Introduction
Acute myeloid leukemia (AML) is one of the most
aggressive types of malignant tumor of the hematopoietic system
affecting the adult population (1).
The incidence of AML increases with age and accounts for ~80% of
cases in patients >18 years (2).
Older patients with AML are often defined as individuals >60
years of age. The median age of adult AML patients in developed
countries is ~67 years (3). The
2018 AML incidence estimates from SEER (4) are <1.23 per 100,000 in the
<40-year-old population, 10.92 per 100,000 in the ≥60-year-old
population and 20.89 per 100,000 in the ≥75-year-old population in
the USA. The adult AML population is comprised largely of the
elderly patients with AML (5).
With the aggravated degree of aging, the incidence
of AML among the elderly in China is also on the rise. In China,
AML is the 7th leading cause of cancer-related death for males and
the 10th for females among all age groups (6–8). AML
is a heterogeneous clonal disorder that is characterized by
hyperplasia of immature cells in the bone marrow (BM) and
peripheral blood, together with a loss of hemopoiesis function
(7,9). Although the overall survival and
complete remission (CR) rates of AML have improved in recent years,
>50% of patients with AML still suffer recurrence, and 20–35% of
adult patients with AML are diagnosed with primary refractory AML.
There has been no significant improvement in treating
relapsed/refractory AML (RR-AML) in the past two decades. The main
treatment strategies for AML include chemotherapy and hematopoietic
stem cell transplantation (HSCT). Allogeneic HSCT following
chemotherapy is the most promising approach for the maintenance of
long-term remission and survival. However, older and less fit
patients are often unsuitable for allogeneic HSCT due to medical
comorbidities (10). The 3-year
survival rate in patients with chemoresistance and relapsed AML is
<10% (11,12), indicating that novel treatment
schedules are urgently needed for these patients (9,13,14).
Several membrane molecules, such as CD33 (15), CD123 (16), CD44 (17), T cell immunoglobulin and mucin
domain-containing 3 (TIM-3) (18),
CD47 (17), CD96 (19), CD99 (20) and CD32 (21), expressed by AML cells may be useful
as tumor-specific markers for targeted therapy (22). The monoclonal antibodies (mAb)
targeting part of these antigens, such as CD33, CD47, have been
shown to exhibit antineoplastic activity in animal models and in
clinical trials (23,24). In addition, the chimeric antigen
receptor (CAR)-directed T cell with CD19 as its specific target
showed good therapeutic effects against relapsed and refractory
leukemia in clinical trials (25).
Subsequent studies have revealed that CAR-T therapy has a
significant cytotoxic effect on B cell tumors (26–28).
Unfortunately, CAR-T therapy for myeloid neoplasms has developed
slowly and off-target effects are often serious because several
myeloid antigens are non-specifically expressed on normal HSCs and
other tumor cells (14,15,29–31).
Therefore, it is important to identify an ideal target antigen for
CAR-T therapy.
C-type lectin-like molecule-1 (CLL-1; also known as
CLEC12A, MICL, or DCAL-2) is a type-II transmembrane glycoprotein
that is selectively expressed on the leukemic stem cell (LSC)
surface and is restricted to the hematopoietic lineage,
particularly to monocytes and granulocytes (32,33).
Wang et al (34) and Tashiro
et al (35) respectively
reported that CLL-1 CAR-T specifically lysed CLL-1+
leukemia cells in vivo and in vitro without severe
hematological toxicity. However, it is not clear whether there are
differences in the anti-leukemia effect of CLL-1 CAR-T derived from
healthy or patient donor T cells. In addition, the co-stimulatory
molecule programmed cell death 1 (PD-1) can be induced following
sustained activation of T cells, PD-1 exhibits inhibitory effects
on T cell function and eventually leads to T cell exhaustion by
binding to its ligands, PD-L1 and PD-L2, PD-1 is also expressed on
the CAR-T cell membrane (36,37).
In the present study, a CLL-1 CAR vector was
constructed, consisting of the following components in-frame from
the 5′ end to the 3′ end: CD8 signaling peptide sequences, anti-CLL
single-chain variable fragment (scFv) (M26), the hinge and
transmembrane regions of the CD8α molecule, the cytoplasmic domain
of CD28 and OX40 and the CD3ζ signaling domain. The cytotoxic
effects of these T cells expressing the CLL-1 CAR were evaluated on
the THP-1 human acute monocytic leukemia cell line, as well as and
primary AML cells. The immunotherapeutic effect of CLL-1 CAR-T was
found to be enhanced when combined with PD-1 silencing.
Materials and methods
Cell lines and primary AML
samples
The THP-1 (cat. no. TIB-202), SUP-B15 (cat. no.
CRL-1929) and K562 (cat. no. CCL-243) cell lines were purchased
from American Type Culture Collection (ATCC). A stably transfected
(st) K562 cell line was generated using a lentiviral plasmid
containing the human CLL-1 gene and cultured in a 5% CO2
atmosphere at 37°C with RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) containing 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 1% Penicillin-Streptomycin solution (cat. no.
15140148; Gibco; Thermo Fisher Scientific, Inc.) 293T cells (cat.
no. CRL-11268; ATCC) were used for lentiviral packaging and were
cultured in DMEM containing 10% FBS and 1% Penicillin-Streptomycin
solution. No cell line was contaminated with Mycoplasma, as
determined using the MycoAlert™ detection kit (Lonza Group,
Ltd.).
BM were obtained from patients with RR-AML (n=10)
enrolled at Huai'an Hospital Affiliated with Xuzhou Medical College
between June 2017 and December 2018. The study was approved by the
hospital's Institutional Review Board (approval no. HEYLL201601).
Written informed consent was obtained from all patients and healthy
donors.
The inclusion criteria were as follows: i) AML
diagnosed according to the Morphology, Immunology, Cytogenetics,
Molecular biology (MICM) classification (38) and categorized as RR-AML; or ii) BM
blasts ≥10%. The exclusion criteria were: i) Extra-medullary
infiltration of relapsed leukemia only; or ii) patients who did not
agree to be included and who did not sign the informed consent.
Patients were diagnosed with refractory AML if: i)
They did not achieve CR after two courses of induction chemotherapy
by standard protocols; ii) they relapsed at 6 months or later
following the first CR and failed at the subsequent induction
chemotherapy; iii) they relapsed within 6 months after the first
CR; or iv) they relapsed more than twice. Patients were diagnosed
with relapsed AML if leukemic cells reappeared in the peripheral
blood or a percentage of BM blasts of >10%.
Lentiviral vector construction and T
cell transduction
The PSE2970 lentiviral vector, was constructed to
express the CLL-1 antigen in the K562 cell line (stK562).
Lentiviral packaging was performed using 293T cells. 293T cells
were seeded at a density of 1×107 cells per 75
cm2 flask 1 day before transfection. Lentivirus was
produced by co-transfecting 293T cells with the CLL-1-expressing
PSE2970 plasmid with the pPac-GP, pPac-R and pEnv-G plasmids
(Shanghai Unicar-Therapy Bio-medicine Technology Co., Ltd.) at a
mass ratio of 20:13:5:20.
The plasmid and Calcium Phosphate transfection
reagent (cat. no. STP07006; Shanghai Sunbio Biotechnology, Co.,
Ltd.) mixture was added to the cells in a dropwise manner. At 48 h
after transfection, the lentivirus was harvested and concentrated
by ultracentrifugation for 16 h at 8,000 × g at 4°C and stored at
−80°C. The lentivirus named PSE2970 was then used to transduce the
K562 cells at a MOI of 20 in the presence of 10 µg/ml polybrene
(Sigma-Aldrich; Merck KGaA; cat no. H9268). After 48 h of
infection, cells were selected with 2 µg/ml puromycin for 7
days.
The scFv was amplified from the CLL-1 antibody using
PCR and was ligated into the pLenti-3G basic lentiviral vector
(Shanghai Sunbio Biotechnology Co., Ltd.) containing the
intramembrane domains of CD28 and OX40, CD8 hinge region, CD8
transmembrane region and CD3ζ intracellular domains. This vector
was named PSE2743, the difference between the lentiviral vector
PSE2744 and PSE2743 vector was the addition of the PD-1 silencing
shRNA sequence. The procedure used for these two lentivirus
packaging was the same as that used for the lentiviral vector of
PSE2970.
