Introduction
Alcohol abuse and alcohol-related problems have been
significant public health issues in a number of countries for
several decades (1). Chronic
alcohol consumption not only affects the central nervous system,
but adversely affects other organs, including the lung (2). Compared with non-alcoholic
individuals, patients with a history of alcohol abuse have enhanced
susceptibility to lung injury. This includes a 2–4-fold risk of
developing acute respiratory distress syndrome (ARDS), which may
affect hospitalization costs, the incidence of intensive care
unit-related mortality and poor patient outcomes (3–5).
During the coronavirus (COVID-19) pandemic, there has been growing
concern over the impact of excessive alcohol consumption in
patients with COVID-19 and alcohol use disorder (6). Chronic alcohol consumption reduces the
immunity of patients to viral and bacterial infections, which may
increase the infection and mortality rates of COVID-19 (7). ARDS is a severe form of respiratory
failure that is life threatening, which has demonstrated an overall
mortality rate of 45% in patients that have been admitted to
hospital since 2010 (8). To the
best of the authors' knowledge, the molecular mechanisms underlying
the association between alcohol abuse and ARDS have yet to be fully
elucidated and no specific therapies are currently available to
treat or decrease the risk of lung injury in patients suffering
from alcoholism.
ARDS, which develops from acute lung injury (ALI),
is characterized by alveolar-capillary barrier injury, resulting in
the accumulation of protein-rich fluid in the alveolar spaces that
impairs gas exchange. This leads to severe hypoxemia and acute
respiratory failure (9). Resolution
of ALI/ARDS requires clearance of excess edema fluid in the
alveolar spaces and repair of the alveolar-capillary barrier
(10). Alveolar edema is resolved
by active salt and water transport through epithelial sodium
channels (ENaC) which provide a driving force to remove edema fluid
from the alveolar spaces (11,12).
ENaC is a multimeric protein composed of three homologous subunits,
α, β and γ (13). ENaC is the
rate-limiting step in the resolution of lung edema; mice lacking
α-ENaC, β-ENaC or γ-ENaC genes are unable to clear alveolar edema
fluid, which indicates the important function of ENaC in alveolar
fluid clearance (AFC) (14–16).
Alcohol ingestion increases the systemic levels of
extracellular adenosine by inhibiting adenosine uptake via the
nucleoside transporter (17,18).
From a previous study, regulation of ENaC expression by adenosine
occurs via the A2 adenosine receptor (AR) pathway (19). However, to the best of the authors'
knowledge, the association between alcohol and AFC and the role of
ENaC in ALI have yet to be fully elucidated. The present study
aimed to determine the role of alcohol in AFC inhibition and the
role of A2 AR signaling through the ENaC in lipopolysaccharide
(LPS)-induced lung injury.
Materials and methods
Animal experiments and materials
Adult female C57BL/6 mice (n=66; Chongqing Medical
University; weight, 20–25 g; age, 8–10 weeks) and male
Sprague-Dawley rats (n=27; Chongqing Medical University; weight,
200–240 g; age, 5–7 weeks) were used in the present study in
accordance with the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health. All animals were
housed under specific pathogen-free conditions in a temperature
(18–25°C)-and humidity (40–60%)-controlled environment with a 12-h
light/dark cycle and given free access to food and water. All
surgery was performed under anesthesia with sodium pentobarbital
and all efforts were made to minimize suffering. The study was
approved by the Ethics Committee of The Second Affiliated Hospital
of Chongqing Medical University (approval no. 2019-009). Ethyl
alcohol (C2H5OH) was purchased from
North-South Chemical, China. LPS (Escherichia coli serotype
O111:B4) was purchased from Sigma-Aldrich (Merck KGaA). The α-ENaC
antibody (cat. no. PA1-920A) was purchased from Thermo Fisher
Scientific, Inc. and β-ENaC (cat. no. YT1551) and γ-ENaC (cat. no.
YT5032) antibodies were purchased from ImmunoWay Biotechnology
Company. AR antibodies (cat. nos. PA1-042 and PA5-72850) were
purchased from Invitrogen (Thermo Fisher Scientific, Inc.).
CGS-21680 (a specific agonist of A2aAR), Bay 60-6583 (a specific
agonist of A2bAR) and cyclic adenosine monophosphate (cAMP)
inhibitor [(R)-Adenosine, cyclic 3′,5′-(hydrogenphosphorothioate)
triethylammonium] were purchased from Tocris Bioscience.
Animal models
Mice were administered alcohol intraperitoneally
(i.p.; 4 g/kg; 5% v/v in sterile water) for the first week of the
experiment until 20% v/v alcohol was reached. Beginning on the
fourth week, mice were administered with 5% incremental doses at
each subsequent week. Mice remained on a 20% v/v alcohol diet for
an additional four weeks to establish a chronic alcohol consumption
model (20,21). This approach had previously been
reported to mimic alcohol abuse in a mouse model for 11 weeks
(22). The control group of mice
were administered an equivalent volume of sterile water. The
alcohol and control mice were given the same diet. Mice were
anesthetized by intraperitoneal administration of sodium
pentobarbital (Sigma-Aldrich; Merck KGaA) at a dose of 50 mg/kg.
The ALI model was established by LPS i.p. injection (10 µg/g) three
days after the establishment of the chronic alcohol consumption
model. The vital signs and behaviors of mice were monitored in
every 1–2 days. Mice were sacrificed and the blood and lung tissue
were collected 24 h after LPS administration. Then, mice were
sacrificed by exsanguination from the bleeding of carotid artery
(mortality was confirmed by disappearance of breath and nerve
reflex as well as muscle relaxation).
Primary cell culture and
treatment
Rats were sacrificed by exsanguination at the
carotid artery. Alveolar epithelial type II (ATII) cells were
isolated from male Sprague-Dawley rats by elastase digestion of
lung tissue and incubated in a humidified atmosphere of 5%
CO2 and 95% air at 37°C as previously described
(23–25). Cells were cultured in DMEM
(Sigma-Aldrich; Merck KGaA) supplemented with 10% fetal bovine
serum (Sigma-Aldrich; Merck KGaA), 100 U/ml penicillin and 0.1
mg/ml streptomycin. Freshly isolated alveolar epithelial cells were
treated with 0.20% v/v ethanol for 72 h. Cells were subsequently
incubated with CGS-21680, Bay 60-6583 or cAMP inhibitor for
subsequent western blotting experiments.
Small interfering (si)RNA
transfection
siRNA targeting A2aAR or A2bAR and the negative
control were obtained from Ambion (Thermo Fisher Scientific, Inc.)
The following pairs of primers were used: A2a AR siRNA forward,
5′-GACGGGAACUCCACGAAGATT-3′ and reverse,
5′-UCUUCGUGGAGUUCCCGUCTT-3′; A2b AR siRNA forward,
5′-ACCTCAACCGAGACTTCCGCT-3′ and reverse,
5′-CAAAAAAACCGAGACTTCCGC-3′; Negative control siRNA forward,
5′-GAAUUAAUUAAAGAUGGCCCG-3′ and reverse,
5′-UCAUCGAAGUUAUAGGGAUAC-3′. Intratracheal delivery of specific
siRNA for A2aAR or A2bAR was performed as previously described
(26). Briefly, 100 µg A2aAR siRNA
or A2bAR siRNA (in 100 µl saline) was administered directly into
the throat of each mouse and the mouth was immediately kept closed
for 2 h to ensure delivery of siRNA to the lung. The control group
were transfected with non-targeting siRNA in an equivalent volume
of sterile water. Cells (9×106) mixed with 100 nmol/l
siRNA/106 cells were transfected using
Lipofectamine® reagent incubated for 15 min at room
temperature (Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's protocol. Following transfection, cells were
incubated with culture medium (DMEM with 10% fetal bovine serum,
100 U/ml penicillin and 0.1 mg/ml streptomycin) and plated onto
cell culture plates at a seeding density of 3×106 cells.
After 48 h transfection, cell lysates were prepared for subsequent
western blotting.
cAMP accumulation assay
Lung tissues were homogenized in 6% trichloroacetic
acid (1 ml trichloroacetic acid/100 mg tissue), centrifuged at
1,000 × g and 4°C for 15 min and extracted with water-saturated
diethyl ether. Cells were seeded into 96-well plates in 100 µl
DMEM. After 24 h, the medium was removed and cells were washed
three times with 100 µl DMEM containing 50 mM HEPES (pH 7.4). Cells
were subsequently incubated with ethanol at 37°C in 5%
CO2. The concentration of cAMP in lung and alveolar
epithelial cells was evaluated using the cAMP Biotrak Enzyme
Immunoassay (Amersham Biosciences; Cytiva). according to the
manufacturer's protocol.
Pulmonary edema evaluation
The ratios of wet lung weight to dry lung weight
were calculated as a measure of pulmonary edema. The lungs of mice
were filled with 100 µl saline via intratracheal instillation. The
lungs were excised and weighed 30 min following instillation. The
lungs were subsequently dried at 60°C for 48 h and weighed again to
obtain the wet:dry weight ratios.
