Introduction
Diabetic nephropathy (DN) is a common diabetic
complication, the incidence of which was 20–40% among patients with
diabetes in 2019 worldwide, which develops from permanent
uncontrolled diabetes. Furthermore, it typically causes end-stage
renal disease (1). DN is
characterized, clinically, by hypertension, progressive proteinuria
and renal disease resulting in multiple diseases, including uremic
symptoms (2). The main
characteristics of DN are increased thickness of the basement
membrane and mesangial expansion in the glomerulus resulting from
the accumulation of the extracellular matrix, followed by
hypercellularity, which ultimately leads to glomerular sclerosis
and renal fibrosis (3–5). As high-glucose (HG)-induced injury of
podocytes has been associated with DN progression (6), it is important to identify novel
strategies for protecting podocytes from injury against HG.
Increased glycolysis in podocytes has been regarded
as a primary cause of DN (7). One
of the key mediators and the first committed factor of glycolytic
flux is 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
(PFKFB3) (8). PFKFB3 serves a role
in the synthesis and degradation of fructose-2,6-bisphosphate,
which has been associated with diabetes (9), cell cycle mediation (10) and drug resistance (11). However, the biological function of
PFKFB3 in podocyte injury remains unclear.
Autophagy is a cellular process that is
characterized by the formation of an isolation membrane called a
phagophore (12). Numerous studies
have indicated that autophagy serves an important role in cell
growth and differentiation (13,14).
Furthermore, autophagy is associated with the progression of
various diseases, including Alzheimer's disease (15), cancer (16) and microorganism infection (17). In addition, it has been previously
reported that induction of autophagy protects podocytes from injury
(18). Therefore, the aim of the
present study was to investigate the association between PFKFB3 and
autophagy. Enolase-1, glyoxalase and aldose reductase (AR) are
considered three important markers in DN, and the regulation of
these three factors may modulate the progression of DN (19,20).
Therefore, AR was used as a positive control to study the effect of
PFKFB3 siRNA on enolase-1 and glyoxalase. The effects of PFKFB3
small interfering (si)RNA on the growth of HG-induced podocytes
in vitro was also determined to identify novel targets for
the treatment of DN.
Materials and methods
Cell culture and treatment
MPC5 mouse podocyte cells were purchased from the
Tongpai Biotechnology Co. Ltd. RPMI-1640 (Gibco; Thermo Fisher
Scientific, Inc.) medium containing 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.), 1% penicillin and streptomycin and 10 IU/ml
recombinant murine IFN-γ (Invitrogen; Thermo Fisher Scientific,
Inc.) was utilized to culture MPC5 cells at 37°C and 5%
CO2. Podocytes were maintained at 37°C in the absence of
IFN-γ for 10–14 days to induce differentiation, and then cultured
for 12 h at 37°C in FBS-free RPMI-1640 medium containing 5.6 mM
D-glucose (Sigma-Aldrich; Merck KGaA) before subsequent experiments
and cell transfection. In addition, MPC5 cells were treated with 25
mM glucose (HG; Sigma-Aldrich; Merck KGaA) or 10 mM glucose [normal
glucose (NG); Sigma-Aldrich; Merck KGaA] at 37°C for 48 h. In order
to maintain the osmotic pressure, MPC5 cells were treated with 19.5
mM mannitol (Sigma-Aldrich; Merck KGaA) at 37°C for 48 h.
Human primary podocytes (cat. no. BNCC340460; BeNa
Culture Collection; Beijing Beina Chunglian Institute of
Biotechnology) were cultured at 33°C in RPMI-1640 medium with 10%
FBS (both Gibco; Thermo Fisher Scientific, Inc.), 100 µg/ml
streptomycin, 100 U/ml penicillin G and 1X
insulin-transferrin-selenium (ITS; Invitrogen) for 48 h. To induce
differentiation, cells were cultured at 37°C for 10–14 days with
ITS-free medium. The differentiated cells were stimulated for 48 h
at 37°C with NG (10 mM) or HG (25 mM).
Reagents
Chloroquine (CQ) and rapamycin are known autophagy
inhibitors; thus, they can be used in rescue experiments to further
confirm the role of PFKFB3 in autophagy. CQ and rapamycin were
obtained from Sigma-Aldrich (Merck KGaA). To investigate the role
of PFKFB3 in cell autophagy, MPC5 cells were treated with CQ (5 mM)
or rapamycin (10 µM) for 48 h at 37°C.
Cell transfection
Cell transfection was performed following HG
treatment. MPC5 cells or primary podocytes were seeded at
3×105 cells/well in a 6-well plate and cultured to 70%
confluence. The cells were subsequently transfected with AR siRNA
(10 nM), PFKFB3 siRNA1 (10 nM), siRNA2 (10 nM) or siRNA3 (10 nM),
or negative control (NC, 10 nM) siRNA using
Lipofectamine® 2000 reagent (Thermo Fisher Scientific,
Inc.) for 24, 48 or 72 h at 37°C. The siRNAs targeting AR, PFKFB3
and NC siRNA were designed and synthesized by Shanghai GenePharma
Co., Ltd.; the sequences were as follows: AR siRNA,
5′-GCAATCTGAGTCTCCAACT-3′; PFKFB3 siRNA1,
5′-GCAAUUGAAAGUUUGGUAA-3′; PFKFB3 siRNA2,
5′-CCCACAACCUUUAGACUGA-3′; PFKFB3 siRNA3,
5′-GAAUCCAUAUCACUACGAA-3′; and NC siRNA, 5′-GUACAGAGUUACUACUUAG-3′.
After 48 h of transfection, the transfected cells were used in
subsequent analysis.
Cell Counting Kit (CCK)-8 assay
MPC5 cells or primary podocytes were seeded in
96-well plates (5×103 cells/well) overnight at 37°C.
Then, the cells were treated with NC siRNA + NG, AR siRNA + NG,
PFKFB3 siRNA + NG, NC siRNA + HG, AR siRNA + HG or PFKFB3 siRNA for
48 h, as aforementioned. A total of 10 µl CCK-8 reagent (Beyotime
Institute of Biotechnology) was added to each well and incubated
for 2 h at 37°C. Finally, the absorbance of MPC5 cells was measured
at 450 nm using a microplate reader (Thermo Fisher Scientific,
Inc.).
Glucose uptake detection
The supernatant of MPC5 cells was collected by
centrifugation at 100 × g at 4°C for 10 min, then the concentration
of glucose and diacylglycerol (DAG) was detected as previously
described (21). The absorbance of
MPC5 cell supernatants was measured at 505 nm using a microplate
reader (Thermo Fisher Scientific, Inc.). The concentration was
calculated as previously described (21).
Oxygen consumption rate (OCR) and
extracellular acidification rate (ECAR) measurement
OCR measurements were performed on a Seahorse XFp
analyzer using the Seahorse XF Cell Mito Stress Test kit (both
Agilent Technologies, Inc.) according to the manufacturer's
instructions. Briefly, podocytes were seeded on XFe96 plates at
5×104 cells/well. OCR was measured following exposure to
mitochondrial modulators as follows: Oligomycin (1 µM), which
inhibits ATP synthase and decreases OCR; carbonyl cyanide
4-trifluoromethoxyphenylhydrazone (1.5 µM), which uncouples oxygen
consumption from ATP production and raises OCR to a maximal value;
and a combination of rotenone and antimycin A (both 0.5 µM), which
decrease OCR to a minimal value. After the OCR readings were
completed, cells were fixed in 4% paraformaldehyde at room
temperature for 10 min and stained with a 0.05% crystal violet
solution at room temperature for 30 min. Next, podocytes were
washed and lysed in 1% SDS. Absorbance of each well (which reflects
cell number) was read at 595 nm using an Infinite 200 Pro plate
reader (Tecan Group Ltd.). OCR values were normalized to cell
number.
For ECAR measurement, podocytes (3×105
per well) were cultured on 8-well culture microplates (Agilent
Technologies, Inc.) coated with Rat Tail Collagen-I Solution
(Sigma-Aldrich; Merck KGaA) overnight. Next, growth medium was
replaced with assay medium (minimal DMEM supplemented with 2 mM
L-glutamine; Gibco; Thermo Fisher Scientific, Inc.) and cells were
allowed to stabilize at 37°C for 1 h. ECAR values were determined
using the Seahorse XFp Analyzer before (baseline state) and after
cells were treated with 10 mM D-glucose, 1 µM oligomycin or 50 mM
2-deoxy-D-glucose. Glycolytic flux parameters were determined from
the slopes of the ECAR values in real-time analysis. Baseline and
post-exposure rates were measured three times every 3 min (total
time, ~80 min). The results were normalized to the protein
concentrations determined for each cell culture plate well using
the Bradford method.
Cell apoptosis analysis
Cell apoptosis was detected using Annexin V-FITC/PI
apoptosis detection kit (cat. no. 556570; BD Biosciences). MPC-5
cells or primary podocytes were seeded in a 6-well plate
(1×106/well) at 37°C overnight. Cells were trypsinized,
then washed, fixed with 70% ethanol at 4°C for 1 h and re-suspended
in Annexin V Binding Buffer. Subsequently, the cells were stained
with 5 µl FITC and PI (BD Biosciences) in the dark at 4°C for 15
min. The rate of early + late-stage cell apoptosis was calculated
using a flow cytometer (BD Biosciences), and the results were
analyzed by flow cytometry (FACSCalibur; BD Biosciences) with
FlowJo (v10.6.2; FlowJo LLC).
Western blot analysis
Total protein was extracted from cell lysate using
RIPA buffer (Beyotime Institute of Biotechnology) and quantified
using a BCA protein kit (Thermo Fisher Scientific, Inc.). Proteins
(40 µg per lane) were separated using SDS-PAGE (10%) and
transferred onto PVDF membranes. Subsequently, the membranes were
blocked with 3% skimmed milk at room temperature for 1 h, incubated
with primary antibodies at 4°C overnight, then incubated with
HRP-conjugated secondary anti-rabbit antibody (1:5,000; cat. no.
ab7090; Abcam) at room temperature for 1 h. The membranes were
scanned using an Odyssey Imaging System and data was analyzed using
ImageJ software (version 1.8.0; National Institutes of Health). The
primary antibodies were as follows: Anti-synaptopodin (cat. no.
