Introduction
ATP-dependent chromatin-remodelling complexes
(remodelers), SWItch/sucrose non-fermentable (SWI/SNF), were
reported to serve a crucial role in gene regulation, where they
affected numerous biological processes (1). However, the molecular architecture of
SWI/SNF complexes was not studied in detail until the previous 5
years (2–4). Nonetheless, to the best of our
knowledge, the role of their core subunit, SWI/SNF related, matrix
associated, actin dependent regulator of chromatin subfamily c
member 2 (SMARCC2), in the development and progression of glioma
remains poorly understood. Over the past two decades, there have
been rapid developments in research regarding chromatin structures
that govern crucial cellular processes, including DNA replication,
transcription and post-transcriptional gene regulation (5). The mammalian SWI/SNF family of
chromatin remodelers, BAF nuclear assembly factor 1 (BAF) and
polybromo-associated BAF, regulate chromatin structure and
transcription via sliding, ejecting and reconstituting the
nucleosomes, and mutations in each have previously been associated
with numerous cancer types, such as breast, colon, liver and
stomach cancer (6–10). Among the four core subunits of the
SWI/SNF complex [SMARC subfamily a member 4 (SMARCA4), BAF155,
SMARCC2 and SMARC subfamily b member 1], SMARCC2 forms a part of
the SWI/SNF base module that displays a compact fold and can be
divided into five closely associated submodules: The head, thumb,
palm, bridge and fingertips. The thumb is formed by the SANT domain
of SMARCC2, the pre-HSA of SMARCA4 and the C-terminal helices of
SMARC subfamily d member 1 (2).
Glioma is the most common type of primary malignant
tumor of the central nervous system and is strongly resistant to
postoperative radiotherapy and chemotherapy (11–13).
Glioblastoma (GBM) is known to be the most severe type (level 4) of
glioma owing to its malignant nature. As the standard Stupp therapy
for glioblastoma, the most common type of intracranial aggressive
tumor, ionizing radiation plus concomitant and adjuvant
temozolomide is widely used following surgical resection.
Temozolomide triggers autophagy--associated cell death to inhibit
tumor growth in GBM (3). However,
the prognosis of patients with GBM remains poor, with a median
survival of 14.6 months. Numerous previous studies focused on
identifying potential molecular targeted therapies and associated
molecular pathways involved in GBM carcinogenesis have shed light
on its pathogenesis, which has improved patient prognosis (14–18).
The present study aimed to investigate the antitumor
effects of SMARCC2 on glioma cells. Furthermore, the possible
underlying mechanisms associated with the effects of SMARCC2 were
also investigated.
Materials and methods
Patient studies
In total, 62 patients (age, 21–67 years; 42 males
and 20 females) with newly diagnosed GBM who had undergone surgery
plus standard chemoradiotherapy (Stupp regimen) were recruited from
October 2012 to March 2019. Patients had no chronic disease, such
as cardiovascular disease, hypertension and hyperglycemia. All
glioma tissue specimens were collected from Nanfang Hospital of
Southern Medical University (Guangzhou, China). Fresh samples were
stored in −196°C liquid nitrogen following surgical removal. All
patients provided written informed prior to participation in the
study, and all study results were stored and analyzed anonymously.
The present study protocol was approved by the Institutional Review
Board at Nanfang Hospital of Southern Medical University (approval
no. 81772656). All research was performed in accordance with the
principles of the Declaration of Helsinki of 1975. Tumor tissue
obtained from the patients was graded according to the World Health
Organization (19) criteria.
Cell lines and culture
U87MG (cat. no. CC-Y1528), which are most probably a
glioblastoma cell line of unknown origin, T98G and LN229 cell lines
were purchased from American Type Culture Collection. Cells were
cultured in DMEM (containing 4.5 g/l glucose; cat. no. 11995065)
supplemented with 10% FBS (cat. no. 16140071) and puromycin (all
Gibco; Thermo Fisher Scientific, Inc.), and maintained in a
humidified incubator at 37°C in a 5% CO2 atmosphere.
Cell transfection and
transduction
The nucleotide sequences of small interfering RNAs
(siRNAs/sis) targeting SMARCC2 were synthesized by Shanghai
GenePharma Co., Ltd. and were as follows: siSMARCC2-1 forward,
5′-CUCGGCAAGAACUACAAGATT-3′ and reverse,
5′-UCUUGUAGUUCUUGCCGAGTT-3′; and siSMARCC2-3 forward,
5′-GGCGUUACGAUUUCCAGAATT-3′ and reverse,
5′-UUCUGGAAAUCGUAACGCCTT-3′. An NC siRNA was also used. The siRNA
negative control sequence was as follows:
5′-TTUUGAACCAAGAAGCCUCCC-3′ (Shanghai GenePharma Co., Ltd.). To
overexpress SMARCC2, SMARCC2 cDNA was subcloned into an adenovirus
expression vector, which also encoded GFP (Shanghai GenePharma Co.,
Ltd.), namely LVS-SMARCC2 (overexpression; oe). Lentiviral vector
non-specific control (LVS-NC) was used as the control. The U87MG
and LN229 glioma cells were seeded 24 h prior to transfection at
room temperature, at 50–60% confluence, then transfected using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
cells were transfected with 50 nM siRNA and 2.5 µg plasmid at room
temperature, according to the manufacturer's protocol. The
transduction efficiency was analyzed by the intensity of GFP
fluorescence at 48 h post-transduction. A confocal microscope
(Olympus CX23; Olympus Corporation) under ×200 magnification was
used to observe the fluorescence intensity of the cells. Subsequent
experiments were performed 48 h post-transfection.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from transfected cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Total RNA (1 µg) was
reverse transcribed into cDNA according to the reverse
transcription kit (RevertAid First Strand cDNA synthesis kit)
(Takara Bio, Inc.) protocol and genomic DNA was also removed using
the gDNA Eraser from the kit. qPCR was subsequently performed using
SYBR Premix Ex Taq II (Takara Bio, Inc.) on a StepOne™ Real-Time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The following primer sequences were used for qPCR: SMARCC2 forward,
5′-ACTGCCGATCAAATGTTTCCT-3′ and reverse,
5′-ACAGGCAATTATTCTGCACCAAG-3′; and GAPDH forward,
5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse, 5′-AGGGGCCATCCACAGTCTTC-3′.
The following thermocycling conditions were used for qPCR: Initial
denaturation at 95°C for 5 min; followed by 40 cycles at 95°C for
15 sec and 60°C for 30 sec. The relative expression levels of
SMARCC2 were quantified using the 2−ΔΔCq method and
normalized to GAPDH, which served as the endogenous control
(20).
Western blotting
The cells were harvested and the total protein was
extracted using RIPA lysis buffer with protease-phosphatase
inhibitor (EMD Millipore). Pierce BCA Protein assay kit (Thermo
Fisher Scientific, Inc.) was used to quantify protein expression.
