Neurodegenerative diseases can cause pain-associated
syndromes and conditions, including musculoskeletal pain, chronic
body pain (central or visceral), fluctuation-related pain,
oro-facial pain and radicular pain, and comprise a heavy medical
burden worldwide. The prevalence and incidence of neurodegenerative
diseases increases with age (1–3).
Therefore, an effective treatment plan is required to aid patients
with neurodegenerative disease and alleviate suffering. The central
nervous system is composed of neurons and glial cells. Glial cells
include astrocytes, microglia and oligodendrocytes (4). Glia have been regarded as support
cells for neurons, and glial cells can be activated by pathological
stimuli, such as neuronal injury or other insults, on the central
nervous system. During these processes, ions and metabolites, such
as Ca2+ and glutamate, are released, which adversely
affect neuronal activity (5–7).
Connexin is expressed on glial cells and neurons,
and different types of this protein, including connexin, pannexin
and innexin, exist according to the nature of the phenotype of the
nerve cells (5,8,9).
Connexins can form gap junctions. These proteins differ between
vertebrates and invertebrates. In vertebrates, the gap junction
protein is termed connexin, whereas the gap junction protein of
invertebrates is termed innexin (10). Connexin is a protein that is
composed of hemichannels and gap junctions (9). It serves a key role in both
physiological and pathological conditions of the human body.
Moreover, connexin opens in response to pathological conditions,
such as cell damage (mechanical stimulation), changes in pH and ion
concentration and induction of ischemia (11,12).
The opening of the hemichannel causes small molecules to be
released from the cell interior to the extracellular space, where
they participate in signal transduction of pro-inflammatory and
pro-cell death members (13,14).
Nerve cell damage and death are pathological features of
neurodegenerative disease. Considering the potential connection
between connexin and neuronal damage, the present review focused on
the role of connexins in neurodegenerative disease.
In the human genome, >20 connexin members are
present in the multigene connexin family (15). Connexins are named according to
their molecular weight in kDa, such as Cx43, Cx30, Cx36, Cx45 and
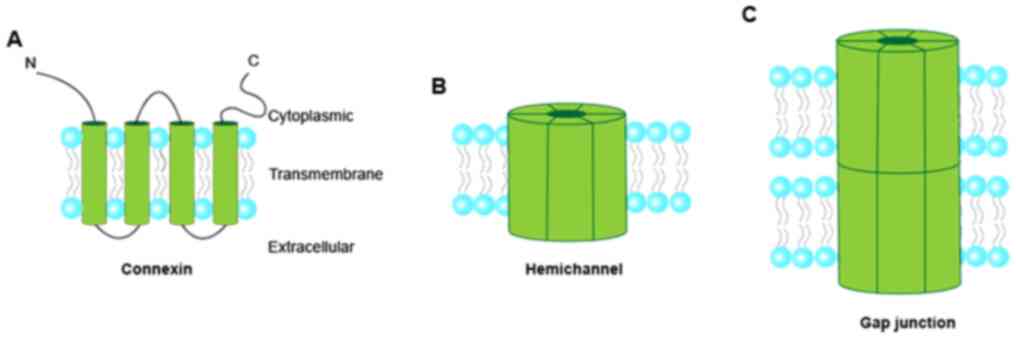
Cx50. Connexin consists of four α-helical transmembrane domains and
two extracellular loops (Fig. 1),
which are highly conserved among family members (9). The N- and C-termini and the
intracellular loop are located in the cytoplasm (16). Connexins are oligomerized into
connexons or hemichannels (a connexin hexamer). The hemichannels
dock with each other to form gap junction channels (17–19).
The body requires cell communication for appropriate
function. As a result, several communication mechanisms have been
developed. Gap junctions are the most direct and fastest
communication channel between cells (16,20).
The opening and closing of gap junction channels are regulated by
various mechanisms, such as changes in connexin, intracellular
Ca2+ levels and pH, as well as phosphorylation and
dephosphorylation reactions (10).
Gap junctions mediate the diffusion of small molecules and ions
between cells and serve a vital role in physiological processes,
such as cell proliferation and development, signal transduction
between nerve cells and hormonal secretion (9,21). Gap
junctions facilitate electrical and metabolic coupling between
adjacent cells and contribute to communication between adjacent
cells (21). Electrical coupling is
key in excitatory tissue, notably in the heart. The transfer of
current between cells occurs via gap junction channels (22). Gap junctions do not need to
recognize receptors and can transmit signals faster than chemical
synapses. This allows multiple neurons to be activated
simultaneously, so that gap junctions are abundant and can activate
mechanisms that require rapid responses, such as escape mechanisms
(20,23).