DF12173; Affinity Biosciences), anti-enolase-1 (cat. no. ab227978;
Abcam), anti-PFKFB3 (cat. no. DF3307; Affinity Biosciences),
anti-glyoxalase 1 (cat. no. DF6700; Affinity Biosciences), anti-AR
(cat. no. DF6554; Affinity Biosciences), anti-Wilms tumor 1
(WT-1;cat. no. 12609-1-AP; ProteinTech Group, Inc.),
anti-E-cadherin (cat. no. ab40772; Abcam), anti-α-smooth muscle
actin (SMA; cat. no. ab7817; Abcam), anti-p62 (cat. no. ab109012;
Abcam), anti-light chain (LC)3 (cat. no. ab192890; Abcam),
anti-phosphorylated (p)-mTOR (cat. no. ab109268; Abcam), anti-mTOR
(cat. no. ab32028; Abcam), anti-sirtuin (SIRT)1 (cat. no. ab32441;
Abcam), anti-AMPKα (cat. no. ab32047; Abcam), anti-p-AMPKα (cat.
no. ab133448; Abcam) and anti-GAPDH (cat. no. ab8245; Abcam) (all
1:1,000). GAPDH was used as an internal control.
Transwell assay
For the migration assay, transfected cells
(3×104 cells/well) in serum-free DMEM (100 µl) were
seeded into the upper chamber of a Transwell plate (24-wells; pore
size, 8 µm; Corning, Inc.). A total of 600 µl DMEM containing 10%
FBS was added into the lower chamber of each well. Following
incubation for 24 h at room temperature, the non-migrated cells on
the upper surface were removed, and the migrated cells on the lower
surface were fixed with 4% paraformaldehyde at 4°C for 15 min,
followed by staining with 0.5% crystal violet at room temperature
for 30 min. The number of migrated cells was calculated using a
light microscope, ×400 magnification; three random fields of view
were selected.
Immunofluorescence staining
Following treatment for 72 h, as aforementioned,
MPC-5 cells were blocked with 4% paraformaldehyde at room
temperature for 15 min, then incubated with anti-phalloidin (cat.
no. ab176753) or anti-LC3 (cat. no. ab192890) antibody (both
1:1,000; both Abcam) overnight at 4°C. Subsequently, the cells were
incubated with the Goat Anti-Rabbit IgG H&L antibody (1:5,000;
cat. no. ab150077; Abcam) at room temperature for 1 h. DAPI
(Beyotime Institute of Biotechnology) was used to stain the nuclei
for 5 min. Finally, the cells were observed under a fluorescence
microscope (magnification, ×200). The fluorescence intensity was
measured using ImageJ software (version 1.8.2; National Institutes
of Health); a total of three random fields of view were
selected.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. The CCK-8 assay was repeated five times;
immunofluorescence staining, RT-qPCR, Transwell and flow cytometry
assays were repeated three times. Comparisons between two groups
were analyzed by unpaired Student's t-test. One-way ANOVA followed
by Tukey's post hoc test was used for multiple group comparisons.
The normality of data was assessed using Shapiro-Wilk test. Data
were analyzed using GraphPad Prism (version 7; GraphPad Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
AR and PFKFB3 siRNAs significantly
decreased the level of AR and PFKFB3 in MPC5 cells,
respectively
To establish an in vitro model of DN, MPC5
cells were treated with HG as previously described (22,23).
As AR inhibitors have been shown to inhibit the progression of DN
(24,25), AR siRNA was used in preliminary
experiments (cell viability, migration and synaptopodin
expression). HG treatment significantly increased the protein
expression levels of enolase-1, PFKFB3 and AR in MPC5 cells at 48
and 72 h (Fig. 1A). By contrast,
the protein expression levels of glyoxalase 1 were significantly
downregulated in the presence of HG. As the aforementioned four
proteins are key markers of DN (26–29),
the results suggested that the in vitro model of DN was
successfully established. Next, western blot analysis was performed
to detect the transfection efficiency of siRNAs. Knockdown of AR
significantly decreased the protein expression level of AR in MPC5
cells (Fig. 1B). Similarly, the
expression level of PFKFB3 protein in MPC5 cells was significantly
inhibited by silencing of PFKFB3 (Fig.
1B); PFKFB3 siRNA3 showed the most significant transfection
efficiency and was selected to be used in the subsequent
experiments. mRNA expression of PFKFB3 in primary podocytes was
significantly decreased by PFKFB3 siRNA3 (Fig. S1A and B). Taken together, these
results indicated that AR and PFKFB3 siRNAs significantly decreased
the expression levels of AR and PFKFB3 in MPC5 cells,
respectively.
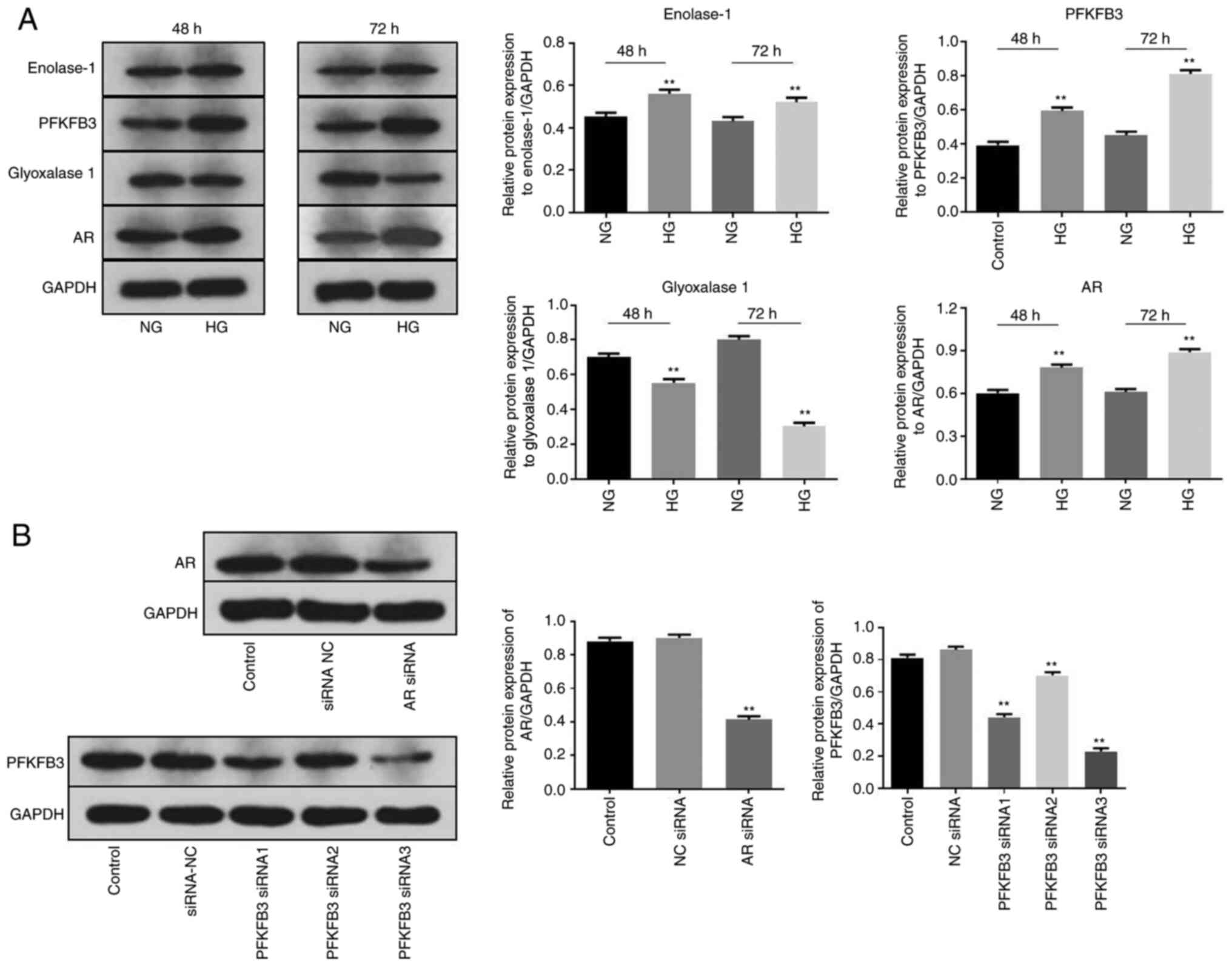 | Figure 1.AR and PFKFB3 siRNAs are successfully
transfected into MPC5 cells. MPC5 cells were treated with HG for 48
or 72 h. (A) Protein expression levels of enolase-1, PFKFB3,
glyoxalase 1 and AR in MPC5 cells were detected by western blot
analysis and normalized to GAPDH. (B) MPC5 cells were transfected
with siRNA NC, siRNA AR or PFKFB3 siRNA1, −2 or −3 for 24 h. Then,
the protein expression levels of PFKFB3 and AR were measured by
western blot analysis and normalized to GAPDH. **P<0.01 vs.
Control (NG). AR, aldose reductase; HG, high glucose; NC, negative
control; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; siRNA, small
interfering RNA. |
HG-induced inhibition of cell
viability is reversed by PFKFB3 siRNA
To investigate the effect of PFKFB3 or AR on the
viability of MPC5 cells, CCK-8 assay was used. Viability of MPC5
cells in the presence of NG was slightly affected by AR siRNA but
was significantly enhanced following PFKFB3 knockdown (Figs. 2A and S2A). In addition, HG significantly
inhibited the viability of MPC5 cells. However, PFKFB3 siRNA
significantly rescued the inhibitory effect of HG on cell
viability. Mannitol did not affect the viability of HG-treated MPC5
cells. Western blot analysis was used to measure the expression
levels of synaptopodin. The protein expression levels of
synaptopodin in MPC5 cells in the presence of NG were significantly
increased by PFKFB3 silencing (Fig.
2B). HG significantly decreased the protein expression levels,
and PFKFB3/AR knockdown partially reversed this inhibitory effect
of HG on synaptopodin protein expression levels (Fig. 2B). Transwell assay was performed to
assess cell migration; migration of the MPC5 cells was
significantly upregulated in the presence of HG compared with NG,
which was partially reversed in the presence of AR or PFKFB3 siRNAs
(Fig. 2C). Taken together, these
results indicated that HG-induced cell viability inhibition,
migration and decrease of synaptopodin expression were reversed by
PFKFB3 siRNA.
