Introduction
Ginkgolide B (GB) is one of the main components of
the extracts of Ginkgo biloba leaves and the pharmacodynamic
component with the strongest activity and the highest specificity
of the Ginkgo diterpenoid lactones (1). GB has been found to possess notable
effects on the central nervous system, such as improving the
cognitive function of patients with Alzheimer's disease (AD) and
promoting the neuroprotective effects on acute hypoxic/ischemic
injury (2,3). The high purity of GB enables it to
pass through the blood-brain barrier and be easily absorbed, which
is beneficial in the treatment of neurodegenerative diseases
(4,5), including AD, which is a destructive
central nervous system lesion characterized by declining learning,
memory loss and cognitive dysfunction (6,7). At
present, the pathogenesis of AD remains to be elucidated, but it is
well known that the abnormal metabolism and deposition of β-amyloid
(Aβ) in the brain tissues contribute to AD (8). Activated astrocytes are found around
Aβ deposition (9,10) and are the most abundant type of
cells in the central nervous system. A previous study indicates
that astrocytes induce Aβ degradation and clearance and the
degradation of Aβ mediated by astrocytes is impeded in the early
stage of AD (11). Thus, astrocytes
have regulatory roles in the onset of AD (12). GB has been found to have clinical
effects on neuroprotection (13),
but the protective effect of GB on astrocytes in AD and its
potential molecular mechanism remain to be elucidated.
Aβ is the main factor leading to AD cognitive
dysfunction and neurodegeneration because the excessive generation
and aggregation of Aβ results in a series of pathological and
physiological changes, including endoplasmic reticulum (ER) stress
(ERS), oxidative stress (OS), inflammatory response, energy
metabolism disorder, tau hyperphosphorylation, synaptic
degeneration, cell dysfunction and even apoptosis, causing abnormal
learning, memory, cognition and behavior (14,15).
The aforementioned pathological processes interact with each other,
stimulating the onset and progression of AD.
ER, which takes part in protein synthesis,
post-translational modification and correct folding of proteins, is
necessary to maintain the normal function of cells (16). ER dysfunction or loss of integrity
causes ERS, which is key to neurodegenerative diseases (17,18).
Previous studies on patients with AD and the brain tissues of
animal models demonstrate that Aβ seriously disturbs the functions
of ER, leading to excessive generation of Aβ, ERS activation
(19,20) and finally cell dysfunction and
apoptosis (21). A previous study
showed that mitochondrial damage was important in the pathogenesis
of AD because mitochondria supplied energy, exchanged information,
antagonized OS and provided energy for various cellular activities
(22). The overaggregation of Aβ
influences the energy metabolism of mitochondria, decreases the
generation of ATP and produces numerous oxygen radicals in the
mitochondria to weaken the ability of cells to provoke oxidation
and cause OS (23), resulting in
apoptosis (24). Meanwhile, oxides
and lipid peroxides increase the levels of the key amylase β
secretase 1 (BACE1) to cause the excessive generation of Aβ
(25). The two interact with each
other to cause malignancy, mitochondrial dysfunction and serious
oxidative injury, thus exacerbating the toxic responses of Aβ
(26). Astrocytes are important in
neurodegenerative diseases and preventing the abnormal changes in
astrocytes may help treat the diseases (27,28).
Therefore, the following hypothesis was proposed in the present
study: GB might antagonize the neurotoxicity of Aβ, prevent ERS and
OS, protect the normal metabolism of astrocytes and interrupt the
excessive generation of Aβ, thus protecting astrocytes and
preventing the progression of AD.
Materials and methods
Reagents
GB was purchased from National Institutes for Food
and Drug Control China, with 98% purity ascertained by
high-performance liquid chromatography. Aβ peptide fragments
(Aβ1-42; cat. no. SCP0038) was purchased from
Sigma-Aldrich (Merck KGaA). Cell counting kit 8 (CCK-8; cat. no.
C0039), annexin V-FITC apoptosis detection kit (cat. no. C1062),
total superoxide dismutase assay kit (SOD; cat. no. S0109), lipid
peroxidation malondialdehyde assay kit (MDA; cat. no. S0131),
glutathione peroxidase assay kit (GSH-Px; cat. no. S0056), reactive
oxygen species assay kit (ROS; cat. no. S0033), ATP assay kit (cat.
no. S0131S), IgG horseradish peroxidase (HRP)-conjugated secondary
antibodies (cat. no. A0208; 1:10,000), BCA Protein Assay kit (cat.
no. P0012S) and BSA (cat. no. ST023) were purchased from Beyotime
Institute of Biotechnology. Minibest universal RNA extraction kit
(cat. no. 9767), PrimeScript™ RT reagent kit with gDNA eraser
(perfect real time; cat. no. RR047) and TB Green® Premix
Ex Taq™ (Tli RNase H Plus; cat. no. RR420) were purchased from
Takara Bio Inc. DMEM/F12 medium (cat. no. C11330500BT) and fetal
bovine serum (cat. no. 10099141C) were purchased from Gibco (Thermo
Fisher Scientific, Inc.). Clarity Western ECL substrate was
purchased from Bio-Rad Laboratories, Inc. Compound C, antibodies
against β-actin (cat. no. 4970; rabbit; 1:1,000), β-secretase
(BACE1; cat. no. 5606; rabbit; 1:1,000), β-tubulin (cat. no. 6181;
rabbit; 1:1,000), protein kinase RNA-like endoplasmic reticulum
kinase (PERK; Phospho Thr980; cat. no. 3179; rabbit; 1:1,000), PERK
(cat. no. 3192; rabbit; 1:1,000), eukaryotic translation initiation
factor 2 subunit α (eIF2α; PhosphoSer51; cat. no. 5199; rabbit;
1:1,000), eIF2α (cat. no. 9079; rabbit; 1:1,000), 5′ adenosine
monophosphate-activated protein kinase (AMPK; Phospho Thr172; cat.
no. 2531; rabbit; 1:1,000), AMPK (cat. no. 2532; rabbit; 1:1,000),
heme oxygenase 1 (HO-1; cat. no. 82206; rabbit; 1:1,000) were
purchased from Cell Signaling Technology, Inc., inositol-requiring
enzyme 1 α (IRE1α; cat. no. 37073; rabbit; 1:1,000),
proliferator-activated receptor γ coactivator 1 α (PGC-1α; cat. no.
54481; rabbit; 1:1,000), C/EBP-homologous protein (CHOP; cat. no.
179823; rabbit; 1:1,000), 78 kDa glucose-regulated protein (GRP78;
cat. no. 108613; rabbit; 1:1,000), activating transcription factor
6 (ATF6; cat. no. 203119; rabbit; 1:1,000), nuclear factor
erythroid 2-related factor 2 (Nrf2; cat. no. 137550; rabbit;
1:1,000), NAD(P)H dehydrogenase (quinone 1) (NQO1; cat. no. 28947;
rabbit; 1:1,000) and peroxisome proliferator-activated receptor α
(PPARα; cat. no. 215270; rabbit; 1:1,000) were obtained from Abcam.
All primers were purchased from Sangon Biotech Co., Ltd.
Animals
A total of 3 pregnant Sprague Dawley (SD) rats (age,
2 months; 2–3 weeks pregnancy; weight, 260–270 g) were purchased
from Experimental Animal Center, Yangzhou University (Yangzhou,
China). The rats were accommodated in an animal room with a 12-h
light/dark cycle and ad libitum access to food and water at
a temperature and humidity of 22±1°C and 50±10%, respectively. The
pregnant rats were fed with basic diet until postpartum. All
experiments were carried out following the guidelines and protocols
approved by the Ethics Committee for the Use of Experimental
Animals of Jiangsu Kanion Pharmaceutical Co. Ltd. and the State Key
Laboratory of New Pharmaceutical Process for Traditional Chinese
Medicine (Lianyungang, China; approval no. 2019012). The
1–2-day-old rats and postpartum SD rats were sacrifice with 5%
isoflurane, followed by cervical dislocation for the confirmation
of mortality.
Astrocyte cultures
The cerebral cortices of 1–2-day-old rats were
digested in 0.25% trypsin solution at 37°C for 30 min and the same
volume of DMEM/F12 containing 10% fetal bovine serum was added to
stop digestion. The cortices were repeatedly blown with a straw
until the tissue mass disappeared completely and the liquid was
turbid. The suspension was filtered through a 200-mesh sterile
screen and the filtrate was collected and centrifuged (300 × g) for
5 min at 20–25°C. The cells were then resuspended in DMEM/F12 [10%
(v/v) fetal bovine serum] and incubated at 37°C in the presence of
5% CO2 and 90% relative humidity. At 90% confluence of
the cells, microglial cells and oligodendrocytes were removed from
astrocyte cultures by shaking (200 rpm) overnight at 37°C (29).
Glial fibrillary acidic protein (GFAP) is a marker
of astrocytes, which can be identified by GFAP immunofluorescence
staining. The identification of purified astrocytes is presented in
Fig. S1 (magnification, ×100).
Following staining with GFAP, green fluorescence was exhibited
under a fluorescence microscope. Following staining with DAPI, blue
fluorescence was exhibited under a fluorescence microscope. The
cell morphology after fused cells stained by GFAP or DAPI and the
purity of astrocytes were >95% (Fig. S1). Astrocytes with purity >95%
were used in subsequent experiments.
Drug treatments
The optimal concentration and time of action of GB
and Aβ1-42 were determined based on a previous study
(30) and the pre-experimental
results of their influence on astrocyte activity (Fig. S2). As demonstrated in Fig. S2, the cells treated with a
concentration of 10 µM Aβ1-42 in DMEM/F12 for 24 h as an
Aβ1-42-damage model and the cell vitality treated with
the concentration of 20 and 40 µM GB in DMEM/F12 for 24 h showed no
difference compared with the normal group although the cell
activity was significantly improved in 80 µM GB. In addition, the
cell viability of 100 and 200 µM GB groups was lower compared with
the 20, 40 and 80 µΜ GB groups. The concentration of compound C, an
effective and reversible AMPK inhibitor, was determined based on
the protein expression of AMPK, as demonstrated in Fig. S2. The preliminary results showed
that 10 µM compound C in DMEM/F12 for 24 h inhibited AMPK
phosphorylation compared with the normal group.
Subsequently, 24 h after of seeding, the medium was
replaced by a fresh medium containing the drugs. The astrocytes
were randomly divided into the following groups: Control (without
any treatment in DMEM/F12), Aβ (treated with the final
concentration of 10 µM Aβ1-42 in DMEM/F12 for 24 h), GB
(treated with the final concentration of 10 µM Aβ1-42
and 20, 40 or 80 µM GB in DMEM/F12 for 24 h) and 80 µM GB +
compound C (treated with the final concentration of 10 µM
Aβ1-42, 80 µM GB and 10 µM compound C in DMEM/F12 for 24
h).
CCK-8 assay
The cell viability was evaluated using the CCK-8
assay. In brief, 100 µl of astrocytes were plated into 96-well
plates (1×104 cells per well) and treated by groups,
with three repeats per group. Then, 10 µl of CCK-8 solution was
added to each well and the plate was further incubated at 37°C for
2 h. The optical density at 450 nm was determined using a
microplate reader (Molecular Devices, LLC).
Annexin V/PI assay
The early and late apoptosis of astrocytes was
detected by flow cytometry. Astrocytes (~2 ml) at the concentration
of 1×105/ml were seeded into 6-well plates and treated
by groups. Then, the astrocytes were washed twice with PBS,
digested with 0.25% trypsin without EDTA, collected in the
centrifuge tube, centrifuged (300 × g) for 5 min at 20–25°C, and
washed three times with PBS. The supernatant was discarded and 195
µl of the binding buffer was added to resuspend the astrocytes.
