Sepsis is a common sequela of severe trauma, burns,
infection and major surgery, often secondary to septic shock and
multiple organ dysfunction syndrome (1). Despite research on the pathogenesis
and treatment of sepsis using a variety of antibiotics and
supportive therapies, its incidence and associated mortality is
still high, as there are an estimated 48.9 million cases of sepsis
and 11 million sepsis-related deaths worldwide (2). An important reason for the high
mortality rate of sepsis is the uncontrolled inflammatory response
leading to damage to multiple organs (3,4). The
pathogenesis of sepsis is complex and involves interaction between
infectious microorganisms and the host. In sepsis, pathogens, such
as bacteria, fungi and viruses, invade the body and release
lipopolysaccharide (LPS), exotoxin and other moieties with
pathogen-associated molecular patterns (PAMPs). The organism
rapidly senses these PAMPs and a series of events are triggered,
culminating in the release of material carrying damage-associated
molecular patterns (DAMPs), such as damaged DNA. These PAMPs and
DAMPs influence fluid control and immune, inflammatory and other
systems, causing complex pathophysiological changes and multi-organ
functional damage (5). In the
course of infection, the activation of various receptors is
essential for the recognition of a variety of microorganisms to
regulate innate and adaptive immunity. In sepsis,
pattern-recognition receptors, such as Toll-like receptors (TLRs)
on the cell membrane and Nod-like receptors, in the cytoplasm
recognize PAMPs and DAMPs, leading to the activation of
intracellular signaling pathways that result in the transcription
and release of pro-inflammatory factors, such as TNFα, IL-18 and
IL-1β (6).
In sepsis, circulating PAMPs, DAMPs and cytokines
activate endothelial cells, cardiac fibroblasts and cardiomyocytes,
and increase the production of inflammatory mediators, thereby
further activating the expression of inducible nitric oxide
synthase and causing myocardial inhibition (7). In the lungs, interstitial and
endothelial barriers are broken down, which may lead to an
imbalance in the alveolar ventilation/blood flow ratio and
decreased lung compliance, resulting in acute respiratory distress
syndrome (8). In the
gastrointestinal tract, the increased permeability of the inner
layer of the mucosa leads to leakage of bacteria from the
intestinal tract, which causes gastrointestinal dysfunction
(9). In the kidneys, decreased
renal perfusion, acute tubular necrosis and microvascular damage
contribute to varying degrees of acute injury (10). These endothelial cell changes also
disrupt the blood-brain barrier, leading to the entry of toxins,
inflammatory cells and cytokines, which in turn cause brain edema,
neurotransmitter destruction and oxidative stress, culminating in
the development of septic encephalopathy. Proinflammatory cytokines
can cause leukocyte activation and proliferation, upregulation of
endothelial adhesion molecules and chemokine expression, production
of tissue factors and amplification of immune responses, which also
leads to host cell and tissue damage. Hence, the ability to treat
sepsis may be significantly improved by inhibiting the inflammatory
response in order to protect against the multi-organ functional
damage caused by this condition (11).
Inflammasomes are multi-protein complexes with a
molecular weight of ~700 kDa that are assembled following
intracytoplasmic pattern recognition receptor ligation (12). They consist primarily of a sensor,
adaptor and pro-caspase-1. Sensors of PAMPs and DAMPs in the
cytoplasm include nucleotide-binding oligomerization domain and
leucine-rich repeat (LRR)-containing receptors (NLRs), absent in
melanoma-2 and pyrin. NLRs include Nod-like receptor family pyrin
domain-containing (NLRP)1, NLRP3, NLRP6, NLRP7, NLRP12 and NLRC4,
which assemble into their own inflammasomes (13). All members of the NLR protein family
contain a central nucleotide-binding domain, and most also have a
variable N-terminal domain and a C-terminal LRR domain. Based on
the presence of an N-terminal pyrin domain (PYD) or caspase
activation and recruitment domain (CARD), the family is further
divided into NLRP or NLRC receptors (14). The NLRP3 inflammasome, one of the
most well-studied inflammasomes, is composed of NLRP3, adaptor
apoptosis-related speck-like protein (ASC) and pro-caspase-1
molecules. When stimulated by infection or other factors, the NLRP3
inflammasome interacts with ASC via the CARD/CARD and PYD/PYD
domains to increase expression levels of pro-caspase-1, which is
self-catalyzed to cleave into two subunits, p20 and p10 (15). Tetramers of p20 and p10 that
constitute active caspase-1, which further cleaves pro-IL-1β and
pro-IL-18, thus promoting the activation and release of the
caspase-1-dependent inflammatory mediators IL-18 and IL-1β
(13). Activated caspase-1 also
activates pore-forming gasdermin D to induce a form of cell death
called pyroptosis (12). Activation
of inflammasomes serves an important role in pathogen defense,
stimulating both innate and adaptive immune responses. However,
dysregulation of inflammasomes activity has been implicated in
numerous human diseases, such as gout, diabetes and
atherosclerosis, amongst others (16–18).
Thus, inflammasome activation is a tightly regulated process which
requires multiple molecular and cellular signals. The present
review summarizes the primary regulatory mechanisms of NLRP3
inflammasome activation and discusses the role of this inflammasome
in the development of sepsis.