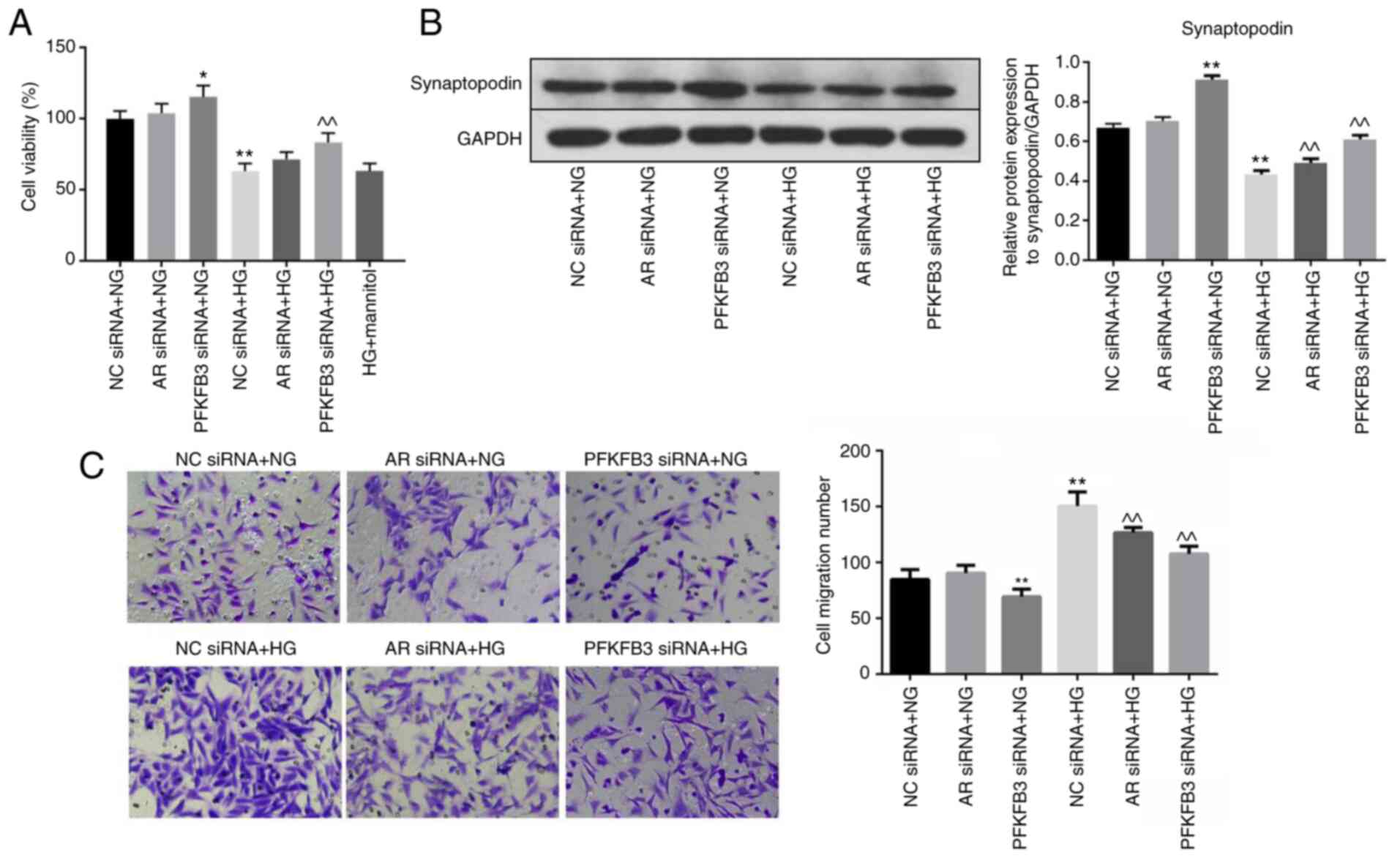 | Figure 2.HG-induced cell viability inhibition
and cell migration is reversed by PFKFB3 siRNA. MPC5 cells were
treated with NC siRNA + NG, AR siRNA + NG, PFKFB3 siRNA + NG, NC
siRNA + HG, AR siRNA + HG, PFKFB3 siRNA + HG or HG + mannitol for
48 h. (A) Cell viability was measured by Cell Counting Kit-8 assay.
(B) Protein expression levels of synaptopodin in MPC5 cells were
detected by western blot analysis and normalized to GAPDH. (C) Cell
migration was examined by Transwell assay. *P<0.05, **P<0.01
vs. NC siRNA + NG; ^^P<0.01 vs. NC siRNA + HG. AR,
aldose reductase; HG, high-glucose; NC, negative control; NG,
normal glucose; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; siRNA, small
interfering RNA. |
Silencing of PFKFB3 reverses
HG-induced podocyte injury
Phalloidin staining was performed to examine the
cytoskeleton. The results revealed that HG treatment significantly
changed the shape and decreased the area of MPC5 cells, whereas
knockdown of PFKFB3 significantly rescued the injury caused by HG
(Fig. 3A). In addition, glucose
uptake by the MPC5 cells was significantly increased after 48 or 72
h of HG treatment, which was partially inhibited by PFKFB3 siRNA
(Fig. 3B). Similarly, PFKFB3 siRNA
significantly reversed the HG-induced apoptosis of MPC-5 cells and
primary podocytes (Figs. 3C and
S2B). Furthermore, DAG
concentration and ECAR in MPC5 cells were significantly upregulated
by HG; this was partially reversed in the presence of PFKFB3 siRNA
(Fig. 3D). By contrast, the
HG-induced decrease of OCR in podocytes was significantly reversed
in the presence of PFKFB3 siRNA (Fig.
3D). Taken together, these results indicated that silencing of
PFKFB3 reversed HG-induced podocyte injury.
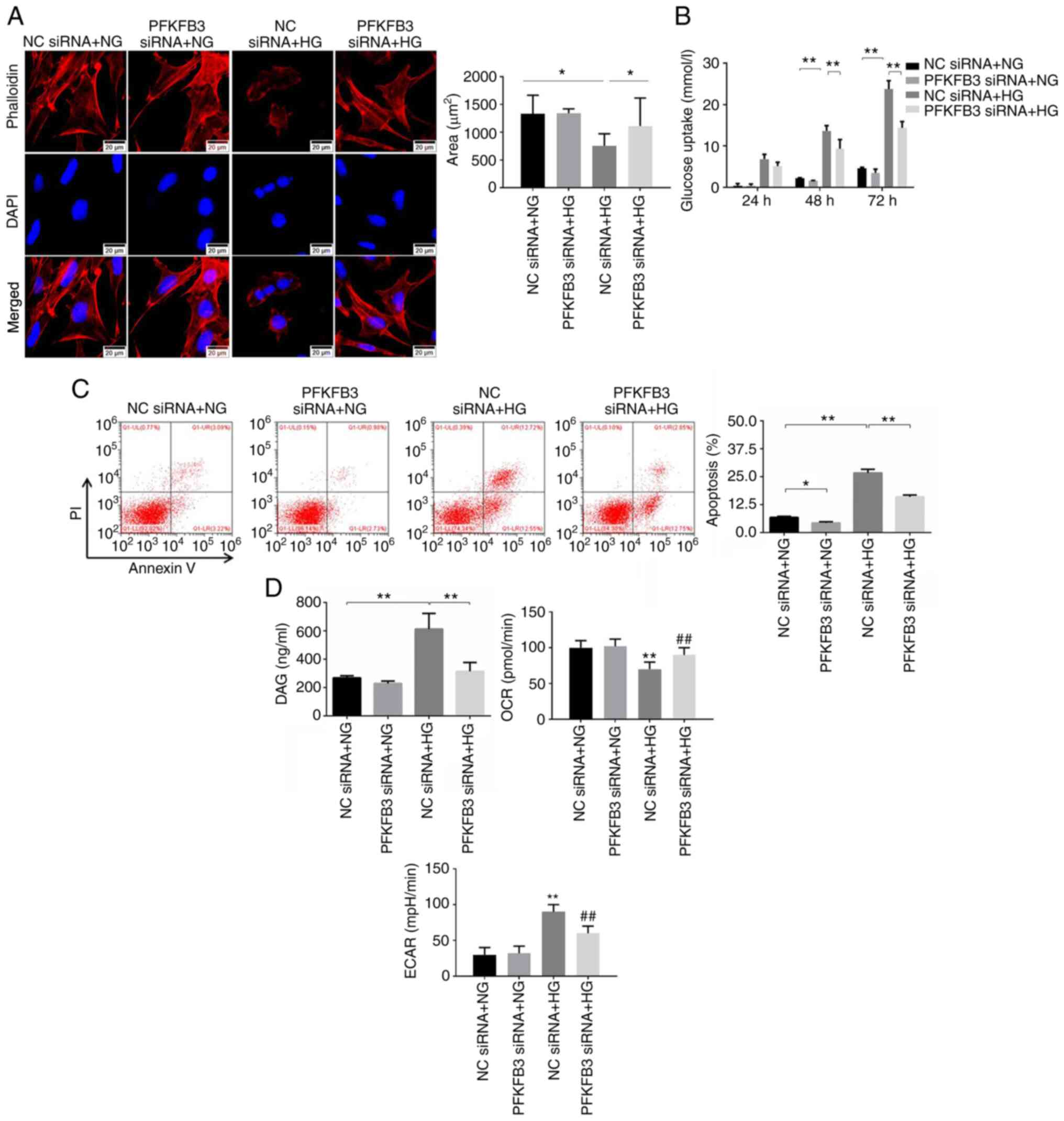 | Figure 3.Silencing of PFKFB3 reverses
HG-induced podocyte injury. (A) Phalloidin immunofluorescence
staining was performed to detect the cytoskeleton, and the area of
MPC5 cells was calculated. (B) Glucose uptake in MPC5 cells was
tested using a microplate reader. (C) Cell apoptosis was detected
by flow cytometry. (D) Levels of DAG in MPC5 cells were detected
using a microplate reader. OCR or ECAR in MPC5 cells was
investigated by Seahorse analyzer. *P<0.05, **P<0.01 vs. NC
siRNA + NG or as indicated; ##P<0.01 vs. NC siRNA +
HG. DAG, diacylglycerol; ECAR, extracellular acidification rate;
HG, high-glucose; NC, negative control; NG, normal glucose; OCR,
oxygen consumption rate; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; siRNA, small
interfering RNA. |