Subsequently, 5 µl of Annexin V/FITC and 10 µl of PI were added to
the astrocytes in the dark, followed by detection using flow
cytometry (ACEA NovoCyte; ACEA Bioscience, Inc.; Agilent
Technologies, Inc.) and NovoExpress 1.2.1 software (ACEA
Biosciences, Inc.; Agilent Technologies, Inc.). SOD, MDA,
GSH-Px, ROS and ATP assay. The cells were collected, washed
once or twice with PBS, precipitated, resuspended, and mixed with
PBS. They were ruptured ultrasonically, and the homogenate was used
for determining the SOD and GSH-Px activities and MDA, ROS and ATP
content using commercially available kits (SOD assay kit with NBT;
GSH-Px assay kit, colorimetric method; MDA assay kit,
thiobarbituric acid method; ROS assay kit with DCFH-DA; and ATP
assay kit, respectively) following the manufacturer's
protocols.
RNA extraction and reverse
transcription-quantitative (RT-q) PCR
In total, 2 ml suspension containing astrocytes
(1×105 cell/ml) was inoculated into the 6-well plates
and treated with the indicated treatments, with three replicates
per treatment. Total RNA was extracted from cells using the
Minibest universal RNA extraction kit, followed by reverse
transcription to synthesize cDNA using PrimeScript RT Master Mix.
The reverse transcription was conducted at 37°C for 15 min and 85°C
for 5 sec. According to the manufacturer's protocols, the gene
transcripts were quantified using the RT-qPCR reaction system (20
µl) with TB Green and PCR was carried out under the following
conditions: Initial denaturation at 95°C for 10 min; followed by 40
cycles of 95°C for 30 sec, 60°C for 30 sec and 95°C for 15 sec; and
a final extension at 60°C for 1 min. The following primer pairs
were used for qPCR: ACTB forward, 5′-TGTCACCAACTGGGACGATA-3′ and
ACTB reverse, 5′-GGGGTGTTGAAGGTCTCAAA-3′; CHOP forward,
5′-CCTCGCTCTCCAGATTCCAGTCAG-3′ and CHOP reverse,
5′-TCTCCTGCTCCTTCTCCTTCATGC-3′; Nrf2 forward,
5′-TGACTCCGGCATTTCACTGA-3′ and Nrf2 reverse,
5′-GTGGGTCTCCGTAAATGGAAGA-3′; HO-1 forward,
5′-TATCGTGCTCGCATGAACACTCTG-3′ and HO-1 reverse,
5′-GTTGAGCAGGAAGGCGGTCTTAG-3′; NQO1 forward,
5′-AGAAGCGTCTGGAGACTGTCTGG-3′ and NQO1 reverse,
5′-GATCTGGTTGTCGGCTGGAATGG-3′; AMPK forward,
5′-ATGATGAGGTGGTGGAGCAGAGG-3′ and AMPK reverse,
5′-GTTCTCGGCTGTGCTGGAATCG-3′; PGC1α forward,
5′-TTGAAGAGCGCCGTGTGAT-3′ and PGC1α reverse,
5′-AAAAACTTCAAAGCGGTCTCTCA-3′; PPARα forward,
5′-TGACTTGGCCATATTTATAGCTGTCA-3′ and PPARα reverse,
5′-GATGTCCTCGATGGGCTTCA-3′. All RT-qPCR experiments were repeated
three times and the relative expression levels were quantified
using the 2−∆∆Cq method (31).
Western blotting
The total protein was extracted from astrocytes with
ice-cold RIPA buffer and the protein content was estimated using a
BCA protein assay kit. The total protein (30 µg) was separated by
10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and
electrotransferred onto a polyvinylidene fluoride membrane for 2 h
in an ice-cold buffer. The membranes were blocked in 5% BSA for 1 h
at room temperature, incubated overnight at 4°C with the
corresponding primary antibodies, washed, incubated with horse
radish peroxidase-conjugated secondary antibody for 1 h at room
temperature and developed using an enhanced chemiluminescence kit.
Gray-scale scanning and quantification were performed using Image
Lab 3.0 software (Bio-Rad Laboratories, Inc.).
Statistical analysis
All data were presented as mean ± standard deviation
from ≥3 independent experiments. Statistical analysis permitted
normalization of the 2−∆∆Cq of RT-qPCR and the value of
protein gray of western blotting in the Aβ group and the cell
activity of CCK8 in control group. Statistical analyses were
performed by one-way ANOVA test evaluating significant differences
between treatments using SPSS v17.0 statistical software (SPSS,
Inc.). The comparison between the two groups was conducted by LSD
method with homogeneous variances and Tamhane method with
inhomogeneous variances. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of GB on
Aβ1-42-induced cell viability in astrocytes
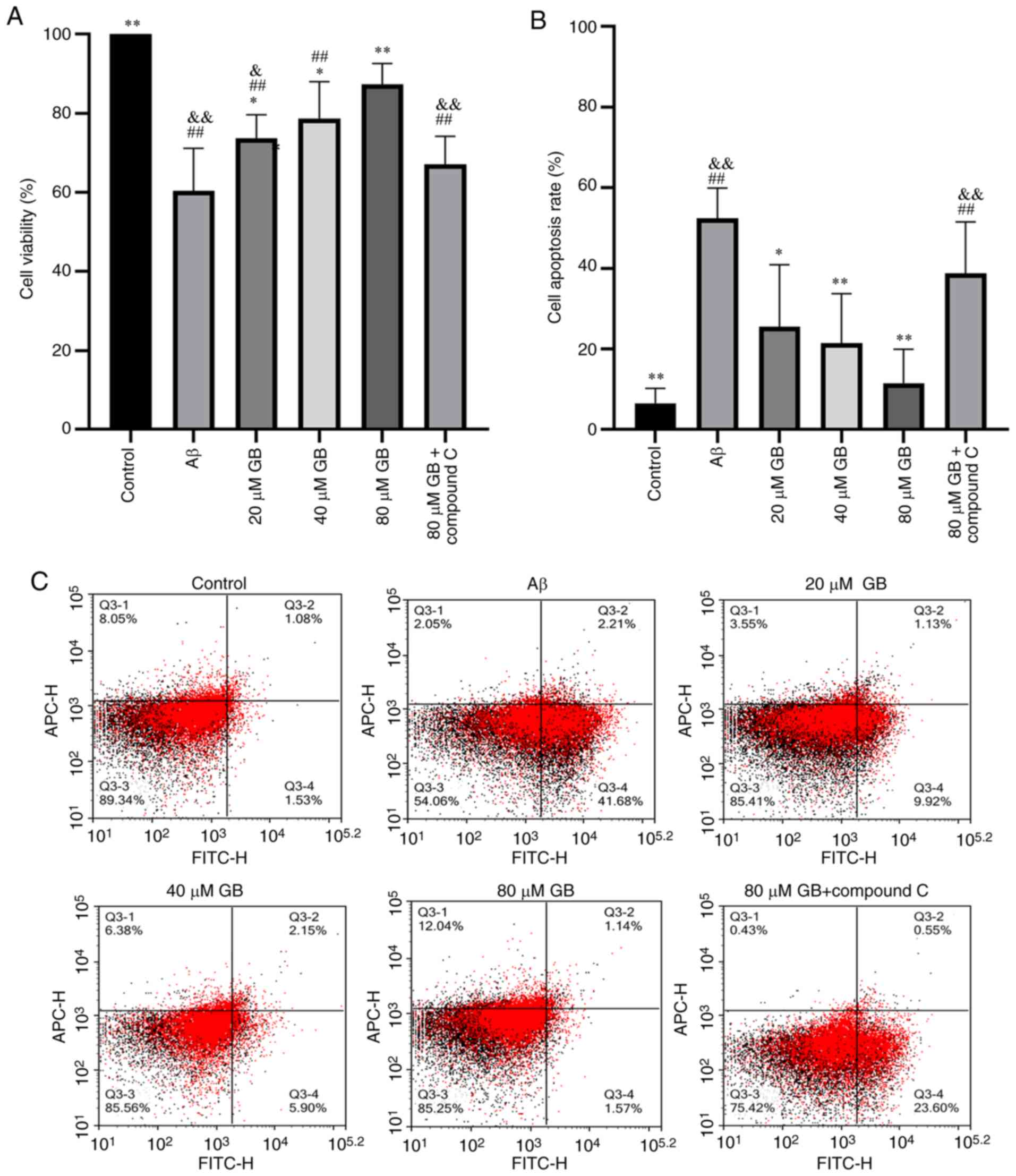
The activity of astrocytes is demonstrated in
Fig. 1A. The activity of astrocytes
clearly decreased in the Aβ group compared with the control group
(P<0.01), while the activity was higher in the 20, 40 and 80 µM
GB groups compared with the Aβ group in a concentration-dependent
manner (P<0.05; P<0.01). However, the cell viability was
significantly lower in the 80 µM GB + compound C group compared
with the 80 µM GB group (P<0.01). The results demonstrated that
Aβ1-42 decreased astrocyte viability and caused cellular
damage, while GB enhanced astrocyte viability and protected the
cells. In addition, the effects of GB on Aβ1-42-induced
decreased astrocyte viability were inhibited by the AMPK inhibitor
compound C.
Effect of GB on
Aβ1-42-induced cell apoptosis in astrocytes
Apoptosis detection was performed on each group of
astrocytes using flow cytometry and the results are demonstrated in
Fig. 1B and C. Compared with the
control group, Aβ1-42 treatment clearly increased the
apoptotic rate of astrocytes (P<0.01). The number of apoptotic
astrocytes was significantly lower in the 20, 40 and 80 µM GB
groups than in the Aβ group (P<0.05, P<0.01), while the
apoptotic rate was significantly higher in the 80 µM GB + compound
C group than in the 80 µM GB group (P<0.01). Hence, GB
effectively reduced the Aβ1-42-induced apoptosis of
astrocytes and compound C inhibited this effect.
Effect of GB on
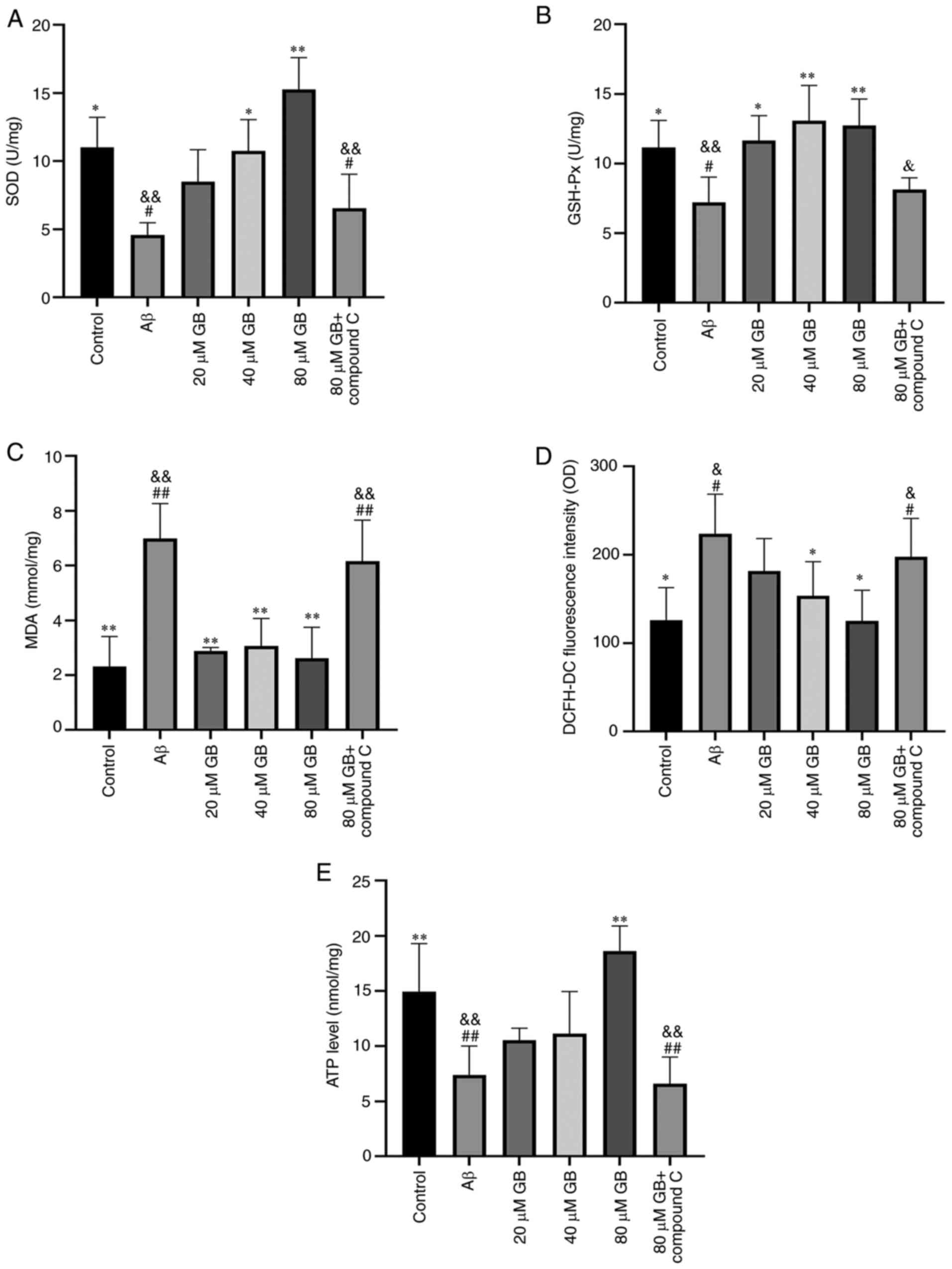
Aβ1-42-induced OS-related factors in astrocytes
As demonstrated in Fig.