Bronchoalveolar epithelial
permeability assessment
Bronchoalveolar epithelial permeability was
determined by bronchoalveolar lavage fluid (BALF)/serum
fluorescence as previously described (27). Briefly, mice were injected with
fluorescein isothiocyanate-conjugated dextran 4000 (FD4;
Sigma-Aldrich; Merck KGaA) solution in PBS (10 mg/kg) via the
internal jugular vein 24 h after LPS-induced lung injury. BALF was
collected by instillation of saline into the lungs three times. The
serum was collected from the internal jugular vein by
centrifugation at 1,500 × g and 4°C for 5 min. The concentrations
of FD4 in BALF and serum were determined by a spectrofluorometer
(Beckman Coulter, Inc.).
Measurement of inflammatory
cytokines
An endotracheal catheter was inserted into the lungs
following instillation of saline five times. BALF was collected
following five rounds of extraction and centrifuged at 4°C for 15
min at 500 × g. The supernatant was used to assay TNF-α (cat. no.
MTA00B) and IL-6 (cat. no. M6000B) using ELISA kits (R&D
Systems, Inc.) according to the manufacturers' protocols.
Lung injury evaluation
The right lungs were harvested and fixed using 10%
neutral buffered formalin for 24 h. The fixed tissues were embedded
in paraffin and sectioned at 5 µm. Sections were mounted onto glass
slides for further staining with hematoxylin and eosin. Briefly,
the slices were soaked with 80–100% ethanol, followed by 0.6%
hematoxylin (15 min at room temperature) and 1.0% eosin (5 min at
room temperature) dyeing, then dehydrated with 80–100% ethanol.
Lung injury was evaluated for intra-alveolar exudate, interstitial
edema, alveolar hemorrhage and inflammatory cell infiltration using
the lung injury scoring system (28). Assessment of histological lung
injury was performed by grading as follows: A, neutrophils in the
alveolar space; B, neutrophils in the interstitial space; C,
hyaline membranes; D, proteinaceous debris filling the airspaces;
and E, alveolar septal thickening. The sum of each of the five
independent variables was calculated and then normalized to the
number of the fields evaluated (total score: 0–2).
AFC measurement
The rate of AFC was measured by Evans's blue-labeled
albumin as previously described (29,30).
Briefly, after mice were completely sedated with diazepam (5 mg/kg;
i.p.) and pentobarbital (50 mg/kg; i.p.), a tracheostomy tube was
inserted. Mice were ventilated at a rate of 200 breaths/min with a
tidal volume of 150 ml and inspired oxygen concentration of 100%.
Physiological saline solution (5 ml/kg) containing 5% albumin and
0.15 mg/ml Evans blue dye (Sigma-Aldrich; Merck KGaA) was instilled
into the tracheostomy tube. Mice were ventilated for 30 min after
alveolar fluid was aspirated. The concentration of Evans
blue-labeled albumin in the injected and aspirated solutions were
measured using a spectrophotometer (Beckman Coulter, Inc.). AFC was
calculated as follows: AFC=[(Vi-Vf)/Vi] ×100, to obtain a
percentage, where Vf is (Vi × Pi)/Pf. V represents the injected (i)
volume and final (f) volume of alveolar fluid. P represents the i
and f concentrations of Evans blue-labeled 5% albumin solution.
Immunohistochemistry
Tissue slices were blocked with BSA (Sigma-Aldrich;
Merck KGaA) for 30 min at room temperature and incubated with
α-ENaC (1:200), β-ENaC (1:300) and γ-ENaC (1:200) antibodies at 4°C
for 24 h. Subsequently, tissues were incubated in biotinylated
anti-rabbit IgG (1:400; cat. no. sc-2491; Santa Cruz Biotechnology,
Inc.) for 30 min at 37°C, followed by avidin-biotin-peroxidase
complex (Sigma-Aldrich; Merck KGaA) for 30 min at 37°C and stained
with 3,3′-diaminobenzidine (Sigma-Aldrich; Merck KGaA) for 5 min at
room temperature. Normal rabbit isotype IgG (1:400; cat. no.
sc-2027; Santa Cruz Biotechnology, Inc.) was used as a negative
control. The number of positive cells was counted using a light
microscopy imaging system (magnification, ×400; Olympus
Corporation).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from lung and alveolar
epithelial cells (3×106) using the RNA isolation kit
(Takara Bio, Inc.) according to the manufacturer's protocol. The
following pairs of primers were used: α-ENaC forward,
5′-GCTCAACCTTGACCTAGACCTTG-3′ and reverse,
5′-CGCCTGTTCTTCACGCTTGT-3′; β-ENaC forward,
5′-GTTCCTGCTTACGCTGCTCTTC-3′ and reverse,
5′-GTCCTGGTGGTGTTGCTGTG-3′; γ-ENaC forward,
5′-TGCTGTGAGTGACCTCCTGAC-3′ and reverse,
5′-TTCCGCTTCCGACCAGTGAA-3′; and GAPDH forward,
5′-CAAGGTCATCCATGACAACTT-3′ and reverse,
5′-GGCCATCCACAGTCTTCTGG-3′. RNA was reverse transcribed into cDNA
using the HiScript 1st Strand cDNA Synthesis kit (Vazyme Biotech
Co., Ltd.). qPCR was performed using a SYBR Green PCR kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The PCR amplification
reaction was conducted at 94°C for 60 sec, followed by 30 cycles at
94°C for 30 sec, then 53°C (α-ENaC), 53°C (β-ENaC), 55°C (γ-ENaC)
or 55°C (β-actin) for 30 sec and finally 72°C for 60 sec. PCR
amplification were performed using a 7500 Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). For each
experiment, the expression levels of the indicated genes were
normalized to endogenous GAPDH expression levels and calculated
using the 2−ΔΔCq method (31). A total of 3–6 repeats was
performed.
Western blotting
Mouse lung tissue was perfused with PBS and placed
on ice with 1 ml lysis buffer and 1 ml extraction buffer (Nanjing
KeyGen Biotech, Co., Ltd.). Alveolar epithelial cells were washed
in ice-cold PBS and solubilized in 1 ml lysis buffer. The lysates
were cleared by centrifugation for 10 min at 4°C and 14,000 × g All
samples were prepared for quantification of the target proteins by
using a protein extraction kit (Nanjing KeyGen Biotech Co., Ltd.)
according to the manufacturer's instructions. The concentration of
each protein sample was determined using a BCA protein assay kit.
Samples containing equal amounts (75–100 µg) of proteins were
separated by 10% SDS-PAGE and transferred to PVDF membranes using a
semi-dry transfer apparatus (Bio-Rad Laboratories, Inc.). After
blocking with 5% skimmed milk in TBS containing 0.05% Tween-20 at
room temperature, the membranes were incubated with α-ENaC antibody
(1:1,000), β-ENaC antibody (1:1,000), γ-ENaC antibody (1:1,000) and
β-actin (1:500) at 4°C. Following the primary incubation, membranes
were incubated with an HRP-conjugated secondary antibody (1:5,000;
cat. no. bs-0295M; BIOSS, Inc.) at room temperature and protein
bands were detected using an Enhanced Chemiluminescence method
(Nanjing KeyGen Biotech, Co., Ltd.). The protein bands were
visualized using a UVP Gel imaging system (Analytik Jena AG) and
analyzed by Quantity One software (version 4.4, Bio-Rad
Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical differences between multiple groups were analyzed using
a one-way ANOVA followed by Tukey post hoc test. Single comparisons
were performed using a Student's unpaired t-test. Statistical
analysis was performed using GraphPad 5.0 Prism software (GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
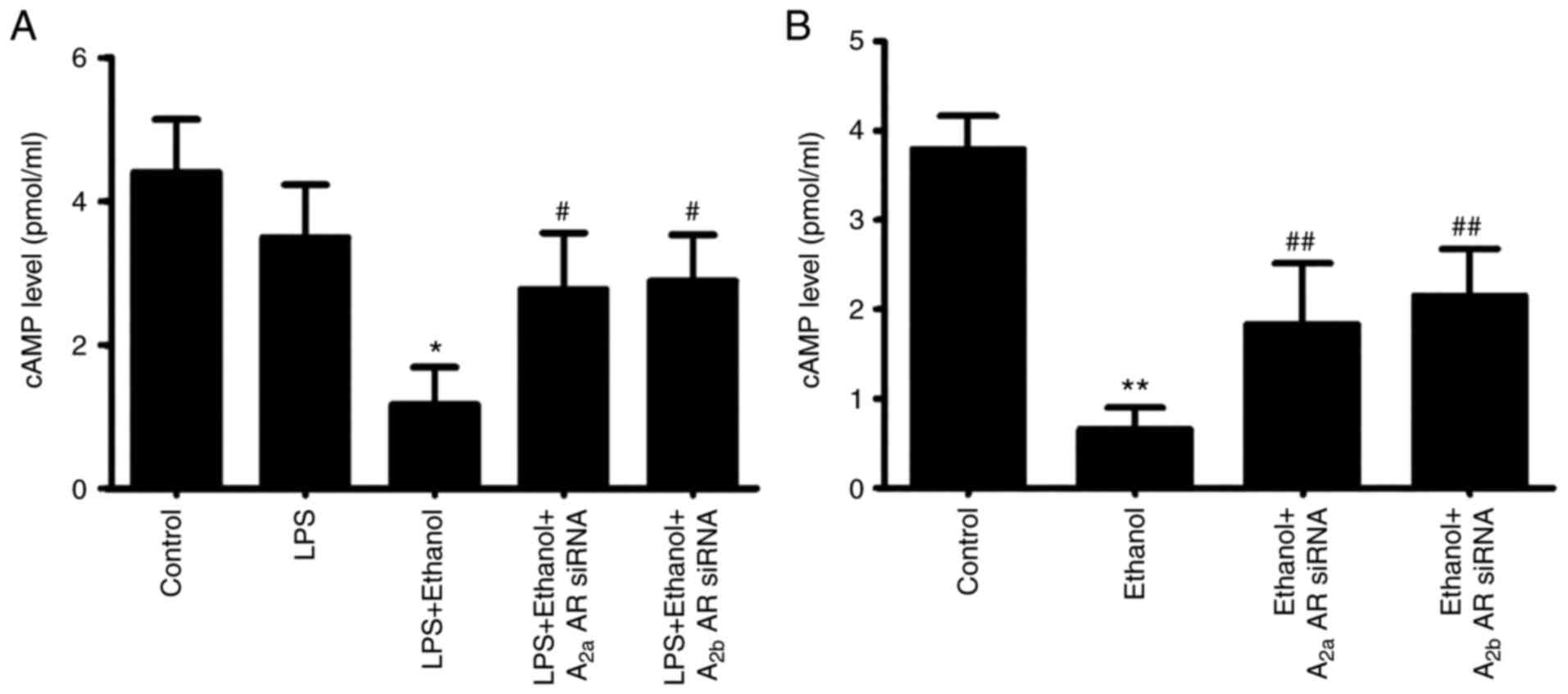
Alcohol inhibits cAMP accumulation in
vivo and in vitro
The concentration of cAMP was significantly
decreased in cells with LPS-induced injury following chronic
alcohol ingestion, but the effect of alcohol on cAMP concentration
was inhibited by co-instillation of A2aAR or A2bAR siRNA (Fig. 1A). Following incubation with
alcohol, the concentration of cAMP was also significantly decreased
in alveolar epithelial cells. The ethanol-induced decrease in cAMP
concentration was significantly blocked by A2aAR or A2bAR siRNA
(Fig. 1B).