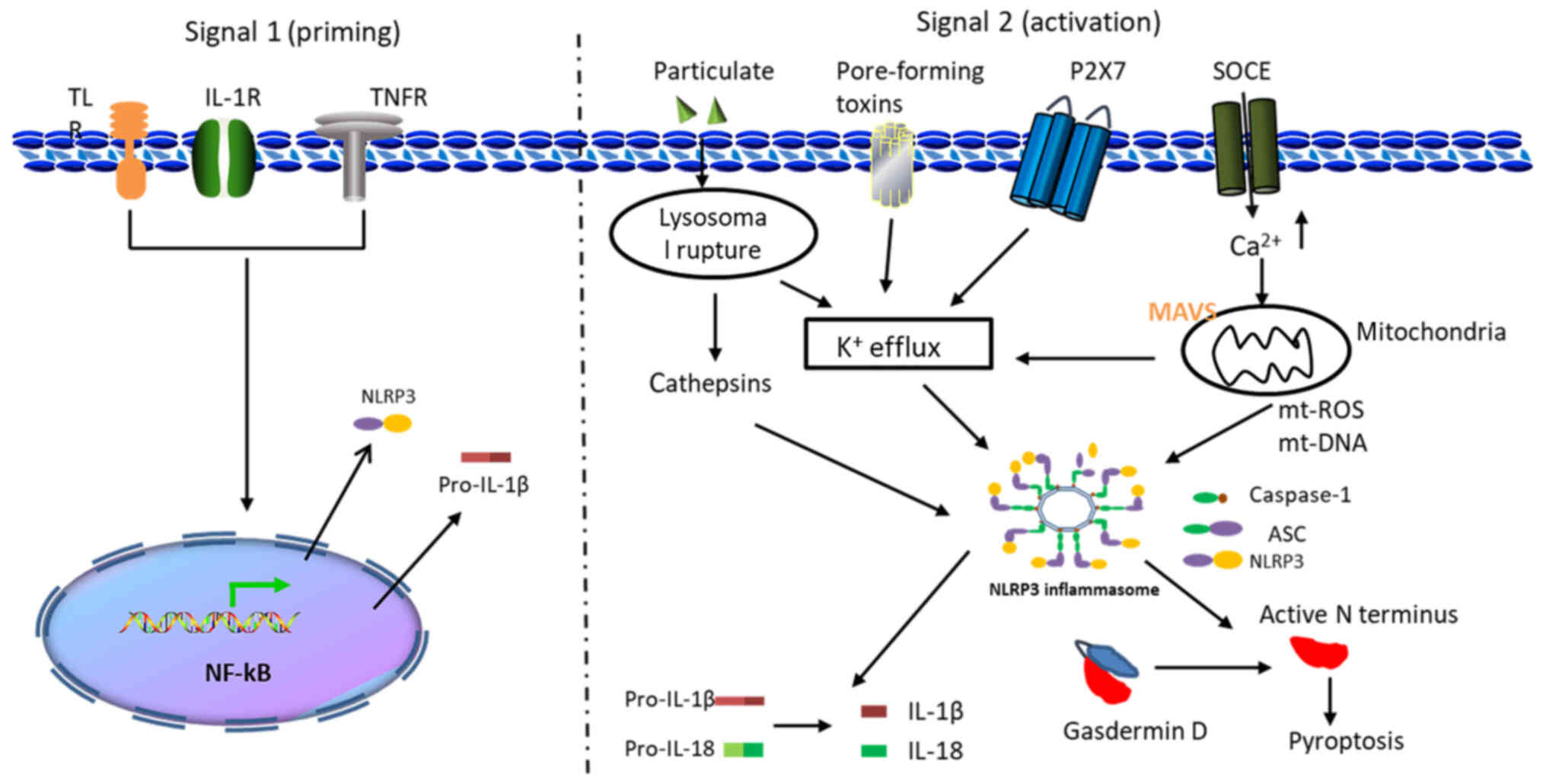
The activation of the NLRP3 inflammasome takes place
in two stages, first priming and then activating. The initial
signal (Signal I) is provided by pro-inflammatory cytokines or
microbial components, leading to the activation of the
transcription factor NF-κB and subsequent upregulation of pro-IL-1β
and NLRP3 (13). The expression of
NLRP3 and pro-IL-1β is regulated at the transcriptional level
during the priming phase. This transcriptional upregulation can be
induced via the recognition of various PAMPs or DAMPs that engage
pattern recognition receptors (PRRs), such as TLRs or IL-1
receptor, or via cytokines, such as TNF and IL-1β, that lead to
NF-κB activation and gene transcription (19). Signal II can be provided by a
variety of different stimuli, including ATP, nigericin, particulate
matter, pore-forming toxins and RNA viruses (13). Several cellular events, including
lysosomal destruction, production of reactive oxygen species (ROS),
mitochondrial dysfunction and changes in ion flux, have all been
shown to activate the NLRP3 inflammasome (20). A two-signal model for NLRP3
inflammasome activation is presented in Fig. 1.
The role of ROS production and mitochondrial
dysfunction in NLRP3 inflammasome activation is widely recognized.
ROS may serve as a common signal for NLRP3 inflammasome activation
because most NLRP3 inflammasome stimuli mediate ROS production and
NADPH oxidase is considered to be one of the main sources of ROS
(27). Recent studies have found
that orexin contributes to vascular disease by inhibiting NLRP3
inflammasome activation via suppressing the expression of NADPH
oxidase 4 in human aortic endothelial cells at high concentrations
of glucose (28). Sterol regulatory
element-binding protein 2 transactivates NADPH oxidase, thereby
activating the NLRP3 inflammasome in vascular endothelial cells and
increasing susceptibility to atherosclerosis (29). However, studies have shown that the
activation of the NLRP3 inflammasome and the release of IL-1β in
human macrophages requires calcium but does not involve NADPH
oxidase (30).
The mitochondrial respiratory chain is another
pathway for ROS production. A growing body of evidence suggests
that mitochondrial dysfunction is involved in the assembly and
conformational alteration of the NLRP3 inflammasome via ROS
production, which results in its activation (13). Studies have shown that MitoQ can
inhibit activation of the NLRP3 inflammasome and release of IL-1β
by blocking the production of mitochondrial ROS (mtROS), thereby
decreasing tubular injury in diabetic nephropathy (31). Shimada et al (32) found that oxidized mitochondrial DNA
(mtDNA) is necessary for the activation of the NLRP3 inflammasome.
NLRP3 secondary signal activators can induce mitochondrial
dysfunction, leading to the release of mtDNA into the cytoplasm,
where it can bind to and activate NLRP3 inflammasomes. Zhong et
al (33) found that
TLR-dependent initiation signals induce the synthesis of new mtDNA,
which is required for NLRP3 inflammasome activation.
Epigallocatechin-3-gallate has been found to inhibit the synthesis
of mtDNA and production of ROS in mouse macrophages, thereby
inhibiting activation of the NLRP3 inflammasome (34). These findings suggest that
mitochondrial dysfunction, mtROS and mtDNA production serve a
critical role in NLRP3 inflammasome activation.
Mitochondria can co-localize with NLRP3
inflammasomes, in addition to producing mtROS and mtDNA.
