Introduction
Germline mutations in the tumor-suppressive BRCA
genes are associated with an increased familial risk of breast and
ovarian cancer (1). Moreover,
loss-of function BRCA mutations are found in ~11% of tumors, most
frequently (25%) in high-grade serous ovarian adenocarcinoma
(2). BRCA1 and −2 mutations have
been discovered to lead to homologous recombination (HR) deficiency
and to an accumulation of chromosomal aberrations that promote
tumorigenesis (3,4).
The treatment of high-grade serous ovarian cancer
with germline or somatic BRCA1 or −2 mutations is predominantly
based on poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi),
such as olaparib (5–7). The PARPi is used in the maintenance
phase following responsiveness to platin-based chemotherapy and has
been demonstrated to increase progression-free survival by >11
months compared with the placebo (5–7).
PARPs are enzymes involved in DNA repair,
particularly in single-strand break repair. The use of PARPis leads
to the accumulation of DNA damage in cells that are HR-deficient
due to BRCA loss-of-function mutations, ultimately resulting in
synthetic lethality (8,9). Synthetic lethality refers to the
effect that certain pharmacological inhibitors can have on a pair
of genes, whereby the pharmacological inhibition of one gene causes
cell death only if the other gene is mutated, as might be the case
in a tumor cell, but not if the other gene is wild-type, as is the
case in a healthy cell (8,9). A number of mechanisms of PARPi
resistance mechanisms have been identified, including the
upregulation of PgP drug transporters (10), loss of p53-binding protein 1
(TP53BP1) function (11) or
secondary mutations within BRCA1 or −2 genes (10).
The TP53 gene is a tumor-suppressive gene encoding
the p53 protein (12). p53
functions as a transcription factor which is involved in the
checkpoint transition between the G1 and S phases of the
cell cycle (13). In the presence
of DNA mutations that cannot be repaired, p53 promotes cell cycle
arrest, induces proapoptotic signals and prevents tumor formation
(11). Notably, the presence of
TP53 mutations has been discovered to indicate resistance to
chemotherapy (11).
In the present study, tumor samples from four
patients with BRCA-mutated cancers treated with olaparib, who
progressed following the PARPi treatment, including three
individuals with ovarian cancers and one with breast cancer, were
analyzed. The exome sequencing profiles of the progressing tumor
were compared with the primary tumor tissue obtained at
diagnosis.
Materials and methods
Exome sequencing
DNA extraction and exome sequencing from
formalin-fixed paraffin-embedded (FFPE) samples were performed as
described previously (14). DNA was
extracted using the Maxwell-16 FFPE Plus LEV DNA purification kit
(cat. no. AS1135; Promega Corporation) according to the
manufacturer's protocol. DNA quality was assessed by
spectrophotometry with absorbance measured at 230, 260 and 280 nm.
DNA was quantified by a Qubit fluorometric assay (cat. no. Q32850;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions.
Libraries were constructed from 200 ng DNA and
captured using the SureSelect Human All Exon v6 kit (Agilent)
following the manufacturer's protocol. Paired-end (2×111 bases)
sequencing was performed on a NextSeq500 device (Illumina, Inc.).
Next, the sequences were aligned and annotated with the human Hg19
genome based on the SureSelect Human all Exon v6 manifest using the
BWA and GATK algorithms. Only sequences with a read depth of 10×, a
mutation allele frequency >5%, and a frequency in the general
population inferior to 1% were retained for further analysis.
Cell culture
HCT116 TP53−/− and BRCA2 mutated
(c.8021dup, p.I2675fsTer6) (15)
cells (kind gift from Professor Olivier Feron, UCLouvain, Brussels,
Belgium) were cultured in high-glucose DMEM supplemented with 10%
(vol/vol) fetal calf serum (Thermo Fisher Scientific, Inc.), 1%
penicillin, 1% streptomycin, 1% amphotericin B and 4 mM HEPES at
37°C with 5% CO2. Transfection of pCMV-Neo-Bam plasmids
containing wild-type p53 (negative control), p53 R175H or p53 R248W
(12) obtained from Addgene, Inc.
was performed as described previously (16). Briefly, cells were transfected with
0.5 µg plasmid using Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
After 6 h, culture medium was replaced by fresh medium. The next
morning, cells were re-plated in new plates.