By contrast to gap junctions, information on the
structure and function of hemichannels is relatively limited. The
hemichannel on the plasma membrane is generally closed and can be
opened under pathological conditions, such as low extracellular
Ca2+, membrane depolarization, mechanical membrane
stress and metabolic inhibition (24–26).
In addition, previous studies have shown that intracellular ATP is
released via hemichannels (27–29).
The release of intracellular ATP is associated with a wide range of
physiological processes that include a major source of energy,
modulation of synaptic transmission, post-translational
modifications and cofactor metabolism (30,31).
In certain neurodegenerative diseases, the death and damage of the
neurons may be directly associated with the opening of connexin in
hemichannels. For example, in Parkinson's disease (PD), α-synuclein
induces the opening of connexin hemichannels (32). The high level of hemichannel
activity causes neurons to be more sensitive to damage caused by
reactive oxygen generation (33).
The harmful effects of hemichannel activation are associated with
long-term Ca2+ influx, leading to the activation of
Ca2+-dependent hydrolase and the depletion of ATP
(34).
PD is a common neurological degenerative disease,
which was first described in detail by British doctor James
Parkinson in 1817 (35). The
overall incidence rate of PD in women who are ≥40 years old was
37.55 per 100,000 individuals/year, and 61.21 in men who are ≥40
years old between 2001 and 2014, in Europe, North America,
Australia and South America (36).
The lesions of PD are primarily located in the substantia nigra and
develop due to degeneration of dopaminergic neurons (37,38).
Dopamine acts on the striatum and directly on the subthalamic
nucleus, globus pallidus and cortex. PD is associated with loss of
dopamine input in these areas, which can cause abnormal firing of
these nuclei (39). Dopamine
differentially regulates the excitability of direct and indirect
pathway spiny projection neurons. The activation of the dopamine 1
(D1) receptor in the direct pathway promotes the potentiation of
excitatory synapses, whereas the activation of the D2 receptor in
the indirect pathway promotes the depression of excitatory synapses
(40). Therefore, degeneration of
dopaminergic neurons in the substantia nigra causes excessive
excitation of internal globus pallidus and substantia nigra
reticulata, which subsequently inhibits the activity of the
thalamus and decreases the excitatory projection of the thalamus to
the cerebral cortex, resulting in PD (41,42).
α-synuclein misfolding and aggregation appears to
have a close association with the majority of PD cases (43); α-synuclein can induce astrocyte
reactivity and increase the synaptic capacity of astrocytes
(44). Gap junctions mediate the
synchronization of neuron activity in several brain regions,
including the amygdala, hippocampus and cerebellum (45,46).
Abundant gap junctions are present between astrocytes, which are
regarded as support cells for neurons and have the ability to
regulate neuronal activity as well as synaptic transmission and
plasticity. These biological processes have received considerable
attention in brain physiology research (47). Neuronal-astrocytic signal
dysfunction is associated with the development of various
neurological and neurodegenerative diseases, including neuropathic
pain and PD (48–50). Alteration or uncoupling of gap
junctions between astrocytes and neurons leads to excessive release
of potassium ions or glutamate (51). As aforementioned, α-synuclein
increases the synaptic capacity of astrocytes (44). Since the recognition of receptor
signals is not required, the electrical synapse between astrocytes
or neuron-astrocytes conducts faster than chemical synapses
(52). When levels of α-synuclein
increase, the conduction of electrical synapses and the synchronous
activity of neurons increase, resulting in the development of PD
(53,54).
Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is
an effective neurotoxin that destroys dopaminergic neurons in the
substantia nigra and induces PD (62). A previous study demonstrated that
expression levels of Cx30 in mice treated with MPTP are
upregulated, whereas knockout of Cx30 expression accelerates
MPTP-induced loss of dopaminergic neurons (63). In total, two different types of
reactive astrocytes have been identified, which are termed harmful
A1 and protective A2 (64).
Previous studies have shown that deficiency of Cx30 decreases
protective A2 levels in an MPTP mouse model (63,65).
Therefore, based on the fact that Cx30 is required for protective
A2, Cx30 may protect astrocytes from the development of PD.
Neurotic plaques formed by the deposition of amyloid
β (Aβ) protein and the loss of brain neurons are considered signs
of AD pathology. Neurotic plaques are the most unique pathological
features of AD (66). The amyloid
hypothesis suggests that Aβ is formed in the brain, triggering
pathological effects, such as the increase in the concentration of
intracellular calcium, to directly or indirectly lead to neuronal
death (67). Since Aβ is the result
of brain aging, rather than the cause, the mitochondrial cascade
hypothesis suggests that Aβ is a sign of brain aging. It is
proposed that during the development of AD, the expression and
processing of amyloid precursor protein and the accumulation of Aβ
protein are affected by mitochondrial function (68).