Mitochondrial antiviral signaling proteins (MAVS) have been shown
to promote recruitment of NLRP3 inflammasomes to mitochondria and
may enhance their oligomerization and activation by bringing them
into close proximity to mtROS (35). Subramanian et al (36) found that the N-terminal of NLRP3
regulates association and mitochondrial recruitment of MAVS, and
MAVS promote the production of NLRP3-mediated mature IL-1β by
facilitating NLRP3 recruitment to mitochondria. Li et al
(37) found that NLRX1 decreases
the inflammatory response and apoptosis of ischemic myocardial
cells by inhibiting the activation of MAVS-dependent NLRP3
inflammasomes. Iyer et al (38) found that mitochondria-specific
lipidocardiol can directly bind to the LRR domain of NLRP3, which
not only serves as a docking station for the co-localization of
NLRP3 and its activated ligands, but also provides activation
signals to the NLRP3 inflammasome itself. Studies have shown that
both NLRP3 and caspase-1 interact with mitochondrial lipids
(34,35). After receiving appropriate
activation signals, cardiolipin eversion into the outer membrane of
mitochondria results in the formation of a calcium-dependent
association between ASC and mitochondria, thus leading to the
assembly and activation of NLRP3 inflammasomes (39). Following infection by RNA viruses,
NLRP3 inflammasome activation is associated with the presence of
mitochondrial protein mitofusin 2 (40). Taken together, the aforementioned
findings suggest that mitochondria may activate inflammasomes by
participating in the assembly of the NLRP3 complex. In addition, it
has been found that phosphatidylinositol 4,5-bisphosphate on the
scattered trans-Golgi network can be used as a scaffold for NLRP3
inflammasome activation (41).
Zhang et al (42) also found
that Golgi can activate NLRP3 inflammasomes via protein kinase D on
the mitochondrial-associated endoplasmic reticulum membrane.
Particulates, such as cholesterol crystals, silica,
and monosodium urate, phagocytosed by macrophages can destroy
lysosomes and cause the lysosomal contents to be released into the
cytoplasm to activate NLRP3 inflammasomes (13). Previous studies have shown that
particle activators induce lysosomal rupture and release of
material into the cytoplasm, which alters plasma membrane
permeability and releases ATP and purine, which in turn activate
the NLRP3 inflammasome (43).
Jessop et al (44) found
that inhaling particles that cause lysosomal membrane permeability
can activate the NLRP3 inflammasome, and that lysosomal
acidification is a prerequisite for particle-induced lysosomal
membrane permeability. Similarly, NLRP3-deficient macrophages show
a considerable decrease in ATP depletion and mitochondrial
function, but maintain lysosomal acidity, suggesting that LMP is
NLRP3-dependent (45).
Previous studies have reported that phagocytosed
particles can destroy lysosomes and release cathepsin B into the
cytoplasm to trigger the activation of the NLRP3 inflammasome, and
that the cathepsin B inhibitor CA-074 Me blocks this response
(46). However, cathepsin
B-knockdown in macrophages does not prevent inflammasome-mediated
cell death, which may be due to off-target effects; alternatively,
CA-074 Me may also act on other members of the cathepsin family
(47). It has been reported that
cathepsins B had redundant effects on the activation of the NLRP3
inflammasome by particulates, while cathepsin X has a non-redundant
effect on the activation of NLRP3 inflammasomes by non-particulates
(48). Cathepsin B is required for
ASC spot formation, IL-1β production and caspase-1 activation in
response to different types of NLRP3 activators (49). Tang et al (50) found that hydroxychloroquine inhibits
cathepsin B, thereby redistributing lysosomal bulk, inhibiting the
activation of the NLRP3 inflammasome and decreasing renal injury.
Oxidative stress activates NLRP3 inflammasomes in microglia by
upregulating the activity of cathepsin B, thereby promoting the
development of neurodegenerative disease (51). Taken together, the aforementioned
studies documented that cathepsin B can activate NLRP3
inflammasomes, but the mechanism still needs further study.
The ubiquitin system is a complex post-translational
modification system, which primarily mediates the addition of
ubiquitin to its substrates via sequential activation of E1-E2-E3
enzymes (52). E3 ubiquitin ligase
may be involved in NLRP3 inflammasome activation by targeting
either NLRP3 or other components of the inflammasome. E3 ubiquitin
ligase regulates NLRP3 expression via autophagy or proteasomal
degradation (33). It has been
reported that the E3 ubiquitin ligase tripartite motif-containing
protein (TRIM)31 binds directly to NLRP3, promotes
polyubiquitination of K48 junctions and proteasomal degradation of
NLRP3, and thereby inhibits NLRP3 inflammasome activation (53). Silencing by small interfering RNA of
the deubiquitinase BRCC36 in macrophages treated with the
proteasome inhibitor MG132 prevented oxidized low-density
lipoprotein-induced NLRP3 inflammasome activation and IL-1β
secretion (54). E3 ubiquitin
ligase also inhibits NLRP3 inflammasome activation by maintaining
NLRP3 in an inactive state not associated with degradation.
Following inflammasome priming, Cullin1 mediates ubiquitination of
NLRP3 causing it to form an inactive inflammasome. Upon exposure to
inflammatory stimuli, Cullin1 dissociates from NLRP3, allowing it
to return to an active state (55).
Ariadne homolog 2 interacts with the NACHT domain of NLRP3 and
induces ubiquitination of K48 and K63, thereby inhibiting NLRP3
inflammasome activation (56). E3
ubiquitin ligase also positively regulates the activation of the
NLRP3 inflammasome. A recent study suggested that the E3 ligase
Pellino2 serves a dual role in the regulation of NLRP3. It can
interact with NLRP3 during LPS priming to promote the
polyubiquitination of NLRP3 K63 and thus inhibit the activation of
the NLRP3 inflammasome (57).
Pellino2 also ubiquitinates IL-1 receptor-associated kinase 1
(IRAK1) and prevents its interaction with NLRP3, thus limiting the
inhibitory effect of IRAK1 on NLRP3 and also promoting the
activation of NLRP3 inflammasomes (57). Xing et al (58) found that lack of TNF
receptor-associated factor 6 (TRAF6) prevents activation of the
NLRP3 inflammasome, which is mediated by the E3 ubiquitin ligase
function of TRAF6. In addition, double-stranded RNA in the
cytoplasm activates the NLRP3 inflammasome, mediated by the
interaction of DHX33, cytoplasmic RNA sensors and NLRP3; this
interaction requires TRIM33-mediated K63 polyubiquitination of
DHX33 (59).