Peripheral blood mononuclear cells (PBMCs) were
obtained from healthy donors or patients with AML using Ficoll
(Beijing DongFang HuaHui Biomedical Technology Co., Ltd.) density
gradient centrifugation. T cells were isolated from PBMCs using CD3
immunomagnetic beads (cat. no. 130-050-101; Miltenyi Biotec, Inc.).
The T cells were cultured in AIM-V T cell medium (Gibco; Thermo
Fisher Scientific, Inc.) containing 100 IU/ml recombinant human
IL-2 (cat. no. AF-200-02; PeproTech, Inc.), 5 ng/ml recombinant
human IL-7 (cat. no. AF-200-07; PeproTech, Inc.) and 5 ng/ml
recombinant human IL-15 (cat. no. AF-200-15; PeproTech, Inc.), and
co-stimulated with anti-CD3 (cat. no. 170-076-116; Miltenyi Biotec,
Inc.) and -CD28 antibodies (cat. no. 170-076-117; Miltenyi Biotec,
Inc.) for 18–24 h in a 5% CO2 atmosphere at 37°C. T
cells were seeded at a density 1×106 cells per 75
cm2 flask 1 day before transduction. Activated T cells
were infected with lentiviral supernatants of PSE2743 or PSE2744 at
an MOI of 10 in the presence of 8 µg/ml polybrene (cat. no. H9268;
Sigma-Aldrich; Merck KGaA) for 48 h. CAR-T cells were cultured and
expanded for 14 days.
Flow cytometry
Antibodies against CD45-phycoerythrin (PE)/Cy7 (cat.
no. 368531), CD34-fluorescein isothiocyanate (FITC; cat. no.
343603), CD33-PE (cat. no. 303403), CD4-APC (cat. no. 357407),
CD8-Percp-cy5.5 (cat. no. 344709), PD-L1-allophycocyanin (APC; cat.
no. 329707), PD-1-PE (cat.no. 367403) and CLL-1-PE (cat. no.
353603) were purchased from BioLegend, Inc. Primary AML cells
derived from the BM of patients with refractory/relapsing AML were
then incubated with red blood cell lysis buffer. Blasts were
identified by gating on CD45/SSC. For CLL-1, CD33 and CD34
expression, staining >5% relative to FMO on CD45/SSC-gated
blasts was considered positive. The expression of PD-L1 on THP-1
cells was examined using flow cytometry before and after 24-h
incubation with the effector cells (untransduced T cells or CLL-1
CAR-T cells) at an effector:target (E:T) ratio of 2.5:1 and
staining >5% relative to FMO was considered positive.
For transduction efficiency analysis, CAR-T cells
were collected and washed twice with 1 ml PBS containing 2% FBS
(Gibco; Thermo Fisher Scientific, Inc.), then labeled with protein
L-PE (ACROBiosystems) for 45 min at 4°C in the dark.
All flow cytometry data were acquired using a BD
Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo
software (version 7.6.5; FlowJo LLC).
AML cells Sorting
CLL-1+ primary AML cells were isolated
using anti-CD33 magnetic cell sorting (cat. no. 130-045-501;
Miltenyi Biotec, Inc.). Patient-derived AML tumor cells were
resuspended with sorting buffer (cat. no. 130-100-008; Miltenyi
Biotec, Inc.), and the ratio of CD33 positive magnetic beads added
was 20 µl magnetic beads per 1×107 cells, then incubated
at 4°C for 15 min, and then sorted by magnetic force.
In vitro cytotoxicity
Cytotoxic activity of untransduced T cells (negative
control, NC) and CAR-T cells was measured using the CytoTox 96
Non-Radioactive Cytotoxicity Assay Kit (Promega Corporation) after
a 16-h incubation with target cells in a 5% CO2
atmosphere at 37°C in RPMI-1640 medium with 4% FBS, according to
the manufacturer's instructions. Target cells (K562 cells or
CLL-1-expressing cells) and effector cells (NC or two types of
CAR-T cells) were first centrifuged at 300 × g for 10 min at room
temperature, then re-suspended in 2 ml RPMI-1640 with 4% FBS
(Gibco; Thermo Fisher Scientific, Inc.). The cell density of the
effector cells was adjusted to 2×106/ml (10:1 group),
1×106/ml (5:1 group) and 5×105/ml (2.5:1
group) using medium. The target cell density was adjusted
2×105 cells/ml. Target cells and effector cells were
added to a 96-well plate at 50 µl per well, respectively.
The 96-well plate was sealed with a sealing film and centrifuged at
250 × g for 5 min at room temperature. The percentage of tumor
lysis was calculated as follows: % tumor lysis=[experimental
value-low control of CART (or NC) cells-low control of target
cells]x 100 / (high control of target cells-low control of target
cells), where the low control was the assay medium+cells and the
high control was the assay medium+2% Triton X-100+cells.
Cytokine release assays
Effector cells (NC T cells or CCL-1 CAR-T cells;
2×106 cells/ml) and target cells (THP-1 cells or
CLL-1-expressing AML cells; 2×105 cells/ml) were
co-cultured in RPMI-1640 at a ratio of 10:1 for 24 h. Cytokine
levels secreted into the culture supernatant were measured using a
human TH1/TH2/TH17 Cytometric Bead
Array kit (BD Biosciences; cat. no. 560484) containing capture
beads specific for IL-2, IL-4, IL-5, IL-10, TNF, IFN-γ and IL-17A
proteins. Briefly, human cytokine standards were prepared, and
samples were mixed with the
TH1/TH2/TH17 cytokine capture
beads at a dilution ratio of 1:4 at room temperature for 3 h in the
dark. Cytokine data were analyzed by flow cytometry using an Attune
NxT Flow Cytometer (Thermo Fisher Scientific, Inc.). Data were
analyzed using FCAP Array Software v3.0 (BD Biosciences).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from CAR-T cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc) and
reverse transcribed into cDNA using HiScript II Q RT SuperMix
(Vazyme Biotech Co., Ltd.), according to the manufacturer's
protocols. The RT-qPCR reaction was performed with AceQ qPCR Probe
Master Mix (Vazyme Biotech Co., Ltd.) at the following
thermocycling conditions: Pre-denaturation at 95°C for 5 min,
followed by a total of 40 cycles that included denaturation at 95°C
for 10 sec, annealing and extension at 60°C for 34 sec. RT-qPCR was
performed using an ABI 7500 system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). Primers against PD-1 (forward,
5′-AGCAGACGGAGTATGCCACCA-3′ and reverse,
3′-ATCCTCAGGCCTCAGTGGCT-5′) and a fluorescent probe
(5′-TGTCTTTCCTAGCGGAATGGGCACCTCATCC-3′). mRNA levels were
quantified using the 2−ΔΔCq method (39) and normalized to the housekeeping
gene GAPDH.
Statistical analysis
SPSS version 17 (SPSS, Inc.) and GraphPad Prism
version 5 (GraphPad Software, Inc.) were used for statistical
analysis. Data are presented as the mean ± SD. Comparisons between
two groups were performed using Student's t-test for normally
distributed data with homogeneous variance. Otherwise,
Mann-Whitney's U-test was used. Comparisons among ≥3 groups were
performed using two-way ANOVA followed by Fisher's Least
Significant Difference for normally distributed data with
homogeneous variance. Otherwise, the Kruskal-Wallis test and a
Dunn's post hoc test with Bonferroni correction were used.
P<0.05 was considered to indicate a statistically significant
difference.