2A-D, Aβ1-42 clearly decreased the activities of SOD
and GSH-Px and distinctly increased the MDA content and ROS levels
in astrocytes compared with the control group (P<0.05;
P<0.01). The activities of SOD and GSH-Px visibly increased
(P<0.01) while the MDA content and ROS levels significantly
decreased (P<0.05, P<0.01) in the 40 and 80 µM GB groups
compared with the Aβ group. The activities of SOD and GSH-Px
significantly decreased (P<0.05; P<0.01) and the MDA content
and ROS level significantly increased (P<0.05; P<0.01) in the
80 µM GB + compound C group compared with the 80 µM GB group. Thus,
the results showed that GB could attenuate the
Aβ1-42-induced decrease in antioxidants in astrocytes;
however, compound C could provoke this effect.
Effect of GB on
Aβ1-42-induced levels of ATP in astrocytes
As demonstrated in Fig.
2E, Aβ1-42 clearly decreased the content of ATP in
astrocytes compared with the control group (P<0.01), whereas the
content of ATP significantly increased (P<0.01) following 80 µM
GB treatment compared with the Aβ treatment. However, the content
of ATP was significantly lower in the 80 µM GB + compound C group
compared with the 80 µM GB group (P<0.01). Thus, GB could
enhance the ATP level of astrocytes following treatment with
Aβ1-42 and compound C could prevent this effect.
Effect of GB on expressions of ERS
signal molecule in astrocytes
The data presented in Fig. 3A and B indicate that the protein
expression levels of CHOP, GRP78, phosphorylated (p)-PERK, p-eIf2α
and ATF6 significantly increased in the Aβ group compared with the
control group (P<0.05, P<0.01). In addition, 20, 40 and 80 µM
GB notably repressed the upregulation of CHOP, GRP78 and p-PERK; 40
and 80 µM GB significantly inhibited the upregulation of p-eIf2α
and IRE1α; and 20 and 80 µM GB notably repressed the upregulation
of ATF6 (P<0.05; P<0.01) compared with Aβ alone. However, 80
µM GB + compound C treatment significantly increased the protein
expression levels of CHOP and ERS marker proteins (P<0.05;
P<0.01) compared with 80 µM GB treatment (Fig. 4A and B). The experimental results
proved that GB protected astrocytes from Aβ1-42-induced
apoptosis via inhibiting ERS, while AMPK inhibitor compound C could
prevent this effect. Therefore, the mechanism of GB inhibiting ERS
may be closely related to the AMPK signaling pathway.
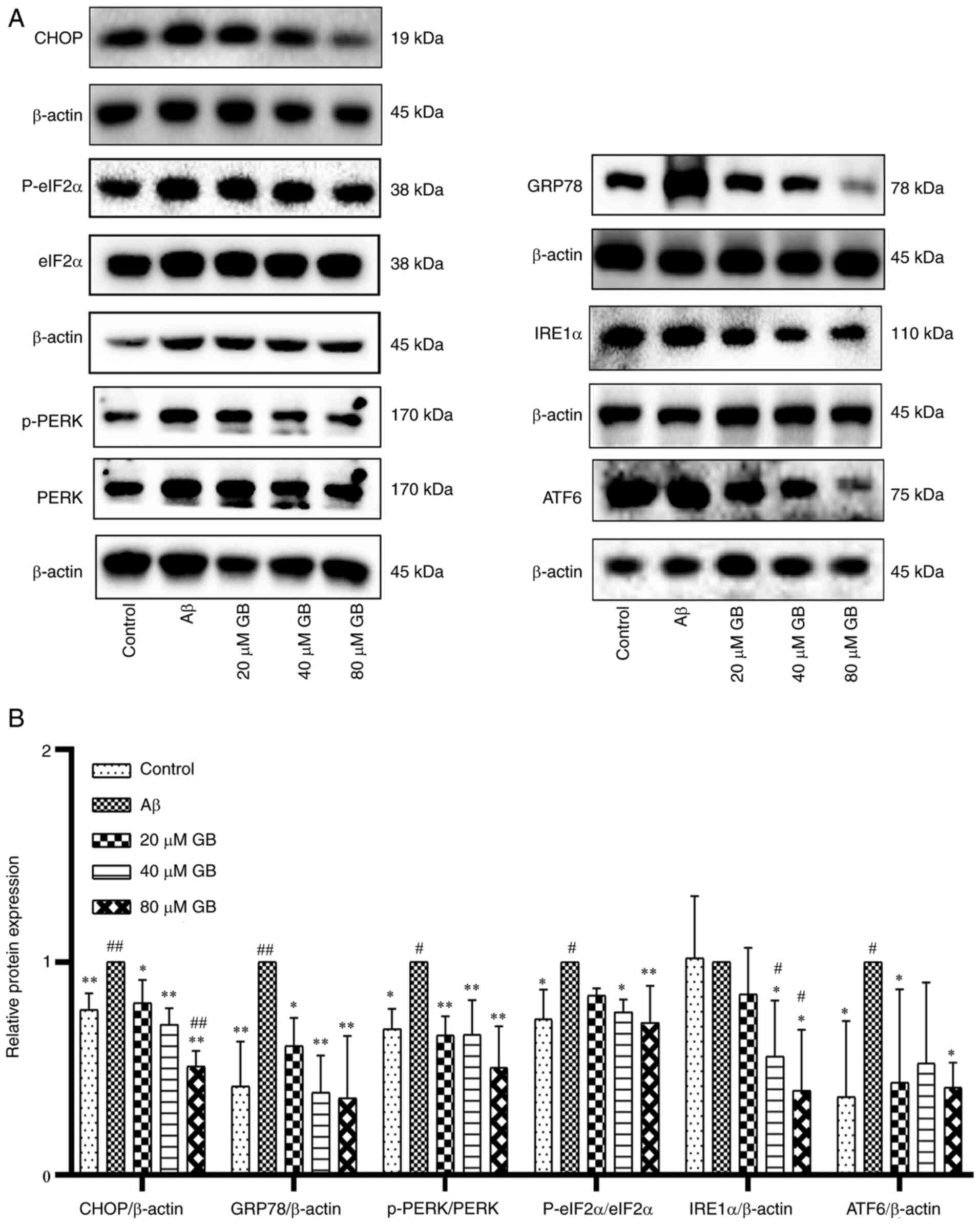 | Figure 3.Effect of GB on expressions of ERS
signal molecule in astrocytes. (A) Western blotting image and (B)