Connexins serve an important role in normal memory,
learning and cognitive function (69,70).
The role of connexins in AD has received extensive attention.
Earlier studies have shown that in samples from an APP/PS1 mouse
model, the expression levels of Cx43 and Cx30 in astrocytes are
increased in the vicinity of Aβ plaques (71,72).
The involvement of connexins in the development of AD is associated
with mitochondrial dysfunction and the production of reactive
oxygen species. Although the human brain only accounts for 2% of
body weight, it is more susceptible to oxidative stress than other
organs (73,74). Oxidative stress is an important
mechanism involved in the pathogenesis of AD. Redox imbalance in
the brain increases the susceptibility of neurons, which contain
high levels of polyunsaturated fatty acids and small amounts of
glutathione (GSH), to oxidative stress (75,76).
GSH is involved in processing of peroxides by brain cells and
protection from reactive oxygen species-induced cell damage
(77). The content of GSH in the
brain area containing reactive astrocyte proliferation is higher
compared with that in the brain area containing neurons (78). The release of GSH in astrocytes has
specific consequences for the synthesis of neuronal GSH and
oxidative status in the brain. The specific consequences include
the decrease of neuroprotective GSH, and the energy and redox
imbalance of neurons, amongst other effects (79). In cases of insufficient GSH
synthesis by neurons, oxidative stress and age-dependent neuronal
degeneration develop (80).
Although the content of neuronal GSH synthesis is lower than that
in astrocytes, oxidative stress significantly increases the amount
of GSH (81). It has been reported
that GSH is released from the connexin hemichannel (82). A previous study also found that Aβ
increases hemichannel activity in glia and neurons (83). Therefore, Aβ not only stimulates the
release of GSH but also increase its release together with
glutamate by increasing the activity of the connexin hemichannel.
As aforementioned, accumulation of large amounts of glutamate
causes excitotoxicity.
ALS is a neurodegenerative disease that causes
degeneration of upper and lower neurons. Its onset is characterized
by minor symptoms, such as muscle weakness or muscle twitching, and
eventually results in paralysis and death (84). The predisposing factors of ALS are
still uncertain. Several pathological causes, including genetic
mutations, excitotoxicity and oxidative stress, have been
identified based on existing research (85–87).
The majority of these processes are accompanied by an imbalance of
Ca2+ homeostasis (88).
In addition, the accumulation of misfolded proteins and
neuroinflammation are common features of ALS (89). Neuroinflammation can develop in the
brainstem and spinal cord of patients with ALS and ALS mouse
models. It is also accompanied by the accumulation of a large
number of activated astrocytes and microglia (90). Astrocytes and microglia contribute
to the degeneration of neurons and exert this effect via gap
junctions and hemichannels between glial cells (91). It has previously been shown that
expression levels of Cx43 are increased in patients with ALS and
mouse models, which contributes to degeneration of motor neurons
(92). Large amounts of ATP are
released from astrocytes via Cx43. Subsequently, ATP binds to P2X
receptors, which increases calcium signaling (93). Prior to stimulation with ATP,
calcium signal decreases when astrocytes are incubated with the
Cx43 mimic peptide Gap26 (92).
This indicates that Cx43 gap junctions and hemichannels contribute
to the transmission of calcium signals. Abnormal expression of Cx43
leads to abnormal transmission of calcium signals. Changes in
intracellular Ca2+ levels serve a prominent role in
regulating fundamental cellular functions, such as neuronal
migration and differentiation, synapse formation and synaptic
plasticity in various cell types, including neurons (94). In neurons, Ca2+ also
participates in the transmission of depolarization signals. Changes
in Ca2+ levels serve an important role in neuronal
degeneration (95). Therefore, Cx43
gap junctions and hemichannel-mediated calcium signaling exert an
important role in ALS. In addition, a delayed decrease in Cx36
expression on spinal cord neurons in ALS has been found. Cx36
expression is downregulated in late ALS (when neuronal degeneration
has already occurred) (96). The
reason for this delayed downregulation may be the primary and
secondary death of Cx36-expressing neurons (96,97).
This part of neurons is a component of overall neuronal damage.
Administration of Cx36 gap junction channel blocker prevents the
ALS-related death of neurons (96).
Therefore, Cx36 is also an important target for the future
treatment of ALS.