Western blotting
Transfection efficiency was verified by western
blotting analysis as described previously (17). Protein concentration was assessed
using the Bio-Rad DC Protein Assay kit (Bio-Rad Laboratories,
Inc.). Then, 50 µg protein/lane was denatured, loaded and separated
via 12% SDS-PAGE and transferred onto nitrocellulose membranes
(Schleicher & Schuell Bioscience GmbH). After blocking with 5%
non-fat milk in phosphate-buffered saline containing 0.1% Tween-20
(PBST) for 1 h at room temperature, membranes were incubated
overnight at 4°C with primary antibody (1 µg/ml) in PBST containing
5% BSA. Afterwards, membranes were washed and incubated for 1 h at
room temperature with secondary antibody diluted in PBST-5% non-fat
milk. After three additional washes with PBST, membranes were
incubated with luminol reagent for 1 min at room temperature (cat.
no. sc2048; Santa Cruz Biotechnology, Inc.). The following primary
antibodies from Santa Cruz Biotechnology, Inc. were used: Mouse
anti-p53 (1:1,000; cat. no. DO-1) and anti-HSC70 (1:1,000; cat. no.
B-6). The following secondary antibodies were used: Peroxidase
AffiniPure Goat polyclonal Peroxidase AffiniPure Goat polyclonal
Anti-Mouse IgG (H+L) (1:10,000; cat. no. 115-035-003; Jackson
ImmunoReseach Europe, Ltd.).
Half maximal inhibitory concentration
(IC50) assessment
HCT116 TP53−/− cells were seeded at 20%
confluence into 96-well plates 24 h after transfection. After 24 h,
cells were treated with 3 µg/ml oxaliplatin (Teva Pharmaceutical
Industries Ltd.) for 72 h to induce DNA damage at 37°C, then with
titrating doses of olaparib (LC Laboratories) (0–2,560 µM) for five
days at 37°C in order to determine the IC50 of this
drug. Cell viability was assessed using crystal violet staining
(15). A total of 50 µl 1% crystal
violet solution (Sigma-Aldrich; Merck KGaA) was added directly to
the cells. After 20 min at room temperature, the crystal violet was
removed and cells were washed with tap water. Then, 100 µl DMSO was
added to dissolve the crystal violet. Absorbance was calculated at
590 nm with a DU370 UV–Vis spectrophotometer (Beckman Coulter,
Inc.)
A two-way ANOVA followed by a Greisser-Greenhouse
correction was performed to determine the statistical differences
between the groups using Graph Pad Prism software (v8.3.0; GraphPad
Software, Inc.). Data are presented as mean ± SD of three
independent repeats. P<0.05 was considered to indicate a
statistically significant difference.
Results
Presentation of cases
All patients were treated at the Georges-François
Leclerc Cancer Center, Dijon, France. Patient 1 (Ovary #1) was a
70–75-year-old woman diagnosed with ovarian cancer in February
2013. She had a past history of malaria and a family history of
breast cancer on her maternal side. In February 2013, she underwent
neoadjuvant chemotherapy (carboplatin-paclitaxel), interval surgery
and adjuvant chemotherapy for an International Federation of
Gynecology and Obstetrics stage-IIIc papillary serous ovarian
bilateral cancer (diagnosed based on clinical pathological
characteristics), and experienced complete remission. She presented
with peritoneal relapse 11 months later diagnosed by CT-scan, which
was treated with Platin-based chemotherapy (carboplatin-paclitaxel
for six treatment cycles). Genomic DNA analysis identified a
constitutional BRCA1 gene mutation (c.3839_3841delCTC;
p.S1280_Q1281delinsTer). An anti-angiogenic (bevacizumab) was added
to the treatment regimen in April 2015, with a partial tumor
response after six courses. In November 2015, olaparib was
introduced as maintenance chemotherapy until March 2016, when the
patient presented with a fast peritoneal and pancreatic relapse
with ascites and biliary compression indicated by CT-scan.
Chemotherapy was administered (weekly carboplatin) but had to be
stopped after one course due to rapid deterioration of the patient,
who died in May 2016. The first next-generation sequencing (NGS)
analysis was performed in April 2015 on the primary tumor. The
second analysis was performed using an ascites effusion in April
2016, after olaparib treatment ended.
Patient 2 (Ovary #2) was a 50–55-year-old woman
treated with six courses of platin-based neoadjuvant chemotherapy
(carboplatin-paclitaxel) and complete resection surgery for a
papillary serous ovary cancer (diagnosed via pathological
examination) in October 2010. She had a family history of breast
cancer and shared a known BRCA2 mutation (c.7845+1G>T) with her
two sisters. She relapsed two years later, in 2012, and was treated
with the same platin-based chemotherapy and complete surgery. In
May 2013, she received a third-line treatment with
carboplatin-gemcitabine-bevacizumab; carboplatin allergy arose at
the third course and treatment was stopped. A new relapse in May
2014 detected by CT-scan was treated by oxaliplatin-gemcitabine for
six courses with a complete tumor response. Finally, in February
2015, she presented with carcinomatous meningitis detected by
CT-scan that was treated by intrathecal methotrexate, brain
radiotherapy and weekly cisplatin. Based on the clinical and
imaging partial response, olaparib maintenance treatment was
started in July 2015 and terminated in August 2016, when the
patient relapsed. She died in September 2016. NGS analyses were
performed in 2015 on the tumor collected on the first surgery and
in August 2016 on cerebrospinal liquid.
