Introduction
Allergic rhinitis (AR) is a common and frequently
recurring disease resulting from exposure to associated allergens,
which promotes allergic reactions that lead to nasal inflammation
(1). Its main manifestation is the
inflammatory reaction of cells and the generation of mucus
(2,3). The clinical manifestations of AR are
sneezing, nasal itching, a runny nose, nasal congestion and itchy
eyes, which seriously affect patient quality of life (4). At present, clinical treatments for AR
include histamine receptor antagonists, hormones and
anticholinergic agents (5).
Although the disease can be alleviated or controlled, currently
available treatments are restricted by considerable side-effects,
drug resistance and high recurrence rates (6). Therefore, the identification of
appropriate treatments for AR has become a focus, as well as a
challenge, in the otolaryngology department.
Psoralen (PSO), derived from the fruit of leguminous
plants, belongs to the group of furanocoumarin compounds, and is
one of the most important active components of the psoralen family
(7). PSO is used in various
prescriptions for tonifying the kidney and strengthening bones, and
modern pharmacology has demonstrated that PSO exerts
anti-inflammatory, antioxidant, antitumor and other pharmacological
effects (8). PSO has been shown to
reduce the expression of TGF-β, IL-1β and TNF-α in pulmonary
fibrosis models (9). Furthermore,
PSO exerts a significant anti-inflammatory response, as well as
protecting and activating chondrocytes to relieve osteoarthritis
(10). Therefore, it was speculated
that PSO may also inhibit the inflammatory response, and thus
alleviate AR.
Activator protein 1 (AP-1) is an intracellular
transcriptional activator composed primarily of
proto-oncogene-encoded proteins Jun and Fos, which bind target DNA
sequences in the form of homologous or heterodimer complexes, thus
regulating the expression of target genes (11). AP-1 is a key regulator of cellular
proliferation, differentiation and apoptosis (12). As AP-1 can act as a molecular switch
at the transcriptional level, the AP-1 signal transduction pathway
can be activated by changes in cellular tension, ionization
effects, DNA damage, oxidative stress and UV irradiation, as well
as bacterial and viral infection (13). Activated AP-1 subsequently binds to
the TPA response element (TRE) to promote the expression of a
variety of inflammatory factors (including IL-2, IL-8, TNF-α,
TGF-β1 and IFN-γ), affecting physiological cell functions and in
turn, influencing the occurrence of certain inflammatory diseases
(14), such as inflammatory skin
disease (15) and chronic
obstructive pulmonary disease (16). According to the Gene Expression
Omnibus (GEO) database, the expression of cystatin-SN (CST1) in
individuals with AR is significantly upregulated compared with that
in healthy controls (GSE19187) (17). Moreover, in patients with AR,
mitogen-activated protein kinases can induce transcription factors
of AP-1, thereby regulating the expression of Charcot-Leyden
crystal protein or CST1 (18).
However, the role of the AP-1 pathway in AR is yet to be reported.
In addition, a literature review indicated that PSO regulates AP-1
pathway activation and promotes osteoclast differentiation and bone
resorption (19). As IL-13 induces
the release of inflammatory cytokines and excessive mucus
secretion, a human nasal epithelial cell model of IL-13-induced AR
has been developed and is widely used for in vitro research
(20,21). Therefore, the aim of the present
study was to investigate the role and underlying mechanisms of PSO
with AR, so as to provide a theoretical basis for the treatment of
AR.
Materials and methods
Cell culture
The JME/CF15 nasal epithelial cell line was obtained
from The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences, and cultured at 37°C (5% CO2) in
DMEM supplemented with 10% FBS (both Gibco; Thermo Fisher
Scientific, Inc.) and 1% (v/v) penicillin/streptomycin
(Sigma-Aldrich; Merck KGaA) at a density of 5×106 cells
per well.
Reagents
PSO (batch no. 110739-201115; National Institutes
for Food and Drug Control) was dissolved in DMSO (Sigma-Aldrich;
Merck KGaA) to prepare a psoralen-conditioned medium stock
solution. Then, 100, 20, 10 and 1 µM working stocks of PSO were
prepared with low-glucose DMEM containing 10% FBS. After 2 h of PSO
pretreatment, JME/CF15 cells were stimulated with 10 ng/ml IL-13
for 24 h at 37°C to generate a cell-based AR model. In this paper,
cells were pretreated with 10 nM PMA for 24 h and 15 µl SP600125
for 24 h at 37°C, as outlined in previous studies (22,23).
After giving different concentrations (1, 10 and 20 µm) of PSO, the
cells were divided into the control, IL-13, 1 µM PSO + IL-13, 10 µM
PSO + IL-13 and 20 µM PSO + IL-13 groups. The control group was
given the same dose of DMEM. In another set of experiments, a
concentration of 20 µM PSO was selected, and the cells were divided
into the control, IL-13, PSO + IL-13, PMA + PSO + IL-13 and
SP600125 + PMA + PSO + IL-13 groups.
Database
According to the GEO (https://www.ncbi.nlm.nih.gov/geo/) database, the
expression level of CST1 in individuals with AR was detected
(GSE19187).
Cell Counting Kit-8 (CCK-8) assay
The CCK-8 system (Dojindo Molecular Technologies,
Inc.) was used to assess cell viability. Cells were seeded into
96-well plates at a density of 5×103 cells per well.
After the relevant treatment, 10 µl CCK-8 solution was added to
each well and incubated for 2 h, after which cell viability was
assessed using a Benchmark microplate spectrometer (Bio-Rad
Laboratories, Inc.).
ELISA
Quantification of granulocyte-macrophage
colony-stimulating factor (GM-CSF; cat. no. SGM00; R&D Systems,
Inc.) and Eotaxin (cat. no. MME00; R&D Systems, Inc.) in cell
supernatants (300 × g; 4°C; 10 min) was performed using an ELISA
kit according to the manufacturer's instructions (24).