In 1872, George Huntington wrote a report on
hereditary chorea, which is now known as HD (98). However, chorea is not the only
dyskinesia characteristic found in this disease. HD can cause a
series of dyskinesia characteristics, such as chorea and rapid
involuntary movements of the face, torso and limbs (99,100).
A previous study demonstrated that neuronal cell death is most
pronounced in the caudate nucleus and pallidum of the basal ganglia
in the brains of patients with HD (101). In recent years, a growing
awareness has been noted with regard to the important role of glial
cells in the nervous system (102). Glial cells and gap junctions are
involved in buffering of potassium ions around active neurons and
protection of nerves from glutamate toxicity (59). Under normal conditions, persistent
increases in glutamate act on N-methyl-D-aspartate receptor-type
glutamate receptors on neurons, resulting in neuronal
excitotoxicity (103). Therefore,
abnormalities in gap junctions between astrocytes may lead to
neuronal cell death in HD. Few studies have investigated the role
of connexins in HD. However, one study demonstrated that Cx43
expression is abnormally increased in the caudate nucleus, whereas
the density of Cx43 increases with development of HD (101). Altered connexin gap junctions
result in the inability of astrocytes to maintain normal neuronal
activity, which may be a factor in neuronal death in HD (101). Therefore, based on the key role of
connexin gap junctions in HD, connexins may be considered an
important addition in the investigation of the pathogenesis and
etiology of HD.
Neurodegenerative diseases are associated with motor
neuron damage and degeneration. It has been shown that the
inhibition of connexin gap junctions and hemichannels can protect
neurons from adverse effects of ion and metabolite homeostasis.
Understanding the role of connexin gap junctions and hemichannels
in neurodegenerative diseases may provide a novel research
direction for the development of potential therapeutic strategies
for neurodegenerative disease.
The authors would like to thank Dr Shandong Liang
(Nanchang University, China) who participated in this study with
his assistance in the copy-editing of the manuscript.
The present study was supported by National Natural
Science Foundation of China (grant nos. 81660199 and 81260187) and
Cultivating Foundation of Young Scientists (Star of Jing Gang) of
Jiangxi Province (grant no. 20153BCB23033).
Not applicable.
JX conducted the literature search, wrote and
revised the manuscript. CX designed the review. Both authors read
and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Checkoway H, Lundin JI and Kelada SN:
Neurodegenerative diseases. IARC Sci Publ. 407–419. 2011.PubMed/NCBI
|
|
2
|
Blanchet PJ and Brefel-Courbon C: Chronic
pain and pain processing in Parkinson's disease. Prog
Neuropsychopharmacol Biol Psychiatry. 87:200–206. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
de Tommaso M, Arendt-Nielsen L, Defrin R,
Kunz M, Pickering G and Valeriani M: Pain assessment in
neurodegenerative diseases. Behav Neurol. 2016:29493582016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nayak D, Roth TL and McGavern DB:
Microglia development and function. Annu Rev Immunol. 32:367–402.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zuchero JB and Barres BA: Glia in
mammalian development and disease. Development. 142:3805–3809.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Subhramanyam CS, Wang C, Hu Q and Dheen
ST: Microglia-mediated neuroinflammation in neurodegenerative
diseases. Semin Cell Dev Biol. 94:112–120. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Skaper SD: Ion channels on microglia:
Therapeutic targets for neuroprotection. CNS Neurol Disord Drug
Targets. 10:44–56. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rash JE, Yasumura T, Dudek FE and Nagy JI:
Cell-specific expression of connexins and evidence of restricted
gap junctional coupling between glial cells and between neurons. J
Neurosci. 21:1983–2000. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beyer EC and Berthoud VM: Gap junction
gene and protein families: Connexins, innexins, and pannexins.
Biochim Biophys Acta Biomembr. 1860:5–8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nielsen MS, Axelsen LN, Sorgen PL, Verma
V, Delmar M and Holstein-Rathlou NH: Gap junctions. Compr Physiol.
2:1981–2035. 2012.PubMed/NCBI
|
|
11
|
Gomes P, Srinivas SP, Van Driessche W,
Vereecke J and Himpens B: ATP release through connexin hemichannels
in corneal endothelial cells. Invest Ophthalmol Vis Sci.
46:1208–1218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H, Liu TF, Lazrak A, Peracchia C,
Goldberg GS, Lampe PD and Johnson RG: Properties and regulation of
gap junctional hemichannels in the plasma membranes of cultured
cells. J Cell Biol. 134:1019–1030. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rhett JM, Fann SA and Yost MJ: Purinergic
signaling in early inflammatory events of the foreign body
response: Modulating extracellular ATP as an enabling technology
for engineered implants and tissues. Tissue Eng Part B Rev.