Ubiquitination of inflammasome-binding protein ASC
also controls the activation of the NLRP3 inflammasome. Following
viral infection, MAVS-mediated E3 ubiquitin ligase TRAF3
recruitment leads to K63 polyubiquitination of ASC via autophagy,
leading to its degradation (60).
Far infrared can cause TRAF6-mediated ASC polyubiquitination via
autophagy degradation in macrophages and has been used in the
treatment of burn wounds (61). The
linear ubiquitin assembly complex regulates the activation of NLRP3
inflammasomes by linear polyubiquitination of ASC. Furthermore,
HOIL-1-deficient macrophages exhibit decreased IL-1β secretion
following LPS stimulation (62).
E3 ubiquitin ligase can also mediate the
ubiquitination of caspase-1 to regulate the activation of NLRP3
inflammasomes. Inhibitor of apoptosis proteins (IAPs) activate the
inflammasome by inducing the polyubiquitination of caspase-1 K63;
accordingly, the activation of caspase-1 is decreased in cellular
(c)IAP1/2 or X-linked (X)IAP-deficient mice (63). cIAP1/2 and XIAP are depleted when
caspase-1 is activated, and the absence of E3 ligase leads to the
production of receptor interacting protein kinase 3-dependent ROS,
which is sufficient to activate the NLRP3 inflammasome (64). The ubiquitination system serves
positive and negative roles in the activation of inflammasomes by
acting on various components of the NLRP3 inflammasome, depending
on cell location, the nature of the substrate and the different
ubiquitin chains that modify it.
Phosphorylation is a common mechanism for
post-translational modification of proteins and is involved in the
signal transduction pathway of NLRP3 inflammasome activation. Basak
et al (65) reported that
PYD of NLRP3 is dephosphorylated by phosphatase 2A at the ser5 site
to activate the NLRP3 inflammasome, and inhibition or knockdown of
phosphatase 2A decreases NLRP3 activation. NLRP3 activators induce
mitochondria-associated endoplasmic reticulum membrane
translocation to the adjacent Golgi to increase diacylglycerol,
which recruits protein kinase D to phosphorylate NLRP3 at ser295,
facilitating the assembly of the NLRP3 inflammasome (42). Li et al (66) found that protein kinase A (PKA)
inhibitor H89 blocks baicalin and induces phosphorylation of NLRP3
on PKA-specific sites and thus inhibit the activation of the NLRP3
inflammasome. However, it is not clear why NLRP3 phosphorylation at
the same site has the opposite effect on the activation of NLRP3
inflammasomes. Bile acids activate the TGR5 receptor pathway,
leading to an increase in cAMP and subsequent PKA activation. PKA
phosphorylates the NOD domain of NLRP3 at ser291, and NLRP3
phosphorylation promotes polyubiquitination of K48 and K63 and
degradation (67). The protein
tyrosine phosphatase non-receptor 22 dephosphorylates NLRP3 at
tyrosine residues Tyr861, thereby promoting activation of the NLRP3
inflammasome (68). Martin et
al (69) found that, during the
activation of NLRP3 inflammasomes, ASC is phosphorylated by IκB
kinase (IKK)i at s58 and promotes the migration of ASC from the
nucleus to the perinuclear region; ASC is also phosphorylated by
IKKα at s16 and s193 and interacts with IKKα. However, Signal II
for NLRP3 inflammasome activation inhibits IKKα kinase activity by
recruiting protein phosphatase 2A, thus enabling ASC to participate
in the assembly of NLRP3 inflammasomes (69). Studies have shown that caspase-1 is
phosphorylated and activated by PI-3K/Rac1/p21-activated kinase at
s376, thus participating in the activation of inflammatory
cortisone (65). Therefore, these
findings suggest that the NLRP3 inflammasome serves a key role in
the development of innate immunity and immune inflammatory disease,
while post-translational modification of NLRP3 inflammasomes,
including ubiquitination and phosphorylation, can precisely
regulate their activation, providing the host with immune
protection against tissue damage.
In sepsis-induced brain injury, recombinant club
cell protein protects the hippocampus from injury by inhibiting p38
protein and the ERK signaling pathway to inhibit the NLRP3
inflammasome (70). The NLRP3
inhibitor MC950 and caspase-1 inhibitor Ac-YVAD-CMK ameliorate
cognitive impairment caused by sepsis-associated encephalopathy via
inhibiting NLRP3/caspase-1 pathway-mediated pyroptosis (71). Melatonin improves spinal cord injury
and protects motor neurons in rats by inhibiting the activation of
the NLRP3 inflammasome (72).
Similarly, melatonin-mediated mitochondrial autophagy prevents
early brain damage following subarachnoid hemorrhage by inhibiting
the activation of NLRP3 inflammasomes (73).
Studies have shown that the NLRP3 inflammasome is
activated in cardiac fibroblasts during sepsis, which induces the
maturation and release of inflammatory factors such as IL-1β.
Inhibiting the activation of the NLRP3 inflammasome in cardiac
fibroblasts can alleviate LPS-induced myocardial dysfunction and
improve the survival of mice with septic peritonitis (74). Carbon monoxide-releasing molecule
treatment inhibits activation of the NLRP3 inflammasome by blocking
interactions between the NLRP3 inflammasome and adaptor protein ASC
and alleviates myocardial dysfunction in septic mice (75). In addition, Tanuseputero et
al (76) found that by blocking
the NLRP3 pathway, LPS-induced inflammatory responses and cell
apoptosis could be alleviated and the function of damaged
myocardial tissue could be restored.
Arginine can protect against acute kidney injury in
sepsis by inhibiting NO-mediated activation of the NLRP3
inflammasome (76). Yang et
al (85) found that transfected
renal tubular epithelial cells over-expressing CD39 exhibit
decreased production of LPS-mediated pro-inflammatory cytokines and
activated NLRP3. Hydrogen sulfide inhibits NLRP3 inflammasome
activation via the TLR4/NLRP3 signaling pathway to protect against
LPS-induced sepsis-associated acute kidney injury (86). In LPS-induced acute kidney injury,
dexmedetomidine inhibits mRNA and protein expression levels of
TLR4, NADPH oxidase-4 and NLRP3, and alleviates LPS-mediated acute
kidney injury by modulating the TLR4/NADPH oxidase-4/NLRP3 pathway
to inhibit the activation of NLRP3 inflammasomes and decrease
oxidative stress damage (87).