Patient 3 (Ovary #3) was a 70–75-year-old female.
She had a family history of ovarian cancer and her mother had
ovarian cancer at 40 years old. She underwent three neoadjuvant
platin-based chemotherapy (carboplatin-paclitaxel) courses in July
2013, interval complete surgery and adjuvant chemotherapy
(carboplatin-paclitaxel), for a papillary serous stage IIIc ovarian
cancer. She first relapsed in November 2014 and was treated with
carboplatin-liposomal doxorubicin for five courses. She presented
with an in situ breast cancer detected by mammography, which
was treated with surgery and radiotherapy in March 2015. Germline
analysis identified a germinal BRCA2 mutation (c.2066_2069delGTAA,
p.S689KfsX11). Chemotherapy resumed with
carboplatin-gemcitabine-bevacizumab in May 2016 due to a new pelvic
progression detected by CT-scan. The tumor responded to the
treatment and olaparib maintenance started in September 2016. In
May 2018, she was still undergoing olaparib, with dose reduction
due to hematologic toxicity. In the interval, she underwent a
mesenteric node resection for a single and localized progression in
March 2018. NGS analyses were performed in May 2016 on the primary
tumor tissue, then on the mesenteric node in March 2018. This
patient died in December 2018.
Patient 4 (Breast #1) was a 50–55-year-old female.
She auto-palpated a left breast lesion in March 2015, which was
later diagnosed via biopsy as a triple-negative infiltrative breast
carcinoma with axillary node infiltration. She underwent
neoadjuvant chemotherapy with three courses of
5-fluorouracil-epirubicin-cyclophosphamide and three courses of
docetaxel, and breast surgery (mastectomy and axillary lymph node
dissection), which demonstrated clear signs of tumor regression.
Treatment was completed in October 2015 with radiotherapy of the
chest wall and lymphatic areas. During radiotherapy, she presented
with a cervical node which identified as triple-negative breast
cancer metastasis. Treatment with capecitabine was introduced in
November 2015 until May 2016, when the cervical node clinically
progressed. NGS analysis identified a BRCA1 somatic mutation
(c.2783G>T, p.G928V), and treatment with
cisplatin-gemcitabine-bevacizumab was initiated in May 2016. In
November 2016, considering the observed complete remission,
maintenance by off-label (olaparib was only indicated for ovarian
cancer at this date) olaparib was introduced. She relapsed in April
2017 with pleural effusion and hepatic metastasis detected by
CT-scan. Olaparib treatment was stopped and chemotherapy with
Halaven® was initiated in April 2017. A total of five
courses were administered, but the patient died in September 2017.
NGS analyses were performed on the primary tumor in September 2015
and on the cervical metastasis node biopsy in May 2017.
NGS analysis