20:392–402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rhett JM and Yeh ES: The potential for
connexin hemichannels to drive breast cancer progression through
regulation of the inflammatory response. Int J Mol Sci.
19:10432018. View Article : Google Scholar
|
|
15
|
Merrifield PA and Laird DW: Connexins in
skeletal muscle development and disease. Semin Cell Dev Biol.
50:67–73. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hervé JC: Membrane channels formed by gap
junction proteins. Biochim Biophys Acta Biomembr. 1860:1–4. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martins-Marques T, Ribeiro-Rodrigues T,
Batista-Almeida D, Aasen T, Kwak BR and Girao H: Biological
functions of connexin43 beyond intercellular communication. Trends
Cell Biol. 29:835–847. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laird DW: Closing the gap on autosomal
dominant connexin-26 and connexin-43 mutants linked to human
disease. J Biol Chem. 283:2997–3001. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vinken M: Connexin hemichannels: Novel
mediators of toxicity. Arch Toxicol. 89:143–145. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hervé JC and Derangeon M:
Gap-junction-mediated cell-to-cell communication. Cell Tissue Res.
352:21–31. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meda P: Gap junction proteins are key
drivers of endocrine function. Biochim Biophys Acta Biomembr.
1860:124–140. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harris AL: Electrical coupling and its
channels. J Gen Physiol. 150:1606–1639. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Traub RD, Whittington MA, Gutiérrez R and
Draguhn A: Electrical coupling between hippocampal neurons:
Contrasting roles of principal cell gap junctions and interneuron
gap junctions. Cell Tissue Res. 373:671–691. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Srinivas M, Calderon DP, Kronengold J and
Verselis VK: Regulation of connexin hemichannels by monovalent
cations. J Gen Physiol. 127:67–75. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Contreras JE, Sáez JC, Bukauskas FF and
Bennett MV: Gating and regulation of connexin 43 (Cx43)
hemichannels. Proc Natl Acad Sci USA. 100:11388–11393. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Quist AP, Rhee SK, Lin H and Lal R:
Physiological role of gap-junctional hemichannels. Extracellular
calcium-dependent isosmotic volume regulation. J Cell Biol.
148:1063–1074. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stout CE, Costantin JL, Naus CC and
Charles AC: Intercellular calcium signaling in astrocytes via ATP
release through connexin hemichannels. J Biol Chem.
277:10482–10488. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Taruno A: ATP release channels. Int J Mol
Sci. 19:8082018. View Article : Google Scholar
|
|
29
|
Xing L, Yang T, Cui S and Chen G: Connexin
hemichannels in astrocytes: Role in CNS disorders. Front Mol
Neurosci. 12:232019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Khakh BS: Molecular physiology of P2X
receptors and ATP signalling at synapses. Nat Rev Neurosci.
2:165–174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rogne P, Andersson D, Grundström C,
Sauer-Eriksson E, Linusson A and Wolf-Watz M: Nucleation of an
activating conformational change by a cation-π interaction.
Biochemistry. 58:3408–3412. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kawasaki A, Hayashi T, Nakachi K, Trosko
JE, Sugihara K, Kotake Y and Ohta S: Modulation of connexin 43 in
rotenone-induced model of Parkinson's disease. Neuroscience.
160:61–68. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sáez JC, Schalper KA, Retamal MA, Orellana
JA, Shoji KF and Bennett MV: Cell membrane permeabilization via
connexin hemichannels in living and dying cells. Exp Cell Res.
316:2377–2389. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Delvaeye T, Vandenabeele P, Bultynck G,
Leybaert L and Krysko DV: Therapeutic targeting of connexin
channels: New views and challenges. Trends Mol Med. 24:1036–1053.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Parkinson J: An essay on the shaking
palsy. 1817. J Neuropsychiatry Clin Neurosci. 14:223–236;
discussion 222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hirsch L, Jette N, Frolkis A, Steeves T
and Pringsheim T: The incidence of Parkinson's disease: A
systematic review and meta-analysis. Neuroepidemiology. 46:292–300.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jankovic J: Parkinson's disease: Clinical
features and diagnosis. J Neurol Neurosurg Psychiatry. 79:368–376.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maatouk L, Yi C, Carrillo-de Sauvage MA,
Compagnion AC, Hunot S, Ezan P, Hirsch EC, Koulakoff A, Pfrieger
FW, Tronche F, et al: Glucocorticoid receptor in astrocytes
regulates midbrain dopamine neurodegeneration through connexin
hemichannel activity. Cell Death Differ. 26:580–596. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
DeLong MR and Wichmann T: Basal ganglia
circuits as targets for neuromodulation in Parkinson disease. JAMA
Neurol. 72:1354–1360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gerfen CR and Surmeier DJ: Modulation of
striatal projection systems by dopamine. Annu Rev Neurosci.