Reverse transcription-quantitative
(RT-q)PCR
Cells (1×103 cells/well) were cultured in
6-well plates and total RNA was extracted using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. Total RNA (1 µg) was
reverse-transcribed into first-strand complementary DNA using the
SuperScript™ III Reverse Transcriptase kit (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol,
and then amplified in triplicate by qPCR (cobas Z 480 system; Roche
Diagnostics) using SYBR Green (final reaction volume, 20 µl; Takara
Biotechnology Co., Ltd.), according to the manufacturer's protocol.
The following thermocycling conditions were used for qPCR: Initial
denaturation at 95°C for 10 min; followed by 40 cycles of 95°C for
10 sec and 60°C for 60 sec. The following primers (GenScript) were
used for qPCR: GM-CSF forward, 5′-CAGCCACTACAAGCAGCACT-3′ and
reverse, 5′-GGGGATGACAAGCAGAAAGT-3′; Eotaxin forward,
5′-TGTCTCGTTCTCCCTCTGCT-3′ and reverse, 5′-CTCCGCTCACAGTCATTTCC-3′;
IL-6 forward, 5′-GGCCCTTGCTTTCTCTTCG-3′ and reverse,
5′-ATAATAAAGTTTTGATTATGT-3′; IL-8 forward,
5′-ATGGCTGCTGAACCAGTAGA-3′ and reverse, 5′-CTAGTCTTCGTTTTGAACAG-3′;
mucin 5AC (MUC5AC) forward, 5′-ATCACCGAAGGCTGCTTCTGTC-3′ and
reverse, 5′-GTTGATGCTGCACACTGTCCAA-3′; and GAPDH forward,
5′-AGCCACATCGCTCAGACAC-3′ and reverse, 5′-GCCCAATACGACCAAATCC-3′.
When evaluating the effects of treatment, the expression level of
each control was assigned an arbitrary value of 1, and those of the
treated cells were evaluated as a fold-change above the control,
and calculated using the 2−ΔΔCq method (25). GAPDH was used as the internal
control gene.
Western blotting
Total cellular protein was extracted using RIPA
buffer, and quantified using a BCA assay (both Beyotime Institute
of Biotechnology). Cell lysates containing 50–100 µg protein were
resolved by electrophoresis on 10% SDS-PAGE gels (Beyotime
Institute of Biotechnology). The separated proteins were
subsequently transferred to PVDF membranes (Thermo Fisher
Scientific, Inc.), which were then blocked in 5% non-fat milk for 1
h at room temperature, and subsequently incubated overnight at 4°C
in 0.25% non-fat milk containing the appropriate primary
antibodies. Primary antibodies against the following targets were