Results
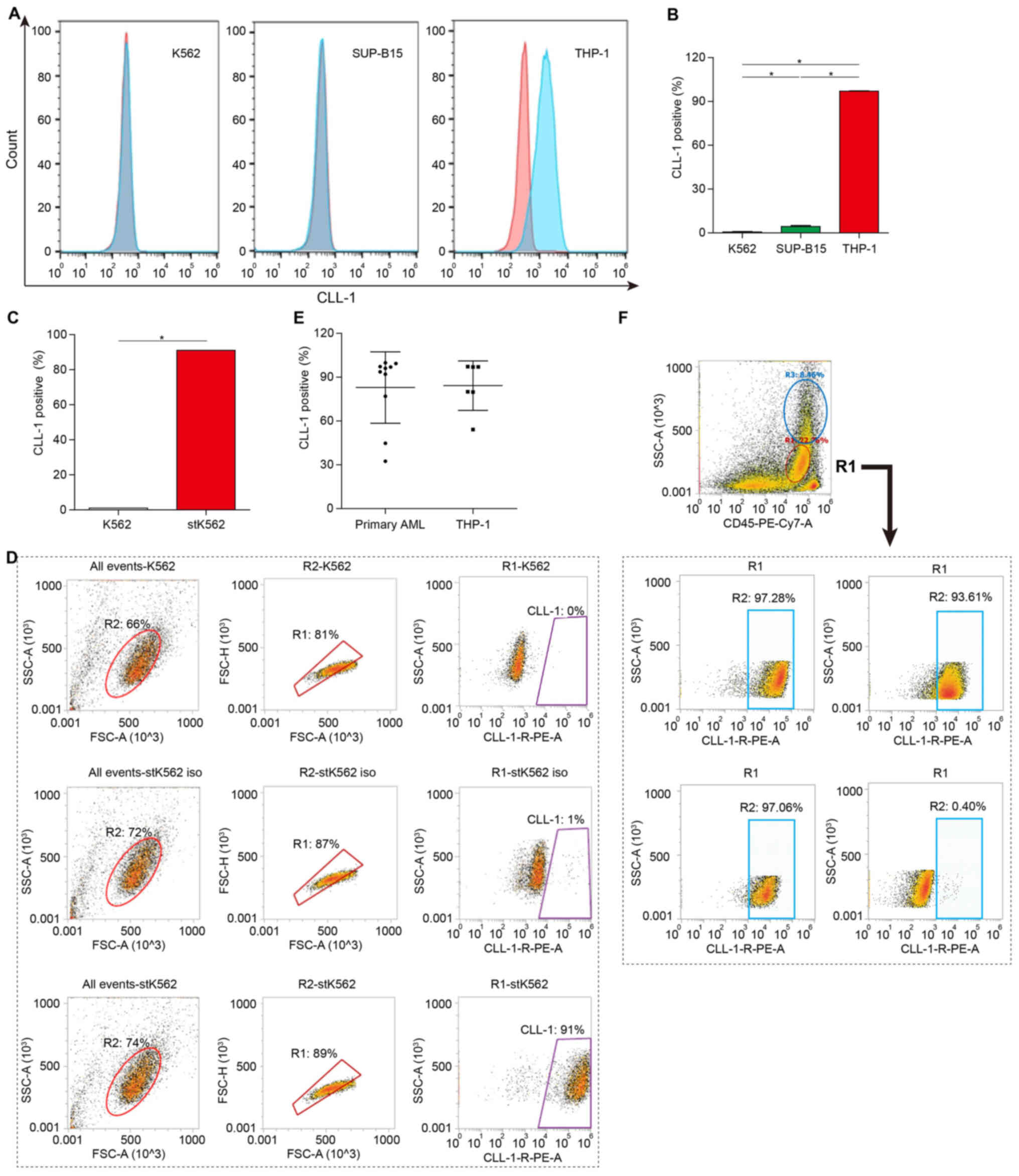
CLL-1 is expressed on the surface of
THP-1 cells and primary AML cells
In a previous study, the CLL-1 protein was reported
to be restrictively expressed in the hematopoietic lineage,
particularly in AML blasts and LSCs (30), suggesting that CLL-1 may be an
optimal target for CAR-T-based cell therapy in the treatment of
AML. The expression of CLL-1 on the cell surface of AML cells was
detected using flow cytometry. CLL-1 was not expressed on the
surface of chronic myeloid leukemia K562 cells and human acute
lymphoblastic SUP-B15 cells; however, it was strongly expressed on
THP-1 cells (Fig. 1A and B).
Moreover, the K562 cell line with CLL-1 specific expression
(stK562) was constructed by stable transfection of the CLL-1
over-expressing plasmid. High CLL-1 expression on stK562 cells was
confirmed using flow cytometry (Fig. 1C
and D).
CLL-1 expression on primary AML blasts
from 10 patients diagnosed with RR-AML was also evaluated using
flow cytometry
The clinical characteristics of these patients are
summarized in Table I. The
frequency of CLL-1-positive cells ranged from 32.43–99.80%
(Fig. 1E and F).
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| Sex | Age, years | Number of relapses,
n | FAB subset | Cytogenetics | Molecular
biology | Gene mutation | CLL-1+, % |
|---|
| Male | 27 | Refractory | AML-M5 | NA | None | None | 92.10 |
| Male | 38 | 3 | AML-M2 | 46,XY | None | CEBPA, TET2,
KMT2D | 99.80 |
| Male | 56 | 1 | AML-M2 | 46,XY | AML1/ETO | None | 97.28 |
| Female | 27 | 1 | AML (MDS
transformation) |
46,XX,t(11;19)(q23;q13)/46,XY | MLL-ELL | SF3A1 | 93.61 |
| Male | 49 | 2 | AML-M2 |
46,XY,t(10;15)(p15;q11)/46,XY | Not available | FLT3-ITD, STAG2,
IDH1, JAK3 | 76.90 |
| Female | 28 | 4 | AML-M4Eo |
46,XX,inv(16)(p13;q22)/46,
idem,del(7)(q22q31) | CBFβ-MYH11 | Kit, BCORL1,
JAK3 | 32.43 |
| Male | 31 | 1 | AML-M2 |
46,XY,t(8;21)(q22;q22) | AML1/ETO | Not available | 99.30 |
| Female | 27 | 2 | AML-M2 |
47,XY,i(8)(q10),der(18),+21 | None | KRAS, NRAS, NPM1,
DNMT3B | 44.73 |
| Male | 51 | 1 | AML-M4 |
45,X,-Y,t(8;21)(q22;q22)/46,XY | AML1/ETO | Kit, FGFR3 | 96.10 |
| Male | 8 | 1 | AML-M5 |
46,XY,t(3;15)(q28;q14)/46,XY | None | FLT3-ITD, IDH1,
STAG2 | 96.94 |
Construction of CLL-1-expressing CAR-T
cells and shRNA PD-1/CLL-1 CAR-T cells
A third-generation CLL-1 CAR was generated using
lentiviral vectors, which were composed of anti-CLL-1 scFv with the
light and heavy chain variable domains, CD8 hinge and TM,
intracellular domain of CD28 and OX40 and the CD3ζ signaling
domain. The schematic structures of the lentiviral vectors are
shown in Fig. 2A-C.