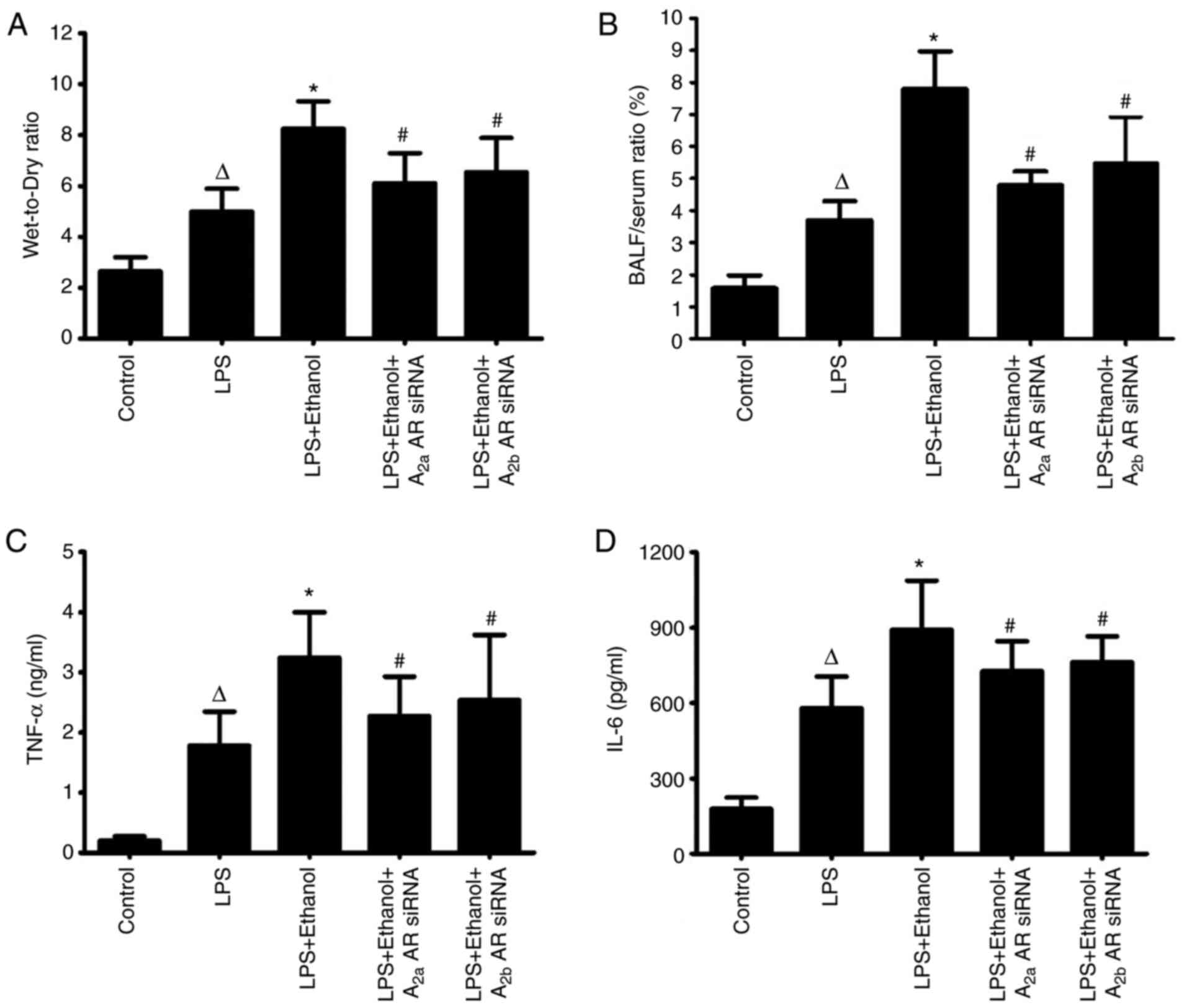
Alcohol aggravates lung pulmonary
edema in LPS-induced lung injury
Wet:dry lung weight ratio was significantly
increased in mice with LPS-induced lung injury following alcohol
ingestion. Following co-instillation of A2aAR or A2bAR siRNA, the
wet: dry lung weight ratio was significantly decreased, indicating
the alleviation of pulmonary edema in LPS-induced lung injury
(Fig. 2A).
Alcohol disrupts the pulmonary
epithelial barrier and increases the release of inflammatory
mediators in LPS-induced lung injury
The pulmonary epithelial barrier was disrupted in
mice with LPS-induced lung injury following alcohol ingestion, with
a significantly high bronchoalveolar epithelial permeability.
However, the bronchoalveolar epithelial permeability was decreased
by A2aAR or A2bAR siRNA in LPS-induced lung injury following
chronic alcohol treatment. This indicated the potential role of
alcohol in altering the function of the pulmonary epithelial
barrier (Fig. 2B). The levels of
TNF-α and IL-6 in the BALF were markedly increased in LPS-induced
lung injury compared with the control group. Alcohol ingestion
significantly contributed to the release of inflammatory mediators
during LPS-induced lung injury. The level of these inflammatory
mediators was decreased following co-instillation of A2aAR or A2bAR
siRNA (Fig. 2C and D).