used in the present study: IL-6 (1:1,000; cat. no. ab6672), IL-8
(1:1,000; cat. no. ab18672), MUC5AC (1:1,000; cat. no. ab3649),
phosphorylated (p)-c-Fos (1:1,000; cat. no. ab27793), c-Fos
(1:1,000; cat. no. ab222699), p-c-Jun (1:1,000; cat. no. ab30620),
c-Jun (1:1,000; cat. no. ab32137), CST1 (1:1,000; cat. no.
ab124281) and GAPDH (1:1,000; cat. no. ab8245). All primary
antibodies were purchased from Abcam. On the second day, the
membranes were incubated with the secondary antibody (1:5,000; cat.
no. ab150077; Abcam) for 2 h at room temperature. The protein bands
were visualized using enhanced chemiluminescence reagents (Thermo
Fisher Scientific, Inc.). Protein band intensity was determined
using ImageJ software (version 146; National Institutes of
Health).
Quantification of reactive oxygen
species (ROS)
Cellular ROS levels were detected using a
fluorescent probe, 2′,7′-dichlorodihydrofluorescein diacetate
(DCFH-DA; Sigma-Aldrich; Merck KGaA), which is rapidly oxidized
into the highly fluorescent 2′,7′-dichlorofluorescein (DCF) in the
presence of intracellular ROS. Fluorescence was monitored with a
laser scanning confocal microscope (magnification, ×200; Leica
Microsystems GmbH) at 488 nm. ROS levels were quantified as the
relative fluorescence intensity of DCF per cell in the scanned
area.
Statistical analysis
All experiments were repeated three times. The data
were analyzed using SPSS version 19.0 (IBM Corp) and GraphPad 6.0
(GraphPad Software, Inc.), and are presented as the mean ± SD.
Comparisons among multiple groups were analyzed using one-way ANOVA
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
PSO inhibits inflammation in
IL-13-induced JME/CF15 cells
JME/CF15 cells were induced using different
concentrations of PSO (0, 1, 10, 20 and 100 µM), and cell viability
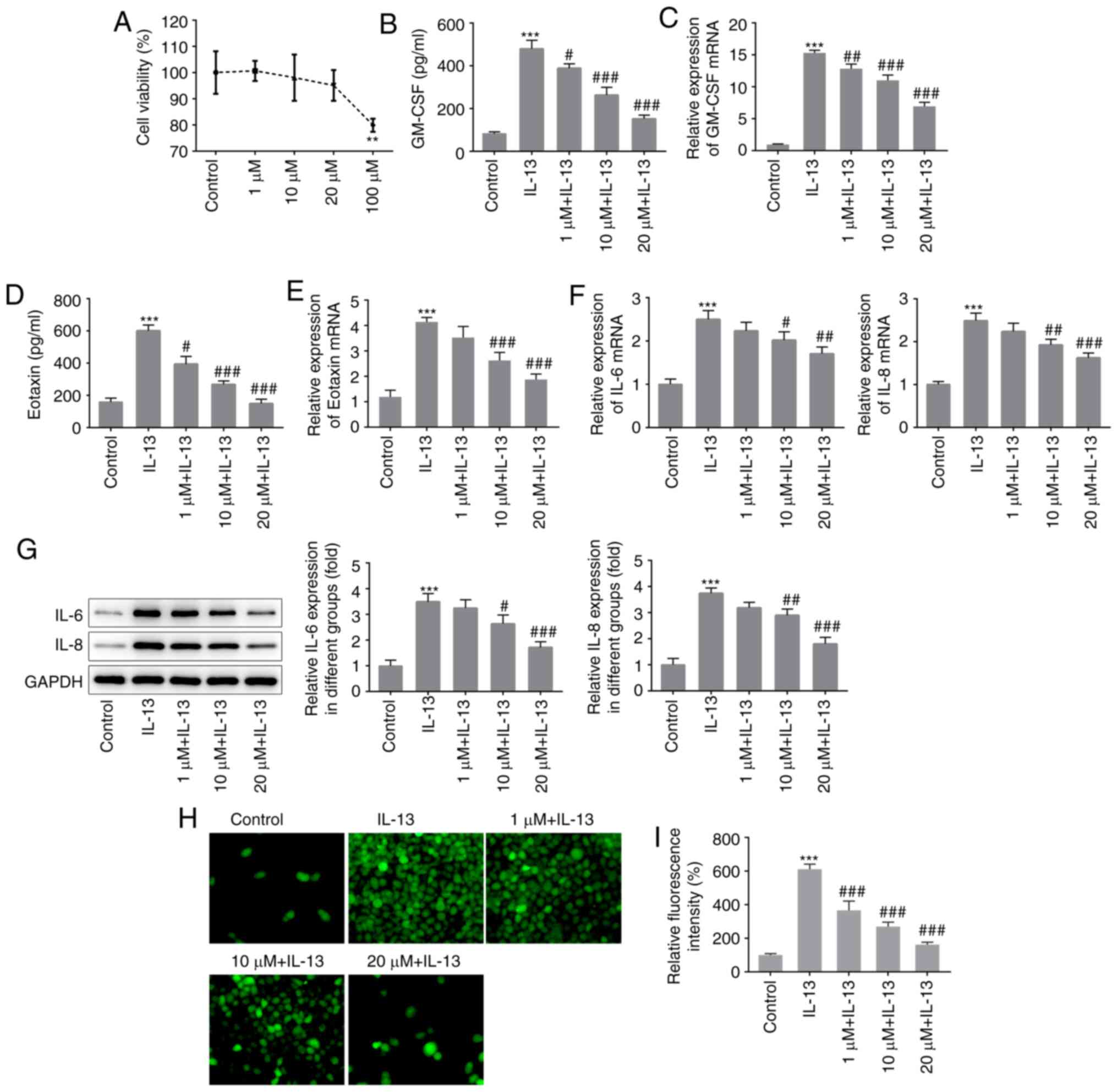
was assessed using an MTT assay. As shown in Fig. 1A, normal cells exhibited marked
damage following treatment with 100 µM PSO; therefore, a
concentration gradient of 1, 10, and 20 µM PSO was used for
subsequent experimentation. The cells were divided into the
control, IL-13, 1 µM PSO + IL-13, 10 µM PSO + IL-13 and 20 µM PSO +
IL-13 groups, and inflammatory responses were detected by ELISA.