Exome analyses were performed on the primary tumor
sample at diagnosis, then on a progressing lesion following
olaparib treatment. Allelic frequencies of all mutations observed
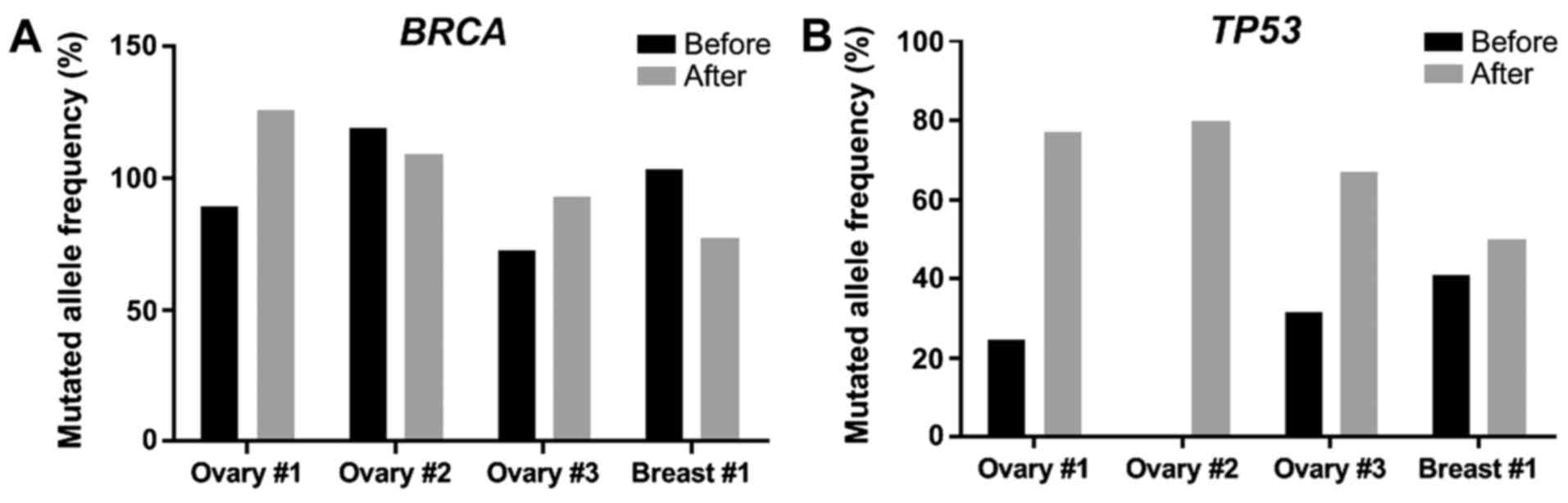
in both lesions of each patient are reported in Tables I–IV. Previously described resistance
mechanisms such as a new BRCA1 or BRCA2 mutation, or TP53BP1
loss-of-function mutations were not observed following olaparib
treatment. The only feature shared by all four patients was an
enrichment in TP53 mutations following treatment. Indeed, all
tumors had a BRCA mutation, whereas 3 out of 4 had low-frequency
TP53 mutations (<50% of tumor cells harboring a TP53
mutation). In the Ovary #2 patient, the tumor only had a BRCA2
mutation without any TP53 mutations prior to olaparib treatment
(Fig. 1; Table II). After progression under
olaparib, the allele frequencies of BRCA1 and BRCA2 mutations
varied between patients. Indeed, some patients (Ovary #1 and 3) had
an increase in the allelic frequency of BRCA mutations, whereas
others (Ovary #2, Breast #1) experienced a decrease. However, in
the case of TP53, de novo mutations were observed (Ovary
#2), as well as increased frequencies of mutations already present
in the primary tumor (Ovary #1 and 3, Breast #1) (Fig. 1; Tables
I–IV). The allelic frequency
of each mutation relative to tumor cell content (assessed by a
pathologist) before and after olaparib treatment for BRCA genes
(Fig. 1A) and for the TP53 gene
(Fig. 1B) was also evaluated. Thus,
the findings suggested that the enrichment in TP53 mutations may be
a marker of resistance to olaparib in these patients.
 | Table I.Allelic frequency of mutations
observed before and after olaparib treatment in Ovary #1. |
Table I.
Allelic frequency of mutations
observed before and after olaparib treatment in Ovary #1.
| Gene | Nucleotide
mutation | Protein mutation | Relativea allelic frequency before
olaparib, % | Relativeb allelic frequency after olaparib,
% |
|---|
| ABL1 | c.2486C>T | p.P829L | 69 | 0 |
| ABL2 | c.47A>C | p.N1860S | 0 | 57 |
| α
thalassemia/mental retardation syndrome X-linked | c.5579A>G | p.N1860S | 77 | 81 |
| BRCA1 |
c.3839_3841delCTC |
p.S1280_Q1281delinsTer | 88 | 126c |
| BRCA1 |
c.3844_3845insCG |
p.E1282AfsTer26 | 82 | 100 |
| BRCA1 interacting
protein C-terminal helicase 1 | c.2876C>A | p.P959Q | 0 | 27 |
| Lysine-specific
demethylase 6A | c.2158C>A | p.P720T | 0 | 18 |
| Low-density
lipoprotein receptor-related 1B | c.9532G>A | p.A3178T | 67 | 60 |
| PI3K catalytic
subunit γ isoform | c.9532G>A | p.N522S | 8 | 76 |
| Equilibrative
nucleoside transporter 1 | c.647T>C | p.I216T | 100 | 79 |
| Tumor p53 | c.916C>T | p.R306T | 24 | 77 |
| Table IV.Allelic frequency of mutations
observed before and after olaparib treatment in Breast #1. |
Table IV.