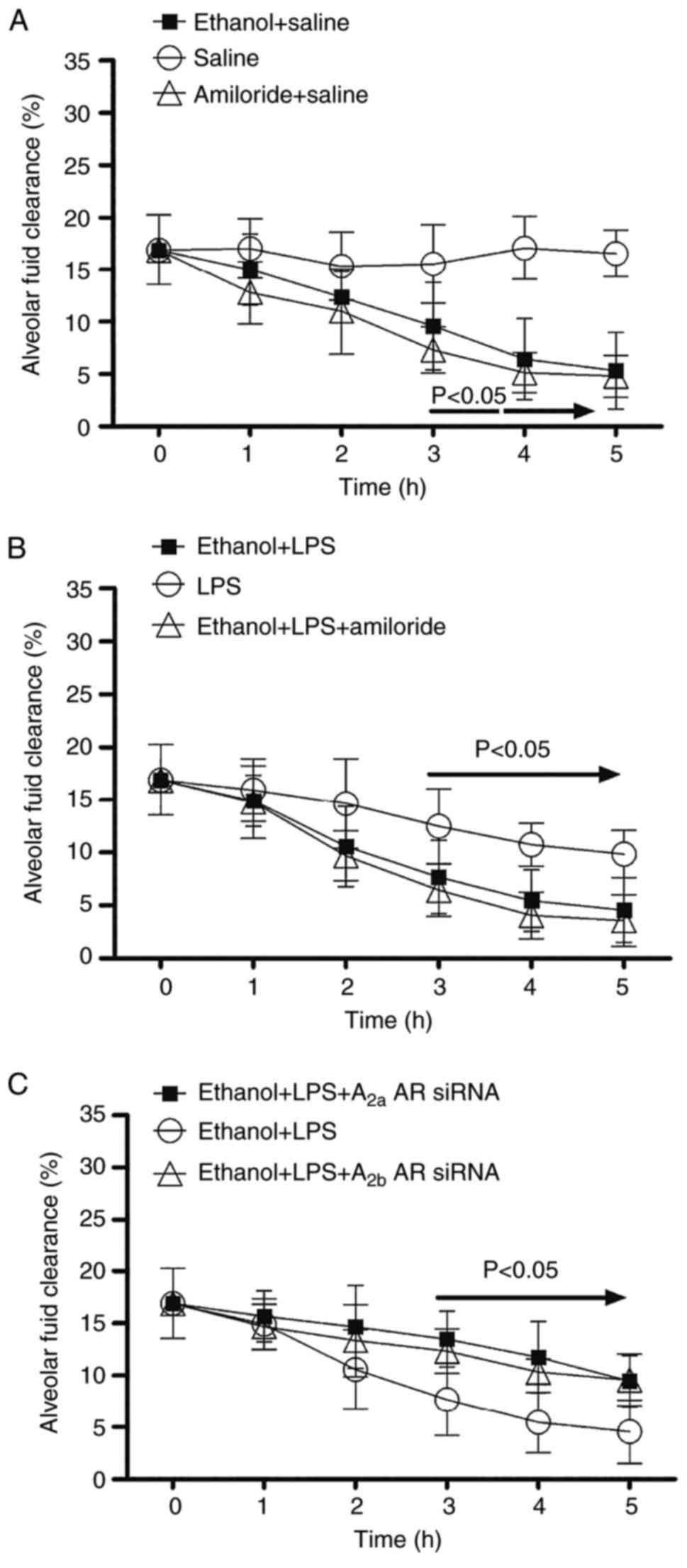
Alcohol inhibits AFC in LPS-induced
lung injury
Following alcohol ingestion or amiloride treatment,
the rate of AFC in mice was significantly decreased at 3 h,
compared with mice with saline treatment (Fig. 3A). Mice with LPS-induced lung injury
treated with alcohol or a combination of alcohol and amiloride
demonstrated a significantly decreased AFC at 3 h compared with the
control group (Fig. 3B). Following
co-administration of A2aAR or A2bAR siRNA, the rate of AFC was
significantly increased at 3 h in mice with LPS-induced lung injury
following alcohol ingestion compared with the control group
(Fig. 3C).
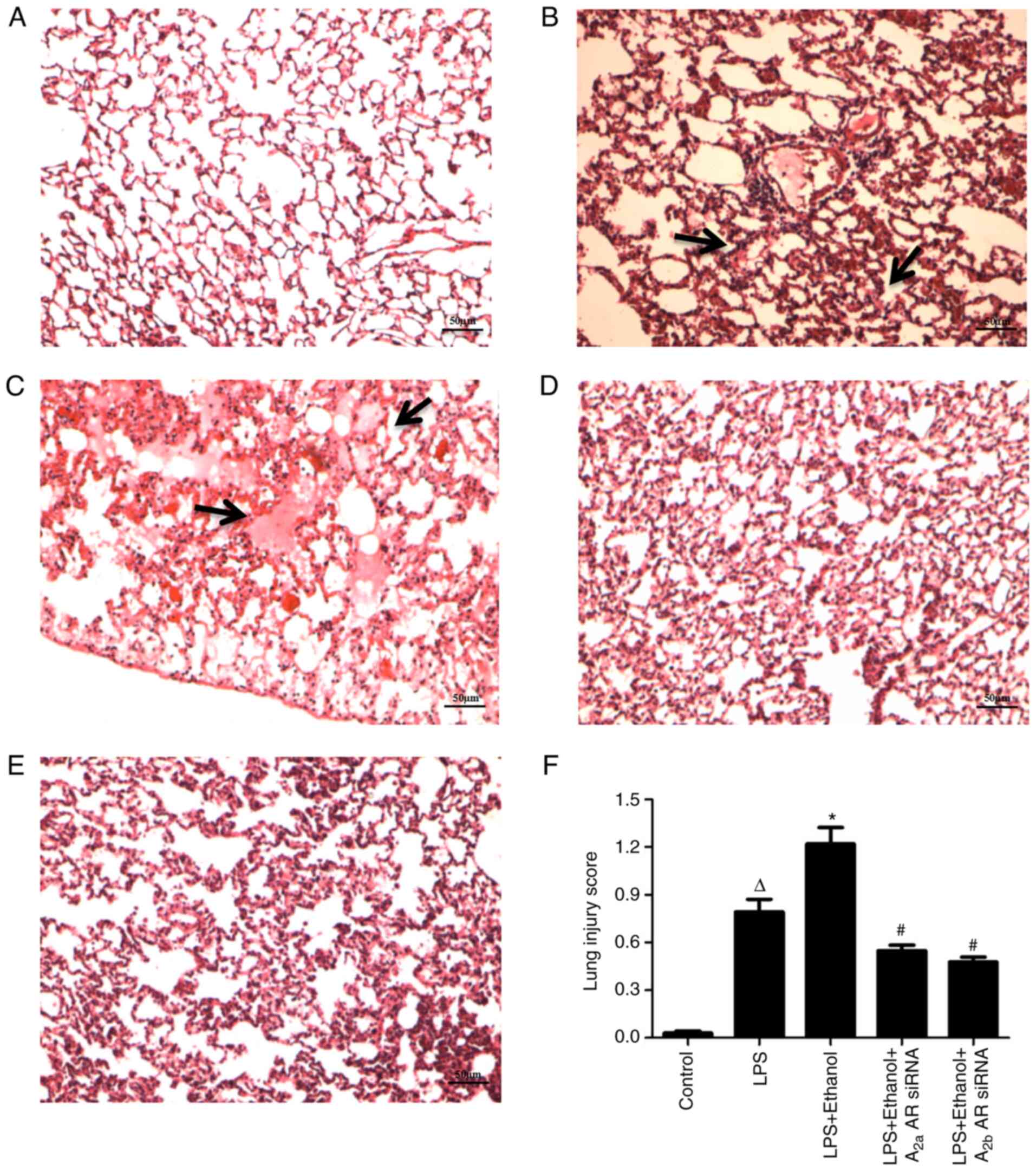
Alcohol aggravates LPS-induced lung
injury
The lung tissue was significantly injured, as
indicated by the presence of intra-alveolar exudate and
inflammatory cell infiltration following LPS treatment (Fig. 4B-F). The presence of marked
intra-alveolar edema with proteinaceous debris filling the
airspaces and inflammation infiltration in the alveolar spaces was
significantly upregulated following alcohol administration in the
LPS-induced ALI model (Fig. 4C-F).
Lung injury was significantly attenuated by A2aAR or A2bAR siRNA
(Fig. 4D-F). The pathobiology of
lung injury was quantified using the lung injury score, which was
determined for each group (Fig.
4F).
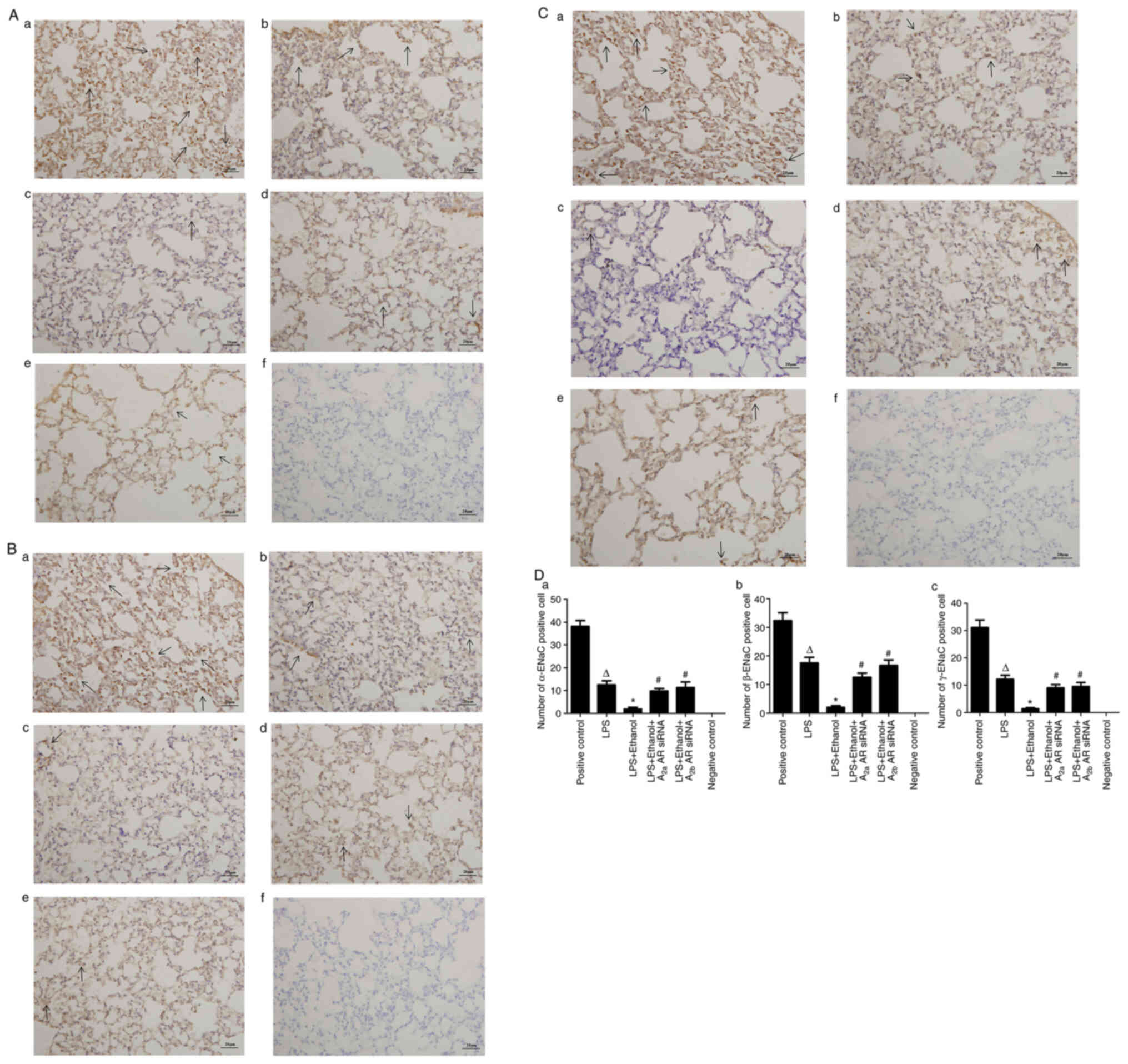
Alcohol decreases ENaC localization in
LPS-induced lung injury
Immunohistochemical analysis was used to determine
the lung distribution of α-ENaC (Fig.