Compared with the control group, the expression levels of GM-CSF
(Fig. 1B and C) and Eotaxin
(Fig. 1D and E) were significantly
increased in the IL-13 group, indicating that an inflammatory
response had occurred. Compared with the IL-13 group, GM-CSF and
Eotaxin expression in the 1 µM PSO + IL-13, 10 µM PSO + IL-13 and
20 µM PSO + IL-13 groups were downregulated in a dose-dependent
manner. RT-qPCR (Fig. 1F) and
western blotting (Fig. 1G) were
used to detect the mRNA and protein expression of IL-6 and −8, and
the expression trends were the same as those observed for GM-CSF
and Eotaxin. Cellular ROS expression was detected using a DCFH-DA
probe, and was found to be increased in the IL-13 group compared
with the control group. Compared with the IL-13 group, ROS
expression in the PSO-treated groups was decreased in a
dose-dependent manner (Fig. 1H and
I). These results indicated that PSO exerted dose-dependent
inhibition of the IL-13-induced inflammatory response in JME/CF15
cells.
PSO inhibits mucus production in
IL-13-induced JME/CF15 cells
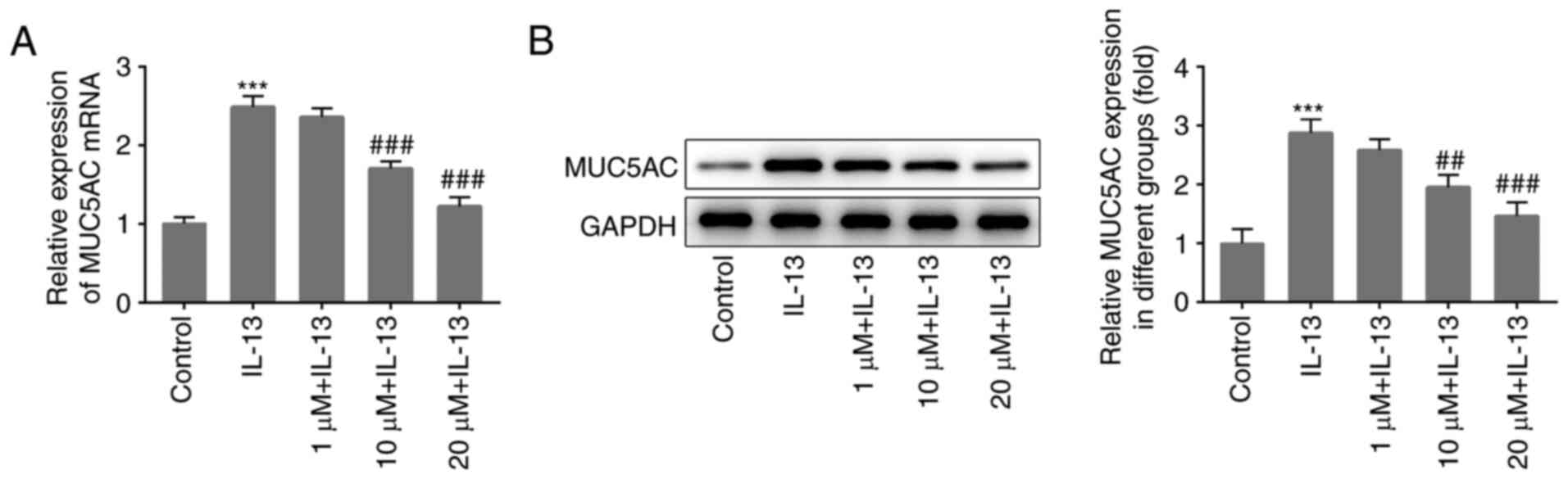
Cellular expression of MUC5AC, a representative
mucus-producing protein, was also detected to determine whether PSO
was able to influence IL-13-induced mucus production. Compared with
the control group, MUC5AC expression was significantly increased in
the IL-13 group, and decreased in a dose-dependent manner following
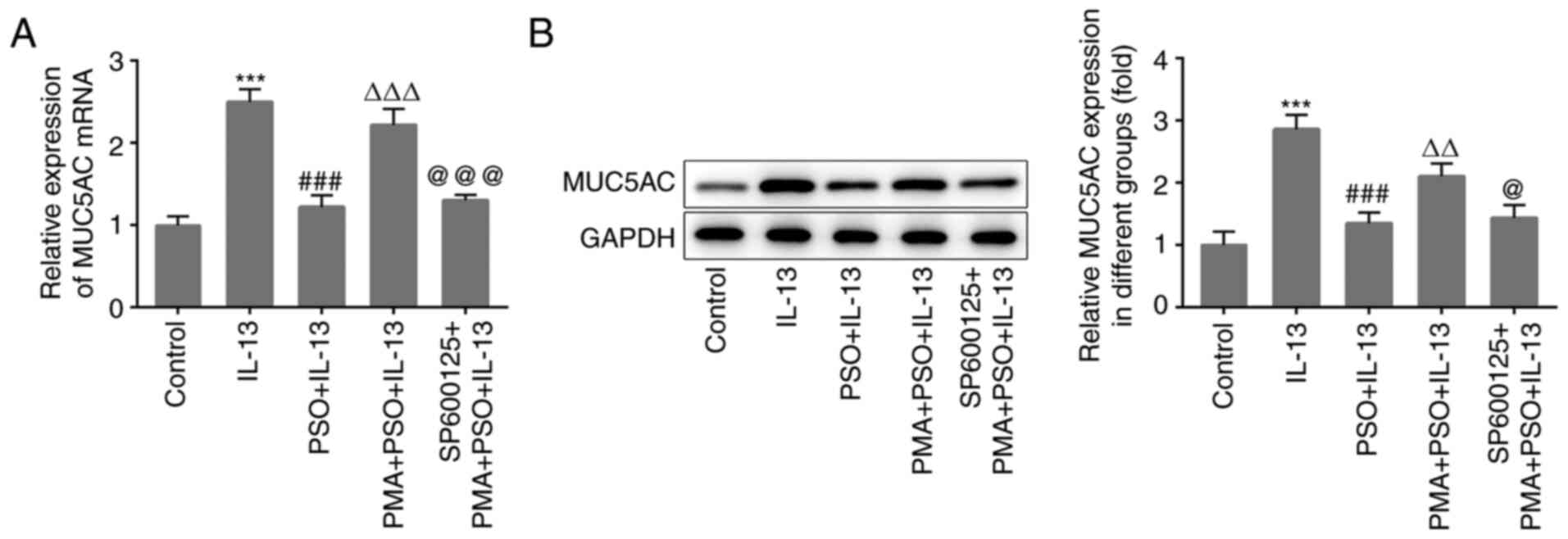
treatment with different concentrations of PSO (Fig. 2A and B). The results confirmed that
IL-13 induced mucus production in JME/CF15 cells, which was
subsequently inhibited by PSO.
PSO inhibits inflammation and mucus
production in IL-13-induced JME/CF15 cells by suppressing the AP-1
signaling pathway
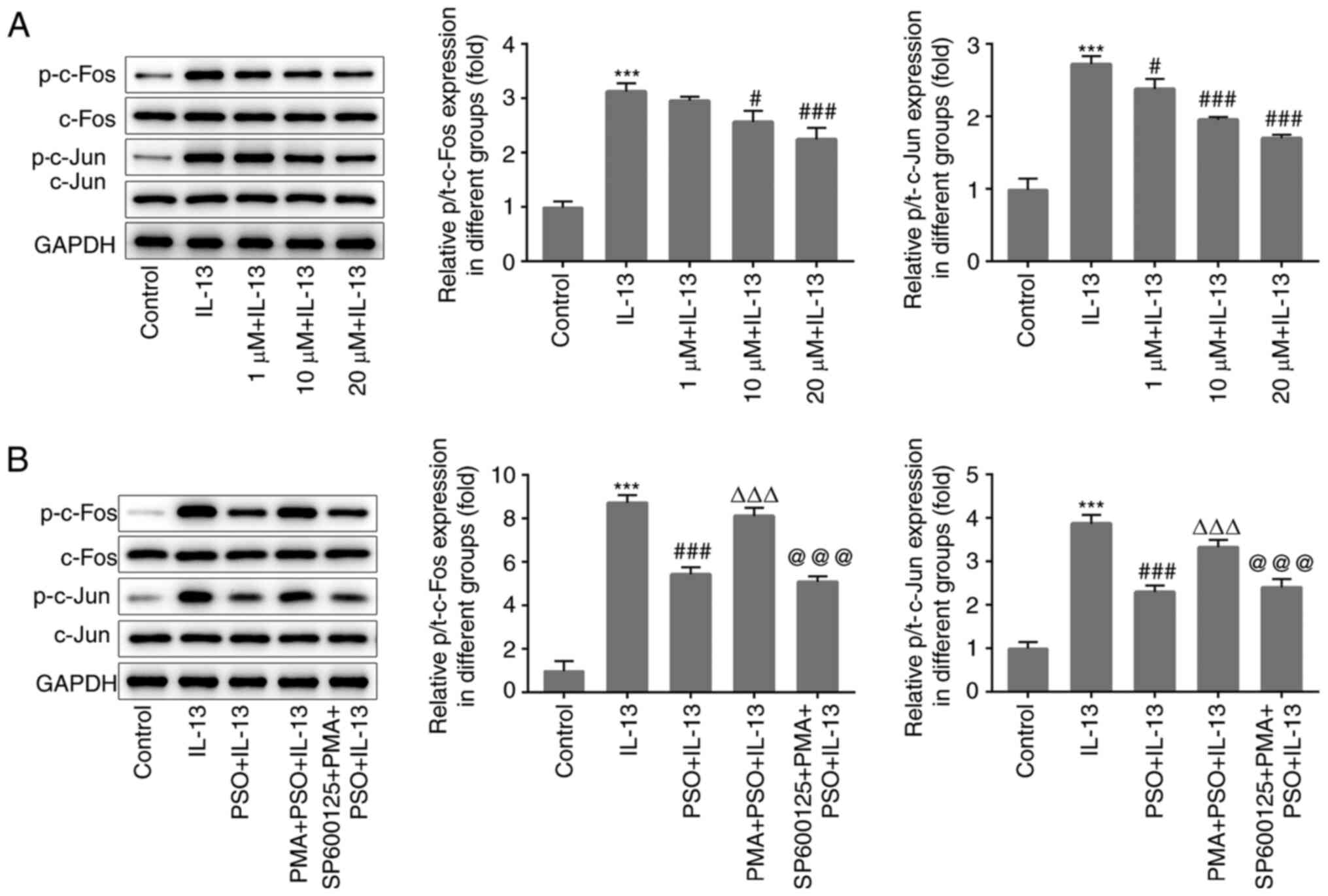
In the present study, phosphorylation levels of the
AP-1 pathway-associated proteins c-Fos and c-Jun were found to be
abnormally altered. Compared with the control group, the levels of
p-c-Fos and p-c-Jun were significantly increased in the IL-13
group, indicating that the AP-1 pathway had been activated. After
the addition of PSO, the levels of p-c-FOS and p-c-Jun were
decreased in a dose-dependent manner, compared with those in the
IL-13 group (Fig. 3A).
