Education Commission (grant no. NGY2015094) and the Natural Science
Foundation of Ningxia (grant no. NZ16142).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JC and TL conceived and designed the study. XH, NL,
BL and JL performed the experiments. JC, WY and ZM analysed the
data. JC, TL and WY provided administrative, technical or material
support. JC and XH wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All mouse studies were performed according to animal
protocols approved by the Institutional Animal Care and Use
Committee of Ningxia Medical University (Yinchuan, China; approval
no. 20170006).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reiner T and Barrios CH: Optimal
management of hormone receptor positive metastatic breast cancer in
2016. Ther Adv Med Oncol. 7:304–320. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vuong D, Simpson PT, Green B, Cummings MC
and Lakhani SR: Molecular classification of breast cancer. Virchows
Arch. 465:1–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mikula-Pietrasik J, Uruski P, Tykarski A
and Ksiazek K: The peritoneal ‘soil’ for a cancerous ‘seed’: A
comprehensive review of the pathogenesis of intraperitoneal cancer
metastases. Cell Mol Life Sci. 75:509–525. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Riggi N, Aguet M and Stamenkovic I: Cancer
metastasis: A reappraisal of its underlying mechanisms and their
relevance to treatment. Annu Rev Pathol. 13:117–140. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lambert AW, Pattabiraman DR and Weinberg
RA: Emerging biological principles of metastasis. Cell.
168:670–691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chiu HC, Li CJ, Yiang GT, Tsai AP and Wu
MY: Epithelial to mesenchymal transition and cell biology of
molecular regulation in endometrial carcinogenesis. J Clin Med.
8:4392019. View Article : Google Scholar
|
|
9
|
Wang B, Liu T, Wu JC, Luo SZ, Chen R, Lu
LG and Xu MY: STAT3 aggravates TGF-β1-induced hepatic
Epithelial-to-mesenchymal transition and migration. Biomed
Pharmacother. 98:214–221. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Akhurst RJ and Derynck R: TGF-beta
signaling in cancer-a Double-edged sword. Trends Cell Biol. 11
(Suppl):S44–S51. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Henke BR and Heyer D: Recent advances in
estrogen receptor modulators. Curr Opin Drug Discov Devel.
8:437–448. 2005.PubMed/NCBI
|
|
12
|
Elkak AE and Mokbel K: Pure antiestrogens
and breast cancer. Curr Med Res Opin. 17:282–289. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Younus J and Vandenberg TA: A practical
overview of aromatase inhibitors in postmenopausal women with
hormone receptor-positive breast cancer. Bull Cancer. 92:E39–E44.
2005.PubMed/NCBI
|
|
14
|
Braga S: Resistance to targeted therapies
in breast cancer. Methods Mol Biol. 1395:105–136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lumachi F, Santeufemia DA and Basso SM:
Current medical treatment of estrogen receptor-positive breast
cancer. World J Biol Chem. 6:231–239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Riddell FG: Structure, conformation, and
mechanism in the membrane transport of alkali metal ions by
ionophoric antibiotics. Chirality. 14:121–125. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gupta PB, Onder TT, Jiang G, Tao K,
Kuperwasser C, Weinberg RA and Lander ES: Identification of
selective inhibitors of cancer stem cells by high-throughput
screening. Cell. 138:645–659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Markowska A, Kaysiewicz J, Markowska J and
Huczynski A: Doxycycline, salinomycin, monensin and ivermectin
repositioned as cancer drugs. Bioorg Med Chem Lett. 29:1549–1554.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang J, Li H, Qaed E, Zhang J, Song Y, Wu
R, Bu X, Wang Q and Tang Z: Salinomycin, as an autophagy
modulator-a new avenue to anticancer: A review. J Exp Clin Cancer
Res. 37:262018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cybulski W, Radko L and Rzeski W:
Cytotoxicity of monensin, narasin and salinomycin and their
interaction with silybin in HepG2, LMH and L6 cell cultures.
Toxicol In Vitro. 29:337–344. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miller SC, Huang R, Sakamuru S, Shukla SJ,
Attene-Ramos MS, Shinn P, Van Leer D, Leister W, Austin CP and Xia
M: Identification of known drugs that act as inhibitors of
NF-kappaB signaling and their mechanism of action. Biochem
Pharmacol. 79:1272–1280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoon MJ, Kang YJ, Kim IY, Kim EH, Lee JA,
Lim JH, Kwon TK and Choi KS: Monensin, a polyether ionophore
antibiotic, overcomes TRAIL resistance in glioma cells via
endoplasmic reticulum stress, DR5 upregulation and c-FLIP
downregulation. Carcinogenesis. 34:1918–1928. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen J, Wang J, Lin L, He L, Wu Y, Zhang
L, Yi Z, Chen Y, Pang X and Liu M: Inhibition of STAT3 signaling
pathway by nitidine chloride suppressed the angiogenesis and growth
of human gastric cancer. Mol Cancer Ther. 11:277–287. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li T, Liu X, Shen Q, Yang W, Huo Z, Liu Q,
Jiao H and Chen J: Salinomycin exerts anti-angiogenic and
anti-tumorigenic activities by inhibiting vascular endothelial
growth factor receptor 2-mediated angiogenesis. Oncotarget.
7:26580–26592. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47:D590–D595. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanehisa M: Toward understanding the
origin and evolution of cellular organisms. Protein Sci.