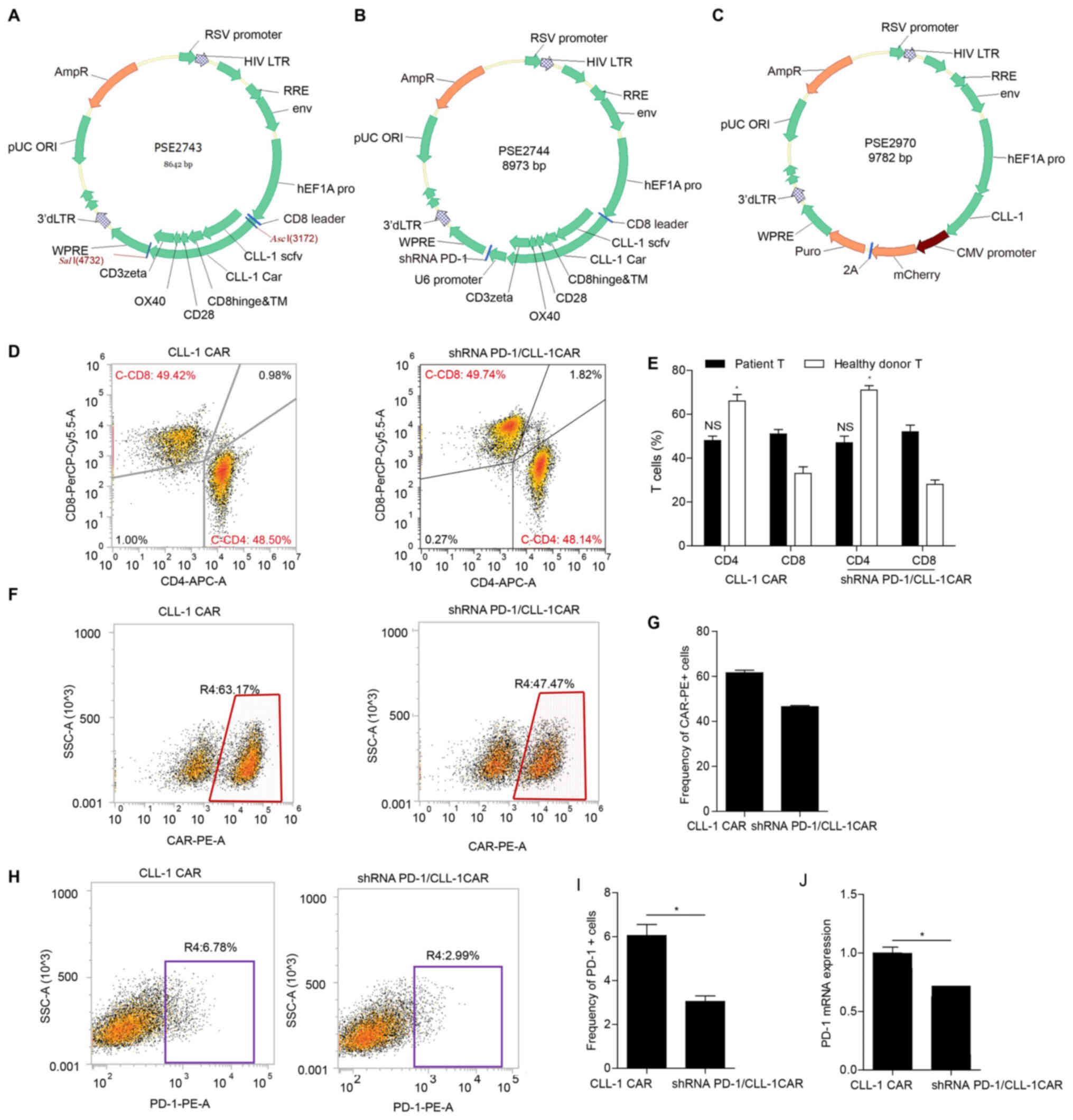 | Figure 2.Construction of CLL-1 CAR, shRNA
PD-1/CLL-1 CAR and CLL-1-overexpressing K562 cells. (A) CLL-1 CAR
vector. The third-generation CAR was composed of the variable-light
domain, followed and variable-heavy domain of the CLL-1 scFv, the
CD8 hinge and TM region, CD28, the OX40 co-stimulatory domains and
the CD3+ signaling domain. (B) shRNA PD-1/CLL-1 CAR
vector. The third-generation shRNA PD-1/CLL-1 CAR was composed of
the variable-light domain, followed and variable-heavy domain of
the CLL-1 scFv, the CD8 hinge and TM region, CD28, the OX40
co-stimulatory domains, the CD3+ signaling domain and
shRNA PD-1. (C) PSE2970 vector. The PSE2970 lentiviral vector, was
constructed to express the CLL-1 antigen in the K562 cell line
(stK562). (D) Flow cytometry plots exhibiting the phenotype of
CAR-T cell subsets. The frequency of CD4+ and
CD8+ T cells was assessed in primary T cells from
patients with AML or healthy donors transduced with CLL-1 CAR or
shRNA PD-1/CLL-1 CAR. (E) Bar graph of flow cytometry data
illustrating the immune phenotype of CLL-1 and shRNA PD-1/CLL-1
CAR-T cells. NS, the proportion of CD4+ vs.
CD8+ CAR-T cells from patients; *P<0.05, the
proportion of CD4+ vs. CD8+ CAR-T cells from
healthy donors. (F) Representative flow cytometry dot plots
demonstrating transduction efficiency. (G) Transfection efficiency
was evaluated in CLL-1 and shRNA PD-1/CLL-1 CAR-T cells. (H)
Representative flow cytometry dot plots for PD-1 expression on the
CLL-1 CAR-T cells and shRNA PD-1/CLL-1 CAR-T cells. (I) PD-1
expression was evaluated in CLL-1 CAR-T and shRNA PD-1/CLL-1 CAR-T
cells using flow cytometry. *P<0.05. (J) PD-1 mRNA expression
was measured in CLL-1 and shRNA PD-1/CLL-1 CAR-T cells using
reverse transcription-quantitative polymerase chain reaction.
*P<0.05. AML, acute myeloid leukemia; CLL-1, C-type lectin-like
molecule-1; scFv, single-chain variable fragment; TM,
transmembrane; PD-1, programmed cell death-1; CAR, chimeric antigen
receptor; APC, allophycocyanin; PerCP, peridinin chlorophyll; NS,
not significant. |
PD-1 is another protein associated with T cell
function. The negative co-stimulatory molecule PD-1 is induced
during T cell activation and is often involved in T cell depletion
by binding to PD-L1 (40). To
understand the role of PD-1 in the activation of CLL-1 CAR T cells,
an shRNA PD-1/CLL-1 CAR was generated. The shRNA PD-1/CLL-1 CAR
encoded the anti-CLL-1 scFv, CD8 hinge and TM, intracellular domain
of CD28 and OX40, the CD3ζ signaling domain and shRNA PD-1
(Fig. 2B). Flow cytometry and
RT-qPCR were used to validate the silencing ability of shRNA PD-1.
Flow cytometry was used to validate transduction efficiency. Both
CLL-1 CAR and shRNA PD-1/CLL-1 CAR had high CAR-T transduction
efficiency (61.57% and 46.55%, respectively; Fig. 2F and G). Moreover, transduction with
shRNA PD-1/CLL-1 CAR significantly decreased PD1 expression,
compared with CLL-1 CAR (Fig.
2H-J). The viral supernatant was used to infect the healthy
donor or patient-derived T cells pre-stimulated with anti-CD3 and
CD28 monoclonal antibodies. The CAR-T cells were harvested to
determine the proportion of CD4+ or CD8+
CAR-T cells using flow cytometry. The frequency of CD8+
cells was slightly higher than CD4+ cells in CLL-1 CAR
or shRNA PD-1/CLL-1 CAR-T cells derived from patients. However, the
frequency of CD4+ T cells was higher than that of
CD8+ T cells in the CLL-1 CAR-T and shRNA PD-1 CAR-T
cells from healthy donors (Fig. 2D and
E) cell populations.
PD-L1 expression in THP-1 cells largely increased
after incubation with T cell or CLL-1 CAR-T at the E:T ratio 2.5:1
(Fig. 3D and E).