Subsequently, the AP-1 pathway activator PMA and the pathway
inhibitor SP600125 were used to further assess whether the
regulatory effects of PSO on AR were achieved through the AP-1
pathway. A concentration of 20 µM PSO was selected, and the cells
were divided into the control, IL-13, PSO + IL-13, PMA + PSO +
IL-13 and SP600125 + PMA + PSO + IL-13 groups. Western blot
analysis was used to detect the expression of AP-1 pathway
proteins. Compared with the PSO + IL-13 group, the levels of
p-c-Fos and p-c-Jun were significantly increased in the PMA + PSO +
IL-13 group (Fig. 3B), and the
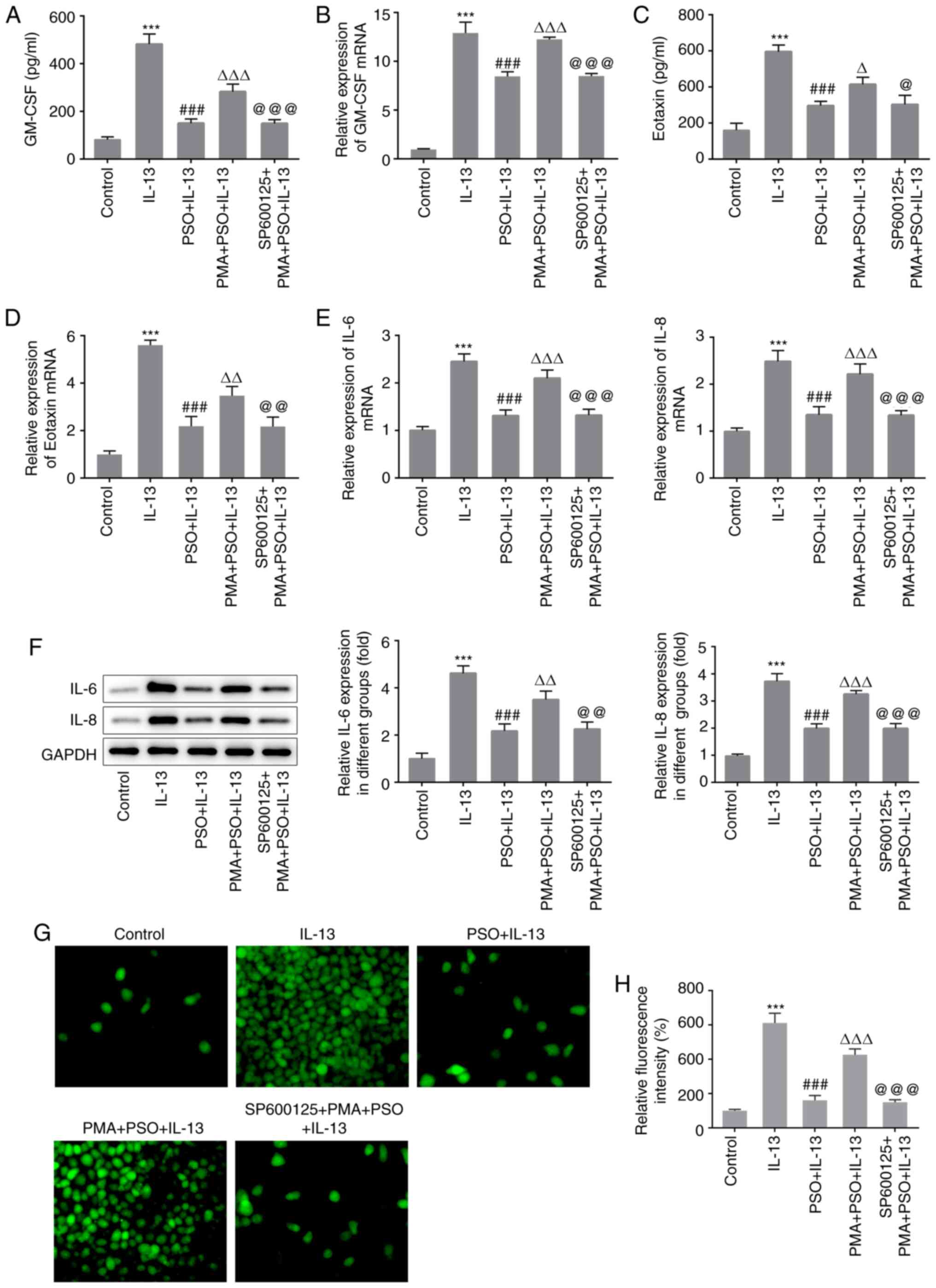
expression of GM-CSF and Eotaxin were also increased (Fig. 4A-D). Furthermore, the expression
levels of IL-6 and −8 (Fig. 4E and
F), ROS (Fig. 4G and H) and
MUC5AC (Fig. 5A and B) were all
increased. These results indicated that PMA reversed the inhibitory
effects of PSO on p-c-FOS, p-c-Jun, IL-13-induced inflammation and
mucus production in JME/CF15 cells. Compared with the PMA + PSO +
IL-13 group, the levels of c-Fos and c-Jun phosphorylation in the
SP600125 + PMA + PSO + IL-13 group were reduced (Fig. 3B), and the expression of GM-CSF,
Eotaxin (Fig. 4A-D), inflammatory
cytokines IL-6 and IL-8 (Fig. 4E and
F), ROS (Fig. 4G and H) and
MUC5AC (Fig. 5A and B) were all
decreased. These results suggested that PSO suppressed inflammation
and mucus production in IL-13-induced JME/CF15 cells by inhibiting
the AP-1 signaling pathway.