Allelic frequency of mutations
observed before and after olaparib treatment in Breast #1.
| Gene | Nucleotide
mutation | Protein
mutation |
Relativea allelic frequency before
olaparib, % |
Relativeb allelic frequency after
olaparib, % |
|---|
| ABL1 | c.3296A>C | p.K1099T | 0 | 56 |
| AT-rich interaction
domain 1A | c.4031C>G | p.S1344C | 0 | 19 |
| BRCA1-associated
RING domain 1 | c.1972C>T | p.R658C | 36 | 81 |
| BRCA1 | c.2783G>T | p.G928V | 104c | 77 |
| CREB-binding
protein | c.2111C>A | p.P704Q | 22 | 0 |
| Catenin α 1 | c.1283A>G | p.N428S | 99 | 69 |
| DNA
methyltransferase 3α | c.89A>C | p.E30A | 107c | 116c |
| E1A binding protein
p300 | c.631G>A | p.G211S | 108c | 113c |
| EPH receptor
A2 | c.2627G>A | p.R876H | 101c | 101c |
| Erb-b2 receptor
tyrosine kinase 3 | c.1981G>A | p.G661S | 106c | 114c |
| Fanconi anemia
group A | c.3427C>G | p.L1143V | 111c | 107c |
| Fms-related
receptor tyrosine kinase 4 | c.1921C>T | p.P641S | 12 | 41 |
| Insulin receptor
substrate 2 | c.2153G>C | p.R718T | 47 | 0 |
| Kinase insert
domain receptor | c.2961A>C | p.E987D | 0 | 23 |
| Myeloid cell
leukemia sequence 1 | c.680C>T | p.A227V | 100 | 0 |
| Multiple endocrine
neoplasia I | c.527G>A | p.R176Q | 63 | 71 |
| Human mutS homolog
6 | c.2633T>C | p.V878A | 0 | 29 |
| mTOR | c.277C>A | p.L93I | 0 | 67 |
| Nibrin | c.628G>T | p.V210F | 0 | 46 |
| NOTCH2 | c.17_18delCC | p.P6RfsTer27 | 63 | 0 |
| PI3K catalytic
subunit γ isoform | c.1025A>G | p.H342R | 0 | 41 |
| ROS proto-oncogene
1 |
c.5483_5486delTCAG | p.F1828Ter | 47 | 0 |
| Equilibrative
nucleoside transporter 1 | c.911T>C | p.I304T | 108c | 107c |
| Tumor p53 |
c.403_421dupTGCCAACTGGCCAAGACCT | p.C141LfsTer14 | 41 | 50 |
| X-ray repair cross
complementing 2 | c.283A>G | p.I95V | 13 | 21 |
| Table II.Allelic frequency of mutations
observed before and after olaparib treatment in Ovary #2. |
Table II.
Allelic frequency of mutations
observed before and after olaparib treatment in Ovary #2.
| Gene | Nucleotide
mutation | Protein
mutation |
Relativea allelic frequency before
olaparib, % |
Relativeb allelic frequency after olaparib,
% |
|---|
| ABL2 | c.2789A>G | p.K930R | 143c | 46 |
| Adenomatous
polyposis coli protein | c.7504G>A | p.G2502S | 110c | 11 |
| Tyrosine-protein
kinase receptor UFO | c.1777G>C | p.D593H | 0 | 44 |
| BRCA2 | c.7617+1G>T | Splicing | 119c | 109c |
|