Sepsis-induced activation of NLRP3 inflammasome in activated
platelets has been shown to be associated with organ damage in
septicemic CLP rat models. Treatment with MCC950, a specific
inhibitor of NLRP3, significantly decreases sepsis-induced platelet
activation and prevents kidney damage and endothelial dysfunction
in CLP (88).
The innate immune system is the first line of host
defense. Pattern recognition receptors are activated in response to
harmful stimuli, such as various pathogenic microorganisms (which
contain PAMPs and result in the release of DAMPs), and trigger
downstream inflammatory responses to eliminate infection and repair
damaged tissue (5). NLRP3
stimulation induces multiple signaling pathways and cellular
events, resulting from NLRP3 inflammasome activation. Among these,
the major activating agents are potassium efflux, ROS production,
mitochondrial dysfunction, lysosomal damage and protein
post-translational modifications (13). However, a unified mechanism of NLRP3
inflammasome activation has not yet been discerned, and further
studies are needed to clarify this.
Sepsis is a disorder in the host response to
infection, resulting in life-threatening organ dysfunction. This
definition emphasizes that infection leads to homeostasis
imbalances in the host and a potentially fatal risk (89). The assembly and activation of the
NLRP3 inflammasome leads to varying degrees of damage to different
systems during sepsis (5,6). So far, a number of studies have shown
that inhibiting the activation of the inflammasome can decrease the
inflammatory response in sepsis (16–18).
Nevertheless, sepsis can cause organ dysfunction, suggesting that
its pathophysiological mechanisms are complex. Further studies are
necessary to elucidate the specific effects of NLRP3 inflammasomes
on the pathophysiological mechanisms of sepsis so that novel
diagnostic and therapeutic measures can be developed.
Not applicable.
The present study was supported by National Natural
Science Foundation of China (grant nos. 81870071, 81671895,
81871610 and 81471897); Natural Science Foundation of Hunan
Province, China (grant no. 2019JJ40393), Changsha Municipal Natural
Science Foundation (grant no. kq2014228) and Fundamental Research
Funds of Central South University for Postgraduate Students (grant
no. 1053320191465).
Not applicable.
XYS drafted the manuscript. XYS and SPT conceived
and designed the framework of this article. XYS and SCT collected
and analyzed the literature. All authors read and approved the
final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Kim M and Li G: Postoperative
complications affecting survival after cardiac arrest in general
surgery patients. Anesth Analg. 126:858–864. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990–2017: Analysis for the Global Burden of Disease
Study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davis FM, Schaller MA, Dendekker A, Joshi
AD, Kimball AS, Evanoff H, Wilke C, Obi AT, Melvin WJ, Cavassani K,
et al: Sepsis Induces Prolonged Epigenetic Modifications in Bone
Marrow and Peripheral Macrophages Impairing Inflammation and Wound
Healing. Arterioscler Thromb Vasc Biol. 39:2353–2366. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu S, Zhou Z, Li H, Liu Z, Pan X, Wang F,
Huang Y, Li X, Xiao Y, Pan J, et al: BMSCs ameliorate septic
coagulopathy by suppressing inflammation in cecal ligation and
puncture-induced sepsis. J Cell Sci. 131:1312018.PubMed/NCBI
|
|
5
|
Gruda MC, Ruggeberg KG, O'Sullivan P,
Guliashvili T, Scheirer AR, Golobish TD, Capponi VJ and Chan PP:
Broad adsorption of sepsis-related PAMP and DAMP molecules,
mycotoxins, and cytokines from whole blood using
CytoSorb® sorbent porous polymer beads. PLoS One.
13:e01916762018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salomão R, Ferreira BL, Salomão MC, Santos
SS, Azevedo LCP and Brunialti MKC: Sepsis: Evolving concepts and
challenges. Braz J Med Biol Res. 52:e85952019. View Article : Google Scholar
|
|
7
|
Lv X and Wang H: Pathophysiology of
sepsis-induced myocardial dysfunction. Mil Med Res.
3:302016.PubMed/NCBI
|
|
8
|
Nieman GF, Andrews P, Satalin J, Wilcox K,
Kollisch-Singule M, Madden M, Aiash H, Blair SJ, Gatto LA and
Habashi NM: Acute lung injury: How to stabilize a broken lung. Crit
Care. 22:1362018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Strate LL and Morris AM: Epidemiology,
pathophysiology, and treatment of diverticulitis. Gastroenterology.
156:1282–1298.e1. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zarbock A, Gomez H and Kellum JA:
Sepsis-induced acute kidney injury revisited: Pathophysiology,
prevention and future therapies. Curr Opin Crit Care. 20:588–595.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Varatharaj A and Galea I: The blood-brain
barrier in systemic inflammation. Brain Behav Immun. 60:1–12. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abderrazak A, Syrovets T, Couchie D, El
Hadri K, Friguet B, Simmet T and Rouis M: NLRP3 inflammasome: From
a danger signal sensor to a regulatory node of oxidative stress and
inflammatory diseases. Redox Biol. 4:296–307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Swanson KV, Deng M and Ting JP: The NLRP3
inflammasome: Molecular activation and regulation to therapeutics.
Nat Rev Immunol. 19:477–489. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu A and Wu H: Structural mechanisms of
inflammasome assembly. FEBS J. 282:435–444. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stutz A, Kolbe CC, Stahl R, Horvath GL,
Franklin BS, van Ray O, Brinkschulte R, Geyer M, Meissner F and
Latz E: NLRP3 inflammasome assembly is regulated by phosphorylation
of the pyrin domain. J Exp Med. 214:1725–1736. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim SR, Lee SG, Kim SH, Kim JH, Choi E,
Cho W, Rim JH, Hwang I, Lee CJ, Lee M, et al: SGLT2 inhibition
modulates NLRP3 inflammasome activity via ketones and insulin in
diabetes with cardiovascular disease. Nat Commun. 11:21272020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tumurkhuu G, Shimada K, Dagvadorj J,
Crother TR, Zhang W, Luthringer D, Gottlieb RA, Chen S and Arditi
M: Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and
Prevents Atherosclerosis. Circ Res. 119:e76–e90. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Renaudin F, Orliaguet L, Castelli F,
Fenaille F, Prignon A, Alzaid F, Combes C, Delvaux A, Adimy Y,
Cohen-Solal M, et al: Gout and pseudo-gout-related crystals promote
GLUT1-mediated glycolysis that governs NLRP3 and interleukin-1β
activation on macrophages. Ann Rheum Dis. 79:1506–1514. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Afonina IS, Zhong Z, Karin M and Beyaert
R: Limiting inflammation-the negative regulation of NF-κB and the
NLRP3 inflammasome. Nat Immunol. 18:861–869. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He Y, Hara H and Núñez G: Mechanism and
regulation of NLRP3 inflammasome activation. Trends Biochem Sci.