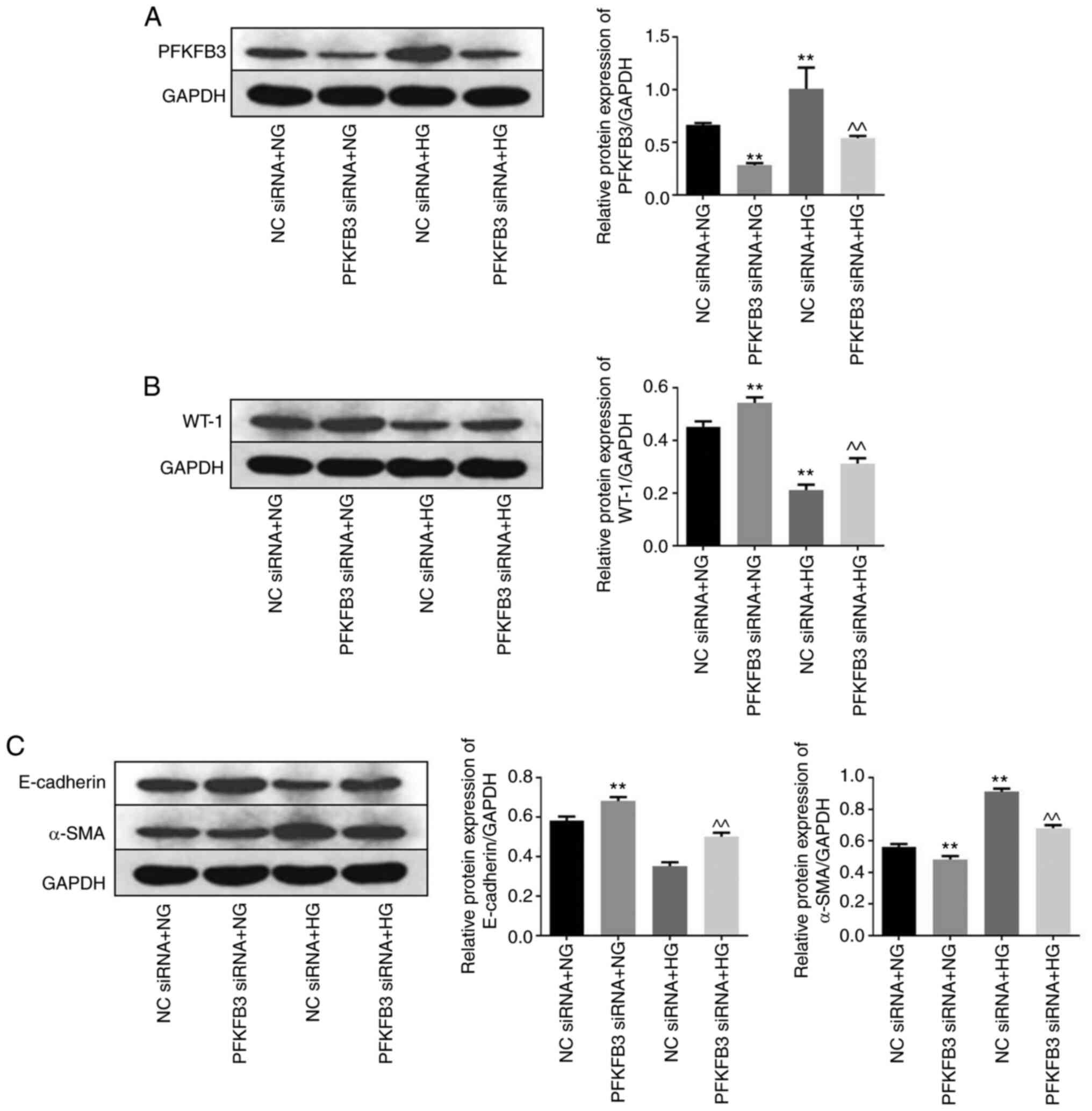
Knockdown of PFKFB3 reverses podocyte
injury by inhibiting the epithelial-mesenchymal transition (EMT)
process
Western blot analysis was performed to measure the
expression levels of PFKFB3, WT-1 and EMT-related proteins in the
MPC5 cells. Protein expression levels of PFKFB3 in MPC5 cells with
NG or HG treatment were significantly decreased by PFKFB3 siRNA
compared with NC siRNA + NG or NC siRNA + HG (Fig. 4A). Meanwhile, the expression of
PFKFB3 in MPC5 cells was notably upregulated by HG, compared with
NG (Fig. 4A). Furthermore,
knockdown of PFKFB3 significantly suppressed the inhibitory effect
of HG on protein expression level of WT-1 (Fig. 4B). In addition, HG significantly
decreased the protein expression levels of E-cadherin and increased
those of α-SMA (Fig. 4C). However,
the effect of HG on these two proteins was significantly reversed
following knockdown of PFKFB3. As E-cadherin and α-SMA are two key
regulators in the EMT process (30,31),
these results indicated that knockdown of PFKFB3 may reversed
podocyte injury through suppression of the EMT process.
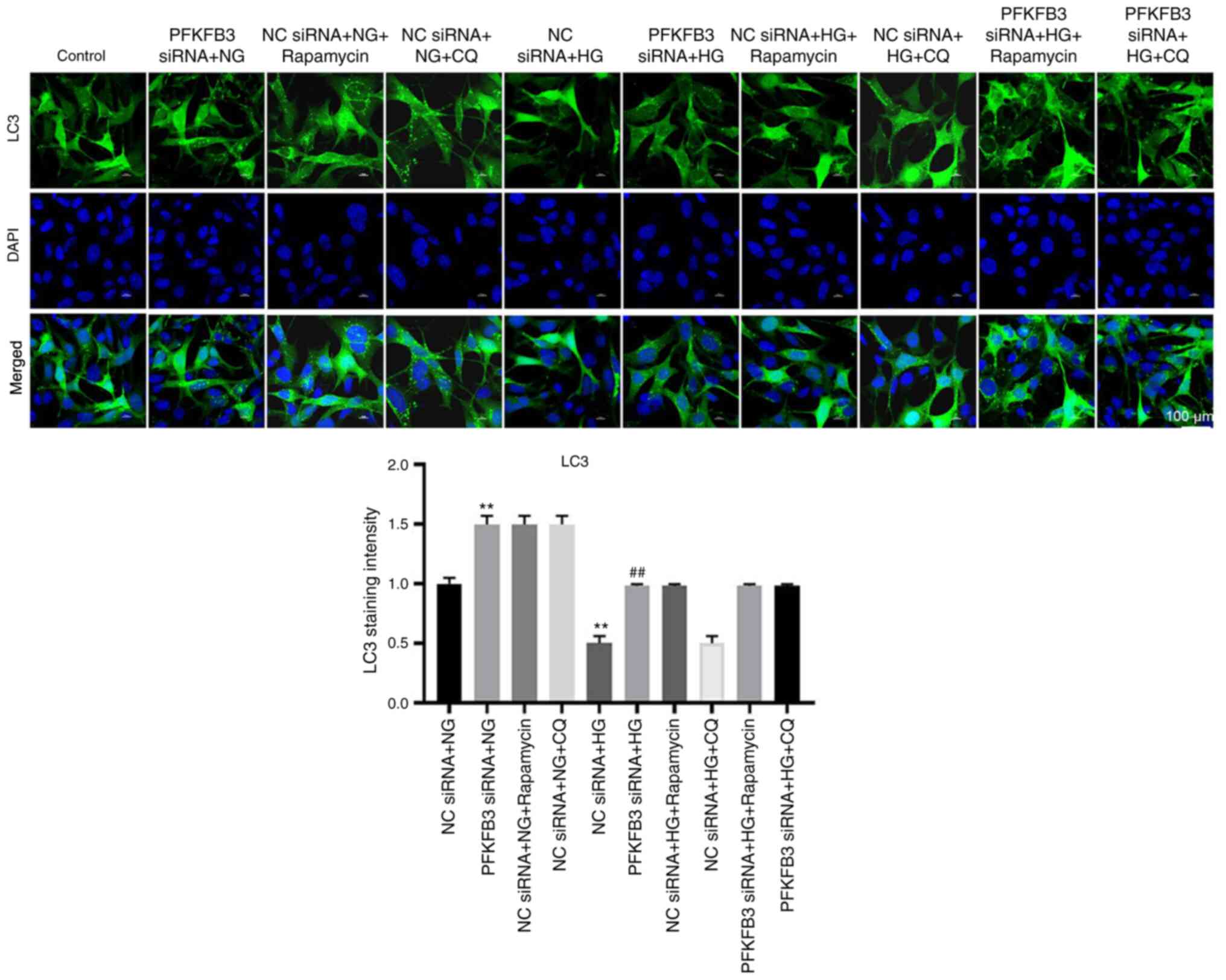
Silencing of PFKFB3 inhibits podocyte
injury through activation of autophagy
To determine the association between PFKFB3 and
autophagy in HG-treated MPC5 cells, autophagy inhibitors (rapamycin
and CQ) were used. Protein expression levels of LC3 II in
HG-treated MPC-5 cells were significantly upregulated in cells
transfected with PFKFB3 siRNA (Fig.
5A-C). Consistently, the ratio of LC3 II/LC3 I in HG-treated
MPC-5 cells was significantly enhanced in the presence of PFKFB3
siRNA compared with in the NC siRNA + HG + rapamycin group. These
data suggested that PFKFB3 may exert an inhibitory effect on
autophagy. By contrast, knockdown of PFKFB3 significantly decreased
the protein expression levels of p62 in MPC5 cells in the presence
of HG, whereas CQ or rapamycin (autophagy inhibitors) partially
reversed the inhibitory effect on p62 (Fig. 5A and D). In addition, CQ or
rapamycin further enhanced the effect of HG on increased protein
expression levels of PFKFB3 in MPC-5 cells (Fig. 5A and E). This showed that
downregulation of PFKFB3 induced autophagy in MPC-5 cells. LC3
staining indicated that the expression of LC3 (represented by
staining intensity) in MPC-5 cells was significantly decreased by
HG, which was reversed by PFKFB3 knockdown (Fig. 6). HG treatment significantly
inhibited the levels of p-AMPKα and SIRT1, and significantly
upregulated the expression of p-mTOR; these effects were reversed
following PFKFB3 knockdown (Fig.