The proteins(30 µg/lane) were separated via 8–10% SDS-PAGE,
transferred onto PVDF membranes and blocked with 0.02 M TBS
containing 5% BSA (Abcam) and 0.1% Tween-20 for 2 h at room
temperature. The membranes were subsequently incubated overnight at
4°C with the following primary antibodies (all from Abcam): Rat
anti-SMARCC2 (1:1,000; cat. no. ab243634), rat anti-c-Myc (1:1,000;
cat. no. ab32072), anti-tumor protein D52 (TPD52) like 2 (TPD52L2;
1:1,000; cat, no. ab234819), anti-snail family transcriptional
repressor 1 (Snail; 1:1,000; cat. no. no. ab216347), rat
anti-N-cadherin, rat anti-T-cadherin (1:1,000; cat. no. ab167407),
rabbit anti-β-catenin (1:1,000; cat. no. ab32572), anti-vimentin
(1:1,000; cat. no. ab92547), anti-GAPDH (1:3,000; cat. no. ab8245)
and mouse anti-β-actin (1:3,000; cat. no. ab8226). Subsequently,
horseradish peroxidase-conjugated secondary goat anti-rabbit IgG
H&L (1:5,000; cat. no. b6721; Abcam), was incubated with the
membrane at room temperature for 1 h. The membranes were visualized
using an ECL kit (Bio-Rad Laboratories, Inc.) according to the
manufacturer's protocol. Protein bands were visualized and the
detection of band gray value was performed using Image Studio
software (version 4.0; LI-COR Biosciences). β-actin and GAPDH were
used as the loading controls.
Immunohistochemistry (IHC)
staining
The tumor samples were collected and fixed in 4%
paraformaldehyde for 24–48 h, then embedded in paraffin.
Paraffin-embedded tissue samples were serially cut (4-µm thick) and
dried overnight at 60°C. The samples were then deparaffinized in
xylene at room temperature and rehydrated using a graded descending
series of ethanol (100, 95 and 80%). Antigen retrieval was
performed in boiling water using 10 mM citrate solution for 5 min,
and endogenous hydrogen peroxidase activity was blocked by
incubation with 10% hydrogen peroxide for 30 min at room
temperature. Each of these aforementioned steps was followed by a
10 min PBS wash. Sections were blocked with 3%
H2O2 (cat. no. SP-9002; OriGene Technologies,
Inc.) at 37°C for 30 min. The samples were subsequently incubated
overnight with an anti-SMARCC2 (1:50; cat. no. ab243634; Abcam)
primary antibody at 4°C. Following the primary antibody incubation,
the sections were incubated with a goat anti-rabbit secondary
antibody (1:1,000; cat. no. SP-9002; OriGene Technologies, Inc.) at
room temperature for 2 h. The sections were then stained with
diaminobenzidine at room temperature for 3–5 min and counterstained
with hematoxylin at room temperature for 5 min. Finally, images
were captured using an Olympus light microscope (Olympus
Corporation) under ×200 magnification. Positive staining of SMARCC2
was scored from 0–3 according to the intensity of the staining: 0
(negative), 1 (weakly positive, light yellow), 2 (moderately
positive, yellowish brown) and 3 (strongly positive, brown). The
percentage of positively-stained cells was also scored using the
following scores: 0 (0%), 1 (1–33%), 2 (34–66%) and 3 (67–100%).
The sum of the intensity and percentage scores was used as the
final staining score.
Co-immunoprecipitation (Co-IP)
assay
The cells were harvested and the total protein was
extracted using lysis buffer for western and IP assays (cat. no.
6505706) with protease-phosphatase inhibitor (EMD Millipore). The
protein lysate was centrifuged at 14,000 × g at 4°C for 15 min and
the supernatant was collected. U87MG and LN229 cells were seeded
into 10-cm dishes at a density of 5×105 cells/dish and
incubated for 24 h. Equal amounts of protein were
coimmunoprecipitated with a rabbit anti-SMARCC2 (1:1,000; cat. no.
ab243634; Abcam), rat anti-c-Myc (1:1,000; cat. no. ab32072;
Abcam), anti-tumor protein D52 (TPD52) like 2 (TPD52L2; 1:1,000;
cat. no. ab234819; Abcam) monoclonal antibody at 4°C overnight and
then incubated with protein A/G (1:1) sepharose magnetic beads (40
µl) (GE Healthcare) at 4°C. The total volume was 400 µl. Following
incubation, lysis buffer was used for washing. After magnetic
separation, the supernatants with unbound free proteins were
separated and the protein complex was resuspended in SDS buffer for
western blotting analyses, which were performed according to the
aforementioned protocols. A rabbit anti-IgG antibody (GeneTex,
Inc.) was used as the negative control.
Wound healing assay
Cell migration was determined using a wound healing
assay. Briefly, U87MG/T98G/LN229 cells were cultured to 100%
confluence in a six-well plate and then the cell monolayer was
scratched with a 1-ml pipette tip to create an artificial wound.
The detached cells were removed with PBS and remaining cells were
cultured in high-sugar serum-free DMEM. Images of the same wound
area were photographed at 0, and 24 or 48 h under a light
microscope (magnification, ×40). The healed scratch area was
calculated using ImageJ 1.8.0 software (National Institutes of
Health) using the following formula: Healed scratch (%)=[(initial
scratch area-final scratch area)/initial scratch area] ×100.
Cell invasion and migration
assays
Cell invasion and migration was investigated using
Transwell plates (Costar; Corning, Inc.) with a pore size of
0.8-µm. Briefly, 5×105 U87MG/T98G/LN229 cells were
seeded in high-sugar serum-free DMEM into the upper chamber of the
Transwell plates, which was precoated with Matrigel at 37°C
overnight (for the invasion assay). Medium supplemented with 10%
FBS was plated into the lower chamber. Following incubation for 6–8
h at 37 °C, non-migratory or non-invasive cells in the upper
chamber were removed, whereas migratory and invasive cells in the
lower chamber were fixed with 20% methanol for 5 min and stained
for 5 min with 0.1% crystal violet. The cells from six randomly
selected fields of view were observed under a confocal microscope
(Olympus CX23; Olympus Corporation) at ×200 magnification, then
ImageJ software (version 1.8.0; National Institutes of Health) was
used to calculate the number of invasive and migrated cells.
Bioinformatics analysis
Level 3 RNA-SeqV2 data (containing data on genes,
isoforms, exons and junction levels), level 3 Agilent microarray
gene expression data and clinical data for LGG and GBM were
downloaded from The Cancer Genome Atlas (TCGA) database (cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga)
using the TCGA biolinks package cgdsr (github.com/cBioPortal/cgdsr) on R platform (version
3.6.3). Data were obtained from the TCGA data sets ‘lgg_tcga’ and
‘gbm_tcga’. The cut-off mode based on median SMARCC2 expression was
selected without specifying a track subset.