PSO downregulates CST1 expression by
inhibiting the AP-1 signaling pathway
CST1 is a targeted regulator of the AP-1 pathway
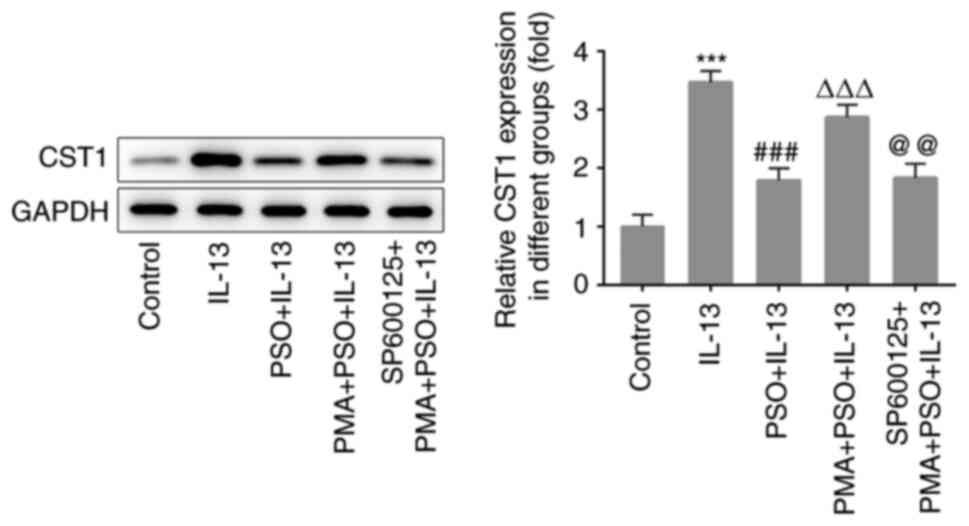
(6). In the present study, the
expression of CST1 in IL-13-induced cells was significantly
increased compared with that in the control group. Compared with
the IL-13 group, the expression of CST1 in the PSO + IL-13 group
was decreased, indicating that PSO inhibited the expression of
CST1. Furthermore, compared with the PSO + IL-13 group, CST1
expression was increased in the PMA + PSO + IL-13 group, and
compared with the PMA + PSO + IL-13 group, CST1 expression was
decreased in the SP600125 + PMA + PSO + IL-13 group (Fig. 6). Collectively, these findings
indicated that PSO downregulated the expression of CST1 by
inhibiting the AP-1 signaling pathway, thus suppressing the
IL-13-induced inflammatory response and mucus production in nasal
mucosal epithelial cells.
Discussion
Since allergic diseases are usually caused by a
variety of inflammatory mediators, the pathological mechanisms
underlying AR are complex. However, IL-13 is a human lymphoid
factor that regulates inflammatory and immune responses, and a
study has reported that IL-4 and −13 produced by T-helper 2 cells
are the primary instigators of AR (26). In addition, IL-13 promotes mucus
secretion and eosinophil production in patients with asthma
(27). Therefore, an IL-13-induced
model has been widely used to conduct basic AR-associated research.
The human JME/CF15 epithelial cell line induced by IL-13 has been
used in previous studies to construct an AR model (28,29).
Therefore, in the current study, IL-13 was used to induce
inflammation and mucus production in human nasal epithelial cells.
In addition, AR mainly refers to the non-infectious inflammatory
disease of nasal mucosa caused by individual exposure to atopic
allergens (1). The primary
manifestation of AR is the inflammatory reaction of cells and the
generation of mucus (2,3). Therefore, the present study focused on
the detection of cellular inflammatory response and mucus
production indicators, in order to determine the severity of
AR.
PSO is widely used in a variety of prescriptions for
tonifying the kidney and strengthening the bones. As such, modern
pharmacological studies have demonstrated the anti-inflammatory,
antibacterial and antioxidant pharmacological effects of PSO, which
exerts therapeutic effects in AR (9,30). In
allergic asthma, PSO can significantly inhibit inflammatory
infiltration and mucus secretion in the lung tissue, and inhibit
cellular IL-13 expression (31). In
mice with periodontitis, PSO dose-dependently reduced mRNA
expression in THP-1 cells, as well as the expression of
inflammatory factors such as IL-8 (7). In the current study, PSO was found to
inhibit the IL-13-induced inflammatory response, oxidative stress
and mucus production. These findings suggested that PSO may have a
significant therapeutic effect on AR.
In the present study, the expression levels of IL-6
and IL-8 were detected in the cells to explore the effect of PSO on
the inflammatory response in IL-13-induced JME/CF15 cells. The IL-6
and IL-8 proteins in the cell supernatant were very small and
difficult to collect, so the expression of IL-6 and IL-8 in the
cells was measured (32). The
expression levels of IL-6 and IL-8 in cells have been detected in
numerous previous studies, in which the expression of IL-6 and IL-8
in the cell supernatant was not detected (32–34).