34:441–466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bryois J, Skene NG, Hansen TF, Kogelman
LJA, Watson HJ, Liu Z; Eating Disorders Working Group of the
Psychiatric Genomics Consortium; International Headache Genetics
Consortium; 23andMe Research Team; Brueggeman L, ; et al: Genetic
identification of cell types underlying brain complex traits yields
insights into the etiology of Parkinson's disease. Nat Genet.
52:482–493. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Orieux G, Francois C, Féger J, Yelnik J,
Vila M, Ruberg M, Agid Y and Hirsch EC: Metabolic activity of
excitatory parafascicular and pedunculopontine inputs to the
subthalamic nucleus in a rat model of Parkinson's disease.
Neuroscience. 97:79–88. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hauser RA: α-Synuclein in Parkinson's
disease: Getting to the core of the matter. Lancet Neurol.
14:785–786. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Diniz LP, Matias I, Araujo APB, Garcia MN,
Barros-Aragão FGQ, Alves-Leon SV, de Souza JM, Foguel D, Figueiredo
CP, Braga C, et al: α-Synuclein oligomers enhance astrocyte-induced
synapse formation through TGF-β1 signaling in a Parkinson's disease
model. J Neurochem. 150:138–157. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Singh-Bains MK, Waldvogel HJ and Faull RL:
The role of the human globus pallidus in Huntington's disease.
Brain Pathol. 26:741–751. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim IS, Ganesan P and Choi DK: Cx43
mediates resistance against MPP+-induced apoptosis in
SH-SY5Y neuroblastoma cells via modulating the mitochondrial
apoptosis pathway. Int J Mol Sci. 17:18192016. View Article : Google Scholar
|
|
47
|
Wu A, Green CR, Rupenthal ID and
Moalem-Taylor G: Role of gap junctions in chronic pain. J Neurosci
Res. 90:337–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pérez-Alvarez A and Araque A:
Astrocyte-neuron interaction at tripartite synapses. Curr Drug
Targets. 14:1220–1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hertz L, Hansson E and Rönnbäck L:
Signaling and gene expression in the neuron-glia unit during brain
function and dysfunction: Holger Hydén in memoriam. Neurochem Int.
39:227–252. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jiang BC, Cao DL, Zhang X, Zhang ZJ, He
LN, Li CH, Zhang WW, Wu XB, Berta T, Ji RR and Gao YJ: CXCL13
drives spinal astrocyte activation and neuropathic pain via CXCR5.
J Clin Invest. 126:745–761. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Durkee CA and Araque A: Diversity and
specificity of astrocyte-neuron communication. Neuroscience.
396:73–78. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Szczupak L: Functional contributions of
electrical synapses in sensory and motor networks. Curr Opin
Neurobiol. 41:99–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Halje P, Brys I, Mariman JJ, da Cunha C,
Fuentes R and Petersson P: Oscillations in cortico-basal ganglia
circuits: Implications for Parkinson's disease and other neurologic
and psychiatric conditions. J Neurophysiol. 122:203–231. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Adamchic I, Hauptmann C, Barnikol UB,
Pawelczyk N, Popovych O, Barnikol TT, Silchenko A, Volkmann J,
Deuschl G, Meissner WG, et al: Coordinated reset neuromodulation
for Parkinson's disease: Proof-of-concept study. Mov Disord.
29:1679–1684. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dauer W and Przedborski S: Parkinson's
disease: Mechanisms and models. Neuron. 39:889–909. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Díaz EF, Labra VC, Alvear TF, Mellado LA,
Inostroza CA, Oyarzún JE, Salgado N, Quintanilla RA and Orellana
JA: Connexin 43 hemichannels and pannexin-1 channels contribute to
the α-synuclein-induced dysfunction and death of astrocytes. Glia.
67:1598–1619. 2019.PubMed/NCBI
|
|
57
|
Sarrouilhe D, Dejean C and Mesnil M:
Connexin43- and pannexin-based channels in neuroinflammation and
cerebral neuropathies. Front Mol Neurosci. 10:3202017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Takeuchi H, Jin S, Wang J, Zhang G,
Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T and Suzumura A: Tumor
necrosis factor-alpha induces neurotoxicity via glutamate release
from hemichannels of activated microglia in an autocrine manner. J
Biol Chem. 281:21362–21368. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lu C, Meng Z, He Y, Xiao D, Cai H, Xu Y,
Liu X, Wang X, Mo L, Liang Z, et al: Involvement of gap junctions
in astrocyte impairment induced by manganese exposure. Brain Res
Bull. 140:107–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sung JY, Lee HJ, Jeong EI, Oh Y, Park J,
Kang KS and Chung KC: Alpha-synuclein overexpression reduces gap
junctional intercellular communication in dopaminergic
neuroblastoma cells. Neurosci Lett. 416:289–293. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Reyes JF, Sackmann C, Hoffmann A,
Svenningsson P, Winkler J, Ingelsson M and Hallbeck M: Binding of
α-synuclein oligomers to Cx32 facilitates protein uptake and
transfer in neurons and oligodendrocytes. Acta Neuropathol.