Statistical analysis
Statistical analyses were performed using SPSS 20.0
software (IBM Corp.). All experiments were performed in triplicate
and data are presented as the mean ± SEM. Comparisons between two
groups were performed using paired Student's t-test or a
Mann-Whitney U test, whereas statistical differences among several
groups were determined using one-way ANOVA followed by Dunnett's
post hoc test or the Kruskal Wallis test followed by Dunn's post
hoc test. Survival plots were generated using the Kaplan-Meier
method and log-rank test; in instances where there was late-stage
crossover between the groups, Cramer-von Mises tests were used.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Expression levels of SMARCC2 in
patients with glioma with different tumor grades
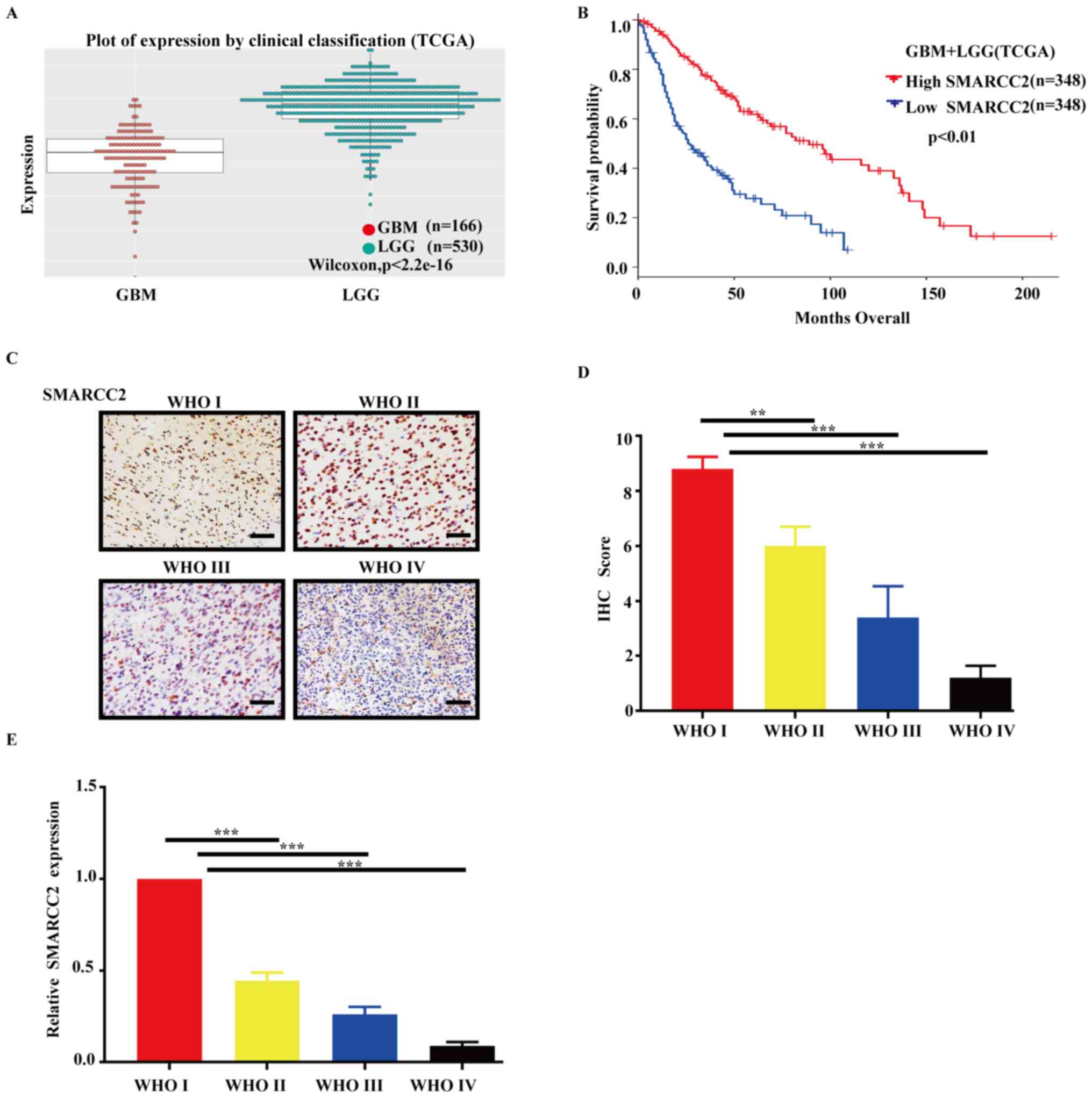
The results of the bioinformatics analyses revealed
that SMARCC2 was differentially expressed in different grades of
glioma (Fig. 1A); as glioma grade
increased, expression levels of SMARCC2 decreased and high
expression levels of SMARCC2 were significantly associated with an
improved prognosis in patients with glioma (Fig. 1B). Furthermore, overall survival
time was separately analyzed in low-grade gliomas (LGG) and
high-grade gliomas (GBM), and the results revealed no significant
difference in overall survival time between GBMs with high or low
SMARCC2 expression (Fig. S1). LGG
shows that high expression of SMARCC2 has a better prognosis.
(Fig. S1). An SMARCC2-specific
antibody was used for the IHC analysis of different glioma grades
(Fig. 1C), and a standardized
scoring method was used to analyze the results of the IHC analysis.
In the WHO III and IV group, the protein expression levels of
SMARCC2 were significantly downregulated compared with the
low-grade group (Fig. 1D). RT-qPCR
analysis revealed that the mRNA expression levels of SMARCC2 were
also significantly downregulated in tissues (WHO I–IV; Fig. 1E), which was consistent with the
results for SMARCC2 protein expression levels. These decreases were
positively associated with severity of the tumor.