G1/S-specific cyclin-D3 | c.759G>T | p.E253D | 130c | 56 |
| Histone-lysine
N-methyltransferase, H3 lysine-79 specific | c.4415C>T | p.P1472L | 57 | 0 |
| Terminal
nucleotidyltransferase 5C | c.201C>G | p.H67Q | 283c |
111c |
| FLT3 | c.970G>A | p.D324N | 161c | 103 |
| FLT4 | c.3962G>A | p.R1321Q | 133c | 98 |
| Transcriptional
activator GLI3 | c.3083G>T | p.S1028I | 127c | 49 |
| MAP2K3 | c.927delC | p.A311QfsTer4 | 0 | 16 |
| PMS1 homolog 2 | c.1531A>G | p.T511A | 109c | 89 |
| Tumor p53 | c.672+1G>T | Splicing | 0 | 44 |
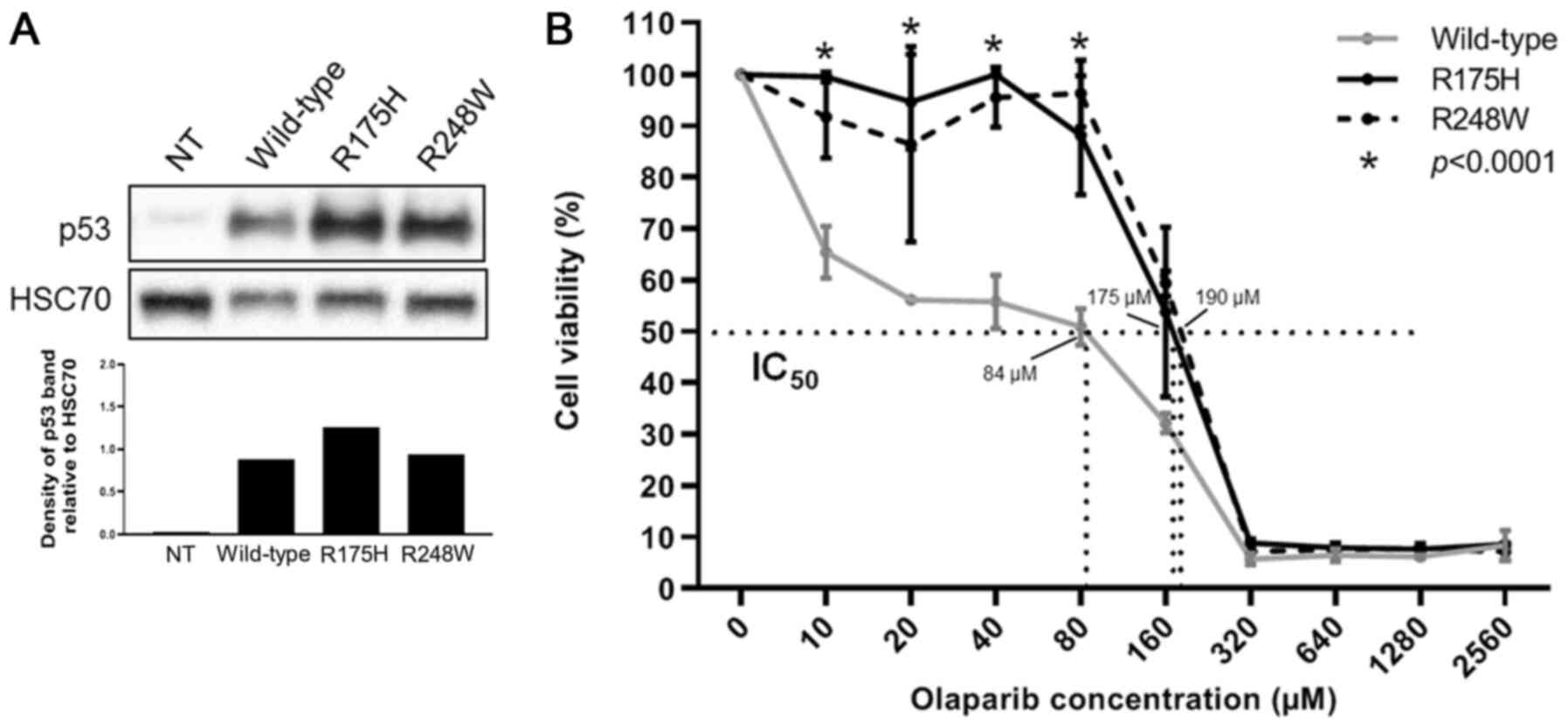
In vitro analysis
The human HCT116 colorectal carcinoma cell line with
a BRCA2 pathogenic mutation (c.8021dup, p.I2675fsTer6) and TP53
deficiency was used to determine whether mutations in TP53 could be
a marker of progression following olaparib treatment in the
presence of BRCA mutations. HCT116 is a cell line with a
TP53-deficient variant and a pathogenic BRCA mutation (15). HCT116 cells transfected with the
mutant R175H and R248W TP53 (Fig.
2A) were significantly more resistant to olaparib compared with
wild-type TP53-transfected cells (P<0.0001; Fig. 2B); the calculated IC50
values were 175, 190 and 84 µM for R175H, R248W and WT,
respectively (Fig. 2B). Thus, these
results indicated that inactivating TP53 mutations may be
associated with olaparib resistance in the presence of BRCA
mutations, supporting the aforementioned clinical observations
(TP53 mutation appearance or enrichment in progressing disease
under olaparib treatment).
Discussion
Patients carrying mutations in BRCA genes are
predisposed to high incidence rates of breast and ovarian cancer
(1). BRCA genes are involved in the
HR pathway, which is required for the repair of spontaneous
double-stranded breaks that arise during DNA replication (18). Defects in HR result in an
accumulation of chromatid breaks and cells that cannot repair
chromatid breaks by HR become dependent on other alternative repair
pathways (e.g. non-homologous end joining) (19). HR-deficient cells have exhibited
acute sensitivity to PARPi-induced cell death (19), and the loss of PARP activity
prevented the repair of single-stranded breaks, which were then
converted into double-stranded breaks during DNA replication. The
ability of PARPi to selectively kill HR-deficient cells is
currently used for the treatment of breast and ovarian cancers with
BRCA1 or −2 mutations (20).