7A-D). Rapamycin significantly enhanced the effect of PFKFB3
siRNA on p-mTOR expression, whereas HG-mediated p-AMPKα and SIRT1
expression was not significantly affected by CQ or rapamycin
(Fig. 7A-D). Taken together, these
results indicated that silencing of PFKFB3 may inhibit podocyte
injury by activating autophagy.
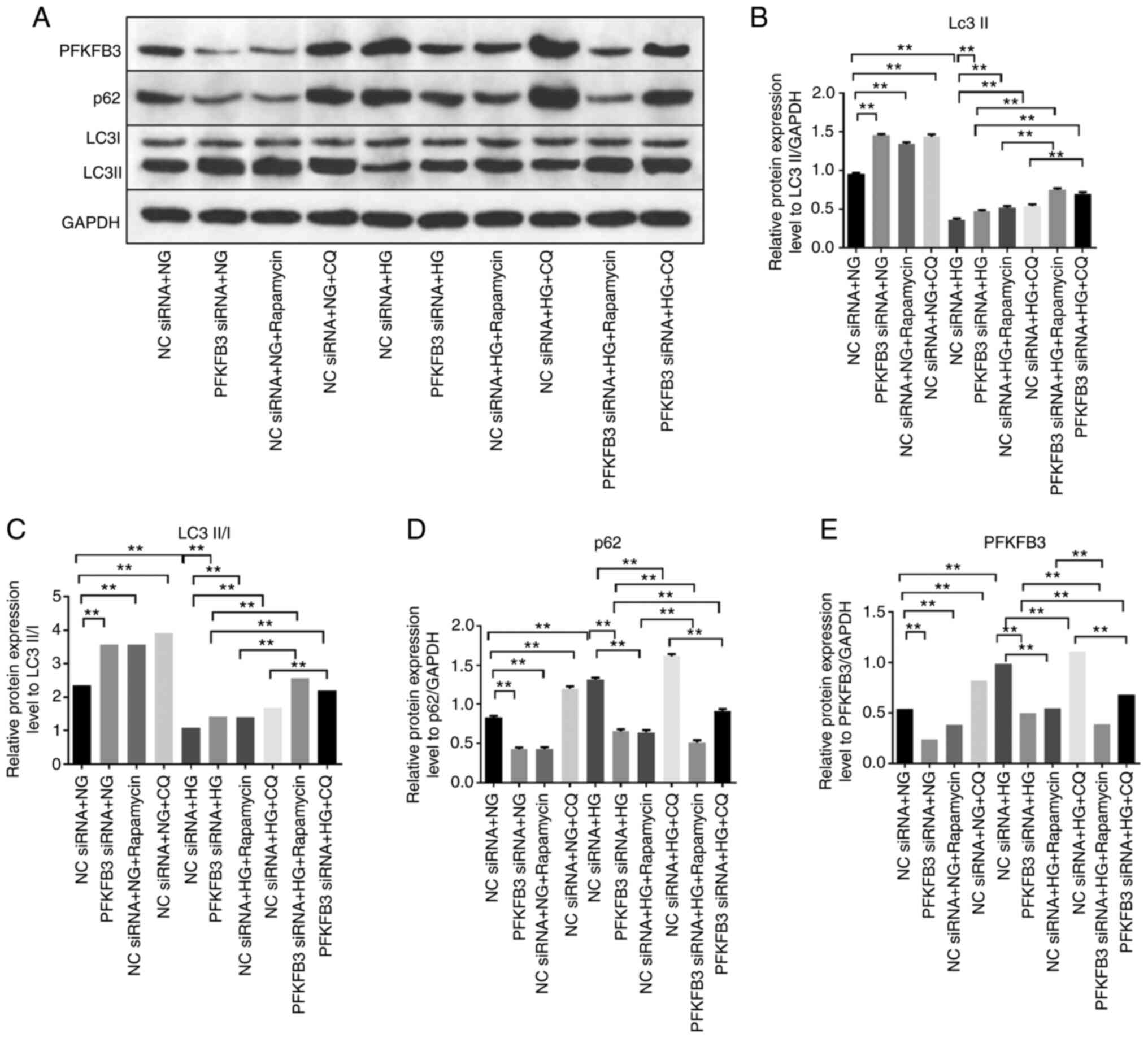 | Figure 5.Silencing of PFKFB3 inhibits podocyte
injury through activation of autophagy. MPC5 cells were treated
with NC siRNA + NG, PFKFB3 siRNA + NG, NC siRNA + NG + rapamycin,
NC siRNA + NG + CQ, NC siRNA + HG, PFKFB3 siRNA + HG, NC siRNA + HG
+ rapamycin, NC siRNA + HG + CQ, PFKFB3 siRNA + HQ + rapamycin or
PFKFB3 siRNA + HQ + CQ. (A) Protein expression levels of PFKFB3,
p62, LC3 I and LC3 II were detected by western blot analysis. (B)
Relative expression levels of LC3 II were normalized to GAPDH. (C)
Ratio of LC3 II/LC3 I was calculated. Relative expression levels of
(D) p62 and (E) PFKFB3 were normalized to GAPDH. **P<0.01. CQ,
chloroquine; HG, high-glucose; LC3, light chain 3; NC, negative
control; NG, normal glucose; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; siRNA, small
interfering RNA. |
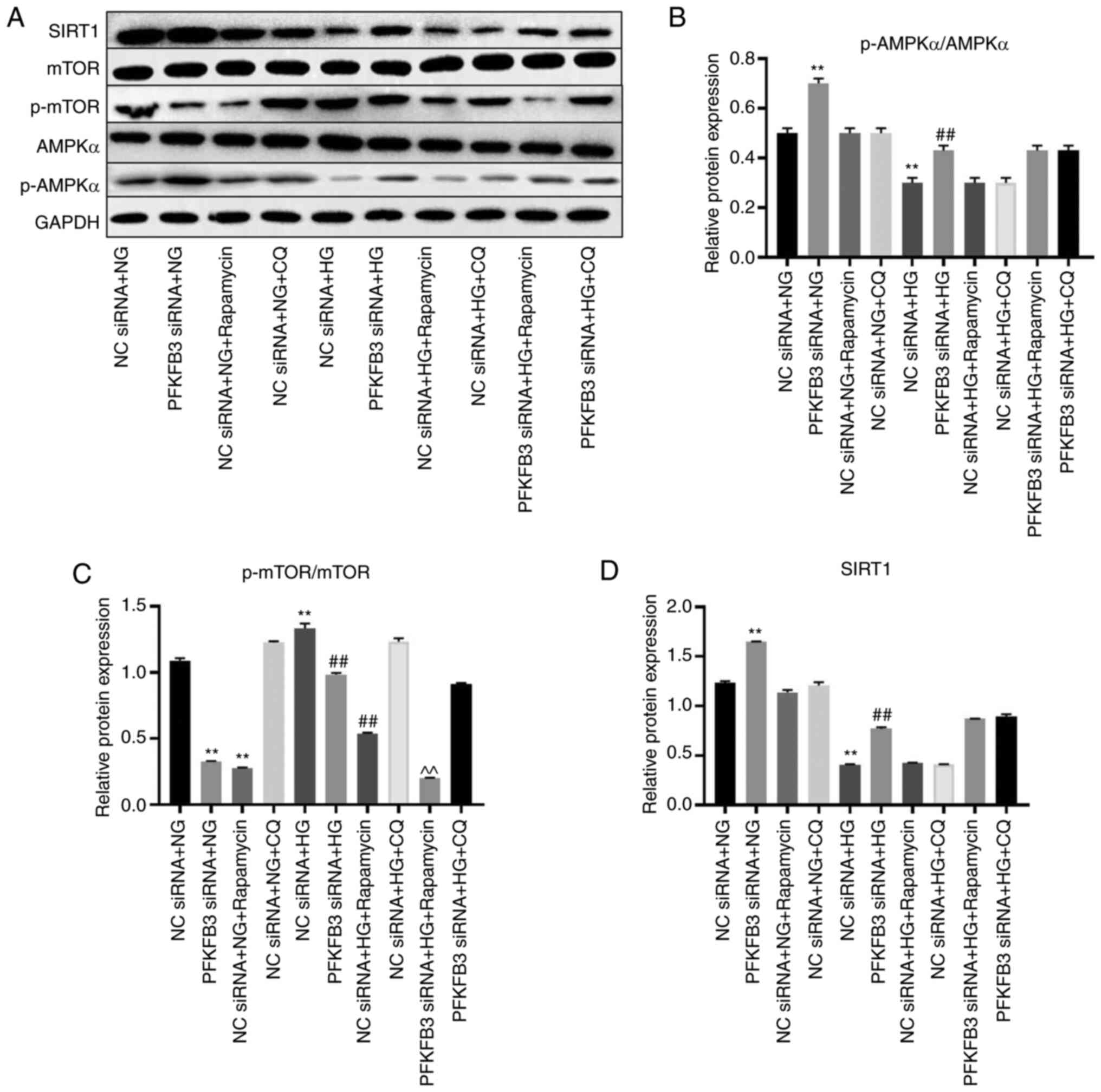 | Figure 7.Silencing of PFKFB3 induces autophagy
by mediating p-mTOR and p-AMPKα. (A) Protein expression levels of
SIRT1, mTOR, p-mTOR, p-AMPKα and AMPKα in MPC5 cells were
investigated by western blotting. (B) Relative expression of
p-AMPKα was normalized to AMPKα. (C) Relative expression of p-mTOR
was normalized to mTOR. (D) Relative expression of SIRT1 was
normalized to GAPDH. **P<0.01 vs. NC siRNA + NG;
##P<0.01 vs. NC siRNA + HG; ^^P<0.01
vs. PFKFB3 siRNA + HG. CQ, chloroquine; HG, high-glucose; NG,
normal glucose; NC, negative control; p-, phosphorylated; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; siRNA, small
interfering RNA; SIRT1, sirtuin 1. |
Effect of PFKFB3 siRNA on apoptosis
and migration of HG-treated podocytes was reversed by autophagy
inhibitor
To verify the association between PFKFB3 and cell
autophagy in MPC-5 cells, CCK8, flow cytometry and Transwell assays
were performed. Inhibition of PFKFB3 significantly increased
primary podocyte viability (Fig.