5Aa-f), β-ENaC (Fig. 5Ba-f) and
γ-ENaC (Fig. 5Ca-f) in the lung
tissue of mice. The number of cells expressing α-ENaC, β-ENaC or
γ-ENaC were significantly decreased in LPS-induced lung injury
following alcohol administration, compared with those with ALI
alone. However, the number of cells expressing α-ENaC, β-ENaC or
γ-ENaC were significantly increased by A2aAR or A2bAR siRNA
(Fig. 5Da, Db and Dc).
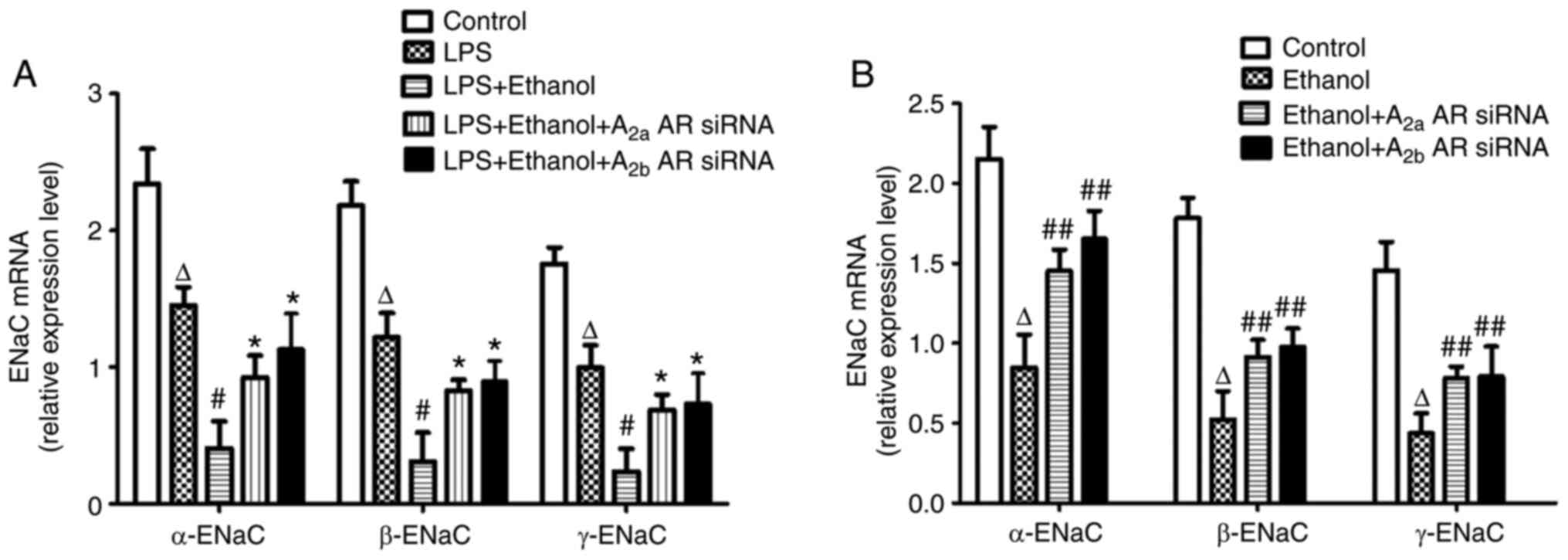
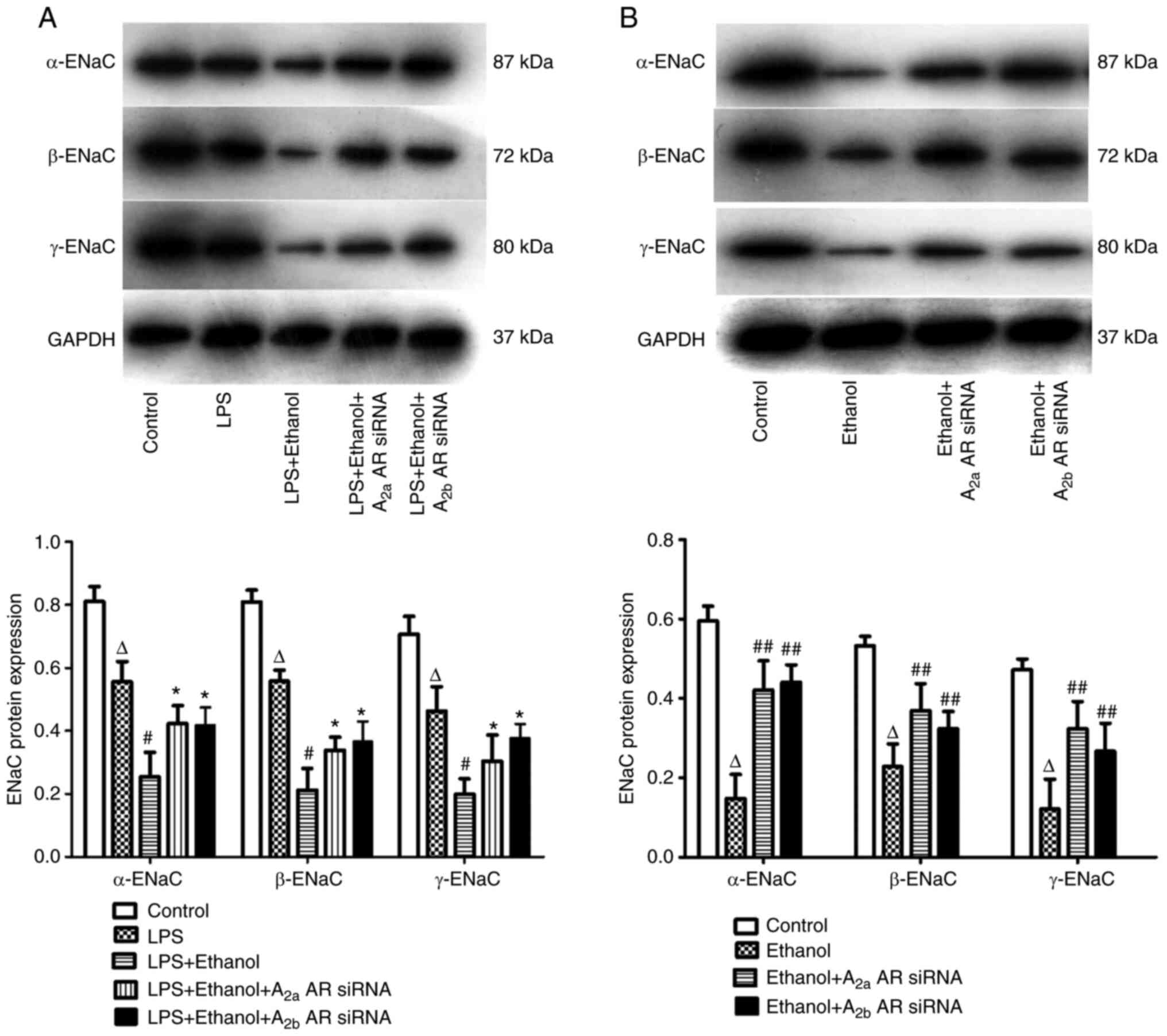
Alcohol inhibits ENaC mRNA and protein
expression levels in LPS-induced lung injury
In vivo, the mRNA and protein expression
levels of α-ENaC, β-ENaC and γ-ENaC in the lung tissue of mice were
significantly decreased in LPS-induced lung injury following
chronic alcohol treatment. However, the mRNA expression levels of
α-ENaC, β-ENaC and γ-ENaC were significantly increased by A2aAR or
A2bAR siRNA (Figs. 6A and 7A). In vitro, the mRNA and protein
expression levels of α-ENaC, β-ENaC and γ-ENaC were significantly
decreased following alcohol treatment, but the effect of alcohol on
ENaC mRNA expression levels was prevented by A2aAR or A2bAR siRNA
(Figs. 6B and 7B).