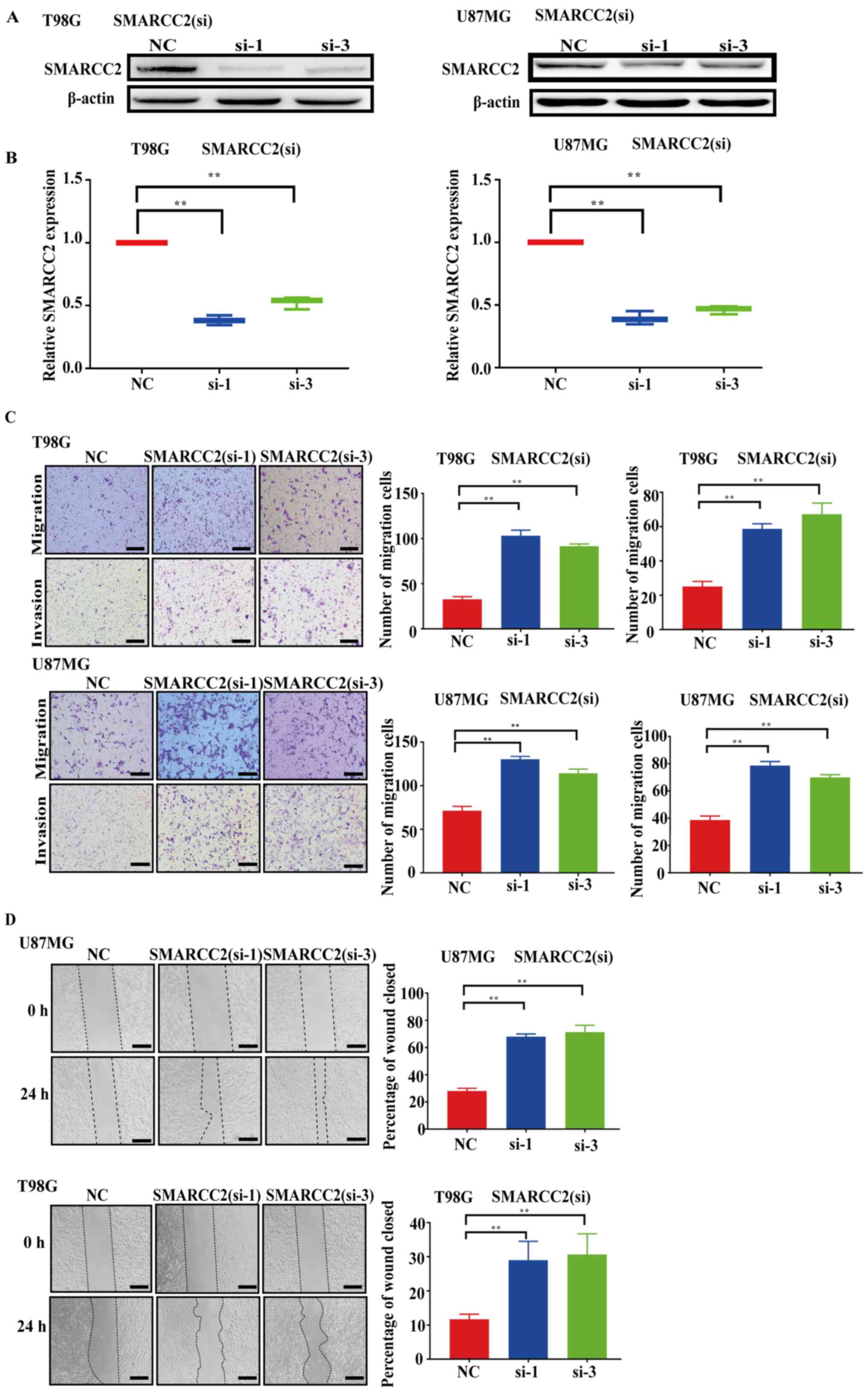
Effects of SMARCC2 knockdown on glioma
cell migration and invasion
siRNAs targeting SMARCC2 were transfected into human
glioma cancer cells, U87MG and T98G, and the protein and mRNA
expression levels of SMARCC2 were analyzed via western blotting
(Fig. 2A) and RT-qPCR (Fig. 2B), respectively, after 48 h. The
expression levels of SMARCC2 were downregulated in the siSMARCC2-1
and siSMARCC2-3 groups compared with the NC group at both the
protein and mRNA levels. Subsequently, Transwell assays were
performed to determine the effect of SMARCC2 on glioma cell
migration and invasion; the results revealed that the knockdown of
SMARCC2 expression significantly increased the number of migratory
and invasive cells compared with the NC group (Fig. 2C). Furthermore, wound healing assays
were performed to further validate the effect of SMARCC2 on cell
migration (Fig. 2D). Compared with
the NC group, GBM cell migration was significantly increased in the
siSMARCC2-1 and siSMARCC2-3 groups.
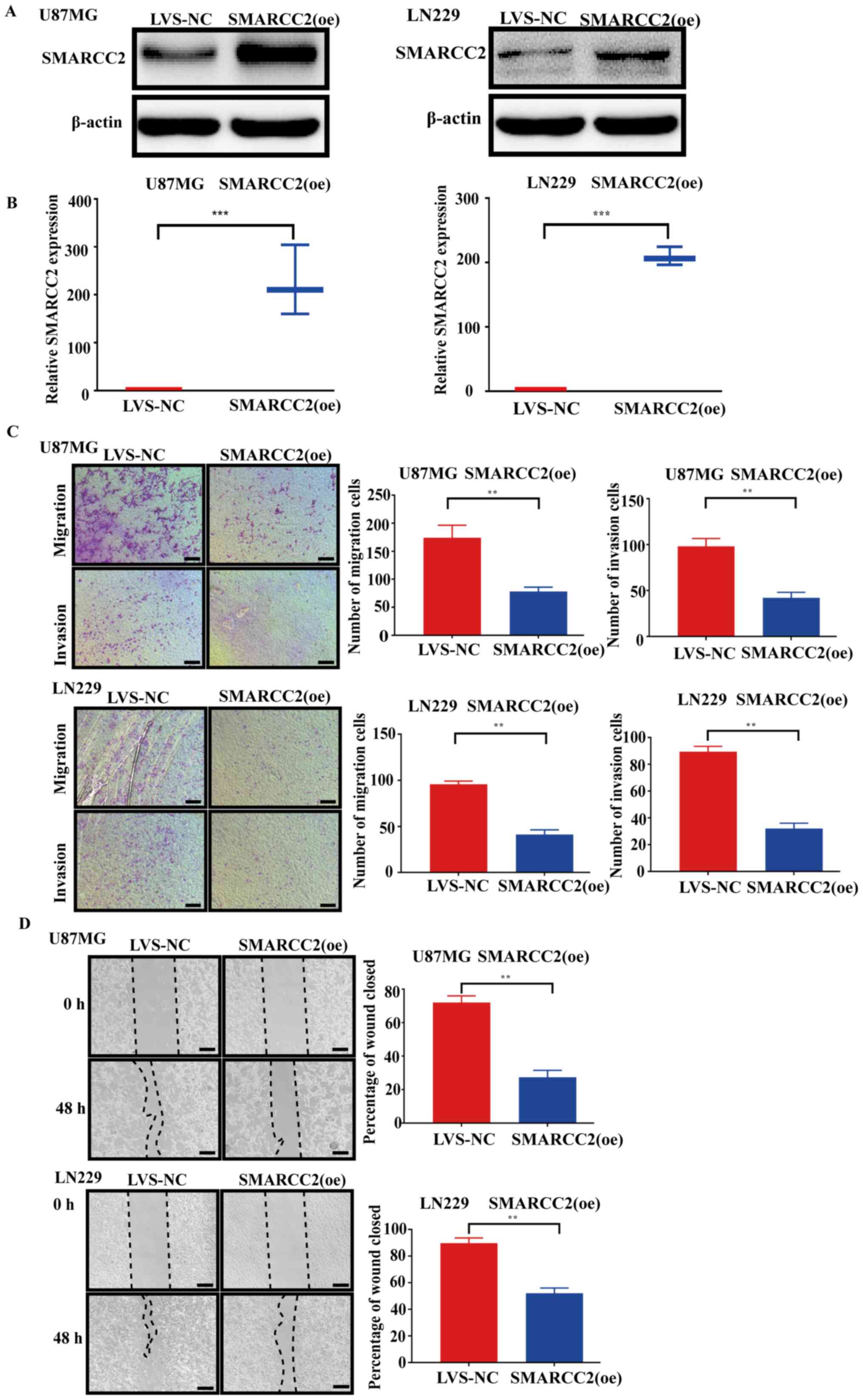
Overexpression of SMARCC2 inhibits
glioma cell migration and invasion
To determine the potential biological functions of
SMARCC2 in glioma cell migration and invasion, U87MG and LN229 cell
lines stably overexpressing SMARCC2 were established by infecting
cells with recombinant adenovirus vectors. The transduction
efficiency was analyzed by the intensity of GFP fluorescence at 48
h post-transduction (data not shown). The transduction efficiency
was determined as 90–95%. The protein and mRNA expression levels of
SMARCC2 in U87MG and LN229 cancer cells following the
overexpression of SMARCC2 were also analyzed via western blotting
(Fig. 3A) and RT-qPCR (Fig. 3B), respectively. Following
transfection and overexpression of lentivirus, the RNA and protein
levels of SMARCC2 increased significantly. Transwell (Fig. 3C) and wound healing assays (Fig. 3D) were conducted to analyze cell
migration and invasion following the overexpression of SMARCC2. The
results demonstrated that the transduction of cells with SMARCC2
(oe) adenovirus significantly decreased U87MG and LN229 cell
migration and invasion compared with the LVS-NC group.