28:1947–1951. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Galili T, O'Callaghan A, Sidi J and
Sievert C: Heatmaply: An R package for creating interactive cluster
heatmaps for online publishing. Bioinformatics. 34:1600–1602. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amaskos C, Garmpi A, Nikolettos K,
Vavourakis M, Diamantis E, Patsouras A, Farmaki P, Nonni A,
Dimitroulis D, Mantas D, et al: Triple-negative breast cancer: The
progress of targeted therapies and future tendencies. Anticancer
Res. 39:5285–5296. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Van Themsche C, Parent S, Leblanc V,
Descôteaux C, Simard AM, Bérubé G and Asselin E: VP-128, a novel
oestradiol-platinum(II) hybrid with selective anti-tumour activity
towards hormone-dependent breast cancer cells in vivo. Endocr Relat
Cancer. 16:1185–1195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi JF, Yang N, Ding HJ, Zhang JX, Hu ML,
Leng Y, Han X and Sun YJ: Rα directly activated the MDR1
transcription to increase paclitaxel-resistance of ERα-positive
breast cancer cells in vitro and in vivo. Int J Biochem Cell Biol.
53:35–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Márquez-Garbán DC, Deng G, Comin-Anduix B,
Garcia AJ, Xing Y, Chen HW, Cheung-Lau G, Hamilton N, Jung ME and
Pietras RJ: Antiestrogens in combination with immune checkpoint
inhibitors in breast cancer immunotherapy. J Steroid Biochem Mol
Biol. 193:1054152019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang
W, Lu W, Liu J, Pang X and Liu M: Suppression of KRas-mutant cancer
through the combined inhibition of KRAS with PLK1 and ROCK. Nat
Commun. 7:113632016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Itoh S, Itoh F, Goumans MJ and Ten Dijke
P: Signaling of transforming growth factor-beta family members
through Smad proteins. Eur J Biochem. 267:6954–6967. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mehra A and Wrana JL: TGF-beta and the
Smad signal transduction pathway. Biochem Cell Biol. 80:605–622.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Heldin CH, Landström M and Moustakas A:
Mechanism of TGF-beta signaling to growth arrest, apoptosis, and
epithelial-mesenchymal transition. Curr Opin Cell Biol. 21:166–176.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dumont N and Arteaga CL: Targeting the TGF
beta signaling network in human neoplasia. Cancer Cell. 3:531–536.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kandasamy M, Reilmann R, Winkler J,
Bogdahn U and Aigner L: Transforming growth factor-beta signaling
in the neural stem cell niche: A therapeutic target for
Huntington's disease. Neurol Res Int. 2011:1242562011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu S, Chen S and Zeng J: TGF-β signaling:
A complex role in tumorigenesis (Review). Mol Med Rep. 17:699–704.
2018.PubMed/NCBI
|
|
41
|
Lucarelli P, Schilling M, Kreutz C, Vlasov
A, Boehm ME, Iwamoto N, Steiert B, Lattermann S, Wasch M, Stepath
M, et al: Resolving the combinatorial complexity of smad protein
complex formation and its link to gene expression. Cell Syst.
6:75–89.e11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lou W, Chen Y, Zhu KY, Deng H, Wu T and
Wang J: Polyphyllin I overcomes EMT-associated resistance to
erlotinib in lung cancer cells via IL-6/STAT3 pathway inhibition.
Biol Pharm Bull. 40:1306–1313. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Akram M, Iqbal M, Daniyal M and Khan AU:
Awareness and current knowledge of breast cancer. Biol Res.
50:332017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Majumder A, Singh M and Tyagi SC:
Post-menopausal breast cancer: From estrogen to androgen receptor.
Oncotarget. 8:102739–102758. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guo W, Zhang S and Liu S: Establishment of
a novel orthotopic model of breast cancer metastasis to the lung.
Oncol Rep. 33:2992–2998. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ghulam J, Stuerken C, Wicklein D, Pries R,
Wollenberg B and Schumacher U: Immunohistochemical analysis of
transcription factors and markers of Epithelial-mesenchymal
transition (EMT) in human tumors. Anticancer Res. 39:5437–5448.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li L, Zhang S, Li H and Chou H: FGFR3
promotes the growth and malignancy of melanoma by influencing EMT
and the phosphorylation of ERK, AKT, and EGFR. BMC Cancer.
19:9632019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Si L, Fu J, Liu W, Hayashi T, Nie Y,
Mizuno K, Hattori S, Fujisaki H, Onodera S and Ikejima T: Silibinin
inhibits migration and invasion of breast cancer MDA-MB-231 cells
through induction of mitochondrial fusion. Mol Cell Biochem.
463:189–201. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang LN, Huang YH and Zhao L: Fusion of
macrophages promotes breast cancer cell proliferation, migration
and invasion through activating epithelial-mesenchymal transition
and Wnt/β-catenin signaling pathway. Arch Biochem Biophys.
676:1081372019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Moustakas A, Souchelnytskyi S and Heldin
CH: Smad regulation in TGF-beta signal transduction. J Cell Sci.
114:4359–4369. 2001.PubMed/NCBI
|
|
52
|
Wang G, Yu Y, Sun C, Liu T, Liang T, Zhan
L, Lin X and Feng XH: STAT3 selectively interacts with Smad3 to
antagonize TGF-β signalling. Oncogene. 35:4388–4398. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Itoh Y, Saitoh M and Miyazawa K:
Smad3-STAT3 crosstalk in pathophysiological contexts. Acta Biochim
Biophys Sin (Shanghai). 50:82–90. 2018. View Article : Google Scholar : PubMed/NCBI
|