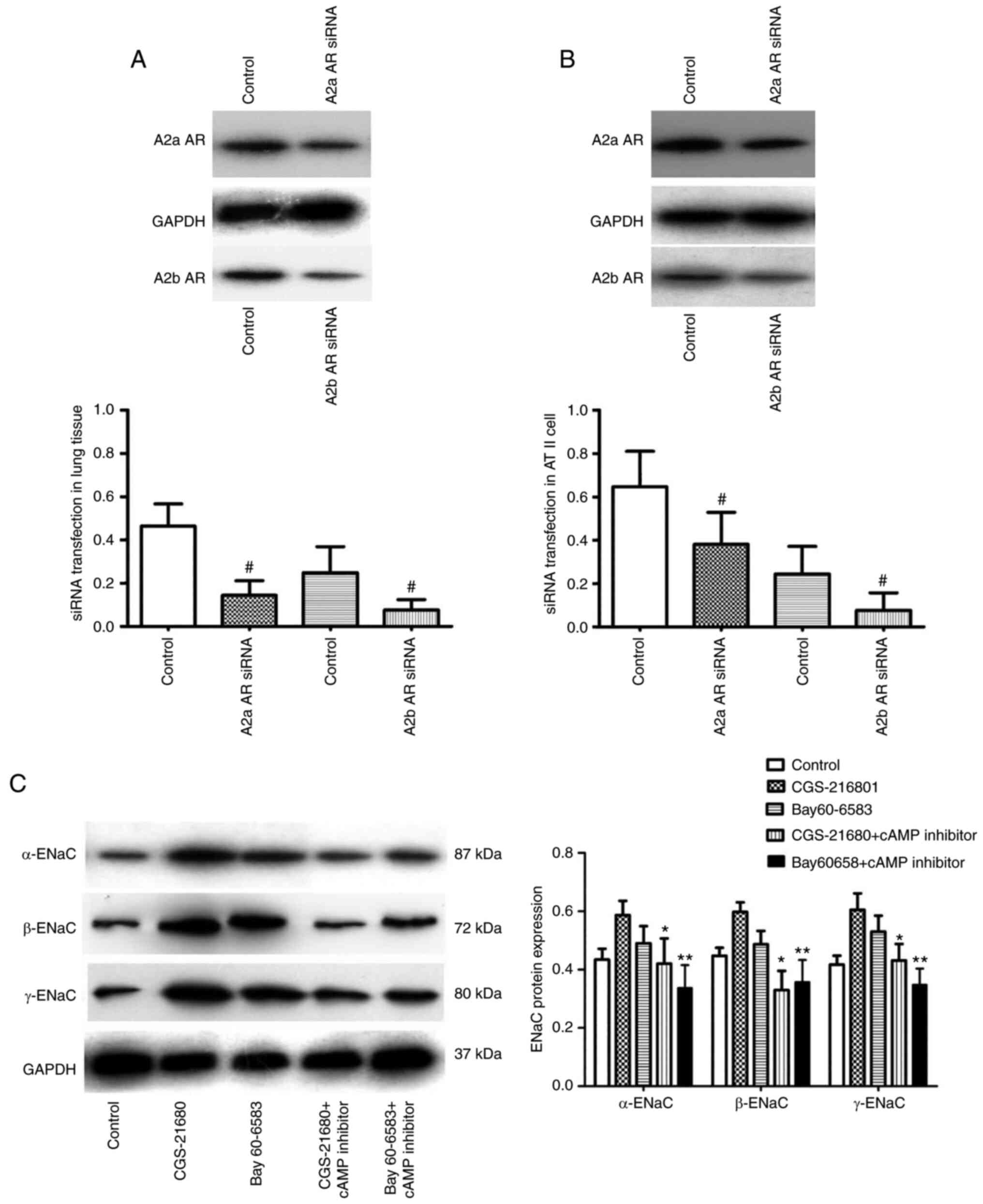
Alcohol regulates ENaC protein
expression levels by the A2aAR or A2bAR-mediated cAMP pathway
The transfection efficiency of A2aAR or A2bAR siRNA
was determined in vivo and in vitro. The successful
transfection of A2aAR and A2bAR siRNA was determined in the lung
tissue of mice and ATII cells by western blot analysis (Fig. 8A and B). The protein expression
level of α-ENaC was significantly increased following treatment
with CGS-21680 or Bay 60–6583 in alveolar epithelial cells
incubated with alcohol. The protein expression levels of α-ENaC,
β-ENaC and γ-ENaC were significantly decreased by the cAMP
inhibitor following incubation with CGS-21680 or Bay 60-6583
(Fig. 8C).
Discussion
A number of clinical studies have demonstrated the
association between chronic alcohol abuse and ARDS (32–35)
and chronic alcohol consumption has been implicated as a positive
risk factor in the development of ARDS. Chronic alcohol abuse is
associated with poor outcomes in patients with ARDS; however, the
mechanisms underlying the effects of alcohol in ARDS remain unclear
(36). The mechanistic associations
between alcohol and ARDS may contribute to alveolar epithelial
dysfunction through a number of mechanisms, including via systemic
effects on vascular tone and fluid retention as well as inducing
the apoptosis of alveolar epithelial cells (37). Furthermore, alveolar epithelial
dysfunction is an important factor in the decrease of AFC in ARDS
(10). In the present study, the
association between chronic alcohol abuse and AFC was the key
focus.
It is well known that alcohol is absorbed and
transported throughout the body in an unbound and unaltered state,
such as in the lungs where it achieves the high concentrations of
alcohol without being immediately metabolized (38). The consequence of chronic ethanol
ingestion was investigated in mice exposed to LPS, a
well-recognized murine model of ALI. The lungs may be particularly
susceptible to damage due to their delicate architecture, numerous
resident inflammatory cells and a high degree of vascularization
(39). It is well known that the
intestine contains an enormous potential reservoir of bacteria and
bacterial products for systemic infections and sepsis following
injury (40,41). The results of previous animal
studies demonstrated that post-burn intestinal permeability was
exacerbated when intoxication preceded the injury, leading to the
greater translocation of bacteria and bacterial products, such as
LPS, into the lung (42,43). LPS stimulation produces
pro-inflammatory cytokines, such as TNF-a and IL-6, that recruit
polymorphonuclear neutrophils into the injured lung and contribute
to the pathogenesis of ALI and ARDS (44,45).
In addition, activated neutrophils transmigrate across the
endothelial surface into the lung by the release of reactive oxygen
species, resulting in alveolar capillary barrier damage and
interstitial and alveolar edema following adherence to the lung
endothelium (46). In the present
study, alcohol administration was accompanied by an increase in
pulmonary edema and a reduction in the AFC. A large number of
inflammatory mediators were released during alcohol administration
following LPS-induced lung injury. The evaluation of the pulmonary
epithelial barrier and the histological lung injury score revealed
the effects of chronic alcohol on ALI. However, inhibition of A2aAR
or A2bAR attenuated alcohol-induced lung injury, which suggested
the involvement of the A2aAR or A2bAR pathway.
Though the ingestion of alcohol was found to
regulate AR via changes in the systemic levels of adenosine, the
association of A2aAR or A2bAR and alcohol had not yet been
elucidated (47). AFC is an
important procedure involving the removal of edema fluid from the
alveolar spaces via the ENaC in ALI/ARDS (48). The AR is made up of
seven-transmembrane spanning receptors, otherwise known as
G-protein coupled receptors and four AR subtypes; A1,
A2a, A2b and A3 have been
identified in lung tissue (49).
The important contribution of A2aAR or A2bAR signaling has been
implied in AFC (49–51). The results of the present study
demonstrated that chronic alcohol consumption decreased AFC in ALI,
while A2aAR or A2bAR siRNA inhibited the effect of alcohol on AFC.
The results illustrated that alcohol impaired AFC via A2aAR or
A2bAR. Furthermore, amiloride, a sodium channel inhibitor, blocked
alcohol-induced AFC, supporting the hypothesis that ENaC
participates in ALI following chronic alcohol ingestion. Therefore,
the association between ENaC and alcohol was further investigated
in the present study. In vivo, the expression levels of
α-ENaC, β-ENaC and γ-ENaC were decreased in ALI following alcohol
administration, but were increased by the intervention with A2aAR
or A2bAR siRNA. In vitro, alcohol administration decreased
the expression levels of α-ENaC, β-ENaC and γ-ENaC and the
aforementioned decrease was inhibited by A2aAR or A2bAR silencing.