In addition, it has been reported that IL-13 can
induce the production of ROS in human bronchial epithelial cell
line 16 (35). In the PM2.5-induced
human nasal mucosa epithelial cell model, ROS production was also
significantly increased (36).
Therefore, in the present study, the expression of ROS in JME7CF15
cells induced by IL-13 was detected, and ROS expression was found
to be significantly increased after IL-13 induction, whereas ROS
decreased in a dose-dependent manner after PSO administration.
Initially, the abnormal expression of AP-1 signaling
pathway-related proteins was identified. Furthermore, following
IL-13 induction, AP-1 pathway proteins p-c-Jun and p-c-Fos were
abnormally activated in JME/CF15 cells. A previous literature
review revealed that the combination of activated AP-1 and TREs
promoted the expression of a variety of inflammatory factors, thus
affecting the physiological functions of cells and influencing the
occurrence of certain diseases (11). In addition, AP-1 regulated the
expression of IL-4, −5 and −13 by central effector cells of airway
inflammation in asthma (37). The
results of the present study indicated that the AP-1 signaling
pathway was activated in AR. PSO has been shown to promote
osteoclast differentiation and bone resorption in osteoporosis by
regulating the AP-1 signaling pathway (13). In the current study, the levels of
c-Jun and c-Fos phosphorylation were dose-dependently decreased
following PSO administration. The mechanism by which PSO regulates
AR was further investigated using the AP-1 agonist PMA and the
inhibitor SP600125. The addition of PMA was found to reverse the
inhibitory effects of PSO on IL-13-induced inflammation and mucus
production, while the further addition of SP600125 inhibited these
processes in JME/CF15 cells. A possible explanation for this
finding is that PMA and SP600125 exert opposing effects on the AP-1
pathway (thus inhibiting the effects of each other), while PSO
inhibits AP-1 pathway activation, and thus suppresses IL-13-induced
cellular inflammation and mucus production.
In addition, the aberrant expression of CST1 protein
was also detected in the current study. Using the GEO database,
CST1 protein expression was revealed to be significantly
upregulated with AR (data not shown), which was also verified by
in vitro experimentation. A study previous study indicated
that the AP-1 signaling pathway regulates CST1 expression in
patients with AR (12). In the
present study, PSO was found to inhibit the expression of CST1,
while PMA reversed the inhibitory effects of PSO on CST1. This
indicated that PSO downregulates the expression of CST1 by
inhibiting AP-1 signaling, thus regulating AR.
The present study only investigated the effect of
PSO on IL-13-induced nasal epithelial inflammation at the juvenile
cellular level, the specific effect of PSO on AR at the animal
level was not studied. This is a major limitation of this study.
Our laboratory will further study the specific effect of PSO on AR
in vivo in future experiments.
In conclusion, PSO was found to inhibit the
inflammatory response and mucus production in AR by inhibiting the
AP-1 pathway and the downstream expression of CST1.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YX and WG wrote the manuscript and analyzed the
data. ZJ and YZ carried out the experiments, supervised the present
study, searched the literature and revised the manuscript. All
authors read and approved the final manuscript. YX and WG confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kakli HA and Riley TD: Allergic rhinitis.
Prim Care. 43:465–475. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ri H, Peiyan Z, Jianqi W, Yunteng Z, Gang
L and Baoqing S: Desmoglein 3 gene mediates epidermal growth
factor/epidermal growth factor receptor signaling pathway involved
in inflammatory response and immune function of anaphylactic
rhinitis. Biomed Pharmacother. 118:1092142019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Song CH, Bui TT, Piao CH, Shin HS, Shon
DH, Han EH, Kim HT and Chai OH: Rosae multiflorae fructus hot water
extract inhibits a murine allergic asthma via the suppression of
Th2 cytokine production and histamine release from mast cells. J
Med Food. 19:853–8539. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khan DA: Allergic rhinitis and asthma:
Epidemiology and common pathophysiology. Allergy Asthma Proc.
35:357–361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoyte FCL and Nelson HS: Recent advances
in allergic rhinitis. F1000Res. 7:F1000 Faculty Rev. 13332018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bernstein DI, Schwartz G and Bernstein JA:
Allergic rhinitis: Mechanisms and treatment. Immunol Allergy Clin
North Am. 36:261–278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seo E, Kang H, Oh YS and Jun HS: Psoralea
corylifolia L. Seed extract attenuates diabetic nephropathy by
inhibiting renal fibrosis and apoptosis in streptozotocin-induced
diabetic mice. Nutrients. 9:8282017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li X, Yu C, Hu Y, Xia X, Liao Y, Zhang J,
Chen H, Lu W, Zhou W and Song Z: New application of psoralen and
angelicin on periodontitis with Anti-bacterial, Anti-inflammatory,
and osteogenesis effects. Front Cell Infect Microbiol. 8:1782018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Du MY, Duan JX, Zhang CY, Yang HH, Guan
XX, Zhong WJ, Liu YZ, Li ZM, Cheng YR, Zhou Y and Guan CX: Psoralen
attenuates bleomycin-induced pulmonary fibrosis in mice through
inhibiting myofibroblast activation and collagen deposition. Cell
Biol Int. Jul 22–2019.doi: 10.1002/cbin.11205 (Epub ahead of
print).
|
|
10
|
Wang C, Al-Ani MK, Sha Y, Chi Q, Dong N,
Yang L and Xu K: Psoralen protects chondrocytes, exhibits
anti-inflammatory effects on synoviocytes, and attenuates
monosodium iodoacetate-induced osteoarthritis. Int J Biol Sci.
15:229–238. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Trop-Steinberg S and Azar Y: AP-1
expression and its clinical relevance in immune disorders and
cancer. Am J Med Sci. 353:474–483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gazon H, Barbeau B, Mesnard JM and
Peloponese JM Jr: Hijacking of the AP-1 signaling pathway during
development of ATL. Front Microbiol. 8:26862018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bejjani F, Evanno E, Zibara K, Piechaczyk
M and Jariel-Encontre I: The AP-1 transcriptional complex: Local
switch or remote command? Biochim Biophys Acta Rev Cancer.