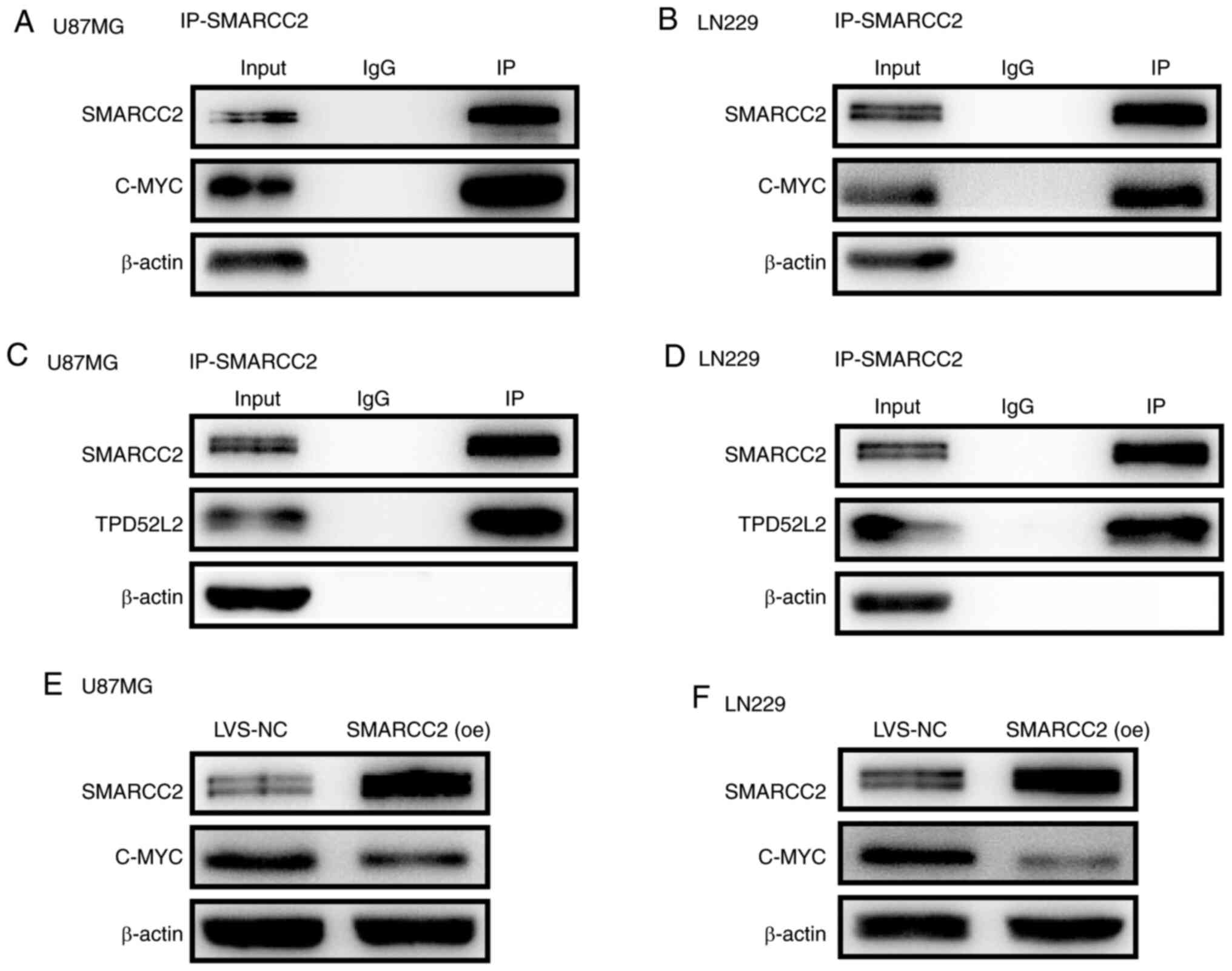
SMARCC2 directly targets c-Myc and
inhibits its oncogenic functions
Co-IP experiments were performed to analyze the
interactions between SMARCC2 and c-Myc (Fig. 4A and B). SMARCC2 was found to
interact with c-Myc. Notably, SMARCC2 also strongly interacted with
TPD52L2 (Fig. 4C and D), which is a
member of the TPD52 family (21).
Furthermore, the western blotting results indicated that the
overexpression of SMARCC2 notably downregulated the expression
levels of c-Myc compared with the LVS-NC group (Fig. 4E and F). These findings suggested
that SMARCC2 may inhibit the oncogenic function of c-Myc by
promoting c-Myc protein degradation.
SMARCC2 induces EMT in glioma cells by
targeting the Wnt/β-catenin signaling pathway
EMT is a key process through which epithelial cells
acquire mesenchymal properties, and it has been closely associated
with cancer invasion and metastasis (22). The expression levels of a number of
EMT markers were analyzed in U87MG and LN229 cells overexpressing
SMARCC2. The present results revealed that the expression levels of
T-cadherin were markedly upregulated, whereas the expression levels
of N-cadherin, β-catenin, vimentin and Snail were notably
downregulated in cells transfected with SMARCC2 (oe) adenovirus
compared with LVS-NC (Fig. 5A and
B). Subsequently, a Wnt/β-catenin signaling pathway agonist,
HLY78, was used for rescue experiments. The results demonstrated
that overexpression of SMARCC2 in combination with HLY78 treatment
markedly reversed the agonistic effect of HLY78 on the
Wnt/β-catenin signaling pathway (Fig.