Overall, ~15–20% of patients with epithelial ovarian cancer harbor
germline or somatic BRCA mutations (21). However, some of these patients do
not respond or eventually develop resistance to PARPis (5–7). The
most common mechanism of resistance to these agents in BRCA-mutated
cancer is secondary intragenic mutation restoring BRCA protein
functionality (22). Additional
mechanisms of resistance have been described, such as loss of
TP53BP1 (11). Nevertheless, the
clinical relevance of these mechanisms has not been demonstrated in
patients with BRCA-mutated cancer.
The p53 protein is an important tumor suppressor
encoded by the TP53 gene (12). p53
is a crucial factor in the cellular response to DNA damage
(12). When an external event (e.g.
mutagenic agent exposure, mutations induced by polymerase errors)
induces genome alterations towards tumorigenesis, the expression of
functional p53 is essential (21).
When DNA damage cannot be repaired, p53 signaling induces
apoptosis. Mutations in the TP53 gene result in failure of the p53
signaling (23). There is evidence
to suggest that homozygous BRCA deficiency induced cell death by
activating a p53-dependent checkpoint. However, the impairment of
this checkpoint resulting from p53 loss removes the cell-lethal
effects of BRCA deficiency (24,25).
Breast cancers from human mutated BRCA carriers have an increased
frequency of TP53 mutations (26)
and high-grade ovarian cancers often harbor TP53 mutations
(27).
The present study documented the case of four
patients for whom progression under PARPi treatment was suggested
to be associated with the enrichment or emergence of TP53
mutations. Moreover, these observations were supported by in
vitro experiments in which deleterious TP53 mutations were
associated with reduced sensitivity to PARPi. In addition, the
frequencies of pathogenic BRCA mutations varied across these
patients after treatment. This phenomenon may be explained by
clonal selection of PARPi-resistant clones. Indeed, the resistance
mutation could be in the same cancer cell as the BRCA mutation, or
in another cancer cell clone containing fewer BRCA mutations.
As the present study was based on clinical
observations with a limited number of cases, it cannot be claimed
that TP53 mutations are a mechanism of resistance or a clinical
marker of progression. In the clinical setting, routine detection
of plasma TP53 mutations could easily be achieved with NGS
analysis. Thus, the emergence of de novo TP53 mutations or
enrichment in pre-existing mutations may represent a marker of
progression in patients under olaparib treatment.
Acknowledgements
The authors thank Dr Isabel Gregoire,
Georges-François Leclerc Cancer Center (Dijon, France), for editing
the manuscript.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TC and JN made substantial contributions to
conception and design of the study and acquisition, analysis and
interpretation of data. SC performed the exome analysis. MC and EH
performed the in vitro analysis. LF, LBL and ID treated the
patients. LA performed the histological analysis and tumor cell
assessment. RB designed the study, supervised the study and wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All patients provided written informed consent for
the use of genomic analysis results in research. Patient
confidentiality was maintained and analysis was performed in
compliance with the Declaration of Helsinki. The present study was
approved by the Institutional Ethics Review Board of The
Georges-François Leclerc Cancer Center (Dijon, France).
Patient consent for publication
The local ethics committee of The Georges-François
Leclerc Center waives the necessity of patient consent for
publication because the patients have died and as the content of
the publication does not provide any identifying data on the
patients, the establishment does not wish to disturb the patient's
family in these unfortunate circumstances.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HR
|
homologous recombination
|
|
IC50
|
half maximal inhibitory
concentration
|
|
PARP
|
poly (ADP-ribose) polymerase
|
|
PARPi
|
PARP inhibitor
|
References
|
1
|
Paul A and Paul S: The breast cancer
susceptibility genes (BRCA) in breast and ovarian cancers. Front
Biosci. 19:605–618. 2014. View
Article : Google Scholar
|
|
2
|
Manchana T, Phoolcharoen N and Tantbirojn
P: BRCA mutation in high grade epithelial ovarian cancers. Gynecol
Oncol Rep. 29:102–105. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bishop AJ and Schiestl RH: Role of
homologous recombination in carcinogenesis. Exp Mol Pathol.
74:94–105. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Helleday T: Homologous recombination in
cancer development, treatment and development of drug resistance.
Carcinogenesis. 31:955–960. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gelmon KA, Tischkowitz M, Mackay H,
Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M,
Gilks B, et al: Olaparib in patients with recurrent high-grade
serous or poorly differentiated ovarian carcinoma or
triple-negative breast cancer: A phase 2, multicentre, open-label,
non-randomised study. Lancet Oncol. 12:852–861. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ledermann J, Harter P, Gourley C,
Friedlander M, Vergote I, Rustin G, Scott C, Meier W,
Shapira-Frommer R, Safra T, et al: Olaparib maintenance therapy in
platinum-sensitive relapsed ovarian cancer. N Engl J Med.