western blotting data of protein expression levels of ERS following
treatment with 10 µM Aβ1-42 with different GB
concentration (20, 40 and 80 µM GB) or treated with 10 µM
Aβ1-42 for 24 h. #P<0.05 and
##P<0.01 vs. control group, *P<0.05 and
**P<0.01 vs. Aβ group. GB, ginkgolide B; ERS, endoplasmic
reticulum stress; Aβ, β-amyloid; p-, phosphorylated; GRP78, binding
immunoglobulin protein; PERK, protein kinase R like endoplasmic
reticulum kinase; eIF2α, eukaryotic translation initiation factor 2
subunit 1; eIF2α, inositol-requiring enzyme 1 α; ATF6, activating
transcription factor 6. |
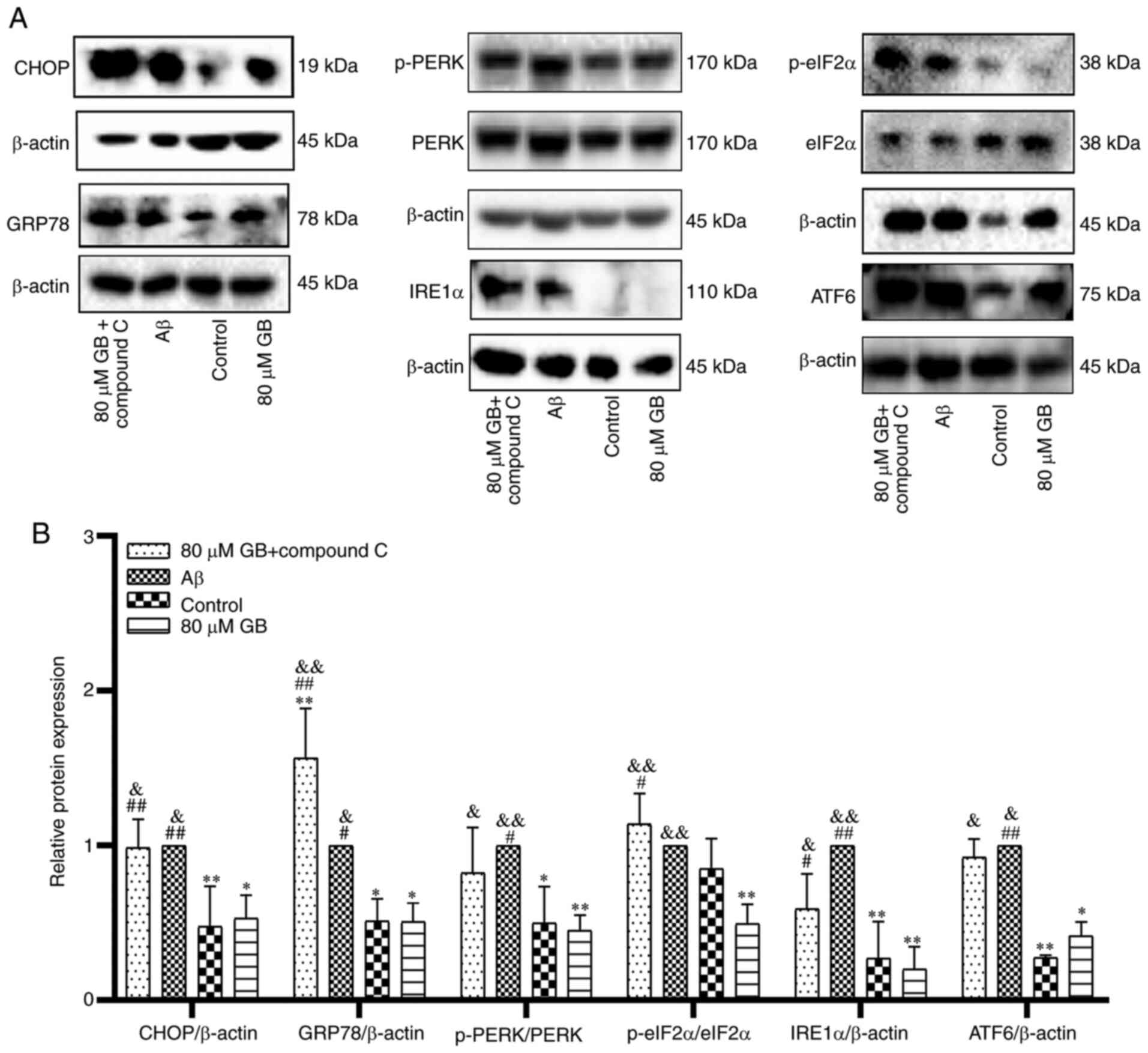 | Figure 4.Compound C prevents GB-inhibits
protein expressions of ERS in astrocytes. (A) Western blotting
image and (B) western blotting data of protein expression levels of
ERS following treatment with 10 µM Aβ1-42 with 80 µM GB
or treated with 10 µM Aβ1-42 or 10 µM Aβ1-42 + 80 µM GB
+ 10 µM compound C for 24 h. #P<0.05 and
##P<0.01 vs. control group, *P<0.05 and
**P<0.01 vs. Aβ group, &P<0.05 and
&&P<0.01 vs. 80 µM GB group. GB, ginkgolide
B; ERS, endoplasmic reticulum stress; Aβ, β-amyloid; p-,
phosphorylated; GRP78, binding immunoglobulin protein; PERK,
protein kinase R like endoplasmic reticulum kinase; eIF2α,
eukaryotic translation initiation factor 2 subunit 1; eIF2α,
inositol-requiring enzyme 1 α; ATF6, activating transcription
factor 6. |
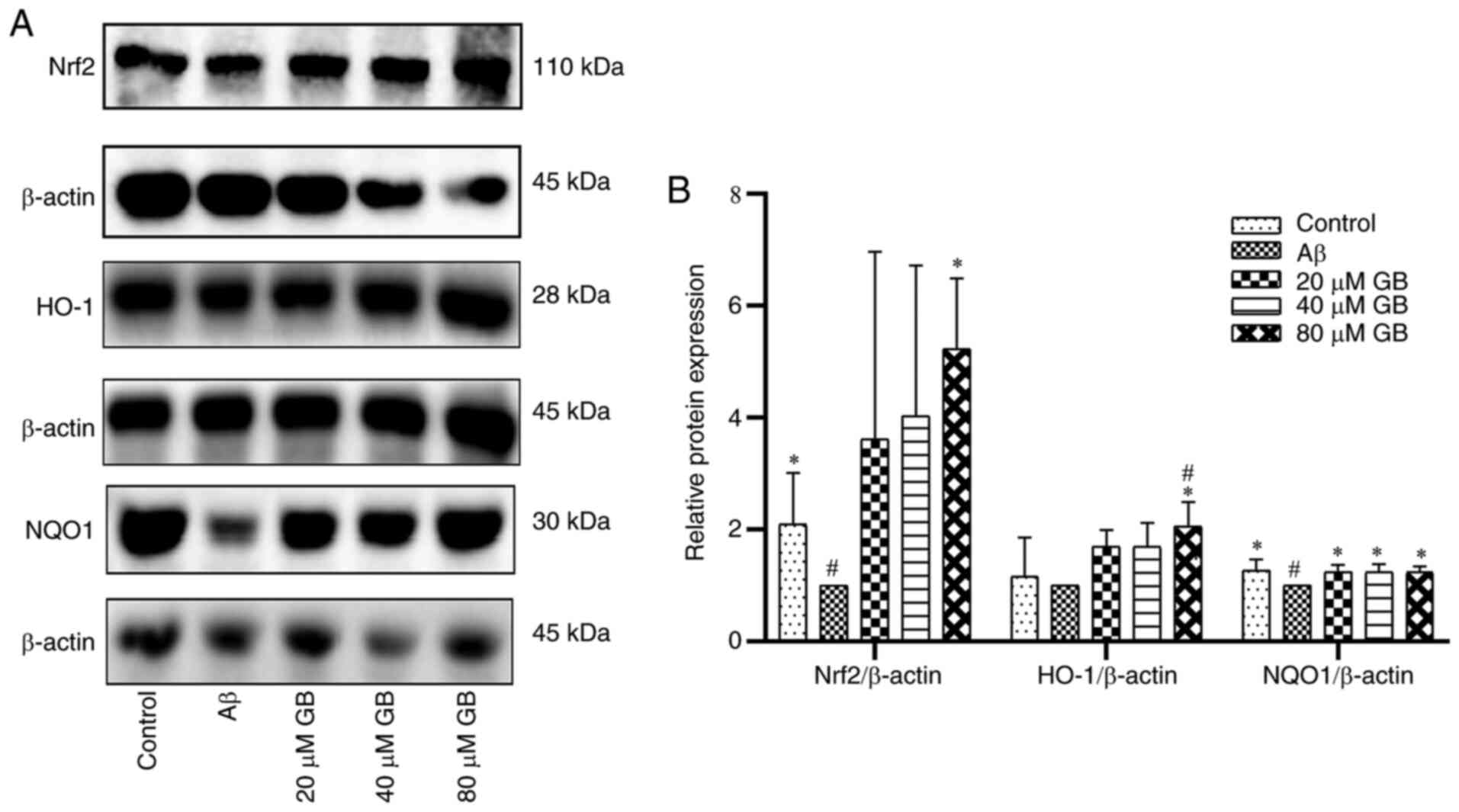
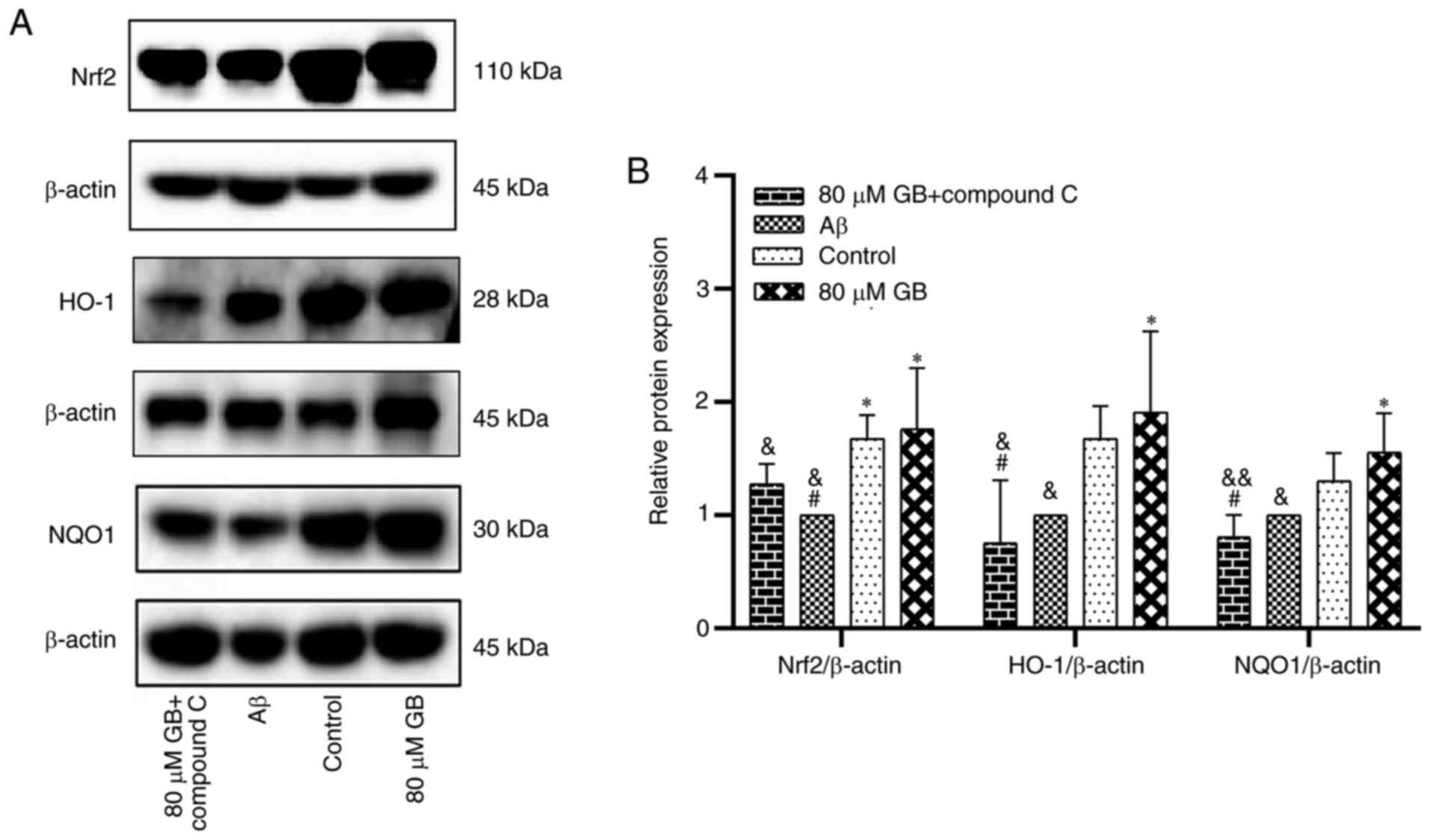
Effect of GB on the expression of
related proteins and genes by Nrf2 pathway
As demonstrated in Fig.
5A and B, the protein expression levels of Nrf2 and NQO1 were
significantly lower in the Aβ group compared with the control group
(P<0.05). However, the expression levels of Nrf2 and HO-1
proteins in the 80 µM GB group and the levels of NQO1 protein in
the 20, 40 and 80 µM GB groups were significantly higher compared
with the Aβ group (P<0.05). As demonstrated in Fig. 6, the expression levels of Nrf2, HO-1
and NQO1 genes were significantly higher in the 40 and 80 µM GB
groups compared with the Aβ group (P<0.05; P<0.01). However,
the mRNA and protein expression levels of Nrf2, HO-1 and NQO1 were
significantly lower in the 80 µM GB + compound C group compared
with the 80 µM GB group (P<0.05; Figs. 6 and 7A
and B). The results indicated that GB could reduce
Aβ1-42-induced OS by activating the Nrf2-HO-1-NQO1
pathway, while compound C prevented this effect. Therefore, the
mechanism of GB inhibiting OS may be closely related to the AMPK
signaling pathway.