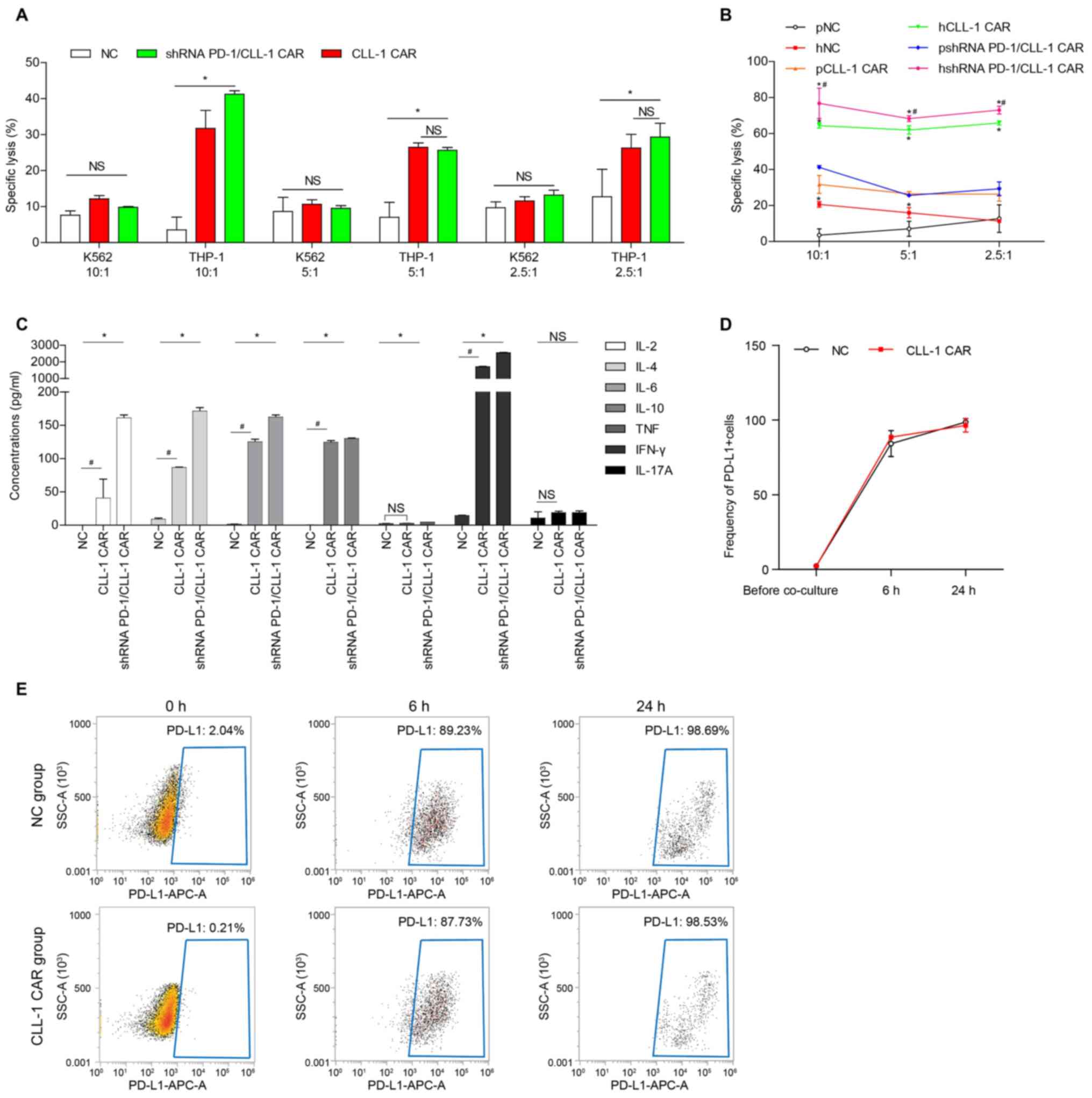 | Figure 3.PD-1 silencing enhanced the
CLL-1-specific CAR-T cytotoxicity against tumor cell lines. (A)
Cytotoxicity of patient-derived CLL-1 CAR-T cells or shRNA
PD-1/CLL-1 CAR-T against THP-1 cells and K562 cells. (B)
Cytotoxicity of patient and healthy donor-derived CLL-1 CAR-T cells
and shRNA PD-1/CLL-1 CAR-T cells against THP-1 cells. *P<0.05,
healthy donor vs. patient CAR; #P<0.05, shRNA
PD-1/CLL-1 CAR vs. CLL-1 CAR from healthy donor. (C) Cytokine
concentrations following co-culture of healthy donor-derived NC,
CLL-1 CAR-T or shRNA PD-1/CLL-1 CAR-T cells with target cells. (D)
Quantification and (E) representative flow cytometry dot plots for
PD-L1 expression on THP-1 before and after 24 h of co-culture with
healthy donor-derived NC and CLL-1 CAR-T cells. Data is presented
as the means ± SD from three patients. *P<0.05;
#P<0.05. CLL-1, C-type lectin-like molecule-1; PD-1,
programmed death 1; PD-L1, programmed death ligand 1; E:T; effector
target; NS, not significant; SSC, side scatter, APC,
allophycocyanin. |
CLL-1 CAR-T has leukemia-specific
cytotoxic ability on CLL-1+ target cells
The cytotoxic effects of healthy donor-derived or
patient-derived CLL-1 CAR-T on CLL-1+ THP-1 target cells
were then evaluated. K562 cells, which do not express CLL-1, were
used as a control. The CLL-1 CAR-T cells were co-incubated with
THP-1 or K562 cells for 16 h at E:T ratios 10:1, 5:1 and 2.5:1.
Both the healthy donor and patient-derived CLL-1 CAR-T cells
displayed significant lysing activity against THP-1 cells, compared
with the NC T cells (Fig. 3B), In
addition, the CLL-1 CAR-T, the shRNA PD-1/CLL-1 CAR-T, and the NC T
cells did not lyse K562 cells, indicating that CLL-1 CAR-T can
specifically recognize CLL-1 as its target (Fig. 3A).
The cytotoxicity of patient-derived shRNA PD-1/CLL-1
CAR-T against THP-1 cells was significantly higher compared with
CLL-1 CAR-T at the E:T ratio of 10:1, but no significant difference
was observed at the ratios of 5:1 and 2.5:1 (Fig. 3A). The cytotoxic efficiency of
healthy donor-derived shRNA PD-1/CLL-1 CAR-T against THP-1 cells
was significantly higher than that of CLL-1 CAR-T at all E:T ratios
(Fig. 3B). Furthermore, healthy
donor-derived CAR-T exhibited more potent cytotoxicity against the
THP-1 cell lines than the patient-derived CAR-T cells at the
aforementioned E:T ratios (Fig.
3B), suggesting that the source of the T cells alone may affect
the killing ability of CAR T cells.
To further examine the effector proteins involved
healthy donor and patient-derived CLL-1 CAR-T cell killing, the
levels of multiple cytokines were analyzed. At an E:T ratio of
10:1, the levels of IL-2, IL-4, IL-6, IL-10 and IFN-γ significantly
increased in THP-1 cell supernatants co-cultured with CLL-1 CAR-T
cells, compared with cells co-cultured with NC T cells (Fig. 3C), indicating that CLL-1 CAR-T cells
can respond to CLL-1+ THP-1 cells by producing
cytokines. Similarly, the levels of IL-2, IL-4, IL-6, IL-10 and
IFN-γ were also significantly increased in THP-1 cell supernatants
co-cultured with shRNA PD-1/CLL-1 CAR-T cells at an E:F ratio of
10:1 (Fig. 3C).
CLL-1 CAR-T cells display
leukemia-specific killing ability of primary AML blasts, especially
following PD-1 silencing
The killing activity of CLL-1 CAR-T cells with and
without PD-1 silencing was evaluated in leukemia cells sorted from
the BM of two patients with AML. AML cells from both patients had
high membrane expression of the CLL-1 antigen. Indeed,
CLL-1+ leukemia cells were as high as 92.10% and 99.80%,
respectively (Fig. 4A). K562 cells,
stK562 cells and primary AML blasts were co-incubated with healthy
donor-derived NC, CLL-1 CAR-T or shRNA PD-1/CLL-1 CAR-T cells. For
the first patient (Fig. 4B), CLL-1
CAR-T and shRNA PD-1/CLL-1 CAR-T cells displayed significantly
stronger cytotoxicity towards stK562 cells AML blasts than the NC T
cells. However, no significant difference between the two groups
was observed. For the second patient, compared with the CLL-1
CAR-T, the shRNA PD-1/CLL-1 CAR-T cells exhibited significantly
higher killing efficiency against the primary AML cells (Fig. 4C). The stK562 cells were killed at
significantly a higher rate by shRNA PD-1/CLL-1 CAR-T cells than
CLL-1 CAR-T at the E:T ratio of 10:1 (Fig. 4C), but not at 5:1 or 2.5:1. In
addition, there was no difference in killing efficiency between the
CLL-1 CAR-T and the NC T cells on K562 cells (Fig. 4B and C).