S3A) and suppressed podocyte apoptosis (Fig. S3B) and MPC-5 cell migration
(Fig. 8A) in the presence of NG,
compared with NG + NC siRNA. In addition, rapamycin inhibited the
migration of MPC5 cells compared with in the NG/HG + NC siRNA
groups (Fig. 8A). HG increased cell
migration compared with the NG + NC siRNA group (Fig. 8A). Besides, HG-induced increase of
cell migration was reversed by PFKFB3 siRNA or rapamycin, whereas
it was enhanced by CQ (Fig. 8A).
Meanwhile, the inhibitory effect of PFKFB3 siRNA on migration of
HG-treated MPC5 cells was enhanced by rapamycin but slightly
rescued by CQ (Fig. 8A).
Furthermore, the HG-induced decrease of cell area was notably
reversed by knockdown of PFKFB3. However, CQ notably reversed the
inhibitory effect of PFKFB3 siRNA on cell morphology (Fig. 8B). In summary, the effect of PFKFB3
siRNA on apoptosis and migration of HG-treated podocytes was
reversed by an autophagy inhibitor.
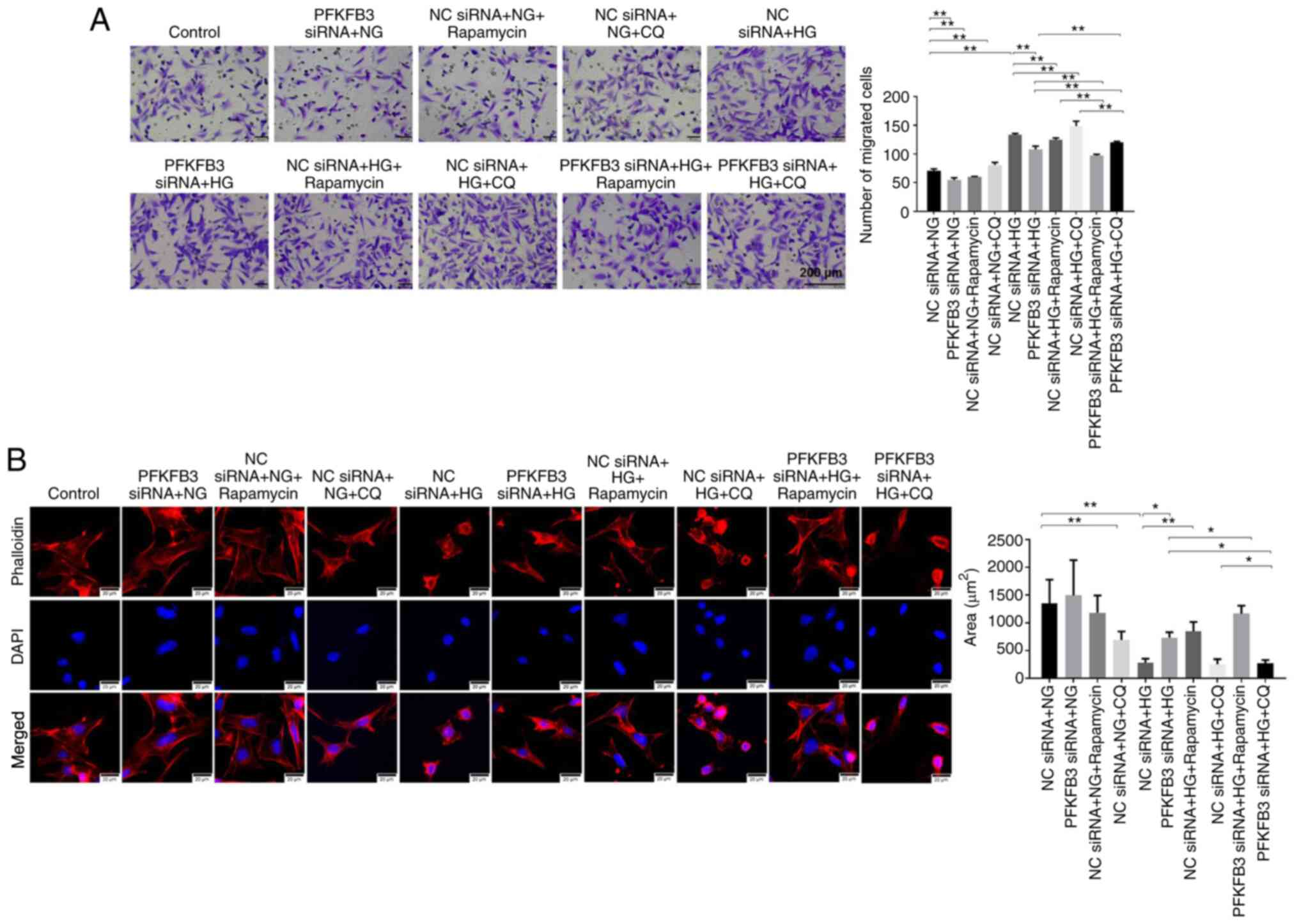 | Figure 8.HG-induced increase in cell migration
is reversed by PFKFB3 siRNA. (A) Cell migration was tested by
Transwell assay. Scale bar, 200 µm. (B) Phalloidin
immunofluorescence staining was performed to detect the
cytoskeleton and the area of MPC5 cells was calculated. Scale bar,
20 µm. *P<0.05, **P<0.01. CQ, chloroquine; HG, high-glucose;
NC, negative control; NG, normal glucose; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; siRNA, small
interfering RNA. |
Autophagy inhibitor reverses the
effect of PFKFB3 siRNA on the viability and DAG of HG-treated
podocytes
Glucose uptake of MPC-5 cells was detected. The
inhibitory effect of PFKFB3 siRNA on glucose uptake of HG-MPC-5
cells was partially reversed by CQ after 24, 48 and 72 h of
incubation (Fig. 9A). Similarly,
knockdown of PFKFB3 significantly reversed the HG-induced increase
in DAG levels in MPC5 cells; this was partially rescued by CQ
(Fig. 9B). CCK-8 assay results
demonstrated that downregulation of PFKFB3 significantly increased
the viability of MPC5 cells in the presence of HG compared with the
NC siRNA + HG group, and the effect was notably inhibited by CQ
(Fig. 9C). Furthermore, western
blot analysis was used to detect the expression levels of
synaptopodin and WT-1 in the MPC5 cells. Protein expression levels
of synaptopodin and WT-1 in the presence of HG were increased
following inhibition of PFKFB3, although this was only significant
for synaptopodin expression levels (Fig. 9D). However, the effect of PFKFB3
siRNA (in the PFKFB3 siRNA + HG group) on WT-1 levels was slightly
rescued by CQ (in the PFKFB3 siRNA + HG + CQ group).
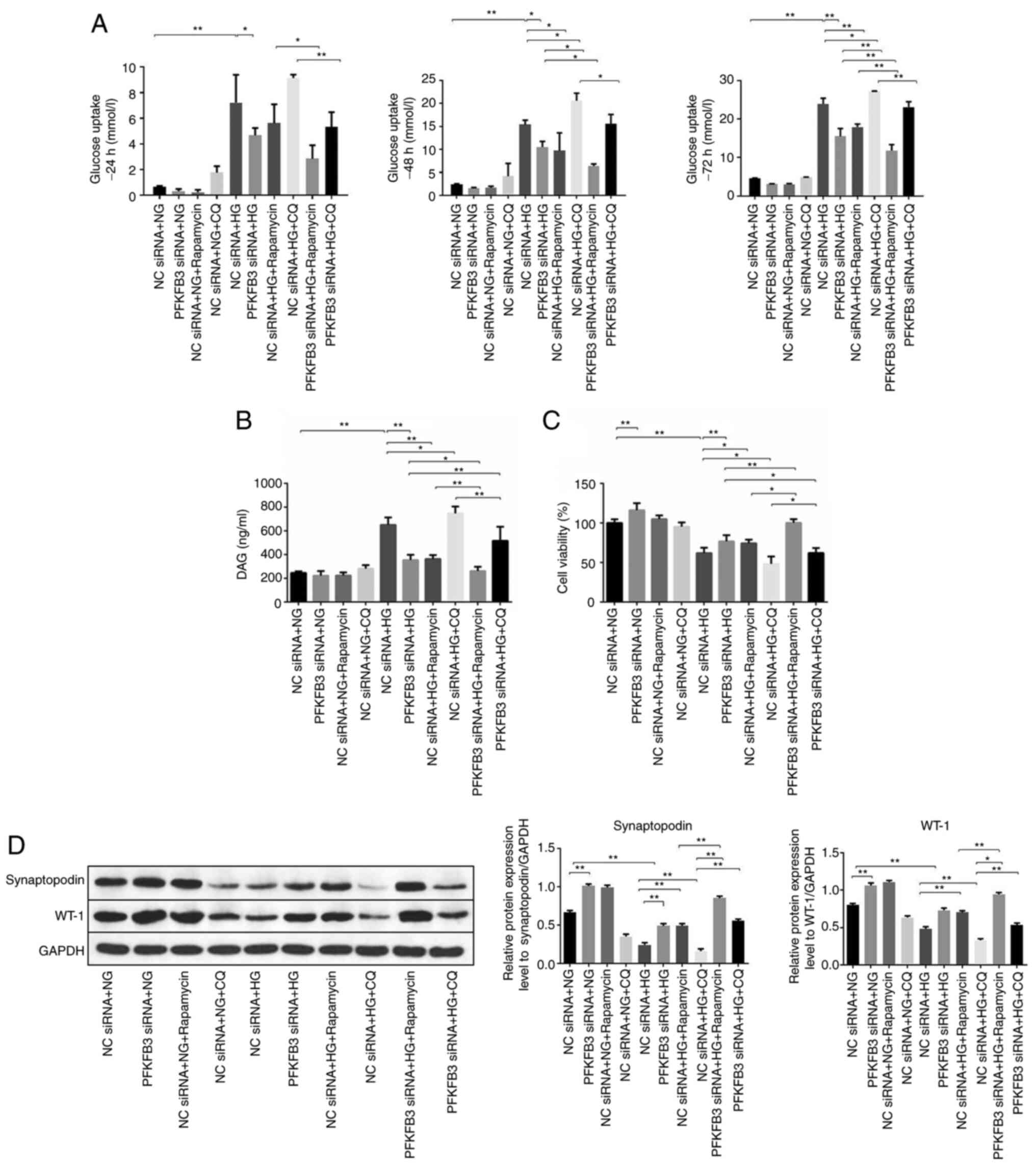 | Figure 9.CQ reverses the effect of PFKFB3
siRNA on podocyte proliferation. (A) Glucose uptake in MPC5 cells
was calculated at 24, 48 or 72 h. (B) Levels of DAG in MPC5 cells
were tested using a microplate reader. (C) Cell viability was
determined by Cell Counting Kit-8 assay. (D) Protein expression
levels of synaptopodin and WT-1 in MPC5 cells were examined by
western blot analysis and normalized to GAPDH. *P<0.05,
**P<0.01. CQ, chloroquine; DAG, diacylglycerol; HG,
high-glucose; NC, negative control; NG, normal glucose; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; siRNA, small
interfering RNA; WT-1, Wilms tumor-1. |
Discussion
A number of previous studies have demonstrated that
mRNA expression levels are dysregulated in a variety of serious
human diseases, including DN (32–34).