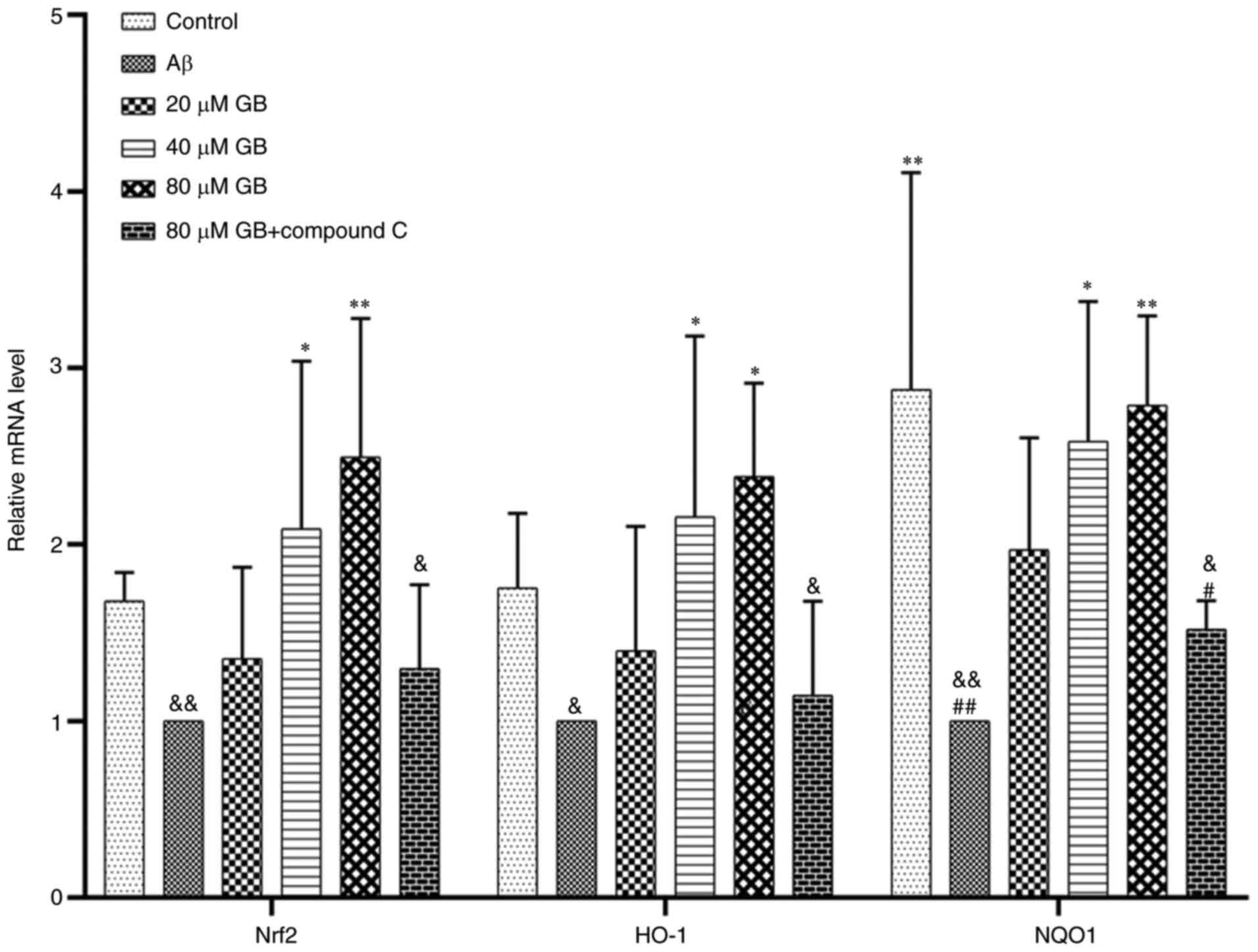 | Figure 6.Effect of GB on gene expressions of
Nrf2-HO-1-NQO1 induced by Aβ1-42 in astrocytes. Reverse
transcription-quantitative PCR data of relative mRNA expression
levels of Nrf2, HO-1 and NQO1 following treatment with 10 µM
Aβ1-42 with different GB concentration (20, 40, 80 µM
GB) or treated with 10 µM Aβ1-42 or treated with 10 µM Aβ1-42 + 80
µM GB + 10 µM compound C for 24 h. #P<0.05 and
##P<0.01 vs. control group, *P<0.05 and
**P<0.01 vs. Aβ group, &P<0.05 and
&&P<0.01 vs. 80 µM GB group. GB, ginkgolide
B; Nrf2, nuclear factor erythroid 2-related factor 2; HO-1, heme
oxygenase 1; NQO1, NAD(P)H dehydrogenase (quinone 1); Aβ,
β-amyloid. |
Effect of GB on the expression of
related genes and proteins by AMPK pathway
As demonstrated in Fig.
8A and B, Aβ1-42 clearly decreased the protein
expression levels of p-AMPK and PPARα and increased the expression
level of BACE1 protein compared with the control group (P<0.05;
P<0.01). In addition, the expression levels of p-AMPK, PGC1α and
PPARα proteins in the 40 and 80 µM GB groups and the expression
level of PPARα proteins in the 20 µM GB group significantly
increased (P<0.05; P<0.01), while the expression level of
BACE1 protein in the 40 and 80 µM GB groups significantly decreased
(P<0.05; P<0.01). As demonstrated in Fig. 9, the expression level of the AMPK
gene in astrocytes significantly decreased and the expression level
of the BACE1 gene significantly increased after treatment with
Aβ1-42 compared with that in the control group
(P<0.05). In contrast, the gene and protein expression levels of
p-AMPK/AMPK, PGC1α and PPARα were significantly lower and the gene
and protein expression level of BACE1 were significantly higher in
the Aβ and 80 µM GB + compound C group compared with the 80 µM GB
group (P<0.05; P<0.01; Figs.
9, 10A and B). The results
showed that GB protected astrocytes from Aβ1-42-induced
apoptosis by regulating energy metabolism, while compound C could
prevent this effect.
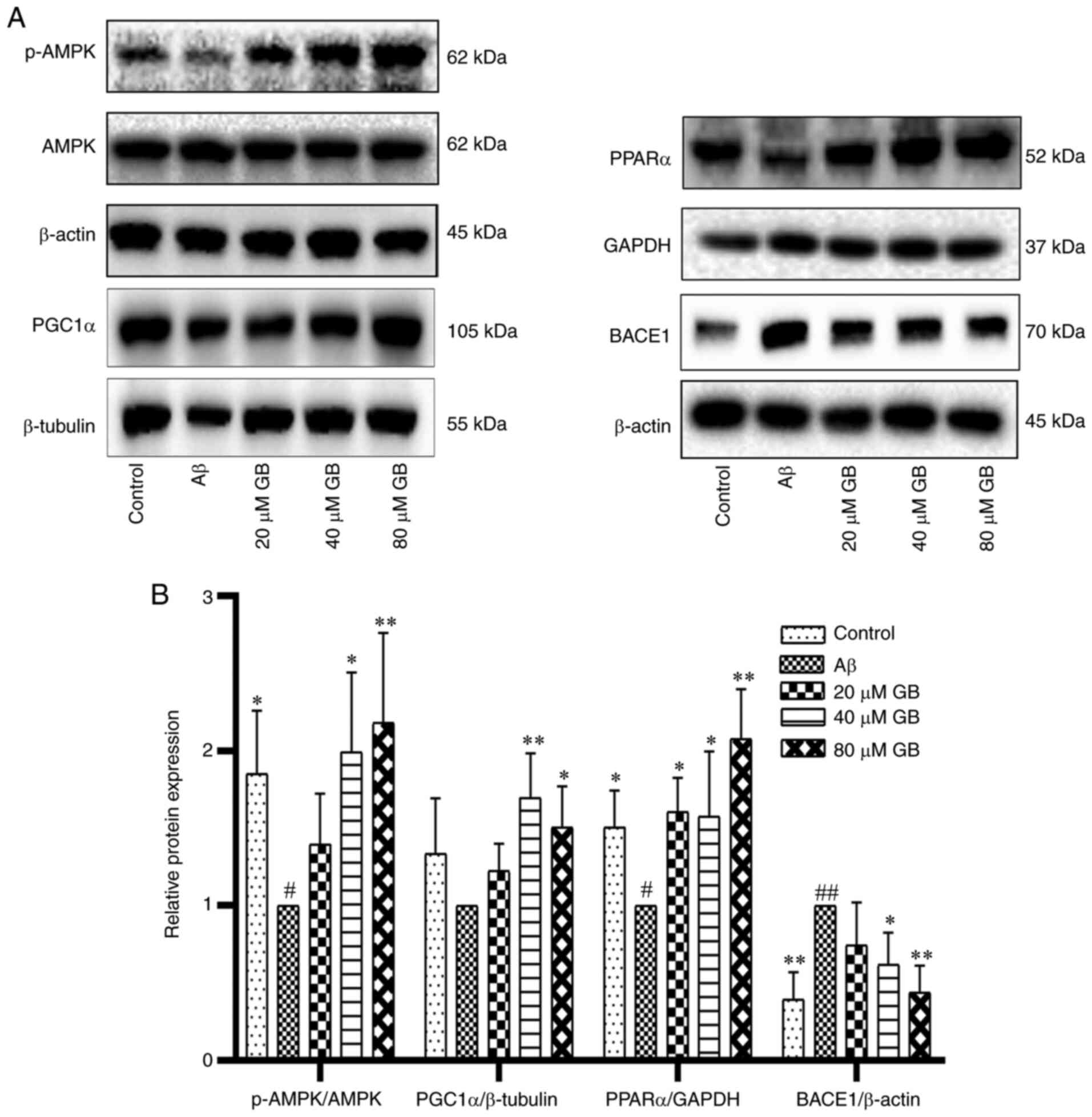 | Figure 8.Effect of GB on the expression of
related proteins by AMPK pathway induced by Aβ1-42 in
astrocytes. (A) Western blotting image and (B) western blotting
data of protein expression levels of p-AMPK, PGC1α, PPARα and BACE1
following treatment with 10 µM Aβ1-42 with different GB
concentration (20, 40 and 80 µM GB) or treated with 10 µM
Aβ1-42 for 24 h. #P<0.05 and
##P<0.01 vs. control group, *P<0.05 and
**P<0.01 vs. Aβ group. GB, ginkgolide B; AMPK, 5′ adenosine
monophosphate-activated protein kinase; Aβ, β-amyloid; p-,
phosphorylated; PGC1α, proliferator-activated receptor γ
coactivator 1α; PPARα, peroxisome proliferator-activated receptor
α; BACE1, amylase β secretase 1. |
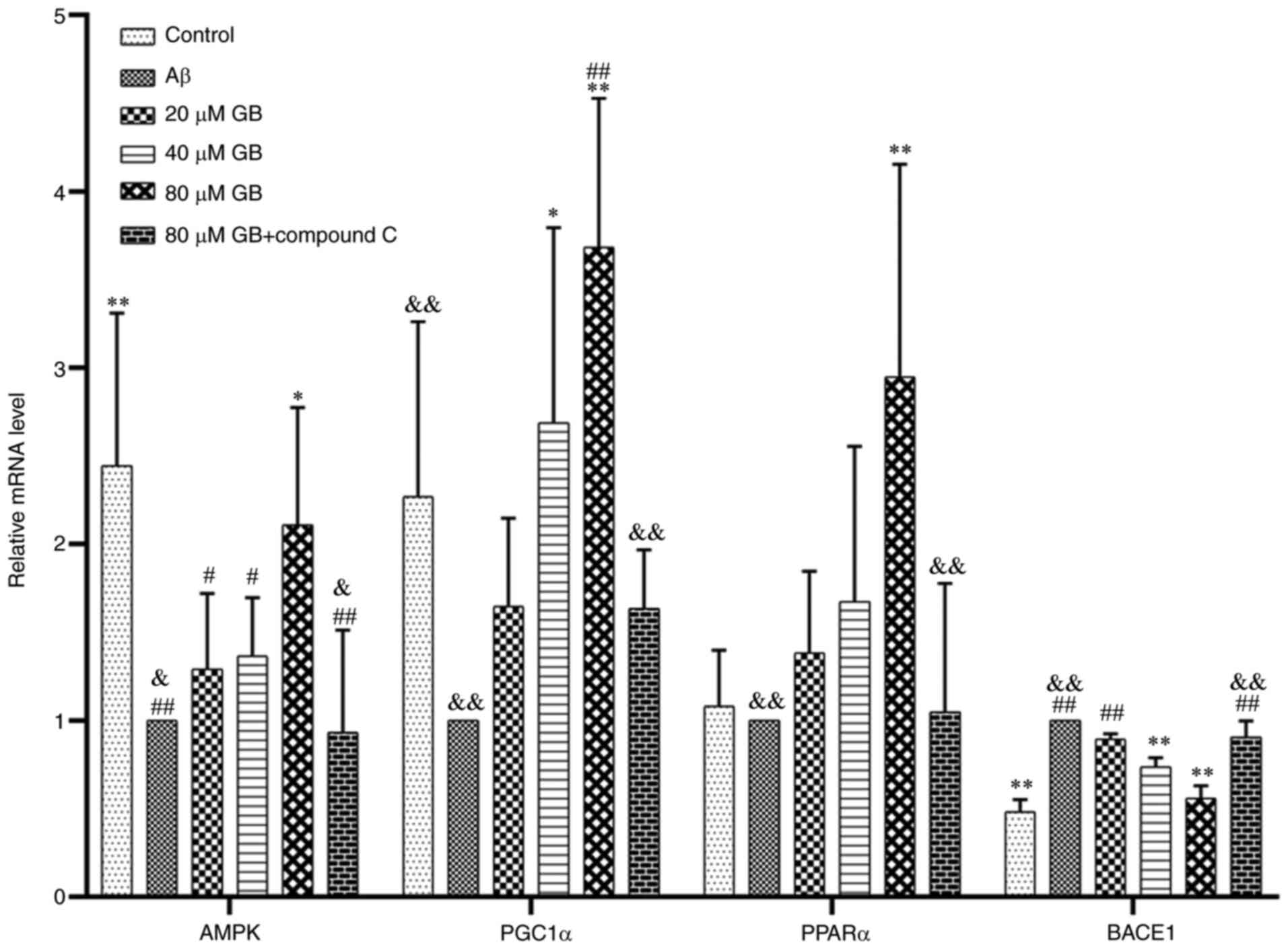 | Figure 9.Effect of GB on gene expressions of
AMPK pathway induced by Aβ1-42 in astrocytes. Reverse
transcription-quantitative PCR data of relative mRNA expression
levels of AMPK, PGC1α, PPARα and BACE1 were treated with 10 µM
Aβ1-42 with different GB concentration (20, 40, 80 µM
GB) or treated with 10 µM Aβ1-42 or treated with 10 µM
Aβ1-42 + 80 µM GB + 10 µM compound C for 24 h.