The results demonstrated that ENaC was inhibited by A2aAR or A2bAR
in ALI following alcohol administration.
cAMP is a ubiquitous second messenger derived from
adenosine triphosphate that serves important regulatory roles in
diverse biological processes, including learning and memory
(52). cAMP is generated via the
binding of a ligand to the membrane-bound G protein-coupled
receptors. Ethanol alters the cAMP signaling pathways by G
protein-related adenylyl cyclase activity in animal models as well
as in model cell culture systems (53). In the present study, alcohol reduced
cAMP levels both in vivo and in vitro, which was
consistent with the findings of previous studies (54,55).
By inhibition of A2aAR or A2bAR, the systemic cAMP levels were
increased in the present study. Furthermore, the expression levels
of α-ENaC, β-ENaC and γ-ENaC were decreased by a cAMP inhibitor
following A2aAR or A2bAR siRNA administration. Activation of A2aAR
or A2bAR enhanced ENaC activity by elevating cAMP levels to promote
AFC in ALI (50,51,56).
Collectively, these results confirmed that alcohol inhibited ENaC
through the A2aAR or A2bAR-mediated cAMP pathway. Therefore, A2aAR
or A2bAR may serve as potential therapeutic targets for the
treatment of ALI following chronic alcohol consumption.
In addition, previous studies have investigated the
mechanism underlying alcohol in pulmonary immune dysfunction
(57,58) and alveolar epithelial dysfunction
(37,59) during the development of ARDS. The
association between chronic alcohol consumption and AFC in ARDS
remains to be elucidated. Dada et al (47) reported that alcohol reduced AFC via
a mechanism that involved the downregulation of the sodium
potassium pump via the AMP-activated protein kinase pathway in
hypoxia-induced lung injury. In another study, chronic alcohol
consumption leads to the altered regulation of α-ENaC in the
alcohol-induced damaged lung without ARDS (60). In the present study, alcohol
inhibited AFC and ENaC through the A2aAR or A2bAR-mediated cAMP
pathway. However, previous studies have reported that ethanol
activated ENaC by increasing the release of intracellular reactive
oxygen species via the increased production of the metabolic
product, acetaldehyde, in A6 distal nephron cells. This stimulation
of ENaC was due to tissue differentiation, acute ethanol exposure
and high concentrations of ethanol (61,62).
Thus, further investigation is required to determine the effects of
ethanol on ENaC in the whole organism.
In conclusion, the results of the present study
demonstrated that alcohol worsened LPS-induced lung injury in mice,
further aggravating pulmonary edema and inhibiting AFC. Alcohol
prevented the expression of ENaC-associated proteins via the
A2AR-mediated cAMP pathway. The results further
indicated that therapies which target A2aAR and/or A2bAR may be
beneficial for the treatment of alcohol abuse-associated ARDS. In
future studies, clinical research will be performed to investigate
the outcome in alcoholic patients with ARDS.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81600058,81800083
and 81670071), Chongqing Natural Science Foundation (grant no.
cstc2020jcyj-msxmX0008) and Kuanren Talents Program of the Second
Affiliated Hospital of Chongqing Medical University (grant no.
202124).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WD, DQ and DXW participated in the design of the
study. WD and JH performed the animal study including the animal
model, cAMP assay, pulmonary edema, bronchoalveolar epithelial
permeability, lung histopathology and AFC measurement. WD and XMT
performed the cell culture and transfection experiment. CYL
performed the immunocytochemistry and RT-qPCR. JT performed the
western blotting. DQ performed the data collection and statistical
analysis. WD interpreted the data and drafted the manuscript. DXW
contributed to the critical revision of the manuscript. WD, DQ and
DXW confirm the authenticity of all the raw data. All authors
reviewed and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Second Affiliated Hospital of Chongqing Medical University
(approval no. 2019-009). All animal experiments were conducted
according to relevant national and international guidelines.
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests
Glossary
Abbreviations
Abbreviations:
|
ALI
|
acute lung injury
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
AFC
|
alveolar fluid clearance
|
|
AR
|
adenosine receptor
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
cAMP
|
cyclic adenosine monophosphate
|
|
COVID-19
|
corona virus disease 2019
|
|
ENaC
|
epithelial sodium channel
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
LPS
|
Lipopolysaccharide
|
|
PCR
|
polymerase chain reaction
|
References
|
1
|
Manthey J, Shield KD, Rylett M, Hasan OSM,
Probst C and Rehm J: Global alcohol exposure between 1990 and 2017
and forecasts until 2030: A modelling study. Lancet. 393:2493–2502.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
GBD 2016 Alcohol Collaborators, . Alcohol
use and burden for 195 countries and territories, 1990–2016: A
systematic analysis for the global burden of disease study 2016.
Lancet. 392:1015–1035. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clark BJ, Williams A, Feemster LM, Bradley
KA, Macht M, Moss M and Burnham EL; NHLBI ARDS Network
Investigators, : Alcohol screening scores and 90-day outcomes in
patients with acute lung injury. Crit Care Med. 41:1518–1525. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarmiento X, Guardiola JJ and Soler M:
Alcohol and acute respiratory distress syndrome: Casuality or
causality? Med Clin (Barc). 140:546–553. 2013.(In Spanish).
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yeligar SM, Chen MM, Kovacs EJ, Sisson JH,
Burnham EL and Brown LA: Alcohol and lung injury and immunity.
Alcohol. 55:51–59. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramalho R: Alcohol consumption and
alcohol-related problems during the COVID-19 pandemic: A narrative
review. Australas Psychiatry. 28:524–526. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chick J: Alcohol and COVID-19. Alcohol
Alcohol. 55:341–342. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Máca J, Jor O, Holub M, Sklienka P, Burša
F, Burda M, Janout V and Ševčík P: Past and present ARDS mortality
rates: A systematic review. Respir Care. 62:113–122. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huppert LA, Matthay MA and Ware LB:
Pathogenesis of acute respiratory distress syndrome. Semin Respir
Crit Care Med. 40:31–39. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Azzam ZS and Sznajder JI: Lung edema
clearance: Relevance to patients with lung injury. Rambam
Maimonides Med J. 6:e00252015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berthiaume Y and Matthay MA: Alveolar
edema fluid clearance and acute lung injury. Respir Physiol
Neurobiol. 159:350–359. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matalon S, Bartoszewski R and Collawn JF:
Role of epithelial sodium channels (ENaC) in the regulation of lung
fluid homeostasis. Am J Physiol Lung Cell Mol Physiol.
309:L1229–L1238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qadri YJ, Rooj AK and Fuller CM: ENaCs and
ASICs as therapeutic targets. Am J Physiol Cell Physiol.
302:C943–C965. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hummler E, Barker P, Gatzy J, Beermann F,
Verdumo C, Schmidt A, Boucher R and Rossier BC: Early death due to
defective neonatal lung liquid clearance in alpha-ENaC-deficient
mice. Nat Genet. 12:325–328. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Randrianarison N, Clerici C, Ferreira C,
Fontayne A, Pradervand S, Fowler-Jaeger N, Hummler E, Rossier BC
and Planès C: Low expression of the beta-ENaC subunit impairs lung
fluid clearance in the mouse. Am J Physiol Lung Cell Mol Physiol.
294:L409–L416. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Elias N, Rafii B, Rahman M, Otulakowski G,
Cutz E and O'Brodovich H: The role of alpha-, beta-, and gamma-ENaC
subunits in distal lung epithelial fluid absorption induced by
pulmonary edema fluid. Am J Physiol Lung Cell Mol Physiol.
293:L537–L545. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagy LE, Diamond I, Casso DJ, Franklin C
and Gordon AS: Ethanol increases extracellular adenosine by
inhibiting adenosine uptake via the nucleoside transporter. J Biol
Chem. 265:1946–1951. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nagy LE, Diamond I, Collier K, Lopez L,
Ullman B and Gordon AS: Adenosine is required for ethanol-induced
heterologous desensitization. Mol Pharmacol. 36:744–748.
1989.PubMed/NCBI
|
|
19
|
Deng W, Wang DX, Zhang W and Li CY:
Regulation of epithelial sodium channel α-subunit expression by
adenosine receptor A2a in alveolar epithelial cells. Chin Med J
(Engl). 124:1551–1555. 2011.PubMed/NCBI
|
|
20
|
Jerrells TR, Pavlik JA, DeVasure J, Vidlak
D, Costello A, Strachota JM and Wyatt TA: Association of chronic
alcohol consumption and increased susceptibility to and pathogenic
effects of pulmonary infection with respiratory syncytial virus in
mice. Alcohol. 41:357–369. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song K, Coleman RA, Zhu X, Alber C, Ballas
ZK, Waldschmidt TJ and Cook RT: Chronic ethanol consumption by mice
resuls in activated splenic T cells. J Leukoc Biol. 72:1109–1116.
2002.PubMed/NCBI
|
|
22
|
Downs CA, Trac D, Brewer EM, Brown LA and
Helms MN: Chronic alcohol ingestion changes the landscape of the
alveolar epithelium. Biomed Res Int. 2013:4702172013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dobbs L, Gonzales G and Williams M: An
improved method for isolating type II cells in high yield and
purity. Am Rev Respir Dis. 134:141–145. 1986.PubMed/NCBI
|
|
24
|
Dobbs LG: Isolation and culture of
alveolar type II cells. Am J Physiol. 258:L134–L147.