1872:11–23. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choi WJ: The heterochromatin-1
phosphorylation contributes to TPA-induced AP-1 expression. Biomol
Ther (Seoul). 22:308–313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Uluçkan Ö, Guinea-Viniegra J, Jimenez M
and Wagner EF: Signalling in inflammatory skin disease by AP-1
(Fos/Jun). Clin Exp Rheumatol. 33 (4 Suppl 92):S44–S49. 2015.
|
|
16
|
Liu X, Yin S, Chen Y, Wu Y, Zheng W, Dong
H, Bai Y, Qin Y, Li J, Feng S and Zhao P: LPSinduced
proinflammatory cytokine expression in human airway epithelial
cells and macrophages via NFκB, STAT3 or AP1 activation. Mol Med
Rep. 17:5484–5491. 2018.PubMed/NCBI
|
|
17
|
Giovannini-Chami L, Marcet B, Moreilhon C,
Chevalier B, Illie MI, Lebrigand K, Robbe-Sermesant K, Bourrier T,
Michiels JF, Mari B, et al: Distinct epithelial gene expression
phenotypes in childhood respiratory allergy. Eur Respir J.
39:1197–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lei Y, Guo P, An J, Guo C, Lu F and Liu M:
Identification of pathogenic genes and upstream regulators in
allergic rhinitis. Int J Pediatr Otorhinolaryngol. 115:97–103.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chai L, Zhou K, Wang S, Zhang H, Fan N, Li
J, Tan X, Hu L and Fan X: Psoralen and bakuchiol ameliorate M-CSF
plus RANKL-induced osteoclast differentiation and bone resorption
via inhibition of AKT and AP-1 pathways in vitro. Cell Physiol
Biochem. 48:2123–2133. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsukura S, Stellato C, Georas SN,
Casolaro V, Plitt JR, Miura K, Kurosawa S, Schindler U and
Schleimer R: Interleukin-13 upregulates eotaxin expression in
airway epithelial cells by a STAT6-dependent mechanism. Am J Respir
Cell Mol Biol. 24:755–761. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wills-Karp M: Interleukin-13 in asthma
pathogenesis. Immunol Rev. 202:175–190. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chun HW, Kim SJ, Pham TH, Bak Y, Oh J, Ryu
HW, Oh SR, Hong JT and Yoon DY: Epimagnolin A inhibits IL-6
production by inhibiting p38/NF-κB and AP-1 signaling pathways in
PMA-stimulated THP-1 cells. Environ Toxicol. 34:796–803. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anuchapreeda S, Rungrojsakul M, Tima S,
Chiampanichayakul S and Krig SR: Co-activation of WT1 and AP-1
proteins on WT1 gene promoter to induce WT1 gene expression in K562
cells. Cell Signal. 53:339–347. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Lv Q, Song X, Jiang K and Zhang J:
ADRB2 suppresses IL-13-induced allergic rhinitis inflammatory
cytokine regulated by miR-15a-5p. Hum Cell. 32:306–315. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wills-Karp M, Luyimbazi J, Xu X, Schofield
B, Neben TY, Karp CL and Donaldson DD: Interleukin-13: Central
mediator of allergic asthma. Science. 282:2258–2261. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuperman DA, Huang X, Koth LL, Chang GH,
Dolganov GM, Zhu Z, Elias JA, Sheppard D and Erle DJ: Direct
effects of interleukin-13 on epithelial cells cause airway
hyperreactivity and mucus overproduction in asthma. Nat Med.
8:885–889. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao Y and Yu Z: MicroRNA16 inhibits
interleukin13induced inflammatory cytokine secretion and mucus
production in nasal epithelial cells by suppressing the IκB kinase
β/nuclear factor-κB pathway. Mol Med Rep. 18:4042–4050.
2018.PubMed/NCBI
|
|
29
|
Wang B, Gao Y, Zheng G, Ren X, Sun B, Zhu
K, Luo H, Wang Z and Xu M: Platycodin D inhibits
interleukin-13-induced the expression of inflammatory cytokines and
mucus in nasal epithelial cells. Biomed Pharmacother. 84:1108–1112.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li JP, Xie BP, Zhang WJ, Shi LY, Li WJ,
Zeng Y, Gan GX and Li YH: Psoralen inhibits RAW264.7
differentiation into osteoclasts and bone resorption by regulating
CD4+T cell differentiation. Zhongguo Zhong Yao Za Zhi.
43:1228–1234. 2018.(In Chinese). PubMed/NCBI
|
|
31
|
Jin H, Wang L, Xu C, Li B, Luo Q, Wu J, Lv
Y, Wang G and Dong J: Effects of Psoraleae fructus and its major
component psoralen on Th2 response in allergic asthma. Am J Chin
Med. 42:665–678. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li K, Zhang F, Wei L, Han Z, Liu X, Pan Y,
Guo C and Han W: Recombinant human elafin ameliorates chronic
hyperoxia-induced lung injury by inhibiting nuclear Factor-Kappa B
signaling in neonatal mice. J Interferon Cytokine Res. 40:320–330.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu S, Dai J and Chen X: Vitamin D reduces
autophagy by regulating NF-κB resistance to Aspergillus fumigatus
infection. Gene. 753:1448192020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu Q, Xu J, Zhang K, Zhong M, Cao H, Wei
R, Jin L and Gao Y: Study on the protective effect and mechanism of
Dicliptera chinensis (L.) Juss (Acanthaceae) polysaccharide on
immune liver injury induced by LPS. Biomed Pharmacother.
134:1111592021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang D, Li Q, Kolosov VP and Zhou X: The
inhibition of aldose reductase on mucus production induced by
interleukin-13 in the human bronchial epithelial cells. Int
Immunopharmacol. 12:588–593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hong Z, Guo Z, Zhang R, Xu J, Dong W,
Zhuang G and Deng C: Airborne fine particulate matter induces
oxidative stress and inflammation in human nasal epithelial cells.
Tohoku J Exp Med. 239:117–125. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Khorasanizadeh M, Eskian M, Gelfand EW and
Rezaei N: Mitogen-activated protein kinases as therapeutic targets
for asthma. Pharmacol Ther. 174:112–1126. 2017. View Article : Google Scholar : PubMed/NCBI
|