#P<0.05 and ##P<0.01 vs. control group,
*P<0.05 and **P<0.01 vs. Aβ group, &P<0.05
and &&P<0.01 vs. 80 µM GB group. GB,
ginkgolide B; AMPK, 5′ adenosine monophosphate-activated protein
kinase; Aβ, β-amyloid; PGC1α, proliferator-activated receptor γ
coactivator 1α; PPARα, peroxisome proliferator-activated receptor
α; BACE1, amylase β secretase 1. |
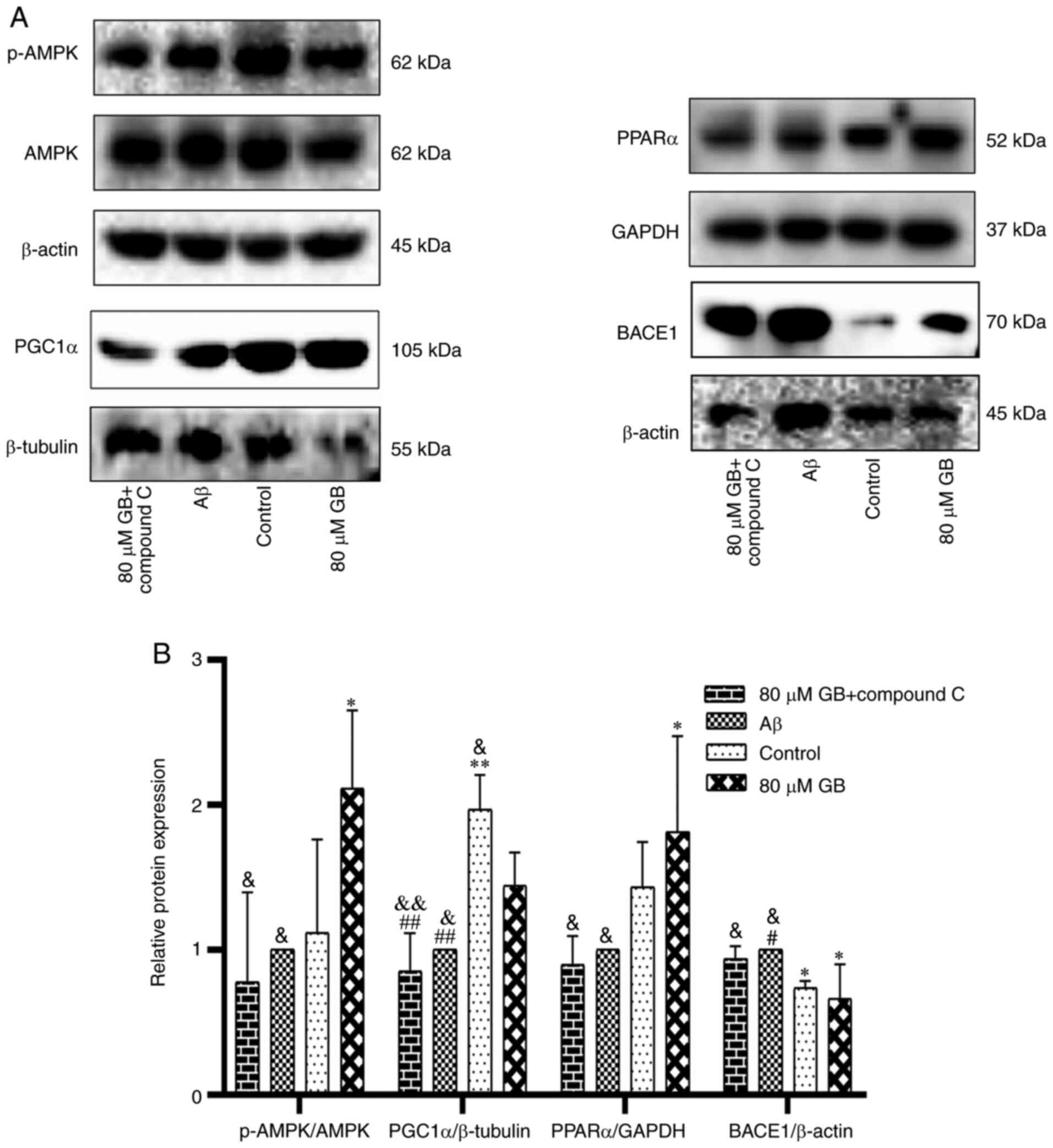 | Figure 10.Compound C inhibits GB-activated
protein expressions of AMPK pathway in astrocytes. (A) Western
blotting image and (B) western blotting data of protein expression
levels of p-AMPK, PGC1α, PPARα and BACE1 following treatment with
10 µM Aβ1-42 with 80 µM GB or treated with 10 µM
Aβ1-42 or Aβ1-42 + 80 µM GB + 10 µM compound
C for 24 h. #P<0.05 and ##P<0.01 vs.
control group, *P<0.05 and **P<0.01 vs. Aβ group,
&P<0.05 and &&P<0.01 vs. 80
µM GB group. GB, ginkgolide B; AMPK, 5′ adenosine
monophosphate-activated protein kinase; p-, phosphorylated; PGC1α,
proliferator-activated receptor γ coactivator 1α; PPARα, peroxisome
proliferator-activated receptor α; BACE1, amylase β secretase 1;
Aβ, β-amyloid. |
Discussion
The Aβ protein cascade hypothesis remains the
dominant theory of AD pathogenesis; it hypothesizes that the main
component of amyloid plaque Aβ is excessively generated to cause
neuronal death, synaptic loss, hyperphosphorylated tau protein and
declining cognitive function (32,33).
Aβ1-42 soluble oligomers are the major form in Aβ and
they aggregate more easily and are more toxic. They surround
necrotic synapses and activated astrocytes (34,35),
the major components of the central nervous system and closely
related to the maintenance and normal operation of brain functions.
The abnormal functions and degeneration of these synapses and
activated astrocytes can induce neurodegenerative diseases.
Physiologically, astrocytes can take up and internalize the Aβ from
cells and degrade them to protect neurons (36,37).
However, pathologically, Aβ causes the overactivation of
astrocytes, leading to ERS, OS, inflammatory response, and finally
apoptosis (19,38). The present study found that
Aβ1-42 (10 µM) caused astrocyte apoptosis and GB (20, 40
and 80 µM) relieved astrocyte apoptosis caused by Aβ1-42
effectively, implying that GB could protect astrocytes from
Aβ1-42-induced apoptosis.
In the progression of AD, the examination of brain
tissues of patients with AD and animal models has demonstrated that
ERS has some association with the toxicity of Aβ (17,20),
thereby disturbing the functions of ER and leading to the
overactivation of ERS in neural cells (19) and deposition of Aβ. In addition, Aβ
and hyperphosphorylated tau protein interact with each other and
move into blood circulation, aggravating the condition of patients
with AD (39,40). Moderate ERS can protect and recover
homeostasis but long-standing ERS induces apoptosis. That is to say
excessive ERS results in apoptosis through CHOP (41). In the present study experiments were
performed to confirm that Aβ1-42 increased the
expression of astrocyte ERS markers, leading to increased
expression of apoptotic protein CHOP. However, GB could protect
astrocytes from apoptosis by halting Aβ1-42-induced ERS.
This finding showed that Aβ1-42 protected astrocytes by
decreasing the expression of ERS-related proteins in AD.
Evidence demonstrates that OS is an incipient factor
that leads to cognitive dysfunction (42). In addition, the levels of OS
products is clearly increased in the brain tissues of patients with
AD (43,44) so that the brains are in a state of
high oxidative stress and oxidative injury occurs earlier than
senile plaques and neurofibrillary tangles (45,46).
In addition, mitochondrial OS, abnormal mitochondrial functions,
their interaction (47,48) and ROS take part in the pathological
progression of AD (49). In
vivo and in vitro studies confirm that Aβ stimulates OS
in the brain tissues via varied pathways (50,51).
The products of OS also induced Aβ deposition to cause abnormal
energy metabolism, mitochondrial dysfunction, even resulting in
apoptosis which induces memory loss and cognitive dysfunction
(52). Astrocytes participate in
the physiological processes of all nervous system diseases and are
critical in protecting neurons (53). The experimental results from the
present study demonstrated that GB improved the activity of
Aβ1-42-induced antioxidant enzymes SOD and GSH-Px and
decreased the content of MDA and ROS, thus inhibiting the increase
in the level of Aβ1-42-induced mitochondrial superoxides
to decrease the generation of ROS. The present study also explored
the involvement of the Nrf2 pathway in the mechanism of GB for
improving Aβ1-42-induced OS. Nrf2 has been verified as a
protective factor in the pathological changes in AD (54). Hence, cognitive dysfunction in
Nrf2-deficient mice was more serious and Aβ deposition and
astrocyte activation clearly increased. In addition, the expression
of the Nrf2 pathway in the brain tissues of animals with AD
distinctly decreased. Activating the Nrf2 signaling pathway
ameliorates OS, actively protects nerves and improves the spatial
learning ability of mice with AD (39,40).
The Nrf2-HO-1-NQO1 pathway is critical in preventing OS and also
serves as an antioxidant transcription factor. When an organism is
stimulated by external factors, Nrf2 dissociates from Kelch-like
ECH-associated protein 1 in the cytoplasm and moves to the nucleus
after activation to recognize antioxidant response elements and
initiate the transcription of downstream antioxidant genes,
including HO-1 and NQO1 (55). The
results of the present study showed that GB increases the gene and
protein expression levels of Nrf2-HO-1-NQO1 in astrocytes, thus
exerting antioxidant activity to prevent OS and apoptosis and
protect astrocytes from Aβ1-42-induced injury.
Energy metabolism disorder is a key pathological
event in the early progression of AD because ATP can inhibit
polymer of Aβ, weaken its neutral toxicity and decrease the
formation of senile plaques, thus antagonizing AD (56,57).