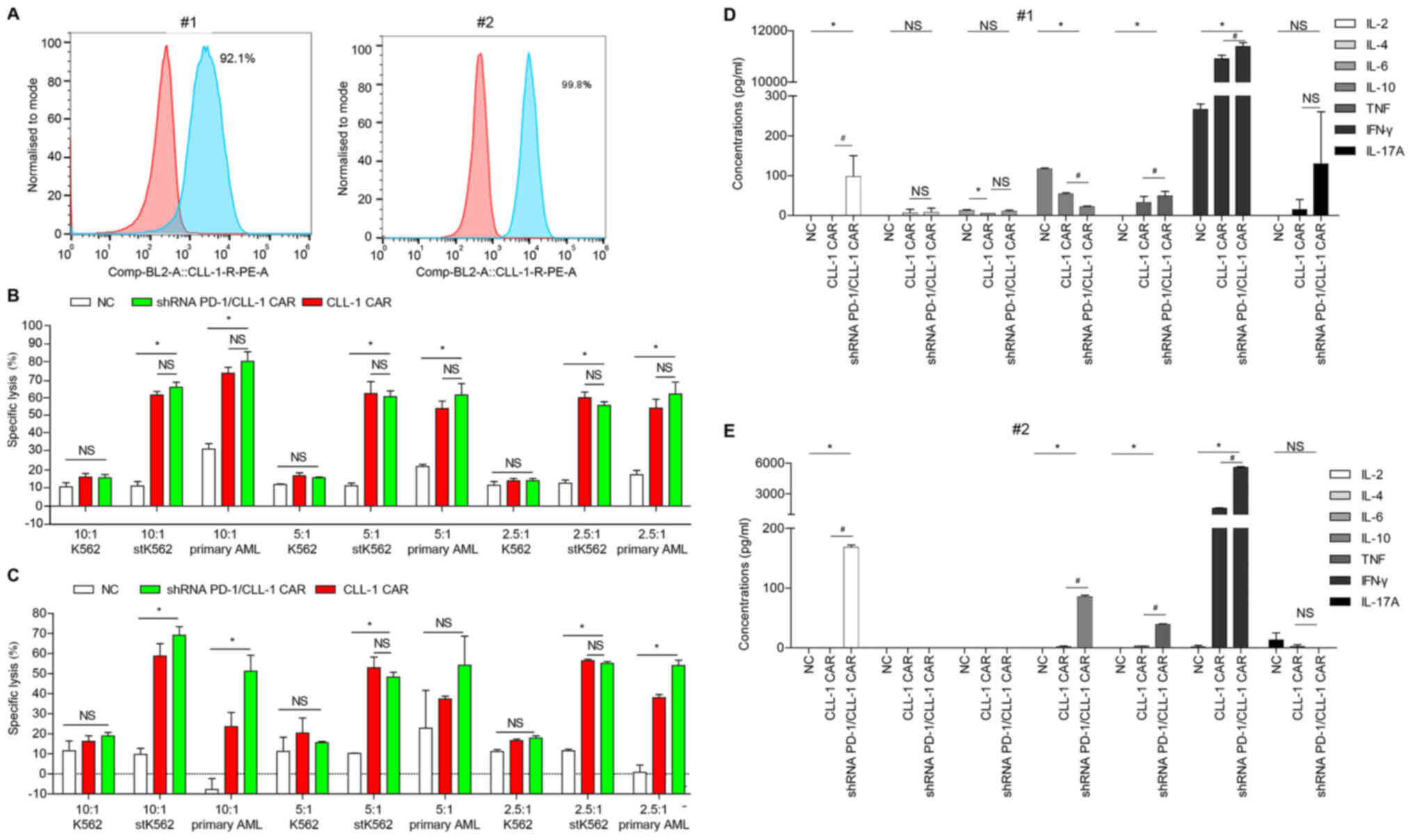 | Figure 4.PD-1 silencing enhances CLL-1 CAR-T
cell cytotoxicity against primary AML cells. (A) CLL-1 was
expressed in the primary AML cells from patients #1 and #2. Red,
isotype control; blue, CLL-1 staining. (B and C) Specific
cytotoxicity of healthy donor-derived CLL-1 CAR-T cells and shRNA
PD-1/CLL-1 CAR-T cells against K562 cells, stK562 cells and primary
AML cells from (B) patient #1 and (C) patient #2. Data represents
the means of triplicate wells ± SD. (D and E) Cytokine
concentrations following 24-h co-culture of healthy donor-derived
NC, CLL-1 CAR-T or shRNA PD-1/CLL-1 CAR-T cells with (D) patient #1
or (E) patient #2 primary AML cells at E:T ratio of 10:1.
#P<0.05; *P<0.05. AML, acute myeloid leukemia;
CLL-1, C-type lectin-like molecule-1; st, stably transfected; LDH,
lactate dehydrogenase; PD-1, programmed death 1; E:T; effector
target; NS, not significant; PE, phycoerythrin. |
The levels of inflammatory cytokines released by
CLL-1 CAR-T cells with and without PD-1 silencing were also
analyzed. The levels of IL-2, TNF-α and IFN-γ were significantly
increased in the shRNA PD-1/CLL-1 CAR-T cells, compared with CLL-1
CAR-T cells and NC T cells (Fig. 4D and
E).
Discussion
Optimal treatment target for CAR-T therapy requires
the identification of a single TM protein that is highly and
specifically expressed on target cells but not on normal cells.
Previous studies have indicated that CLL-1 selective expression on
the LSC surface, rather than on other HSCs, is restricted to the
hematopoietic lineage, especially monocytes and granulocytes
(33,41). The present study demonstrated that
CLL-1 was highly expressed on THP-1 cells and primary AML cells
from refractory or relapse patients, which is consistent with
previous reports (19,30,41,42).
Darwish et al (43)
suggested that overexpression of LSC markers, such as CLL-1 and
TIM-3, in clinical AML specimens was significantly associated with
poor prognosis. However, Wang et al (44) demonstrated that low expression of
CLL-1 independently predicted a low CR rate in 123 patients with
de novo CD34+ AML. Thus, these studies suggested
that CLL-1 was a predictable marker of AML that could distinguish
normal HSCs from LSCs. Furthermore, stable expression of CLL-1
during the initial diagnosis and recurrence (33,41,42) of
AML makes CLL-1 an optimal marker for diagnosis and evaluation of
its curative effect of AML. However, further investigation is
needed to confirm whether CLL-1-targeted therapy is an effective
supplement to the current AML prognostic risk stratification system
or whether it is suitable for the treatment of AML.
CLL-1 CAR-T cells have been reported to have
specific killing activity against CLL-1+ AML cell lines,
as well as primary AML blasts, and to reduce the colony forming
ability of CLL-1+ AML cells (31,45).
In the present study, a third-generation CLL-1 CAR was developed,
that could effectively lyse CLL-1+ AML cell lines and
primary AML blasts and release numerous inflammatory cytokines,
especially at a E:T ratio of 10:1.
The immune system plays an important role in
eliminating malignant cells through immune surveillance (46). Several studies have reported
abnormal changes in the relative frequency and function of
lymphocytes, especially T cells, in AML (47,48).
CAR-T cells can specifically kill tumor cells without the
limitations of major histocompatibility complex. The function of
engineered T cells depends to some extent on the source of the
donor T cells. In this study, the CAR-T cells derived from healthy
donors displayed improved killing efficiency than those derived
from patients, indicating that CAR function was affected by the
source of the T cells.
Although the engineered T cells can specific
recognize the target antigens and kill tumor cells, the
hypofunction or even immune depletion of the modified T cells often
re-emerged when the cells entered the tumor microenvironment
containing PD-1 or other immune checkpoint proteins (49). A recent study demonstrated that a
similar phenomenon also occurred in the bone morrow
microenvironment of AML patients (50). Therefore, given the complexity of
immunoregulation and multiple factors involved in the immune
system, it is often unsatisfactory to treat most tumors with a
single modified T cell antigen recognition site. It is necessary to
optimize the function of the modified CAR with a combination of
multiple approaches. In the present study, PD-L1 expression also
increased in THP-1 cells following co-culture with effector T
cells. Thus, an shRNA PD-1/CLL-1 CAR was generated, which enhanced
antitumor ability and the ability to lyse AML cells. The CAR-T
cells derived from healthy donors displayed more cytotoxicity
against THP-1 cells than patient-derived T cells. The therapeutic
effect of shRNA PD-1/CLL-1 CAR-T cells for RR-AML should be tested
in clinical trials.