138:23–47. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hare DJ, Adlard PA, Doble PA and
Finkelstein DI: Metallobiology of
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity.
Metallomics. 5:91–109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fujita A, Yamaguchi H, Yamasaki R, Cui Y,
Matsuoka Y, Yamada KI and Kira JI: Connexin 30 deficiency
attenuates A2 astrocyte responses and induces severe
neurodegeneration in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
hydrochloride Parkinson's disease animal model. J
Neuroinflammation. 15:2272018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liddelow SA, Guttenplan KA, Clarke LE,
Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS,
Peterson TC, et al: Neurotoxic reactive astrocytes are induced by
activated microglia. Nature. 541:481–487. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pannasch U, Freche D, Dallérac G, Ghézali
G, Escartin C, Ezan P, Cohen-Salmon M, Benchenane K, Abudara V,
Dufour A, et al: Connexin 30 sets synaptic strength by controlling
astroglial synapse invasion. Nat Neurosci. 17:549–558. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Evin G and Hince C: BACE1 as a therapeutic
target in Alzheimer's disease: Rationale and current status. Drugs
Aging. 30:755–764. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hardy JA and Higgins GA: Alzheimer's
disease: The amyloid cascade hypothesis. Science. 256:184–185.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Swerdlow RH, Burns JM and Khan SM: The
Alzheimer's disease mitochondrial cascade hypothesis: Progress and
perspectives. Biochim Biophys Acta. 1842:1219–1231. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Jammal L, Whalley B and Barkai E:
Learning-induced modulation of the effect of neuroglial
transmission on synaptic plasticity. J Neurophysiol. 119:2373–2379.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Walrave L, Vinken M, Albertini G, De
Bundel D, Leybaert L and Smolders IJ: Inhibition of connexin43
hemichannels impairs spatial short-term memory without affecting
spatial working memory. Front Cell Neurosci. 10:2882016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Nagy JI, Li W, Hertzberg EL and Marotta
CA: Elevated connexin43 immunoreactivity at sites of amyloid
plaques in Alzheimer's disease. Brain Res. 717:173–178. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mei X, Ezan P, Giaume C and Koulakoff A:
Astroglial connexin immunoreactivity is specifically altered at
β-amyloid plaques in β-amyloid precursor protein/presenilin1 mice.
Neuroscience. 171:92–105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sokoloff L: Energetics of functional
activation in neural tissues. Neurochem Res. 24:321–329. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tholey G and Ledig M: Neuronal and
astrocytic plasticity: Metabolic aspects. Ann Med Interne (Paris).
141 (Suppl 1):S13–S18. 1990.(In French).
|
|
75
|
Nunomura A, Castellani RJ, Zhu X, Moreira
PI, Perry G and Smith MA: Involvement of oxidative stress in
Alzheimer disease. J Neuropathol Exp Neurol. 65:631–641. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Pocernich CB and Butterfield DA: Elevation
of glutathione as a therapeutic strategy in Alzheimer disease.