1990.PubMed/NCBI
|
|
25
|
Goodson P, Kumar A, Jain L, Kundu K,
Murthy N, Koval M and Helms MN: Nadph oxidase regulates alveolar
epithelial sodium channel activity and lung fluid balance in vivo
via O2− signaling. Am J Physiol Lung Cell Mol
Physiol. 302:L410–L419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lomas-Neira JL, Chung CS, Wesche DE, Perl
M and Ayala A: In vivo gene silencing (with siRNA) of pulmonary
expression of MIP-2 versus KC results in divergent effects on
hemorrhage-induced, neutrophil-mediated septic acute lung injury. J
Leukoc Biol. 77:846–853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
You K, Xu X, Fu J, Xu S, Yue X, Yu Z and
Xue X: Hyperoxia disrupts pulmonary epithelial barrier in newborn
rats via the deterioration of occludin and ZO-1. Respir Res.
13:362012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matute-Bello G, Downey G, Moore BB,
Groshong SD, Matthay MA, Slutsky AS and Kuebler WM; Acute Lung
Injury In Animals Study Group, : An official American thoracic
society workshop report: Features and measurements of experimental
acute lung injury in animals. Am J Respir Cell Mol Biol.
44:725–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mutlu GM, Adir Y, Jameel M, Akhmedov AT,
Welch L, Dumasius V, Meng FJ, Zabner J, Koenig C, Lewis ER, et al:
Interdependency of beta-adrenergic receptors and CFTR in regulation
of alveolar active Na+ transport. Circ Res. 96:999–1005. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mutlu GM, Dumasius V, Burhop J, McShane
PJ, Meng FJ, Welch L, Dumasius A, Mohebahmadi N, Thakuria G,
Hardiman K, et al: Upregulation of alveolar epithelial active Na+
transport is dependent on beta2-adrenergic receptor signaling. Circ
Res. 94:1091–1100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moss M, Bucher B, Moore FA, Moore EE and
Parsons PE: The role of chronic alcohol abuse in the development of
acute respiratory distress syndrome in adults. JAMA. 275:50–54.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moss M, Parsons PE, Steinberg KP, Hudson
LD, Guidot DM, Burnham EL, Eaton S and Cotsonis GA: Chronic alcohol
abuse is associated with an increased incidence of acute
respiratory distress syndrome and severity of multiple organ
dysfunction in patients with septic shock. Crit Care Med.
31:869–877. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gajic O, Dabbagh O, Park PK, Adesanya A,
Chang SY, Hou P, Anderson H III, Hoth JJ, Mikkelsen ME, Gentile NT,
et al: Early identification of patients at risk of acute lung
injury: Evaluation of lung injury prediction score in a multicenter
cohort study. Am J Respir Crit Care Med. 183:462–470. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Toy P, Gajic O, Bacchetti P, Looney MR,
Gropper MA, Hubmayr R, Lowell CA, Norris PJ, Murphy EL, Weiskopf
RB, et al: Transfusion-related acute lung injury: Incidence and
risk factors. Blood. 119:1757–1767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moazed F and Calfee CS: Environmental risk
factors for acute respiratory distress syndrome. Clin Chest Med.
35:625–637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang R, Ramos C, Joshi I, Zagariya A,
Pardo A, Selman M and Uhal BD: Human lung myofibroblast-derived
inducers of alveolar epithelial apoptosis identified as angiotensin
peptides. Am J Physiol. 277:L1158–L1164. 1999.PubMed/NCBI
|
|
38
|
Dubowski KM: Absorption, distribution and
elimination of alcohol: Highway safety aspects. J Stud Alcohol
Suppl. 10:98–108. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Guidot DM and Hart CM: Alcohol abuse and
acute lung injury: Epidemiology and pathophysiology of a recently
recognized association. J Investig Med. 53:235–245. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Deitch EA: Role of the gut lymphatic
system in multiple organ failure. Curr Opin Crit Care. 7:92–98.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hassoun HT, Kone BC, Mercer DW, Moody FG,
Weisbrodt NW and Moore FA: Post-injury multiple organ failure: The
role of the gut. Shock. 15:1–10. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kavanaugh MJ, Clark C, Goto M, Kovacs EJ,
Gamelli RL, Sayeed MM and Choudhry MA: Effect of acute alcohol
ingestion prior to burn injury on intestinal bacterial growth and
barrier function. Burns. 31:290–296. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zahs A, Bird MD, Ramirez L, Choudhry MA
and Kovacs EJ: Anti-IL-6 antibody treatment but not IL-6 knockout
improves intestinal barrier function and reduces inflammation after
binge ethanol exposure and burn injury. Shock. 39:373–379. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Toews GB: Cytokines and the lung. Eur
Respir J Suppl. 34:3s–17s. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Idell S: Anticoagulants for acute
respiratory distress syndrome: Can they work? Am J Respir Crit Care
Med. 164:517–520. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fialkow L, Wang Y and Downey GP: Reactive
oxygen and nitrogen species as signaling molecules regulating
neutrophil function. Free Radic Biol Med. 42:153–164. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dada L, Gonzalez AR, Urich D, Soberanes S,
Manghi TS, Chiarella SE, Chandel NS, Budinger GR and Mutlu GM:
Alcohol worsens acute lung injury by inhibiting alveolar sodium
transport through the adenosine A1 receptor. PLoS One.
7:e304482012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eaton DC, Helms MN, Koval M, Bao HF and
Jain L: The contribution of epithelial sodium channels to alveolar
function in health and disease. Annu Rev Physiol. 71:403–423. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fan M, Qin W and Mustafa SJ:
Characterization of adenosine receptor(s) involved in
adenosine-induced bronchoconstriction in an allergic mouse model.
Am J Physiol Lung Cell Mol Physiol. 284:L1012–L1019. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Eckle T, Grenz A, Laucher S and Eltzschig
HK: A2B adenosine receptor signaling attenuates acute lung injury
by enhancing alveolar fluid clearance in mice. J Clin Invest.
118:3301–3315. 2008.PubMed/NCBI
|
|
51
|
Factor P, Mutlu GM, Chen L, Mohameed J,
Akhmedov AT, Meng FJ, Jilling T, Lewis ER, Johnson MD, Xu A, et al:
Adenosine regulation of alveolar fluid clearance. Proc Natl Acad
Sci USA. 104:4083–4088. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kleinboelting S, van den Heuvel J and
Steegborn C: Structural analysis of human soluble adenylyl cyclase
and crystal structures of its nucleotide complexes-implications for
cyclase catalysis and evolution. FEBS J. 281:4151–4164. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tabakoff B and Hoffman PL: Adenylyl
cyclases and alcohol. Adv Second Messenger Phosphoprotein Res.
32:173–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gupta R, Qualls-Creekmore E and Yoshimura
M: Real-time monitoring of intracellular cAMP during acute ethanol
exposure. Alcohol Clin Exp Res. 37:1456–1465. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu Z, Liu Y, Gao R, Li H, Dunn T, Wu P,
Smith RG, Sarkar PS and Fang X: Ethanol suppresses PGC-1α
expression by interfering with the cAMP-CREB pathway in neuronal
cells. PLoS One. 9:e1042472014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ohta A and Sitkovsky M: Role of
G-protein-coupled adenosine receptors in downregulation of
inflammation and protection from tissue damage. Nature.
414:916–920. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Greenberg SS, Zhao X, Hua L, Wang JF,
Nelson S and Ouyang J: Ethanol inhibits lung clearance of
pseudomonas aeruginosa by a neutrophil and nitric oxide-dependent
mechanism, in vivo. Alcohol Clin Exp Res. 23:735–744. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Burnham EL, Kovacs EJ and Davis CS:
Pulmonary cytokine composition differs in the setting of alcohol
use disorders and cigarette smoking. Am J Physiol Lung Cell Mol
Physiol. 304:L873–L882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Brown LA, Harris FL, Bechara R and Guidot
DM: Effect of chronic ethanol ingestion on alveolar type II cell:
Glutathione and inflammatory mediator-induced apoptosis. Alcohol
Clin Exp Res. 25:1078–1085. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Downs CA, Trac DQ, Kreiner LH, Eaton AF,
Johnson NM, Brown LA and Helms MN: Ethanol alters alveolar fluid
balance via Nadph oxidase (NOX) signaling to epithelial sodium
channels (ENaC) in the lung. PLoS One. 8:e547502013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bao HF, Song JZ, Duke BJ, Ma HP, Denson DD
and Eaton DC: Ethanol stimulates epithelial sodium channels by
elevating reactive oxygen species. Am J Physiol Cell Physiol.
303:C1129–C1138. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Snyder PM: Intoxicated Na(+) channels.
Focus on ‘ethanol stimulates epithelial sodium channels by
elevating reactive oxygen species’. Am J Physiol Cell Physiol.
303:C1125–C1126. 2012. View Article : Google Scholar : PubMed/NCBI
|