This study on CLL-1 CAR-T and shRNA PD-1/CLL-1 CAR-T
cells has limitations. First, CLL-1 is also expressed on mature
granulocytes and monocytes. A study by Tashiro et al
(35) indicated that the toxicity
of CLL-1 CAR-Ts is confined to mature myeloid cells. The side
effects of the immune therapy included neutropenia and
agranulocytosis, because the HSCs and primitive myeloid precursors
were spared by the CLL-1 CAR-T cells. However, these side effects
could be reduced after symptomatic treatment, including raising
white blood cell count by granulocyte transfusion or injecting
granulocyte colony-stimulating factor. Second, CLL-1 CAR alone may
not be able to cope with complex tumor microenvironments. Thus,
combinatorial therapies, such as a PD-1 monoclonal antibody or
multi-target CARs, should be considered. Third, similarly to other
CAR therapies, sustained remission status cannot be maintained over
longer periods of time. Therefore, further research is needed to
maintain CAR treatment efficiency and to effectively use CAR as a
bridge to HSC transplantation.
In conclusion, a CLL-1 CAR-T was generated that
specifically targeted AML cells, particular following silencing
PD-1 using shRNA technology. Clinical trials are required to
further evaluate CLL-1 CAR-T cell clinical efficacy and safety.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 81730003), National Science
and Technology Major Project (grant no. 2017ZX09304021), National
Key R&D Program of China (grant no. 2017YFA0104502), Priority
Academic Program Development of Jiangsu Higher Education
Institutions (PAPD), Jiangsu Medical Outstanding Talents Project
(grant no. JCRCA2016002), Jiangsu Provincial Key Medical Center
(grant no. YXZXA2016002), Research Project of Natural Science
Foundation of Huai'an Jiangsu (grant no. HAB201814) and Xuzhou
Medical College (grant no. 2018KJ11).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GL conceived the study, designed and performed
research, analyzed data and participated in manuscript writing; YZ
provided patient samples and assisted with data collection. GL and
YZ confirm the authenticity of all the raw data. LY provided advice
and instructed the construction of the CAR; DW conceived the study
and designed the research. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by the Institutional Review
Board of Huai'an Hospital. All procedures were in accordance with
the ethical standards of the responsible committee on human
experimentation (institutional and national) and with the Helsinki
Declaration of 1975. Informed consent was obtained from all
patients included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CAR
|
chimeric antigen receptor
|
|
scFv
|
single-chain variable fragment
|
|
AML
|
acute myeloid leukemia
|
|
RR-AML
|
relapsed/refractory AML
|
|
PD-1
|
programmed cell death 1
|
|
CLL-1
|
C-type lectin-like molecule-1
|
|
CR
|
complete remission
|
|
HSCT
|
hematopoietic stem cell
transplantation
|
|
mAb
|
monoclonal antibody
|
|
PBMC
|
peripheral blood mononuclear cell
|
|
LSC
|
leukemic stem cell
|
|
TIM-3
|
T cell immunoglobulin and mucin
domain-containing 3
|
References
|
1
|
Juliusson G, Lazarevic V, Hörstedt AS,
Hagberg O and Höglund M; Swedish Acute Leukemia Registry Group, :
Acute myeloid leukemia in the real world: Why population-based
registries are needed. Blood. 119:3890–3899. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shallis RM, Wang R, Davidoff A, Ma X and
Zeidan AM: Epidemiology of acute myeloid leukemia: Recent progress
and enduring challenges. Blood Rev. 36:70–87. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
National Cancer Institute: Surveillance,
Epidemiology, and End Results (SEER) Program Cancer Stat Facts:
Leykemia-Acute myeloid leukemia (AML). https://seer.cancer.gov/statfacts/html/amyl.htmlApril
18–2019
|
|
5
|
Chen C, Wang P and Wang C: Prognostic
nomogram for adult patients with acute myeloid leukemia: A SEER
database analysis. Medicine (Baltimore). 98:e158042019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen W, Zheng R, Zhang S, Zeng H, Xia C,
Zuo T, Yang Z, Zou X and He J: Cancer incidence and mortality in
China, 2013. Cancer Lett. 401:63–71. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rodriguez-Abreu D, Bordoni A and Zucca E:
Epidemiology of hematological malignancies. Ann Oncol. 18 (Suppl
1):i3–i8. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sant M, Allemani C, Tereanu C, De Angelis
R, Capocaccia R, Visser O, Marcos-Gragera R, Maynadié M, Simonetti
A, Lutz JM, et al HAEMACARE Working Group, : Incidence of
hematologic malignancies in Europe by morphologic subtype: Results
of the HAEMACARE project. Blood. 116:3724–3734. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang X, Xiao Q, Wang Z and Feng WL: CAR-T
therapy for leukemia: Progress and challenges. Transl Res.
182:135–144. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tasian SK, Kenderian SS, Shen F, Ruella M,
Shestova O, Kozlowski M, Li Y, Schrank-Hacker A, Morrissette JJD,
Carroll M, et al: Optimized depletion of chimeric antigen receptor
T-cells in murine xenograft models of human acute myeloid leukemia.
Blood. 129:2395–2407. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Clarke CA and Glaser SL: Acute myeloid
leukemia. N Engl J Med. 342:358–359. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rowe JM and Tallman MS: How I treat acute
myeloid leukemia. Blood. 116:3147–3156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stahl M, Kim TK and Zeidan AM: Update on
acute myeloid leukemia stem cells: New discoveries and therapeutic
opportunities. World J Stem Cells. 8:316–331. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lichtenegger FS, Krupka C, Haubner S,
Köhnke T and Subklewe M: Recent developments in immunotherapy of
acute myeloid leukemia. J Hematol Oncol. 10:1422017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Taussig DC, Pearce DJ, Simpson C,
Rohatiner AZ, Lister TA, Kelly G, Luongo JL, Danet-Desnoyers GA and
Bonnet D: Hematopoietic stem cells express multiple myeloid
markers: Implications for the origin and targeted therapy of acute
myeloid leukemia. Blood. 106:4086–4092. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tettamanti S, Marin V, Pizzitola I,
Magnani CF, Giordano Attianese GM, Cribioli E, Maltese F,
Galimberti S, Lopez AF, Biondi A, et al: Targeting of acute myeloid
leukaemia by cytokine-induced killer cells redirected with a novel
CD123-specific chimeric antigen receptor. Br J Haematol.
161:389–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ghaffari S, Smadja-Joffe F, Oostendorp R,
Lévesque JP, Dougherty G, Eaves A and Eaves C: CD44 isoforms in
normal and leukemic hematopoiesis. Exp Hematol. 27:978–993. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kikushige Y, Shima T, Takayanagi S, Urata
S, Miyamoto T, Iwasaki H, Takenaka K, Teshima T, Tanaka T, Inagaki
Y, et al: TIM-3 is a promising target to selectively kill acute
myeloid leukemia stem cells. Cell Stem Cell. 7:708–717. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hosen N, Park CY, Tatsumi N, Oji Y,
Sugiyama H, Gramatzki M, Krensky AM and Weissman IL: CD96 is a
leukemic stem cell-specific marker in human acute myeloid leukemia.