Biochim Biophys Acta. 1822:625–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Dringen R: Metabolism and functions of
glutathione in brain. Prog Neurobiol. 62:649–671. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ong WY, Hu CY, Hjelle OP, Ottersen OP and
Halliwell B: Changes in glutathione in the hippocampus of rats
injected with kainate: Depletion in neurons and upregulation in
glia. Exp Brain Res. 132:510–516. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bolaños JP: Bioenergetics and redox
adaptations of astrocytes to neuronal activity. J Neurochem. 139
(Suppl 2):S115–S125. 2016. View Article : Google Scholar
|
|
80
|
Aoyama K, Suh SW, Hamby AM, Liu J, Chan
WY, Chen Y and Swanson RA: Neuronal glutathione deficiency and
age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat
Neurosci. 9:119–126. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hohnholt MC and Dringen R: Short time
exposure to hydrogen peroxide induces sustained glutathione export
from cultured neurons. Free Radic Biol Med. 70:33–44. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Rana S and Dringen R: Gap junction
hemichannel-mediated release of glutathione from cultured rat
astrocytes. Neurosci Lett. 415:45–48. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Orellana JA, Shoji KF, Abudara V, Ezan P,
Amigou E, Sáez PJ, Jiang JX, Naus CC, Sáez JC and Giaume C: Amyloid
β-induced death in neurons involves glial and neuronal
hemichannels. J Neurosci. 31:4962–4977. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Hardiman O, Al-Chalabi A, Chio A, Corr EM,
Logroscino G, Robberecht W, Shaw PJ, Simmons Z and van den Berg LH:
Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 3:170712017.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Riva N, Agosta F, Lunetta C, Filippi M and
Quattrini A: Recent advances in amyotrophic lateral sclerosis. J
Neurol. 263:1241–1254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ohta Y, Nomura E, Shang J, Feng T, Huang
Y, Liu X, Shi X, Nakano Y, Hishikawa N, Sato K, et al: Enhanced
oxidative stress and the treatment by edaravone in mice model of
amyotrophic lateral sclerosis. J Neurosci Res. 97:607–619. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Holecek V and Rokyta R: Possible etiology
and treatment of amyotrophic lateral sclerosis. Neuro Endocrinol
Lett. 38:528–531. 2018.PubMed/NCBI
|
|
88
|
Tedeschi V, Petrozziello T and Secondo A:
Calcium dyshomeostasis and lysosomal Ca2+ dysfunction in
amyotrophic lateral sclerosis. Cells. 8:12162019. View Article : Google Scholar
|
|
89
|
Mandrioli J, D'Amico R, Zucchi E, Gessani
A, Fini N, Fasano A, Caponnetto C, Chiò A, Dalla Bella E, Lunetta
C, et al: Rapamycin treatment for amyotrophic lateral sclerosis:
Protocol for a phase II randomized, double-blind,
placebo-controlled, multicenter, clinical trial (RAP-ALS trial).
Medicine (Baltimore). 97:e111192018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
McGeer PL and McGeer EG: Inflammatory
processes in amyotrophic lateral sclerosis. Muscle Nerve.
26:459–470. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Spitale FM, Vicario N, Rosa MD, Tibullo D,
Vecchio M, Gulino R and Parenti R: Increased expression of connexin
43 in a mouse model of spinal motoneuronal loss. Aging (Albany NY).
12:12598–12608. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Almad AA, Doreswamy A, Gross SK, Richard
JP, Huo Y, Haughey N and Maragakis NJ: Connexin 43 in astrocytes
contributes to motor neuron toxicity in amyotrophic lateral
sclerosis. Glia. 64:1154–1169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hamilton N, Vayro S, Kirchhoff F,
Verkhratsky A, Robbins J, Gorecki DC and Butt AM: Mechanisms of
ATP- and glutamate-mediated calcium signaling in white matter
astrocytes. Glia. 56:734–749. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Sheng L, Leshchyns'ka I and Sytnyk V: Cell
adhesion and intracellular calcium signaling in neurons. Cell
Commun Signal. 11:942013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Brini M, Calì T, Ottolini D and Carafoli
E: Neuronal calcium signaling: Function and dysfunction. Cell Mol
Life Sci. 71:2787–2814. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Belousov AB, Nishimune H, Denisova JV and
Fontes JD: A potential role for neuronal connexin 36 in the
pathogenesis of amyotrophic lateral sclerosis. Neurosci Lett.
666:1–4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Decrock E, Vinken M, De Vuyst E, Krysko
DV, D'Herde K, Vanhaecke T, Vandenabeele P, Rogiers V and Leybaert
L: Connexin-related signaling in cell death: To live or let die?
Cell Death Differ. 16:524–536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
McColgan P and Tabrizi SJ: Huntington's
disease: A clinical review. Eur J Neurol. 25:24–34. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Goetz CG: The history of Parkinson's
disease: Early clinical descriptions and neurological therapies.
Cold Spring Harb Perspect Med. 1:a0088622011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Wichmann T and Dostrovsky JO: Pathological
basal ganglia activity in movement disorders. Neuroscience.
198:232–244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Vis JC, Nicholson LF, Faull RL, Evans WH,
Severs NJ and Green CR: Connexin expression in Huntington's
diseased human brain. Cell Biol Int. 22:837–847. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Allen NJ and Lyons DA: Glia as architects
of central nervous system formation and function. Science.
362:181–185. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Scheefhals N and MacGillavry HD:
Functional organization of postsynaptic glutamate receptors. Mol
Cell Neurosci. 91:82–94. 2018. View Article : Google Scholar : PubMed/NCBI
|