5C and D). The expression of EMT-associated proteins
(N-cadherin/snail/vimentin/β-catenin) increased significantly
following treatment with HLY78-alone, but decreased when SMARCC2
was simultaneously overexpressed and treated with HLY78.
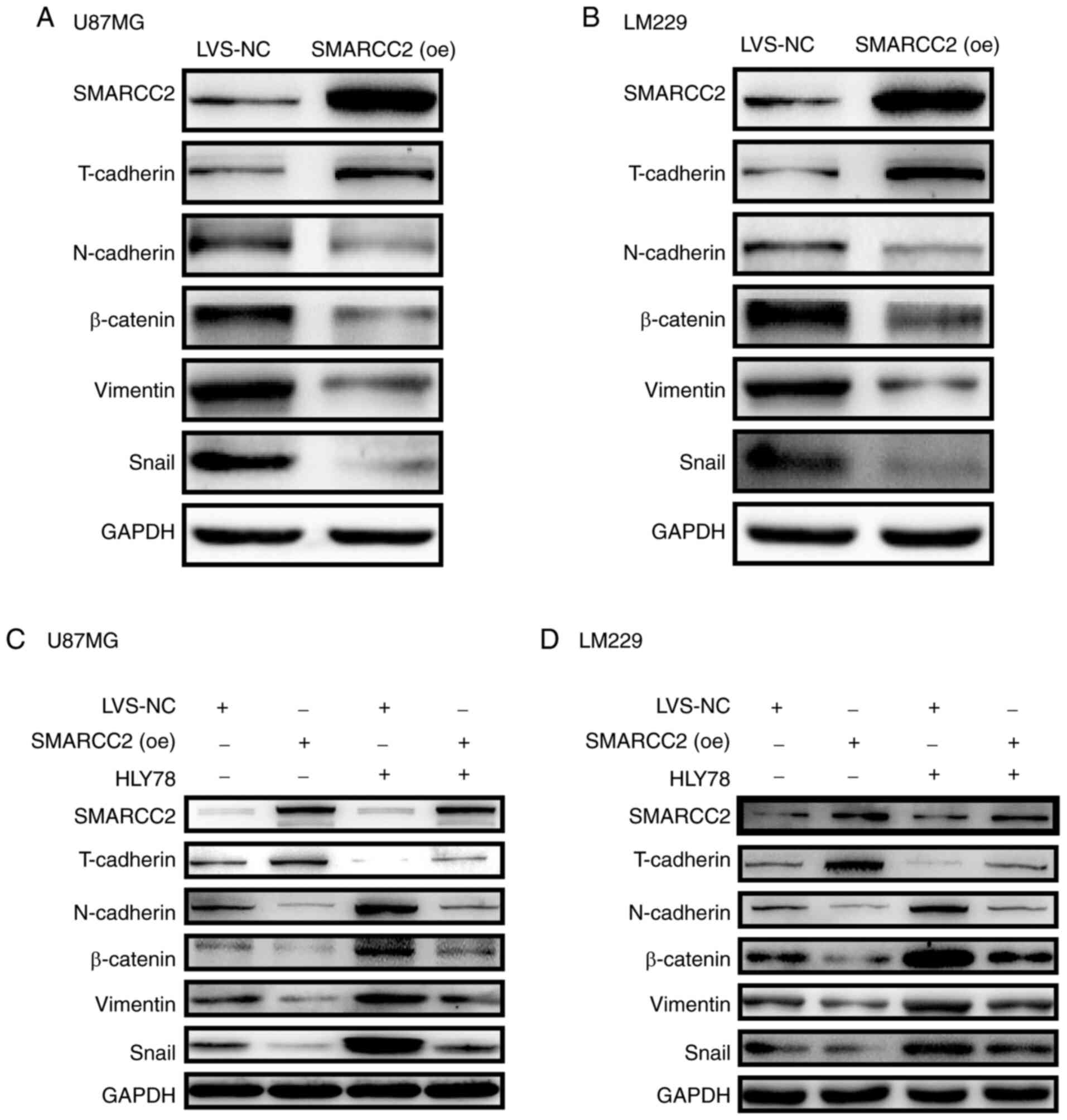 | Figure 5.Analysis of the expression levels of
EMT-related proteins in cells overexpressing SMARCC2. Expression
levels of EMT-associated markers T-cadherin, N-cadherin, β-catenin,
vimentin and Snail were analyzed via western blotting in (A) U87MG
and (B) LM229 cells overexpressing SMARCC2. GAPDH was used as the
internal control. Western blotting was performed to determine the
expression levels of EMT-related proteins in (C) U87MG and (D)
LM229 cells following overexpression of SMARCC2 and treatment with
the Wnt/β-catenin signaling pathway agonist, HLY78. EMT,
epithelial-mesenchymal transition; SMARCC2, SWItch/sucrose
non-fermentable related, matrix associated, actin dependent
regulator of chromatin subfamily c member 2; oe, overexpression;
LVS-NC, lentiviral vector-non-specific control; NC, negative
control; Snail, snail family transcriptional repressor 1. |
Discussion
The molecular structure of the ATP-dependent
chromatin-remodeling complex and its effect on the growth and
development of individuals have been extensively studied (23–25).
Chromatin remodelers are specialized multi-protein machines that
enable access to nucleosomal DNA by altering the structure,
composition and positioning of nucleosomes (26). Mutations in the SWI/SNF family of
proteins have been shown to be involved in the development of a
number of cancer cell types (including breast, colon, liver and
stomach cancer); however, to the best of our knowledge, the
underlying mechanism remains unknown (27–29).
The present study aimed to determine whether the core subunit of
SWI/SNFs, SMARCC2, served a tumor suppressive or oncogenic role in
glioma cells.
The expression of SMARCC2 is associated with glioma
grade. High-grade gliomas show lower expression of SMARCC2, and the
higher the expression of SMARCC2, the better the prognosis. SMARCC2
may have a tumor suppressor function. When the interference rna
fragment was used to knock down expression of SMARCC2, GBM showed
stronger migration and invasion capabilities. When oe lentiivirus
was used to increase expression of SMARCC2, the migration and
invasion capabilities of GBM and expression levels of
EMT-associated proteins (N-cadherin/snail/vimentin/β-catenin)
significantly decreased.
The results of the present study suggested that
SMARCC2 may serve as a tumor suppressor gene in glioma, inhibiting
glioma cell migration and invasion. Moreover, SMARCC2 was shown to
interact with c-Myc, which reportedly serves a role in the
Wnt/β-catenin signaling pathway (30–33).