In the present study, the molecular mechanisms by which PFKFB3
siRNA alleviated HG-induced podocyte injury were investigated.
PFKFB3 expression levels were upregulated in MPC5 cells following
HG treatment. In addition, it has been reported that induction of
autophagy alleviates podocyte injury (35), which is consistent with the results
of the present study. This may be because activation of autophagy
may inhibit cell apoptosis (22).
A recent study demonstrated that multiple distinct
mRNAs (SIRT1, DNMT1, etc.) modulate the progression of DN by
targeting a wide range of signaling pathways associated with HG
(36,37). A range of interrelated factors,
including genetics, ischemia, inflammation and hypoxia, have been
shown to promote DN progression (38–40).
Numerous mRNAs (SIRT3, LC3, etc.) modulate DAG and glucose uptake
in cells by targeting the AMPK signaling pathway (41,42).
The present study explored the detailed mechanism by which PFKFB3
regulates the progression of DN.
In the present study, the effect of PFKFB3 on
autophagy was investigated. Autophagy is the biological cellular
process by which intracellular components undergo lysosome-mediated
self-digestion and recycling (43).
Autophagy is associated with the progression of numerous diseases,
such as inflammation and cancer (44–46).
Previous reports have indicated that activation of autophagy
ameliorates the symptoms of DN (47–49),
which was similar to the results of the present in vitro
study; knockdown of PFKFB3 activated autophagy. Results from the
present study also indicated that PFKFB3 siRNA may mediate the
expression of LC3, p-mTOR, p-AMPKα and SIRT1 in podocytes. As these
proteins are crucial factors during cell autophagy (50,51),
it may be concluded that PFKFB3 knockdown induced cell autophagy
through the modulation of these protein expressions. By contrast,
La Belle Flynn et al (52)
revealed that inhibition of PFKFB3 significantly inhibits autophagy
in the progression of breast cancer. This discrepancy may be due to
different cell types being investigated.
During the activation of autophagy flux, LC3 II
accumulation increases, while soluble p62 is decreased (53,54).
In addition, certain reports have indicated that p62 downregulation
does not always activate autophagic flux as LC3 I may not be
converted to LC3 II (55,56). In the present study, LC3 II
accumulation was increased in the PFKFB3 siRNA + HG + CQ group
compared with the NC siRNA + HG + CQ group. Moreover, p62
accumulation was notably inhibited in the PFKFB3 siRNA + HG + CQ
group compared with the NC siRNA + HG + CQ group. Thus, these data
suggested that PFKFB3 siRNA activated autophagic flux in MPC5
cells. Moreover, the present results suggested that PFKFB3
silencing reversed HG-induced podocyte apoptosis. It has been
confirmed that autophagy serves an important role in cell apoptosis
(57). Despite their distinct
mechanisms and functions, apoptosis and autophagy are closely
associated. Autophagy usually mediates the cell apoptosis (58). Thus, it can be concluded that PFKFB3
knockdown reversed HG-induced podocyte apoptosis by inducing
autophagy.
It was observed in the present study that CQ and
rapamycin did not always have the same effect on PFKFB3
siRNA-mediated DN. Rapamycin is known to function as an inhibitor
of the mTOR pathway, whereas CQ can suppress autophagy via
inhibiting the binding between autophagosomes and lysosomes
(59,60). Thus, the different mechanism by
which CQ and rapamycin regulate autophagy may result in different
results. Notably, CQ and rapamycin were shown to have no effect on
LC3 levels. LC3 is involved in the formation of autophagosomes, and
it is not associated with the mTOR pathway or the binding between
autophagosomes and lysosomes (55,56);
therefore, this may explain why CQ and rapamycin had no effect on
LC3 levels.
There are certain limitations in the present study.
First, in vivo experiments are required to further support
the results. In addition, the association between TGF-β and the EMT
process in podocytes is unclear. Therefore, more investigations are
required in the future.
In conclusion, knockdown of PFKFB3 may protect
podocytes from HG injury by inducing autophagy. Thus, PFKFB3 may
serve as a novel target for the treatment of DN.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZZ and QL conceived and supervised the study. QL and
JS designed the study. ZB and WW performed the experiments and
analyzed the data. QL and ZZ confirm the authenticity of all the
raw data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
These authors declare they have no competing
interests.
References
|
1
|
Cankurtaran V, Inanc M, Tekin K and Turgut
F: Retinal microcirculation in predicting diabetic nephropathy in
type 2 diabetic patients without retinopathy. Ophthalmologica.
243:271–279. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elbassuoni EA, Аziz NM and Habeeb WN: The
role of activation of KATP channels on hydrogen sulfide
induced renoprotective effect on diabetic nephropathy. J Cell
Physiol. 235:5223–5228. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shao J, Xu H, Wu X and Xu Y: Epigenetic
activation of CTGF transcription by high glucose in renal tubular
epithelial cells is mediated by myocardin-related transcription
factor A. Cell Tissue Res. 379:549–559. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al Shawaf E, Abu-Farha M, Devarajan S,
Alsairafi Z, Al-Khairi I, Cherian P, Ali H, Mathur A, Al-Mulla F,
Al Attar A, et al: ANGPTL4: A predictive marker for diabetic
nephropathy. J Diabetes Res. 2019:49431912019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Du L, Wang J, Chen Y, Li X, Wang L, Li Y,
Jin X, Gu X, Hao M, Zhu X, et al: Novel biphenyl diester derivative
AB-38b inhibits NLRP3 inflammasome through Nrf2 activation in
diabetic nephropathy. Cell Biol Toxicol. 36:243–260. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu L, Chen H, Yun J, Song L, Ma X, Luo S
and Song Y: miRNA-483-5p targets HDCA4 to regulate renal tubular
damage in diabetic nephropathy. Horm Metab Res. 2021.(Epub ahead of
print). doi: 10.1055/a-1480-7519. View Article : Google Scholar
|
|
7
|
Clem BF, O'Neal J, Tapolsky G, Clem AL,
Imbert-Fernandez Y, Kerr DA II, Klarer AC, Redman R, Miller DM,
Trent JO, et al: Targeting 6-phosphofructo-2-kinase (PFKFB3) as a
therapeutic strategy against cancer. Mol Cancer Ther. 12:1461–1470.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu L, Chen Y and Zhu Y: The molecular
basis of targeting PFKFB3 as a therapeutic strategy against cancer.
Oncotarget. 8:62793–62802. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sarkar Bhattacharya S, Thirusangu P, Jin
L, Roy D, Jung D, Xiao Y, Staub J, Roy B, Molina JR and Shridhar V:
PFKFB3 inhibition reprograms malignant pleural mesothelioma to
nutrient stress-induced macropinocytosis and ER stress as
independent binary adaptive responses. Cell Death Dis. 10:7252019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tao L, Yu H, Liang R, Jia R, Wang J, Jiang
K and Wang Z: Rev-erbα inhibits proliferation by reducing
glycolytic flux and pentose phosphate pathway in human gastric
cancer cells. Oncogenesis. 8:572019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wade SM, Ohnesorge N, McLoughlin H,
Biniecka M, Carter SP, Trenkman M, Cunningham CC, McGarry T,
Canavan M, Kennedy BN, et al: Dysregulated miR-125a promotes
angiogenesis through enhanced glycolysis. EBioMedicine. 47:402–413.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36:522017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ravanan P, Srikumar IF and Talwar P:
Autophagy: The spotlight for cellular stress responses. Life Sci.
188:53–67. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang L: Pharmacokinetics and drug
delivery systems for puerarin, a bioactive flavone from traditional
Chinese medicine. Drug Deliv. 26:860–869. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin X, Chen Y, Zhang P, Chen G, Zhou Y and
Yu X: The potential mechanism of postoperative cognitive
dysfunction in older people. Exp Gerontol. 130:1107912020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Son YO: Molecular mechanisms of
nickel-induced carcinogenesis. Endocr Metab Immune Disord Drug
Targets. 20:1015–1023. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang Q, Chen Z, Zhao L and Xu H: Circular
RNA hsa_circ_0000515 acts as a miR-326 sponge to promote cervical
cancer progression through up-regulation of ELK1. Aging (Albany
NY). 11:9982–9999. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Q, Li R, Xiao Z and Hou C: Lycopene
attenuates high glucose-mediated apoptosis in MPC5 podocytes by
promoting autophagy via the PI3K/AKT signaling pathway. Exp Ther
Med. 20:2870–2878. 2020.PubMed/NCBI
|
|
19
|
Sawada N and Arany Z: Metabolic regulation
of angiogenesis in diabetes and aging. Physiology (Bethesda).