41:1012–1021. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karmakar M, Katsnelson M, Malak HA, Greene
NG, Howell SJ, Hise AG, Camilli A, Kadioglu A, Dubyak GR and
Pearlman E: Neutrophil IL-1β processing induced by pneumolysin is
mediated by the NLRP3/ASC inflammasome and caspase-1 activation and
is dependent on K+ efflux. J Immunol. 194:1763–1775.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Di A, Xiong S, Ye Z, Malireddi RKS,
Kometani S, Zhong M, Mittal M, Hong Z, Kanneganti T-D, Rehman J, et
al: The TWIK2 potassium efflux channel in macrophages mediates
NLRP3 inflammasome-induced inflammation. Immunity. 49:56–65.e4.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gianfrancesco MA, Dehairs J, L'homme L,
Herinckx G, Esser N, Jansen O, Habraken Y, Lassence C, Swinnen JV,
Rider MH, et al: Saturated fatty acids induce NLRP3 activation in
human macrophages through K+ efflux resulting from
phospholipid saturation and Na, K-ATPase disruption. Biochim
Biophys Acta Mol Cell Biol Lipids. 1864:1017–1030. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang W, Hu D, Feng Y, Wu C, Song Y, Liu W,
Li A, Wang Y, Chen K, Tian M, et al: Paxillin mediates ATP-induced
activation of P2X7 receptor and NLRP3 inflammasome. BMC Biol.
18:1822020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rühl S and Broz P: Caspase-11 activates a
canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J
Immunol. 45:2927–2936. 2015. View Article : Google Scholar
|
|
26
|
Katsnelson MA, Rucker LG, Russo HM and
Dubyak GR: K+ efflux agonists induce NLRP3 inflammasome
activation independently of Ca2+ signaling. J Immunol.
194:3937–3952. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abais JM, Xia M, Zhang Y, Boini KM and Li
PL: Redox regulation of NLRP3 inflammasomes: ROS as trigger or
effector? Antioxid Redox Signal. 22:1111–1129. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang C, Abdukerim M, Abilailieti M, Tang
L, Ling Y and Pan S: The protective effects of orexin a against
high glucose-induced activation of NLRP3 inflammasome in human
vascular endothelial cells. Arch Biochem Biophys. 672:1080522019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang
W-C, Marin T, Shentu TP, Wen L, Gongol B, et al: Sterol regulatory
element binding protein 2 activation of NLRP3 inflammasome in
endothelium mediates hemodynamic-induced atherosclerosis
susceptibility. Circulation. 128:632–642. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rada B, Park JJ, Sil P, Geiszt M and Leto
TL: NLRP3 inflammasome activation and interleukin-1β release in
macrophages require calcium but are independent of
calcium-activated NADPH oxidases. Inflamm Res. 63:821–830. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han Y, Xu X, Tang C, Gao P, Chen X, Xiong
X, Yang M, Yang S, Zhu X, Yuan S, et al: Reactive oxygen species
promote tubular injury in diabetic nephropathy: The role of the
mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol.
16:32–46. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shimada K, Crother TR, Karlin J, Dagvadorj
J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et
al: Oxidized mitochondrial DNA activates the NLRP3 inflammasome
during apoptosis. Immunity. 36:401–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhong B, Liu X, Wang X, Liu X, Li H,
Darnay BG, Lin X, Sun SC and Dong C: Ubiquitin-specific protease 25
regulates TLR4-dependent innate immune responses through
deubiquitination of the adaptor protein TRAF3. Sci Signal.
6:ra352013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee HE, Yang G, Park YB, Kang HC, Cho YY,
Lee HS and Lee JY: Epigallocatechin-3-gallate prevents acute gout
by suppressing NLRP3 inflammasome activation and mitochondrial DNA
synthesis. Molecules. 24:21382019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park S, Juliana C, Hong S, Datta P, Hwang
I, Fernandes-Alnemri T, Yu JW and Alnemri ES: The mitochondrial
antiviral protein MAVS associates with NLRP3 and regulates its
inflammasome activity. J Immunol. 191:4358–4366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Subramanian N, Natarajan K, Clatworthy MR,
Wang Z and Germain RN: The adaptor MAVS promotes NLRP3
mitochondrial localization and inflammasome activation. Cell.
153:348–361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li H, Zhang S, Li F and Qin L: NLRX1
attenuates apoptosis and inflammatory responses in myocardial
ischemia by inhibiting MAVS-dependent NLRP3 inflammasome
activation. Mol Immunol. 76:90–97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Iyer SS, He Q, Janczy JR, Elliott EI,
Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, et
al: Mitochondrial cardiolipin is required for Nlrp3 inflammasome
activation. Immunity. 39:311–323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Elliott EI, Miller AN, Banoth B, Iyer SS,
Stotland A, Weiss JP, Gottlieb RA, Sutterwala FS and Cassel SL:
Cutting edge: Mitochondrial assembly of the NLRP3 inflammasome
complex is initiated at priming. J Immunol. 200:3047–3052. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ichinohe T, Yamazaki T, Koshiba T and
Yanagi Y: Mitochondrial protein mitofusin 2 is required for NLRP3
inflammasome activation after RNA virus infection. Proc Natl Acad
Sci USA. 110:17963–17968. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen J and Chen ZJ: PtdIns4P on dispersed
trans-Golgi network mediates NLRP3 inflammasome activation. Nature.
564:71–76. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Z, Meszaros G, He WT, Xu Y, de
Fatima Magliarelli H, Mailly L, Mihlan M, Liu Y, Puig Gámez M,
Goginashvili A, et al: Protein kinase D at the Golgi controls NLRP3
inflammasome activation. J Exp Med. 214:2671–2693. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Riteau N, Baron L, Villeret B, Guillou N,
Savigny F, Ryffel B, Rassendren F, Le Bert M, Gombault A and
Couillin I: ATP release and purinergic signaling: A common pathway
for particle-mediated inflammasome activation. Cell Death Dis.