SMARCC2 inhibited the EMT process of GBM cell lines by
downregulating T-cadherin and upregulating
N-cadherin/β-catenin/snail/vimentin. SMARCC2 was also discovered to
regulate EMT via the Wnt/β-catenin signaling axis; therefore, it
was hypothesized that c-Myc may participate in SMARCC2-mediated
regulation of EMT. These results were consistent with the results
of previous studies investigating SWI/SNF mutations in a number of
types of cancer (34–36). The present results showed
differential expression of SMARCC2 in glioma of different
grades.
The SWI/SNF complex has been demonstrated to exert
essential roles in the life cycle of cells and was found to
participate in the developmental maturation of various types of
neural cell (37,38). As the core subunit of SWI/SNF,
SMARCC2 may be responsible for these functions in neural cells,
thus indicating its role as a possible tumor suppressor gene. In
the absence of SMARCC2, cells cannot develop into mature functional
neuronal cells. The present study transfected two GBM cell lines
(U87MG and LN229) with SMARCC2 (oe) adenoviruses, which resulted in
significantly increased expression levels of SMARCC2 compared with
the LVS-NC group, as demonstrated via RT-qPCR and western blotting.
In U87MG and LN229 cells, the expression of SMARCC2 was
significantly lower than in T98G cells. It was hypothesized that
there is a SMARCC2 gene mutation in the U87MG and LN229 cell line,
resulting in low expression of SMARCC2. These results also
validated that there were mutations in the U87MG and LN229 cell
lines.
The results of the present study indicated that
SMARCC2 may interact with c-Myc to inhibit its oncogenic functions
in U87MG and LN229 cells. Nevertheless, the underlying mechanism of
action remains unclear. TPD52L2 is a member of the TPD52 family
that has been implicated in multiple types of human cancer
(39–41). Further research has demonstrated
that the TPD52 gene encodes regulators of cancer cell
proliferation, indicating that TPD52 may be important for
maintaining tumorigenesis and metastasis of cancer cells (42). Our previous study found that TPD52L2
is associated with the EMT process of the GBM cell line; the
present study demonstrated interaction between SMARCC2 and TPD52L2,
further suggesting that SMARCC2 affects the EMT process of the GBM
cell line. However, one possible mechanism is that SMARCC2 may
serve as a post-translationally modified protease to promote c-Myc
protein degradation, which will be the focus of future
research.
In conclusion, the findings of the present study
suggested that SMARCC2, as the core subunit of SWI/SNF, may be
involved in the occurrence, development and prognosis of GBM..
These results may provide novel insight into the molecular
mechanisms underlying glioma formation and progression, and may
provide a new potential target for molecular-targeted therapies and
prognostic markers for glioma.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81802830).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CL, YL and QZ made substantial contributions to the
study conception. CL designed the study. CF performed the
experiments and analyzed and interpreted the data. HW and RR
drafted and critically revised the manuscript for important
intellectual content. RR made substantial contributions to data
analysis. JL, HW, LC, HL, LS, CS and JG made substantial
contributions to acquisition of data. CL and CF confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study protocol was approved by the
Institutional Review Board at Nanfang Hospital of Southern Medical
University. All research was performed in accordance with the
principles of the Declaration of Helsinki of 1975. All patients
provided written informed prior to participation in the study, and
all study results were stored and analyzed anonymously.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wilson BG and Roberts CW: SWI/SNF
nucleosome remodellers and cancer. Nat Rev Cancer. 11:481–492.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, Li
J, Liu B and Xu Y: Structure of nucleosome-bound human BAF complex.
Science. 367:875–881. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou Y, Johnson SL, Gamarra NI and
Narlikar GJ: Mechanisms of ATP dependent chromatin remodeling
motors. Annu Rev Biophys. 45:153–181. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clapier R, Iwasa J, Cairns BR and Peterson
CL: Mechanisms of action and regulation of ATP-dependent
chromatin-remodelling complexes. Nat Rev Mol Cell Biol. 18:407–422.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saha A, Wittmeyer J and Cairns BR:
Chromatin remodelling: The industrial revolution of DNA around
histones. Nat Rev Mol Cell Biol. 7:437–447. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kassabov SR, Zhang B, Persinger J and
Bartholomew B: SWI/SNF unwraps, slides, and rewraps the nucleosome.
Mol Cell. 11:391–403. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sati S and Cavalli G: Chromosome
conformation capture technologies and their impact in understanding
genome function. Chromosoma. 126:33–44. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Masliah-Planchon J, Bièche I,
Guinebretière JM, Bourdeaut F and Delattre O: SWI/SNF chromatin
remodeling and human malignancies. Ann Rev Pathol. 10:145–171.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kadoch C, Hargreaves DC, Hodges C, Elias
L, Ho L, Ranish J and Crabtree GR: Proteomic and bioinformatic
analysis of mammalian SWI/SNF complexes identifies extensive roles
in human malignancy. Nat Genet. 45:592–601. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Almeida R, Fernández-Justel JM,
Santa-María C, Cadoret JC, Cano-Aroca L, Lombraña R, Herranz G,
Agresti A and Gómez M: Chromatin conformation regulates the
coordination between DNA replication and transcription. Nat Commun.
9:15902018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qiang Z, Jun-Jie L, Hai W, Hong L, Bing-Xi
L, Lei C, Wei X, Ya-Wei L, Huang A, Song-Tao Q and Yun-Tao L:
TPD52L2 impacts proliferation, invasiveness and apoptosis of
glioblastoma cells via modulation of wnt/β-catenin/snail signaling.
Carcinogenesis. 39:214–224. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H, Chen L, Qi S, Yu S, Weng Z, Hu Z,
Zhou Q, Xin Z, Shi L, Ma L, et al: HERC3-mediated SMAD7
ubiquitination degradation promotes autophagy-induced EMT and
chemoresistance in glioblastoma. Clin Cancer Res. 25:3602–3616.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jackson CM, Choi J and Lim M: Mechanisms
of immunotherapy Resistance: Lessons from glioblastoma. Nat
Immunol. 20:1100–1109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tomiyama A and Ichimura K: Signal
transduction pathways and resistance to Targeted therapies in
glioma. Semin Cancer Biol. 58:118–129. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan MS, Sandanaraj E, Chong YK, Lim SW,
Koh LW, Ng WH, Tan NS, Tan P, Ang BT and Tang C: A STAT3-based gene
signature stratifies glioma patients for targeted therapy. Nat
Commun. 10:36012019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang K, Liu X, Li Y, Wang Q, Zhou J, Wang
Y, Dong F, Yang C, Sun Z, Fang C, et al: Genome-Wide CRISPR-Cas9
screening identifies NF-κB/E2F6 responsible for EGFRvIII-associated
temozolomide resistance in glioblastoma. Adv Sci (Weinh).