Insufficient levels of ATP accelerate the pathogenesis of AD
(58,59). β-amyloid precursor protein (APP) and
Aβ accumulate on the mitochondrial membranes and cause functional
and structural injury to mitochondria and a metabolic decline in
mitochondrial energy, thus decreasing the generation of ATP
(60) and disrupting the normal
functions of neural cells. The experimental results from the
present study identified that GB improved the levels of
Aβ1-42-induced ATP, thereby improving mitochondrial
damage and hence stabilizing energy metabolism. However, AMPK
inhibitor compound C inhibited the GB-induced improvement, which
was probably associated with AMPK. AMPK is vital in metabolic
homeostasis (61). A previous study
found that the levels of Aβ increase when the levels of hippocampal
p-AMPK decrease in 6-month-old APP/PS1 mice. That is, decreasing
AMPK activity exacerbates the pathology of AD (62). BACE1 mediates the first cleavage of
APP and is the key and speed-limiting enzyme in the process of Aβ
generation (63). AMPK inhibits the
expression and activity of BACE1 and then regulates APP cleavage to
reduce Aβ production (64). AMPK
activates PGC-1α (59), which is
the transcriptional co-activator regulating the expression of
energy metabolism-related genes. PGC-1α improves the synthesis and
metabolism in mitochondria, increases the number of mitochondria
and increases the content of ATP (65). PGC-1 family members possess
multifunctional transcriptional co-activation effects. They combine
with PPARs and act as a ‘molecular switch’ in a number of energy
metabolic signaling pathways, which regulate glycolipid metabolism
and energy metabolism (66,67). A previous study demonstrates that
the expression of PGC1α in the brain tissues of patients with AD
declines, leading to abnormal functioning of mitochondria (68). PGC1α inhibits the generation of Aβ
and a decrease in the expression of PGC1α may be involved in the
pathological progression of AD (69). PPARα does not influence the
generation of Aβ, but its excessive expression decreases the
expression level of Aβ (70,71).
In the present study, Aβ1-42 inhibited the expression of
the AMPK-PGC1α-PPARα pathway and increased the expression of BACE1
in astrocytes, which was in accordance with the aforementioned
findings. That is, Aβ1-42 induced the dysfunction of
mitochondrial energy metabolism and also Aβ generation. However, GB
increased the expression of the AMPK-PGC1α-PPARα signaling pathway,
indicating that GB protected astrocytes via suppressing the
inhibition of energy metabolism of Aβ1-42 to reduce the
generation of Aβ. However, AMPK inhibitor compound C inhibited the
functions of GB, implying that GB regulated energy metabolism and
Aβ generation via likely activating the AMPK-PGC1α-PPARα
pathway.
Energy metabolism causes environmental disorders of
the endoplasmic reticulum to result in ERS (72). However, the persistence of metabolic
injury causes neural damage resulting from prolonged activation of
ERS (73,74). AMPK is the key sensor of the state
of energy in eukaryotic cells and has strong effects on ERS,
insulin resistance and lipid metabolism (75,76).
Activating AMPK can help prevent hypoxic injury, atherosclerosis
and heart injury caused by ERS (77,78).
In the present study, Aβ1-42 increased the expression of
astrocyte ERS markers and resulted in higher expression levels of
the apoptotic protein CHOP. GB inhibited ERS and apoptosis, while
AMPK inhibitor compound C inhibited GB-induced ERS and apoptosis.
These findings indicated that GB impeded the activation of ERS
caused by energy metabolism disorders and suppressed ERS-related
signaling pathways through the AMPK signaling pathway to inhibit
Aβ1-42-induced astrocyte injury and protect astrocytes.
ERS can increase the generation of ROS. Conversely, the generation
of ROS can induce ERS. Hence, a strong correlation exists between
ROS and ERS (79,80). A previous study noted that activated
AMRK is associated with the regulation of the Nrf2 signaling
pathway (81). The present study
demonstrated that the antioxidant activity of GB prevented OS and
apoptosis via increasing the expression of Nrf2-HO-1-NQO1 in
astrocytes to protect astrocytes from Aβ1-42-induced
injury. AMPK inhibitor compound C inhibited the improvement in
GB-induced OS and apoptosis. Hence, the GB probably acted by
promoting the activation of AMPK and Nrf2 to increase the gene and
protein expression levels of HO-1 and NQO1. In other words, GB
ameliorated Aβ1-42-induced OS responses, which was
probably related to the activation of AMPK-Nrf2-HO-1-NQO1 signaling
pathways.
In conclusion, GB protects against
Aβ1-42-induced cell apoptosis by inhibiting ERS, OS and
energy metabolism disorders via activating AMPK signaling pathways.
These findings might provide an innovative insight into the
treatment of Aβ-related AD using GB.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was sponsored by the National
Natural Science Foundation of China (grant no. U1603285). The
funders had no role in study design and collection, analysis and
interpretation of data, or the decision to submit the work for
publication.
Availability of data and materials
The data used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW and LZ confirm the authenticity of all the raw
data. JW designed the experiments, performed the experimental
procedures, analyzed the data, and wrote the manuscript. YD and LZ
performed experimental procedures, and analyzed the data. ZW
designed experiments and reviewed the manuscript. WX and JZ
conceived the project, designed experiments, and reviewed the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures related to the use and care of
animals in the present study were approved by the Ethics Committee
for the Use of Experimental Animals of Jiangsu Kanion
Pharmaceutical Co. Ltd. State Key Laboratory of New Pharmaceutical
Process for Traditional Chinese Medicine (approval no.
2019012).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maclennan KM, Darlington CL and Smith PF:
The CNS effects of Ginkgo biloba extracts and ginkgolide B. Prog
Neurobiol. 67:235–257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang P, Cai X, Zhou K, Lu C and Chen W: A
novel oil-body nanoemulsion formulation of Ginkgolide B:
Pharmacokinetics study and in vivo pharmacodynamics evaluations. J
Pharm Sci. 103:1075–1084. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bate C, Tayebi M and Williams A:
Ginkgolides protect against amyloid-β1-42-mediated synapse damage
in vitro. Mol Neurodegener. 3:12008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Werneke U, Turner T and Priebe S:
Complementary medicines in psychiatry: Review of effectiveness and
safety. Br J Psychiatry. 188:109–121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jean NS, Kathy GW, Pauline M and Stacy H:
Dysgraphia in Alzheimer's disease: A review for clinical and
research purposes. J Speech Lang Hear Res. 49:1313–1330. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nobuyuki K: Diabetes mellitus induces
Alzheimer's disease pathology: Histopathological evidence from
animal models. Int J Mol Sci. 17:5032016. View Article : Google Scholar
|
|
7
|
Ballard C, Gauthier S, Corbett A, Brayne C
and Jones E: Alzheimer's disease. Lancet. 377:1019–1031. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shankar GM, Li S, Mehta TH, Garcia-Munoz
A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere
CA, et al: Amyloid-β protein dimers isolated directly from
Alzheimer's brains impair synaptic plasticity and memory. Nat Med.
14:837–842. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wyss-Coray T, Loike JD, Brionne TC, Lu E
and Husemann J: Adult mouse astrocytes degrade amyloid-beta in
vitro and in situ. Nat Med. 9:453–457. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nielsen HM, Veerhuis R, Bo H and
Janciauskiene S: Binding and uptake of Abeta1-42 by
primary human astrocytes in vitro. Glia. 57:978–988. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Basak JM, Verghese PB, Yoon H, Kim J and
Holtzman DM: Low-density lipoprotein receptor represents an
apolipoprotein E-independent pathway of Aβ uptake and degradation
by astrocytes. J Biol Chem. 287:13959–13971. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thal DR, Schultz C, Dehghani F, Yamaguchi
H, Braak H and Braak E: Amyloid beta-protein (Abeta)-containing
astrocytes are located preferentially near N-terminal-truncated
Abeta deposits in the human entorhinal cortex. Acta Neuropathol.
100:608–617. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang ZZ, Li J, Li SX, Feng W and Wang H:
Effect of ginkgolide B on striatal extracellular amino acids in
middle cerebral artery occluded rats. J Ethnopharmacol.
136:117–122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Poon WW, Carlos AJ, Aguilar BL, Berchtold
NC, Kawano CK, Zograbyan V, Yaopruke T, Shelanski M and Cotman CW:
β-Amyloid (Aβ) oligomers impair brain-derived neurotrophic factor
retrograde trafficking by down-regulating ubiquitin C-terminal
hydrolase, UCH-L1. J Biol Chem. 288:16937–16948. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ihara Y, Morishima-Kawashima M and Nixon
R: The ubiquitin-proteasome system and the autophagic-lysosomal
system in Alzheimer disease. Cold Spring Harb Perspect Med.
2:a0063612012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Iurlaro R and Muñoz-Pinedo C: Cell death
induced by endoplasmic reticulum stress. FEBS J. 283:2640–2652.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li HH, Lu FJ, Hung HC, Liu GY, Lai TJ and
Lin CL: Humic acid increases amyloid β-induced cytotoxicity by
induction of ER stress in human SK-N-MC neuronal cells. Int J Mol
Sci. 16:10426–10442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li JQ, Tai YJ, Jiang T and Tan L:
Endoplasmic reticulum dysfunction in Alzheimer's disease. Mol
Neurobiol. 51:383–395. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Umeda T, Tomiyama T, Sakama N, Tanaka S,
Lambert MP, Klein WL and Mori H: Intraneuronal amyloid-beta
oligomers cause cell death via endoplasmic reticulum stress,
endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo.
J Neurosci Res. 89:1031–1042. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoozemans JJ, van Haastert ES, Nijholt DA,
Rozemuller AJ, Eikelenboom P and Scheper W: The unfolded protein
response is activated in pretangle neurons in Alzheimer's disease
hippocampus. Am J Pathol. 110:165–172. 2005.PubMed/NCBI
|
|
21
|
Araki E, Oyadomari S and Mori M:
Endoplasmic reticulum stress and diabetes mellitus. Intern Med.
42:7–14. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Verri M, Pastoris O, Dossena M, Aquilani R
and Bongiorno AI: Mitochondrial alterations, oxidative stress and
neuroinflammation in Alzheimer's disease. Int J Immunopathol
Pharmacol. 25:345–353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Su X, Wu W, Huang Z, Hu J, Lei P, Yu C,
Zhao YF and Li Y: Hydrogen peroxide can be generated by tau in the
presence of Cu(II). Biochem Biophys Res Commun. 358:661–665. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cha M, Han S, Son S, Hong H, Cha Y, Byun J
and Inhee M: Mitochondria-specific accumulation of amyloid β
induces mitochondrial dysfunction leading to apoptotic cell death.
PLoS One. 7:e349292012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Borghi R, Patriarca S, Traverso N, Piccini
A, Storace D, Garuti A, Cirmena G, Odetti P and Tabaton M: The
increased activity of BACE1 correlates with oxidative stress in
Alzheimer's disease. Neurobiol Aging. 28:1009–1014. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ribeiro MF, Genebra T, Rego AC, Rodrigues
CMP and Solá S: Amyloid β peptide compromises neural stem cell fate
by irreversibly disturbing mitochondrial oxidative state and
blocking mitochondrial biogenesis and dynamics. Mol Neurobiol.
56:3922–3936. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jahanshahi M, Sadeghi Y, Hosseini A,
Naghdi N and Marjani A: The effect of spatial learning on the
number of astrocytes in the CA3 subfield of the rat hippocampus.
Singapore Med J. 49:388–391. 2008.PubMed/NCBI
|
|
28
|
Kawano H, Oyabu K, Yamamoto H, Eto K,
Adaniya Y, Kubota K, Watanabe T, Hiranoiwata A, Nabekura J,
Katsurabayashi S and Iwasaki K: Astrocytes with previous chronic
exposure to amyloid β-peptide fragment 1–40 suppress excitatory
synaptic transmission. J Neurochem. 143:624–634. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang N, Xiong WW, Yuan X and Zhang W:
Culture method of rat fetal hippocampal neurons and astrocytes.