3:e4032012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jessop F, Hamilton RF Jr, Rhoderick JF,
Fletcher P and Holian A: Phagolysosome acidification is required
for silica and engineered nanoparticle-induced lysosome membrane
permeabilization and resultant NLRP3 inflammasome activity. Toxicol
Appl Pharmacol. 318:58–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Heid ME, Keyel PA, Kamga C, Shiva S,
Watkins SC and Salter RD: Mitochondrial reactive oxygen species
induces NLRP3-dependent lysosomal damage and inflammasome
activation. J Immunol. 191:5230–5238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu A, Gao X, Zhang Q and Cui L: Cathepsin
B inhibition attenuates cardiac dysfunction and remodeling
following myocardial infarction by inhibiting the NLRP3 pathway.
Mol Med Rep. 8:361–366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Newman ZL, Leppla SH and Moayeri M:
CA-074Me protection against anthrax lethal toxin. Infect Immun.
77:4327–4336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Orlowski GM, Colbert JD, Sharma S, Bogyo
M, Robertson SA and Rock KL: Multiple cathepsins promote Pro-IL-1β
synthesis and NLRP3-mediated IL-1β activation. J Immunol.
195:1685–1697. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chevriaux A, Pilot T, Derangère V, Simonin
H, Martine P, Chalmin F, Ghiringhelli F and Rébé C: Cathepsin B is
required for NLRP3 inflammasome activation in macrophages, through
NLRP3 interaction. Front Cell Dev Biol. 8:1672020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tang TT, Lv LL, Pan MM, Wen Y, Wang B, Li
ZL, Wu M, Wang FM, Crowley SD and Liu BC: Hydroxychloroquine
attenuates renal ischemia/reperfusion injury by inhibiting
cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis.
9:3512018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bai H, Yang B, Yu W, Xiao Y, Yu D and
Zhang Q: Cathepsin B links oxidative stress to the activation of
NLRP3 inflammasome. Exp Cell Res. 362:180–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang D, Bu F and Zhang W: The role of
ubiquitination in regulating embryonic stem cell maintenance and
cancer development. Int J Mol Sci. 20:26672019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Song H, Liu B, Huai W, Yu Z, Wang W, Zhao
J, Han L, Jiang G, Zhang L, Gao C, et al: The E3 ubiquitin ligase
TRIM31 attenuates NLRP3 inflammasome activation by promoting
proteasomal degradation of NLRP3. Nat Commun. 7:137272016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Singh M, Kumari B and Yadav UCS:
Regulation of oxidized LDL-induced inflammatory process through
NLRP3 inflammasome activation by the deubiquitinating enzyme
BRCC36. Inflamm Res. 68:999–1010. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhuo Y, Li D, Cui L, Li C, Zhang S, Zhang
Q, Zhang L, Wang X and Yang L: Treatment with
3,4-dihydroxyphenylethyl alcohol glycoside ameliorates
sepsis-induced ALI in mice by reducing inflammation and regulating
M1 polarization. Biomed Pharmacother. 116:1090122019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kawashima A, Karasawa T, Tago K, Kimura H,
Kamata R, Usui-Kawanishi F, Watanabe S, Ohta S, Funakoshi-Tago M,
Yanagisawa K, et al: ARIH2 ubiquitinates NLRP3 and negatively
regulates NLRP3 inflammasome activation in macrophages. J Immunol.
199:3614–3622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Humphries F, Bergin R, Jackson R, Delagic
N, Wang B, Yang S, Dubois AV, Ingram RJ and Moynagh PN: The E3
ubiquitin ligase Pellino2 mediates priming of the NLRP3
inflammasome. Nat Commun. 9:15602018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xing Y, Yao X, Li H, Xue G, Guo Q, Yang G,
An L, Zhang Y and Meng G: Cutting edge: TRAF6 mediates TLR/IL-1R
signaling-induced nontranscriptional priming of the NLRP3
inflammasome. J Immunol. 199:1561–1566. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Weng L, Mitoma H, Trichot C, Bao M, Liu Y,
Zhang Z and Liu YJ: The E3 ubiquitin ligase tripartite motif 33 is
essential for cytosolic RNA-induced NLRP3 inflammasome activation.
J Immunol. 193:3676–3682. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Guan K, Wei C, Zheng Z, Song T, Wu F,
Zhang Y, Cao Y, Ma S, Chen W, Xu Q, et al: MAVS promotes
inflammasome activation by targeting ASC for K63-linked
ubiquitination via the E3 ligase TRAF3. J Immunol. 194:4880–4890.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chiu HW, Chen CH, Chang JN, Chen CH and
Hsu YH: Far-infrared promotes burn wound healing by suppressing
NLRP3 inflammasome caused by enhanced autophagy. J Mol Med (Berl).
94:809–819. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Rodgers MA, Bowman JW, Fujita H, Orazio N,
Shi M, Liang Q, Amatya R, Kelly TJ, Iwai K, Ting J, et al: The
linear ubiquitin assembly complex (LUBAC) is essential for NLRP3
inflammasome activation. J Exp Med. 211:1333–1347. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Labbé K, McIntire CR, Doiron K, Leblanc PM
and Saleh M: Cellular inhibitors of apoptosis proteins cIAP1 and
cIAP2 are required for efficient caspase-1 activation by the
inflammasome. Immunity. 35:897–907. 2011. View Article : Google Scholar
|
|
64
|
Vince JE, Wong WW-L, Gentle I, Lawlor KE,
Allam R, O'Reilly L, Mason K, Gross O, Ma S, Guarda G, et al:
Inhibitor of apoptosis proteins limit RIP3 kinase-dependent
interleukin-1 activation. Immunity. 36:215–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Basak C, Pathak SK, Bhattacharyya A,
Mandal D, Pathak S and Kundu M: NF-kappaB- and C/EBPbeta-driven
interleukin-1beta gene expression and PAK1-mediated caspase-1
activation play essential roles in interleukin-1beta release from
Helicobacter pylori lipopolysaccharide-stimulated
macrophages. J Biol Chem. 280:4279–4288. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Li C-G, Yan L, Mai F-Y, Shi Z-J, Xu L-H,
Jing Y-Y, Zha Q-B, Ouyang D-Y and He X-H: Baicalin inhibits
NOD-like receptor family, pyrin containing domain 3 inflammasome
activation in murine macrophages by augmenting protein kinase A
signaling. Front Immunol. 8:14092017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang
L, Zheng M, Zhang X, Xia D, Ke Y, et al: Bile acids control
inflammation and metabolic disorder through inhibition of NLRP3
inflammasome. Immunity. 45:802–816. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Spalinger MR, Kasper S, Gottier C, Lang S,
Atrott K, Vavricka SR, Scharl S, Raselli T, Frey-Wagner I, Gutte
PM, et al: NLRP3 tyrosine phosphorylation is controlled by protein
tyrosine phosphatase PTPN22. J Clin Invest. 126:1783–1800. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Martin BN, Wang C, Willette-Brown J,
Herjan T, Gulen MF, Zhou H, Bulek K, Franchi L, Sato T, Alnemri ES,
et al: IKKα negatively regulates ASC-dependent inflammasome
activation. Nat Commun. 5:49772014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhou R, Yang X, Li X, Qu Y, Huang Q, Sun X
and Mu D: Recombinant CC16 inhibits NLRP3/caspase-1-induced
pyroptosis through p38 MAPK and ERK signaling pathways in the brain
of a neonatal rat model with sepsis. J Neuroinflammation.