Proc Natl Acad Sci USA. 104:11008–11013. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bonardi F, Fusetti F, Deelen P, van
Gosliga D, Vellenga E and Schuringa JJ: A proteomics and
transcriptomics approach to identify leukemic stem cell (LSC)
markers. Mol Cell Proteomics. 12:626–637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saito Y, Kitamura H, Hijikata A,
Tomizawa-Murasawa M, Tanaka S, Takagi S, Uchida N, Suzuki N, Sone
A, Najima Y, et al: Identification of therapeutic targets for
quiescent, chemotherapy-resistant human leukemia stem cells. Sci
Transl Med. 2:17ra92010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Moshaver B, van Rhenen A, Kelder A, van
der Pol M, Terwijn M, Bachas C, Westra AH, Ossenkoppele GJ,
Zweegman S and Schuurhuis GJ: Identification of a small
subpopulation of candidate leukemia-initiating cells in the side
population of patients with acute myeloid leukemia. Stem Cells.
26:3059–3067. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Wang L, Zhao F, Tseng S, Narayanan
C, Shura L, Willingham S, Howard M, Prohaska S, Volkmer J, et al:
Pre-clinical development of a humanized anti-CD47 antibody with
anti-cancer therapeutic potential. PLoS One. 10:e01373452015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amadori S, Suciu S, Selleslag D, Aversa F,
Gaidano G, Musso M, Annino L, Venditti A, Voso MT, Mazzone C, et
al: Gemtuzumab ozogamicin versus best supportive care in older
patients with newly diagnosed acute myeloid leukemia unsuitable for
intensive chemotherapy: Results of the randomized phase III
EORTC-GIMEMA AML-19 trial. J Clin Oncol. 34:972–979. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Park JH, Rivière I, Gonen M, Wang X,
Sénéchal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, et
al: Long-term follow-up of CD19 CAR therapy in acute lymphoblastic
leukemia. N Engl J Med. 378:449–459. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cao J, Wang G, Cheng H, Wei C, Qi K, Sang
W, Zhenyu L, Shi M, Li H, Qiao J, et al: Potent anti-leukemia
activities of humanized CD19-targeted chimeric antigen receptor T
(CAR-T) cells in patients with relapsed/refractory acute
lymphoblastic leukemia. Am J Hematol. 93:851–858. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sotillo E, Barrett DM, Black KL, Bagashev
A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et
al: Convergence of acquired mutations and alternative splicing of
CD19 enables resistance to CART-19 immunotherapy. Cancer Discov.
5:1282–1295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fischer J, Paret C, El Malki K, Alt F,
Wingerter A, Neu MA, Kron B, Russo A, Lehmann N, Roth L, et al:
CD19 isoforms enabling resistance to CART-19 immunotherapy are
expressed in B-ALL patients at initial diagnosis. J Immunother.
40:187–195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fan M, Li M, Gao L, Geng S, Wang J, Wang
Y, Yan Z and Yu L: Chimeric antigen receptors for adoptive T cell
therapy in acute myeloid leukemia. J Hematol Oncol. 10:1512017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Beavis PA, Sek K and Darcy PK: A novel
target antigen for the treatment of acute myeloid leukemia by CAR-T
cells. Mol Ther. 25:1997–1998. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Prommersberger S, Jetani H, Danhof S,
Monjezi R, Nerreter T, Beckmann J, Einsele H and Hudecek M: Novel
targets and technologies for CAR-T cells in multiple myeloma and
acute myeloid leukemia. Curr Res Transl Med. 66:37–38. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu H, Zhou Q, Deshmukh V, Phull H, Ma J,
Tardif V, Naik RR, Bouvard C, Zhang Y, Choi S, et al: Targeting
human C-type lectin-like molecule-1 (CLL1) with a bispecific
antibody for immunotherapy of acute myeloid leukemia. Angew Chem
Int Ed Engl. 53:9841–9845. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bakker AB, van den Oudenrijn S, Bakker AQ,
Feller N, van Meijer M, Bia JA, Jongeneelen MA, Visser TJ, Bijl N,
Geuijen CA, et al: C-type lectin-like molecule-1: A novel myeloid
cell surface marker associated with acute myeloid leukemia. Cancer
Res. 64:8443–8450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang J, Chen S, Xiao W, Li W, Wang L, Yang
S, Wang W, Xu L, Liao S, Liu W, et al: CAR-T cells targeting CLL-1
as an approach to treat acute myeloid leukemia. J Hematol Oncol.
11:72018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tashiro H, Sauer T, Shum T, Parikh K,
Mamonkin M, Omer B, Rouce RH, Lulla P, Rooney CM, Gottschalk S, et
al: Treatment of acute myeloid leukemia with T-cells expressing
chimeric antigen receptors directed to C-type lectin-like
molecule-1. Mol Ther. 25:2202–2213. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Heczey A, Louis CU, Savoldo B, Dakhova O,
Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, et al: CAR-T cells
administered in combination with lymphodepletion and PD-1
inhibition to patients with neuroblastoma. Mol Ther. 25:2214–2224.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ankri C, Shamalov K, Horovitz-Fried M,
Mauer S and Cohen CJ: Human T-cells engineered to express a
programmed death 1/28 costimulatory retargeting molecule display
enhanced antitumor activity. J Immunol. 191:4121–4129. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the World Health Organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jin HT, Ahmed R and Okazaki T: Role of
PD-1 in regulating T cell immunity. Curr Top Microbiol Immunol.
350:17–37. 2011.PubMed/NCBI
|
|
41
|
van Rhenen A, van Dongen GA, Kelder A,
Rombouts EJ, Feller N, Moshaver B, Stigter-van Walsum M, Zweegman
S, Ossenkoppele GJ and Jan Schuurhuis G: The novel AML stem cell
associated antigen CLL-1 aids in discrimination between normal and
leukemic stem cells. Blood. 110:2659–2666. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Larsen HO, Roug AS, Just T, Brown GD and
Hokland P: Expression of the hMICL in acute myeloid leukemia-a
highly reliable disease marker at diagnosis and during follow-up.
Cytometry B Clin Cytom. 82:3–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Darwish NH, Sudha T, Godugu K, Elbaz O,
Abdelghaffar HA, Hassan EE and Mousa SA: Acute myeloid leukemia
stem cell markers in prognosis and targeted therapy: Potential
impact of BMI-1, TIM-3 and CLL-1. Oncotarget. 7:57811–57820. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang YY, Chen WL, Weng XQ, Sheng Y, Wu J,
Hao J, Liu ZY, Zhu YM, Chen B, Xiong SM, et al: Low CLL-1
expression is a novel adverse predictor in 123 patients with de
novo CD34+ acute myeloid leukemia. Stem Cells Dev.
26:1460–1467. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Laborda E, Mazagova M, Shao S, Wang X,
Quirino H, Woods AK, Hampton EN, Rodgers DT, Kim CH, Schultz PG, et
al: Development of a chimeric antigen receptor targeting C-type
lectin-like molecule-1 for human acute myeloid leukemia. Int J Mol
Sci. 18:182017. View Article : Google Scholar
|
|
46
|
Dunn GP, Old LJ and Schreiber RD: The
immunobiology of cancer immunosurveillance and immunoediting.
Immunity. 21:137–148. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Park Y, Lim J, Kim S, Song I, Kwon K, Koo
S and Kim J: The prognostic impact of lymphocyte subsets in newly
diagnosed acute myeloid leukemia. Blood Res. 53:198–204. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Alcasid M, Ma L, Gotlib JR, Arber DA and
Ohgami RS: The clinicopathologic significance of lymphocyte subsets
in acute myeloid leukemia. Int J Lab Hematol. 39:129–136. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gray KD, Vedvyas Y, Kalloo O, Shevlin E
and Min IM: Abstract 2738: PD-L1/PD-1 checkpoint inhibition in
anaplastic thyroid cancer and enhancement of ICAM-1-targeted
chimeric antigen receptor (CAR)-T cell tumor lysis. Cancer Res.
78:2738. 2018.
|
|
50
|
Williams P, Basu S, Garcia-Manero G,
Hourigan CS, Oetjen KA, Cortes JE, Ravandi F, Jabbour EJ, Al-Hamal
Z, Konopleva M, et al: The distribution of T cell subsets and the
expression of immune checkpoint receptors and ligands in patients
with newly diagnosed and relapsed acute myeloid leukemia. Cancer.
125:1470–1481. 2019. View Article : Google Scholar : PubMed/NCBI
|