6:19007822019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Annibali D, Whitfield JR, Favuzzi E,
Jauset T, Serrano E, Cuartas I, Redondo-Campos S, Folch G,
Gonzàlez-Juncà A, Sodir NM, et al: Myc inhibition is effective
against glioma and reveals a role for Myc in profificient mitosis.
Nat Commun. 5:46322014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sang Y, Li Y, Zhang Y, Alvarez AA, Yu B,
Zhang W, Hu B, Cheng SY and Feng H: CDK5-dependent phosphorylation
and nuclear Translocation of TRIM59 promotes macroH2A1
ubiquitination and tumorigenicity. Nat Commun. 10:40132019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weller M, Wick W, Aldape K, Brada M,
Berger M, Pfister SM, Nishikawa R, Rosenthal M, Wen PY, Stupp R and
Reifenberger G: Glioma. Nat Rev Dis Primers. 1:150172015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Method. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang M, Wang X, Jia J, Gao H, Chen P, Sha
X and Wu S: Tumor protein D52-like 2 contributes to proliferation
of breast cancer cells. Cancer Biother Radiopharm. 30:1–7. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Cai H, Sun L, Zhan P, Chen M,
Zhang F, Ran Y and Wan J: LGR5, a novel functional glioma stem cell
marker, promotes EMT by activating the Wnt/β-catenin pathway and
predicts poor survival of glioma patients. J Exp Clin Cancer Res.
37:2252018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Savas S and Skardasi G: The SWI/SNF
complex subunit genes: Their functions, variations, and links to
risk and survival outcomes in human cancers. Crit Rev Oncol
Hematol. 123:114–131. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bögershausen N and Wollnik B: Mutational
landscapes and phenotypic spectrum of SWI/SNF-related intellectual
disability disorders. Front Mol Neurosci. 11:2522018. View Article : Google Scholar
|
|
25
|
Alver BH, Kim KH, Lu P, Wang X, Manchester
HE, Wang W, Haswell JR, Park PJ and Roberts CW: The SWI/SNF
chromatin remodelling complex is required for maintenance of
lineage specific enhancers. Nat Commun. 8:146482017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pillidge Z an Bray SJ, . SWI/SNF chromatin
remodeling controls Notch-responsive enhancer accessibility. EMBO
Rep. 20:e469442019.PubMed/NCBI
|
|
27
|
Wang W, Friedland SC, Guo B, O'Dell MR,
Alexander WB, Whitney-Miller CL, Agostini-Vulaj D, Huber AR, Myers
JR, Ashton JM, et al: ARID1A, a SWI/SNF subunit, is critical to
acinar cell homeostasis and regeneration and is a barrier to
transformation and epithelial-mesenchymal transition in the
pancreas. Gut. 68:1245–1258. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang X, Lee RS, Alver BH, Haswell JR, Wang
S, Mieczkowski J, Drier Y, Gillespie SM, Archer TC, Wu JN, et al:
SMARCB1-mediated SWI/SNF complex function is essential for enhancer
regulation. Nat Genet. 49:289–295. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nagarajan S, Rao SV, Sutton J, Cheeseman
D, Dunn S, Papachristou EK, Prada JG, Couturier DL, Kumar S,
Kishore K, et al: ARID1A influences HDAC1/BRD4 activity, intrinsic
proliferative capacity and breast cancer treatment response. Nat
Genet. 52:187–197. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Helming KC, Wang X, Wilson BG, Vazquez F,
Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ,
et al: ARID1B is a specific vulnerability in ARID1A-mutant cancers.
Nat. Med. 20:251–254. 2014.PubMed/NCBI
|
|
31
|
Chang L, Azzolin L, Di Biagio D, Zanconato
F, Battilana G, Lucon Xiccato R, Aragona M, Giulitti S, Panciera T,
Gandin A, et al: The SWI/SNF complex is a mechanoregulated
inhibitor of YAP and TAZ. Nature. 563:265–269. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ruijtenberg S and van den Heuvel S: G1/S
inhibitors and the SWI/SNF complex control cell-cycle exit during
muscle differentiation. Cell. 162:300–313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Q, Zhou Y, Rychahou P, Harris JW,
Zaytseva YY, Liu J, Wang C, Weiss HL, Liu C, Lee EY and Evers BM:
Deptor is a novel target of Wnt/β-Catenin/c-Myc and contributes to
colorectal cancer cell growth. Cancer Res. 78:3163–3175. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lissanu Deribe Y, Sun Y, Terranova C, Khan
F, Martinez-Ledesma J, Gay J, Gao G, Mullinax RA, Khor T, Feng N,
et al: Mutations in the SWI/SNF complex induce a targetable
dependence on oxidative phosphorylation in lung cancer. Nat Med.
24:1047–1057. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsurusaki Y, Okamoto N, Ohashi H, Kosho T,
Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, et al:
Mutations affecting components of the SWI/SNF complex cause
Coffin-Siris syndrome. Nat Genet. 44:376–378. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim KH, Kim W, Howard TP, Vazquez F,
Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, et
al: SWI/SNF-mutant cancers depend on catalytic and non-catalytic
activity of EZH2. Nat Med. 21:1491–1496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rogers HA, Sousa S, Salto C, Arenas E,
Coyle B and Grundy RG: WNT/β-catenin pathway activation in Myc
immortalised cerebellar progenitor cells inhibits neuronal
differentiation and generates tumours resembling medulloblastoma.
Br J Cancer. 107:1144–1452. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patel R, Brzezinska EA, Repiscak P, Ahmad
I, Mui E, Gao M, Blomme A, Harle V, Tan EH, Malviya G, et al:
Activation of β-catenin cooperates with loss of pten to drive
ar-independent castration-resistant prostate cancer. Cancer Res.
80:576–590. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu J, Wu SW and Wu WP: A tumor-suppressive
microRNA, miRNA-485-5p, inhibits glioma cell proliferation and
invasion by down-regulating TPD52L2. Am J Transl Res. 9:3336–3344.
2017.PubMed/NCBI
|
|
40
|
Chen Q, Wang P, Fu Y, Liu X, Xu W, Wei J,
Gao W, Jiang K, Wu J and Miao Y: MicroRNA-217 inhibits cell
proliferation, invasion and migration by targeting Tpd52l2 in human
pancreatic adenocarcinoma. Oncol Rep. 38:3567–3573. 2017.PubMed/NCBI
|
|
41
|
Xu J, Wang W, Zhu Z, Wei Z, Yang D and Cai
Q: Tumor protein D52-like 2 accelerates gastric cancer cell
proliferation in vitro. Cancer Biother Radiopharm. 30:111–116.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou J, Lin Y, Shi H, Huo K and Li Y:
hABCF3, a TPD52L2 interacting partner, enhances the proliferation
of human liver cancer cell lines in vitro. Mol Biol Rep.
40:5759–5767. 2013. View Article : Google Scholar : PubMed/NCBI
|