32:290–307. 2017.PubMed/NCBI
|
|
20
|
Mizukami H, Osonoi S, Takaku S, Yamagishi
SI, Ogasawara S, Sango K, Chung S and Yagihashi S: Role of
glucosamine in development of diabetic neuropathy independent of
the aldose reductase pathway. Brain Commun. 2:fcaa1682020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu B, He X, Li S, Xu B, Birnbaumer L and
Liao Y: Deletion of diacylglycerol-responsive TRPC genes attenuates
diabetic nephropathy by inhibiting activation of the TGFβ1
signaling pathway. Am J Transl Res. 9:5619–5630. 2017.PubMed/NCBI
|
|
22
|
Chen JN, Li T, Cheng L, Qin TS, Sun YX,
Chen CT, He YZ, Liu G, Yao D, Wei Y, et al: Synthesis and in vitro
anti-bladder cancer activity evaluation of quinazolinyl-arylurea
derivatives. Eur J Med Chem. 205:1126612020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen Y, Tong ZW, Zhou Y, Sun Y, Xie Y, Li
R and Liu H: Inhibition of lncRNA-PAX8-AS1-N directly associated
with VEGF/TGF-β1/8-OhdG enhances podocyte apoptosis in diabetic
nephropathy. Eur Rev Med Pharmacol Sci. 24:6864–6872.
2020.PubMed/NCBI
|
|
24
|
He J, Gao HX, Yang N, Zhu XD, Sun RB, Xie
Y, Zeng CH, Zhang JW, Wang JK, Ding F, et al: The aldose reductase
inhibitor epalrestat exerts nephritic protection on diabetic
nephropathy in db/db mice through metabolic modulation. Acta
Pharmacol Sin. 40:86–97. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Albers JW and Pop-Busui R: Diabetic
neuropathy: Mechanisms, emerging treatments, and subtypes. Curr
Neurol Neurosci Rep. 14:4732014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qian X, Xu W, Xu J, Shi Q, Li J, Weng Y,
Jiang Z, Feng L, Wang X, Zhou J and Jin H: Enolase 1 stimulates
glycolysis to promote chemoresistance in gastric cancer.
Oncotarget. 8:47691–47708. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Almacellas E, Pelletier J, Manzano A,
Gentilella A, Ambrosio S, Mauvezin C and Tauler A:
Phosphofructokinases axis controls glucose-dependent mTORC1
activation driven by E2F1. iScience. 20:434–448. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
ElGamal H and Munusamy S: Aldose reductase
as a drug target for treatment of diabetic nephropathy: Promises
and challenges. Protein Pept Lett. 24:71–77. 2017.PubMed/NCBI
|
|
29
|
Liu YW, Cheng YQ, Liu XL, Hao YC, Li Y,
Zhu X, Zhang F and Yin XX: Mangiferin upregulates glyoxalase 1
through activation of Nrf2/ARE signaling in central neurons
cultured with high glucose. Mol Neurobiol. 54:4060–4070. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wong SHM, Fang CM, Chuah LH, Leong CO and
Ngai SC: E-cadherin: Its dysregulation in carcinogenesis and
clinical implications. Crit Rev Oncol Hematol. 121:11–22. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Saitoh M: Involvement of partial EMT in
cancer progression. J Biochem. 164:257–264. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao C, Chen J, Fan F, Long Y, Tang S,
Jiang C, Wang J and Xu Y and Xu Y: RIPK2-mediated autophagy and
negatively regulated ROS-NLRP3 inflammasome signaling in GMCs
stimulated with high glucose. Mediators Inflamm. 2019:62075632019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gong J, Zhan H, Li Y, Zhang W, Jin J and
He Q: Kruppel-like factor 4 ameliorates diabetic kidney disease by
activating autophagy via the mTOR pathway. Mol Med Rep.
20:3240–3248. 2019.PubMed/NCBI
|
|
34
|
Sankrityayan H, Oza MJ, Kulkarni YA, Mulay
SR and Gaikwad AB: ER stress response mediates diabetic
microvascular complications. Drug Discov Today. 24:2247–2257. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tu Q, Li Y, Jin J, Jiang X, Ren Y and He
Q: Curcumin alleviates diabetic nephropathy via inhibiting podocyte
mesenchymal transdifferentiation and inducing autophagy in rats and
MPC5 cells. Pharm Biol. 57:778–786. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hou Y, Lin S, Qiu J, Sun W, Dong M, Xiang
Y, Wang L and Du P: NLRP3 inflammasome negatively regulates
podocyte autophagy in diabetic nephropathy. Biochem Biophys Res
Commun. 521:791–798. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Syed AA, Reza MI, Garg R, Goand UK and
Gayen JR: Cissus quadrangularis extract attenuates diabetic
nephropathy by altering SIRT1/DNMT1 axis. J Pharm Pharmacol. Jun
15–2021.(Epub ahead of print). doi: 10.1093/jpp/rgab078. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo L, Tan K, Luo Q and Bai X:
Dihydromyricetin promotes autophagy and attenuates renal
interstitial fibrosis by regulating miR-155-5p/PTEN signaling in
diabetic nephropathy. Bosn J Basic Med Sci. 20:372–380.
2020.PubMed/NCBI
|
|
39
|
Bu J, Shi S, Wang HQ, Niu XS, Zhao ZF, Wu
WD, Zhang XL, Ma Z, Zhang YJ, Zhang H and Zhu Y: Acacetin protects
against cerebral ischemia-reperfusion injury via the NLRP3
signaling pathway. Neural Regen Res. 14:605–612. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhuang L, Jin G, Hu X, Yang Q and Shi Z:
The inhibition of SGK1 suppresses epithelial-mesenchymal transition
and promotes renal tubular epithelial cell autophagy in diabetic
nephropathy. Am J Transl Res. 11:4946–4956. 2019.PubMed/NCBI
|
|
41
|
Wang Y, Zhang X, Wang P, Shen Y, Yuan K,
Li M, Liang W and Que H: Sirt3 overexpression alleviates
hyperglycemia-induced vascular inflammation through regulating
redox balance, cell survival, and AMPK-mediated mitochondrial
homeostasis. J Recept Signal Transduct Res. 39:341–349. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Woo CY, Kc R, Kim M, Kim HS, Baek JY and
Koh EH: Autophagic flux defect in diabetic kidney disease results
in megamitochondria formation in podocytes. Biochem Biophys Res
Commun. 521:660–667. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dasgupta S: Mitochondrion: I am more than
a fuel server. Ann Transl Med. 7:5942019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu Q, Tian AL, Li B, Leduc M, Forveille S,
Hamley P, Galloway W, Xie W, Liu P, Zhao L, et al: IGF1 receptor
inhibition amplifies the effects of cancer drugs by autophagy and
immune-dependent mechanisms. J Immunother Cancer. 9:e0027222021.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kommalapati VK, Kumar D and Tangutur AD:
Quisinostat mediated autophagy is associated with differentiation
in neuroblastoma SK-N-SH cells. Mol Biol Rep. 48:4973–4979. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xu J, Liu LQ, Xu LL, Xing Y and Ye S:
Metformin alleviates renal injury in diabetic rats by inducing
Sirt1/FoxO1 autophagic signal axis. Clin Exp Pharmacol Physiol.
47:599–608. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ye X, Zhou XJ and Zhang H: Autophagy in
immune-related renal disease. J Immunol Res. 2019:50716872019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Alvarez-Cilleros D, Lopez-Oliva ME, Martin
MA and Ramos S: Cocoa ameliorates renal injury in Zucker diabetic
fatty rats by preventing oxidative stress, apoptosis and
inactivation of autophagy. Food Funct. 10:7926–7939. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen L, Zhao L, Samanta A, Mahmoudi SM,
Buehler T, Cantilena A, Vincent RJ, Girgis M, Breeden J, Asante S,
et al: STAT3 balances myocyte hypertrophy vis-a-vis autophagy in
response to Angiotensin II by modulating the AMPKα/mTOR axis. PLoS
One. 12:e01798352017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang P, Liu X, Li H, Chen Z, Yao X, Jin J
and Ma X: TRPC5-induced autophagy promotes drug resistance in
breast carcinoma via CaMKKβ/AMPKα/mTOR pathway. Sci Rep.
7:31582017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
La Belle Flynn A, Calhoun BC, Sharma A,
Chang JC, Almasan A and Schiemann WP: Autophagy inhibition elicits
emergence from metastatic dormancy by inducing and stabilizing
Pfkfb3 expression. Nat Commun. 10:36682019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chu CW, Ko HJ, Chou CH, Cheng TS, Cheng
HW, Liang YH, Lai YL, Lin CY, Wang C, Loh JK, et al: Thioridazine
enhances P62-Mediated autophagy and apoptosis through Wnt/β-catenin
signaling pathway in glioma cells. Int J Mol Sci. 20:4732019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Guo FX, Wu Q, Li P, Zheng L, Ye S, Dai XY,
Kang CM, Lu JB, Xu BM, Xu YJ, et al: The role of the
LncRNA-FA2H-2-MLKL pathway in atherosclerosis by regulation of
autophagy flux and inflammation through mTOR-dependent signaling.
Cell Death Differ. 26:1670–1687. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guo H, Ding H, Yan Y, Chen Q, Zhang J,
Chen B and Cao J: Intermittent hypoxia-induced autophagy via
AMPK/mTOR signaling pathway attenuates endothelial apoptosis and
dysfunction in vitro. Sleep Breath. 2021.(Epub ahead of print).
View Article : Google Scholar
|
|
56
|
Chen J, Wang L, Liu WH, Shi J, Zhong Y,
Liu SJ and Liu SM: Aspirin protects human coronary artery
endothelial cells by inducing autophagy. Physiol Int. 107:294–305.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang Z, Liu N, Liu K, Zhou G, Gan J, Wang
Z, Shi T, He W, Wang L, Guo T, et al: Autophagy mediated CoCrMo
particle-induced peri-implant osteolysis by promoting osteoblast
apoptosis. Autophagy. 11:2358–2369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hale AN, Ledbetter DJ, Gawriluk TR and
Rucker EB III: Autophagy: Regulation and role in development.
Autophagy. 9:951–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Corral-Ramos C, Barrios R, Ayté J and
Hidalgo E: TOR and MAP kinase pathways synergistically regulate
autophagy in response to nutrient depletion in fission yeast.
Autophagy. Jun 23–2021.(Epub ahead of print). doi:
10.1080/15548627.2021.1935522. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gray JP, Uddin MN, Chaudhari R, Sutton MN,
Yang H, Rask P, Locke H, Engel BJ, Batistatou N, Wang J, et al:
Directed evolution of cyclic peptides for inhibition of autophagy.
Chem Sci. 12:3526–3543. 2021. View Article : Google Scholar : PubMed/NCBI
|