Acta Neuropharmacol. 7:24–28. 2017.
|
|
30
|
Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro
M, Iradi A, Obrador E, Ortega A, Mauricio MD, Vila JM and Valles
SL: Astrocytes protect neurons from Aβ1-42 peptide-induced
neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and
SIRT-1. International Journal of Medical ences. 12:48–56.
2015.PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hardy J and Higgins G: Alzheimer's
disease: The amyloid cascade hypothesis. Science. 256:184–185.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jan A, Gokce O, Luthi-Carter R and Lashuel
HA: The ratio of monomeric to aggregated forms of Abeta40 and
Abeta42 is an important determinant of amyloid-beta aggregation,
fibrillogenesis, and toxicity. J Biol Chem. 283:28176–28189. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mulder SD, Veerhuis R, Blankenstein MA and
Nielsen HM: The effect of amyloid associated proteins on the
expression of genes involved in amyloid-β clearance by adult human
astrocytes. Exp Neurol. 233:373–379. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Paris D, Beaulieu-Abdelahad D, Bachmeier
C, Reed J, Ait-Ghezala G, Bishop A, Chao J, Mathura V, Crawford F
and Mullan M: Anatabine lowers Alzheimer's Aβ production in vitro
and in vivo. Eur J Pharmacol. 670:384–391. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mohamed A and Chaves EPd: Aβ
internalization by neurons and glia. Int J Alzheimers Dis.
2011:1279842011.PubMed/NCBI
|
|
37
|
Fan J, Donkin J and Wellington C: Greasing
the wheels of Abeta clearance in Alzheimer's disease: The role of
lipids and apolipoprotein E. Biofactors. 35:239–248. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Phillips EC, Croft CL, Kurbatskaya K,
O'Neill MJ, Hutton ML, Hanger DP, Garwood CJ and Noble W:
Astrocytes and neuroinflammation in Alzheimer's disease. Biochem
Soc Trans. 42:1321–1325. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Unterberger U, Höftberger R, Gelpi E,
Flicker H, Budka H and Voigtländer T: Endoplasmic reticulum stress
features are prominent in Alzheimer disease but not in prion
diseases in vivo. J Neuropathol Exp Neurol. 65:348–357. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Doyle KM, Kennedy D, Gorman AM, Gupta S,
Healy S and Samali A: Unfolded proteins and endoplasmic reticulum
stress in neurodegenerative disorders. J Cell Mol Med.
15:2025–2039. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lindholm D, Wootz H and Korhonen L: ER
stress and neurodegenerative diseases. Cell Death Differ.
3:385–392. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Torres L, Quaglio NB, de Souza GT, Garcia
RT, Dati LM, Moreira WL, Loureiro AP, de Souza-Talarico JN, Smid J,
Porto CS, et al: Peripheral oxidative stress biomarkers in mild
cognitive impairment and Alzheimer's disease. J Alzheimer's Dis.
26:59–68. 2011. View Article : Google Scholar
|
|
43
|
Smith CD, Carney JM, Tatsumo T, Stadtman
ER, Floyd RA and Markesbery WR: Protein oxidation in aging brain.
Ann N Y Acad Sci. 663:110–119. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Choi D, Lee Y, Hong JT and Lee H:
Antioxidant properties of natural polyphenols and their therapeutic
potentials for Alzheimer's disease. Brain Res Bull. 87:144–153.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Silva DF, Selfridge JE, Lu J, Lezi E and
Swerdlow RH: Mitochondrial abnormalities in Alzheimer's Disease.
Possible targets for therapeutic intervention. Adv Pharmacol.
64:83–126. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Darvesh AS, Carroll RT, Bishayee A,
Geldenhuys WJ and Van der Schyf CJ: Oxidative stress and
Alzheimer's disease: Dietary polyphenols as potential therapeutic
agents. Expert Rev Neurother. 10:729–745. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Aliev G, Palacios HH, Walrafen B, Lipsitt
AE, Obrenovich ME and Morales L: Brain mitochondria as a primary
target in the development of treatment strategies for Alzheimer
disease. Int J Biochem Cell Biol. 41:1989–2004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Milton NG: Role of hydrogen peroxide in
the aetiology of Alzheimer's disease: Implications for treatment.
Drugs Aging. 21:81–100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kontush A: Amyloid-beta: An antioxidant
that becomes a pro-oxidant and critically contributes to
Alzheimer's disease. Free Radic Biol Med. 31:1120–1131. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Perry G, Cash AD and Smith MA: Alzheimer
disease and oxidative stress. J Biomed Biotechnol. 2:120–123. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Honda K, Smith MA, Zhu X, Baus D, Merrick
WC, Tartakoff AM, Hattier T, Harris PL, Siedlak SL, Fujioka H, et
al: Ribosomal RNA in Alzheimer disease is oxidized by bound
redox-active iron. J Biol Chem. 280:20978–20986. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Casley CS, Canevari L, Land JM, Clark JB
and Sharpe MA: Beta-amyloid inhibits integrated mitochondrial
respiration and key enzyme activities. J Neurochem. 80:91–100.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gegg ME, Clark JB and Heales SJ:
Co-culture of neurones with glutathione deficient astrocytes leads
to increased neuronal susceptibility to nitric oxide and increased
glutamate-cysteine ligase activity. Brain Res. 1036:1–6. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tian Y, Wang W, Xu L, Li H, Wei Y, Wu Q
and Jia J: Activation of Nrf2/ARE pathway alleviates the cognitive
deficits in PS1V97L-Tg mouse model of Alzheimer's disease through
modulation of oxidative stress. J Neurosci Res. 97:492–505. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mccubrey JA, Lahair MM and Franklin RA:
Reactive oxygen species-induced activation of the MAP kinase
signaling pathways. Antioxid Redox Signal. 8:1775–1789. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Eckert A, Hauptmann S, Scherping I, Rhein
V, Mullerspahn F, Gotz J and Muller WE: Soluble beta-amyloid leads
to mitochondrial defects in amyloid precursor protein and tau
transgenic mice. Neurodegener Dis. 5:157–159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schmidt C, Lepsverdize E, Chi SL, Das AM
and Schachner M: Amyloid precursor protein and amyloid beta-peptide
bind to ATP synthase and regulate its activity at the surface of
neural cells. Mol Psychiatry. 13:953–969. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Coskuner O and Murray IV: Adenosine
triphosphate (ATP) reduces amyloid-β protein misfolding in vitro. J
Alzheimers Dis Jad. 41:561–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Müller WEG, Wang S, Ackermann M, Neufurth
M, Steffen R, Mecja E, Muñoz-Espí R, Feng Q, Schröder HC and Wang
X: Rebalancing β-amyloid-induced decrease of ATP level by amorphous
nano/micro polyphosphate: Suppression of the neurotoxic effect of
amyloid β-protein fragment 25–35. Int J Mol Sci. 18:21542017.
View Article : Google Scholar
|
|
60
|
Du H and Yan SS: Mitochondrial
permeability transition pore in Alzheimer's disease: Cyclophilin D
and amyloid beta. Biochim Biophys Acta. 1802:198–204. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ke R, Xu Q, Li C, Luo L and Huang D:
Mechanisms of AMPK in the maintenance of ATP balance during energy
metabolism. Cell Biol Int. 42:384–392. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ou Z, Kong X, Sun X, He X, Zhang L, Gong
Z, Huang J, Xu B, Long D, Li J, et al: Metformin treatment prevents
amyloid plaque deposition and memory impairment in APP/PS1 mice.
Brain Behav Immun. 69:351–363. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sun X, He G and Song W: BACE2, as a novel
APP theta-secretase, is not responsible for the pathogenesis of
Alzheimer's disease in Down syndrome. FASEB J. 20:1369–1376. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Coimbra JRM, Marques DFF, Baptista SJ,
Pereira CMF, Moreira PI, Dinis TCP, Santos AE and Salvador JAR:
Highlights in BACE1 inhibitors for Alzheimer's disease treatment.
Front Chem. 6:1782018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jeon SM: Regulation and function of AMPK
in physiology and diseases. Exp Mol Med. 48:e2452016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Finck BN and Kelly DP: PGC-1 coactivators:
Inducible regulators of energy metabolism in health and disease. J
Clin Invest. 116:615–622. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tiraby C and Langin D: Conversion from
white to brown adipocytes: A strategy for the control of fat mass?
Trends Endocrinol Metab. 14:439–441. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Austin S and St-Pierre J: PGC1α and
mitochondrial metabolism-emerging concepts and relevance in ageing
and neurodegenerative disorders. J Cell Sci. 125:4963–4971. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ettcheto M, Petrov D, Pedros I, Alva N,
Carbonell T, Beaszarate C, Pallas M, Auladell C, Folch J and Camins
A: Evaluation of neuropathological effects of a high-fat diet in a
presymptomatic Alzheimer's disease stage in APP/PS1 mice. J
Alzheimer's Dis. 54:233–251. 2016. View Article : Google Scholar
|
|
70
|
Li Z, Li H, Zhao CH, Lv C, Zhong CJ, Xin
WF and Zhang WS: Protective effect of Notoginsenoside R1 on an
APP/PS1 mouse model of Alzheimer's disease by up-regulating insulin
degrading enzyme and inhibiting Aβ accumulation. CNS Neurol Disord
Drug Targets. 14:360–369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Camacho IE, Serneels L, Spittaels K,
Merchiers P, Dominguez D and De Strooper B: Peroxisome
proliferator-activated receptor gamma induces a clearance mechanism
for the amyloid-beta peptide. J Neurosci. 24:10908–10917. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mamelak M: Energy and the Alzheimer brain.
Neurosci Biobehav Rev. 75:297–313. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Biswas J, Gupta S, Verma DK and Singh S:
Streptozotocin alters glucose transport, connexin expression and
endoplasmic reticulum functions in neurons and astrocytes.
Neuroscience. 356:151–166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Park-York MJ, Kim Y and York DA: Cage food
location alters energy balance and endoplasmic reticulum stress in
the brain of mice. Physiol Behav. 106:158–163. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Grahame Hardie D: AMP-activated protein
kinase: A key regulator of energy balance with many roles in human
disease. J Intern Med. 276:543–559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Boß M, Newbatt Y, Gupta S, Collins I,
Brüne B and Namgaladze D: AMPK-independent inhibition of human
macrophage ER stress response by AICAR. Sci Rep. 6:321112016.
View Article : Google Scholar
|
|
77
|
Yeh CH, Chen TP, Wang YC, Lin YM and Fang
SW: AMP-activated protein kinase activation during
cardioplegia-induced hypoxia/reoxygenation injury attenuates
cardiomyocytic apoptosis via reduction of endoplasmic reticulum
stress. Mediators Inflamm. 2010:1306362010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gao F, Chen J and Zhu H: A potential
strategy for treating atherosclerosis: Improving endothelial
function via AMP-activated protein kinase. Sci China Life Sci.
61:1024–1029. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Cao SS and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress in cell fate decision and
human disease. Antioxid Redox Signal. 21:396–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zheng W, Wang B, Si M, Zou H, Song R, Gu
J, Yuan Y, Liu X, Zhu G, Bai J, et al: Zearalenone altered the
cytoskeletal structure via ER stress-autophagy-oxidative stress
pathway in mouse TM4 Sertoli cells. Sci Rep. 8:33202018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wu P, Yan Y, Ma L, Hou B, He Y, Zhang L,
Niu Z, Song J, Pang X and Yang X: Effects of the Nrf2 modulator
salvianolic acid A alone or combined with metformin on
diabetes-associated macrovascular and renal injury. J Biol Chem.
291:22288–22301. 2016. View Article : Google Scholar : PubMed/NCBI
|