366:1382–1392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaufman B, Shapira-Frommer R, Schmutzler
RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G,
Stemmer SM, Hubert A, et al: Olaparib monotherapy in patients with
advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol.
33:244–250. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ashworth A: A synthetic lethal therapeutic
approach: Poly(ADP) ribose polymerase inhibitors for the treatment
of cancers deficient in DNA double-strand break repair. J Clin
Oncol. 26:3785–3790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi YE, Meghani K, Brault ME, Leclerc L,
He YJ, Day TA, Elias KM, Drapkin R, Weinstock DM, Dao F, et al:
Platinum and PARP inhibitor resistance due to overexpression of
microRNA-622 in BRCA1-mutant ovarian cancer. Cell Rep. 14:429–439.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bunting SF, Callén E, Wong N, Chen HT,
Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao
L, et al: 53BP1 inhibits homologous recombination in
Brca1-deficient cells by blocking resection of DNA breaks. Cell.
141:243–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baker SJ, Markowitz S, Fearon ER, Willson
JK and Vogelstein B: Suppression of human colorectal carcinoma cell
growth by wild-type p53. Science. 249:912–915. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang M, Zhuang G, Sun X, Shen Y, Wang W,
Li Q and Di W: TP53 mutation-mediated genomic instability induces
the evolution of chemoresistance and recurrence in epithelial
ovarian cancer. Diagn Pathol. 12:162017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alsadoun N, MacGrogan G, Truntzer C,
Lacroix-Triki M, Bedgedjian I, Koeb MH, El Alam E, Medioni D,
Parent M, Wuithier P, et al: Solid papillary carcinoma with reverse
polarity of the breast harbors specific morphologic,
immunohistochemical and molecular profile in comparison with other
benign or malignant papillary lesions of the breast: A comparative
study of 9 additional cases. Mod Pathol. 31:1367–1380. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dong Z, Dong H, Zhong X, Peng Z, Zhu X,
Sun Y, Chen Y, Liu C, Yin X, Zhu G, et al: Development of a
compehensive NGS workflow for the analysis of tumor BRCA1 and BRCA2
mutations and large rearrangements. J Genet Genome Res. Sept
28–2015.(Epub ahead of print). doi: 10.23937/2378-3648/1410019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Végran F, Boidot R, Solary E and
Lizard-Nacol S: A short caspase-3 isoform inhibits
chemotherapy-induced apoptosis by blocking apoptosome assembly.
PLoS One. 6:e290582011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Végran F, Mary R, Gibeaud A, Mirjolet C,
Collin B, Oudot A, Charon-Barra C, Arnould L, Lizard-Nacol S and
Boidot R: Survivin-3B potentiates immune escape in cancer but also
inhibits the toxicity of cancer chemotherapy. Cancer Res.
73:5391–5401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Andreassen PR, Ho GP and D'Andrea AD: DNA
damage responses and their many interactions with the replication
fork. Carcinogenesis. 27:883–892. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et
al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA
mutation carriers. N Engl J Med. 361:123–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cancer Genome Atlas Research Network, .
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Norquist B, Wurz KA, Pennil CC, Garcia R,
Gross J, Sakai W, Karlan BY, Taniguchi T and Swisher EM: Secondary
somatic mutations restoring BRCA1/2 predict chemotherapy resistance
in hereditary ovarian carcinomas. J Clin Oncol. 29:3008–3015. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Levine AJ: Reviewing the future of the P53
field. Cell Death Differ. 25:1–2. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hakem R, de la Pompa JL, Elia A, Potter J
and Mak TW: Partial rescue of Brca1 (5–6) early embryonic lethality
by p53 or p21 null mutation. Nat Genet. 16:298–302. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jonkers J, Meuwissen R, van der Gulden H,
Peterse H, van der Valk M and Berns A: Synergistic tumor suppressor
activity of BRCA2 and p53 in a conditional mouse model for breast
cancer. Nat Genet. 29:418–425. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Greenblatt MS, Chappuis PO, Bond JP, Hamel
N and Foulkes WD: TP53 mutations in breast cancer associated with
BRCA1 or BRCA2 germ-line mutations: Distinctive spectrum and
structural distribution. Cancer Res. 61:4092–4097. 2001.PubMed/NCBI
|
|
27
|
Bernardini MQ, Baba T, Lee PS, Barnett JC,
Sfakianos GP, Secord AA, Murphy SK, Iversen E, Marks JR and
Berchuck A: Expression signatures of TP53 mutations in serous
ovarian cancers. BMC Cancer. 10:2372010. View Article : Google Scholar : PubMed/NCBI
|