16:2392019. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Fu Q, Wu J, Zhou XY, Ji MH, Mao QH, Li Q,
Zong MM, Zhou ZQ and Yang JJ: NLRP3/Caspase-1 Pathway-induced
pyroptosis mediated cognitive deficits in a mouse model of
sepsis-associated encephalopathy. Inflammation. 42:306–318. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Xu G, Shi D, Zhi Z, Ao R and Yu B:
Melatonin ameliorates spinal cord injury by suppressing the
activation of inflammasomes in rats. J Cell Biochem. 120:5183–5192.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cao S, Shrestha S, Li J, Yu X, Chen J, Yan
F, Ying G, Gu C, Wang L and Chen G: Melatonin-mediated mitophagy
protects against early brain injury after subarachnoid hemorrhage
through inhibition of NLRP3 inflammasome activation. Sci Rep.
7:24172017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tong Z, Jiang B, Zhang L, Liu Y, Gao M,
Jiang Y, Li Y, Lu Q, Yao Y and Xiao X: HSF-1 is involved in
attenuating the release of inflammatory cytokines induced by LPS
through regulating autophagy. Shock. 41:449–453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhang W, Tao A, Lan T, Cepinskas G, Kao R,
Martin CM and Rui T: Carbon monoxide releasing molecule-3 improves
myocardial function in mice with sepsis by inhibiting NLRP3
inflammasome activation in cardiac fibroblasts. Basic Res Cardiol.
112:162017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Tanuseputero SA, Lin MT, Yeh SL and Yeh
CL: Intravenous arginine administration downregulates NLRP3
inflammasome activity and attenuates acute kidney injury in mice
with polymicrobial sepsis. Mediators Inflamm. 2020:32016352020.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang YC, Liu QX, Zheng Q, Liu T, Xu XE,
Liu XH, Gao W, Bai XJ and Li ZF: Dihydromyricetin alleviates
sepsis-induced acute lung injury through inhibiting NLRP3
inflammasome-dependent pyroptosis in mice model. Inflammation.
42:1301–1310. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lyubasyuk V, Ouyang H, Yu FX, Guan KL and
Zhang K: YAP inhibition blocks uveal melanogenesis driven by GNAQ
or GNA11 mutations. Mol Cell Oncol. 2:e9709572014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhou Y, Zhang CY, Duan JX, Li Q, Yang HH,
Sun CC, Zhang J, Luo XQ and Liu SK: Vasoactive intestinal peptide
suppresses the NLRP3 inflammasome activation in
lipopolysaccharide-induced acute lung injury mice and macrophages.
Biomed Pharmacother. 121:1095962020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ying Y, Mao Y and Yao M: NLRP3
inflammasome activation by microRNA-495 promoter methylation may
contribute to the progression of acute lung injury. Mol Ther
Nucleic Acids. 18:801–814. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang L, Mosoian A, Schwartz ME, Florman
SS, Gunasekaran G, Schiano T, Fiel MI, Jiang W, Shen Q, Branch AD,
et al: HIV infection modulates IL-1β response to LPS stimulation
through a TLR4-NLRP3 pathway in human liver macrophages. J Leukoc
Biol. 105:783–795. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chen X, Liu G, Yuan Y, Wu G, Wang S and
Yuan L: NEK7 interacts with NLRP3 to modulate the pyroptosis in
inflammatory bowel disease via NF-κB signaling. Cell Death Dis.
10:9062019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Bai RX, Xu YY, Qin G, Chen YM, Wang HF,
Wang M and Du SY: Repression of TXNIP-NLRP3 axis restores
intestinal barrier function via inhibition of myeloperoxidase
activity and oxidative stress in nonalcoholic steatohepatitis. J
Cell Physiol. 234:7524–7538. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Han J, Bae J, Choi CY, Choi SP, Kang HS,
Jo EK, Park J, Lee YS, Moon HS, Park CG, et al: Autophagy induced
by AXL receptor tyrosine kinase alleviates acute liver injury via
inhibition of NLRP3 inflammasome activation in mice. Autophagy.
12:2326–2343. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yang M, Lu L, Kang Z, Ma T and Wang Y:
Overexpressed CD39 mitigates sepsis-induced kidney epithelial cell
injury via suppressing the activation of NLR family pyrin domain
containing 3. Int J Mol Med. 44:1707–1718. 2019.PubMed/NCBI
|
|
86
|
Chen Y, Jin S, Teng X, Hu Z, Zhang Z, Qiu
X, Tian D and Wu Y: Hydrogen sulfide attenuates LPS-induced acute
kidney injury by inhibiting inflammation and oxidative stress. Oxid
Med Cell Longev. 2018:67172122018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yao Y, Hu X, Feng X, Zhao Y, Song M, Wang
C and Fan H: Dexmedetomidine alleviates lipopolysaccharide-induced
acute kidney injury by inhibiting the NLRP3 inflammasome activation
via regulating the TLR4/NOX4/NF-κB pathway. J Cell Biochem.
120:18509–18523. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cornelius DC, Travis OK, Tramel RW,
Borges-Rodriguez M, Baik CH, Greer M, Giachelli CA, Tardo GA and
Williams JM: NLRP3 inflammasome inhibition attenuates
sepsis-induced platelet activation and prevents multi-organ injury
in cecal-ligation puncture. PLoS One. 15:e02340392020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|