Introduction
Hyperbilirubinemia, one of the most common disorders
in newborns, is characterized by an imbalance between bilirubin
production and elimination (1).
Aberrant accumulation of bilirubin may cause bilirubin
encephalopathy, which is the most serious complication of neonatal
hyperbilirubinemia that can lead to neonatal death (2–4).
Survivors often have neurological sequelae, including learning
disabilities, movement disorders, mental retardation and cerebral
palsy (5). Neonatal bilirubin
encephalopathy causes 4.8% of all pediatric hospitalization and its
mortality is estimated to be 16.1% in a multicenter study in China
(6). Although a number of
treatment options have been employed for the treatment of neonatal
hyperbilirubinemia, including phototherapy and exchange transfusion
(7), neurological damage may still
occur, suggesting that several other factors contribute to
increased neurotoxicity (8).
Hemolysis and its major byproduct, ferrous ion (Fe2+),
are among the factors that serve important roles in the development
of neurotoxicity (9,10); however, the underlying mechanism
remains unclear.
In both developing and developed countries,
hemolysis is one of the risks factors for hyperbilirubinemia
(11). During excessive hemolysis,
increased amounts of iron are released from damaged red blood
cells. Iron enters the endothelial cells that form the blood-brain
barrier via transferrin receptor-mediated endocytosis, or it is
independently transported as non-protein-bound iron (12). A significant increase in
non-protein-bound iron levels and an imbalance between pro-oxidant
and antioxidant systems is observed in neonatal hemolytic diseases
(10,13).
Ferroptosis, a recently identified form of
programmed cell death that differs from apoptosis and necrosis, is
driven by iron-dependent accumulation of lipid hydroperoxides
(14). Dysregulation of iron
homeostasis, glutathione (GSH) depletion and lipid peroxidation are
closely linked to cell sensitivity to ferroptosis (15). Iron homeostasis imbalance has been
implicated in ferroptosis, which may lead to pathological
conditions in the central nervous system (16,17).
Studies have reported that ferroptosis is involved in numerous
neurodegenerative diseases, such as Alzheimer's disease (16,18),
Parkinson's disease (19),
ischemic stroke (20),
intracerebral hemorrhage (21) and
traumatic brain injury (22).
These conditions lead to increased hemoglobin release and iron
overload, resulting in an abnormal increase in lipid peroxidation,
which is the main cause of secondary injury and neuronal
ferroptosis (23). Hemolytic
hyperbilirubinemia also results in an increased release of
hemoglobin and iron (11).
Moreover, specific features of ferroptosis, such as
increased products of lipid peroxidation, are similar to those that
appear in iron-mediated neurological dysfunction caused by neonatal
hemolytic diseases (24). In a rat
model of hemolytic hyperbilirubinemia-induced brain damage (HHIBD),
comparing to the control group, the level of malondialdehyde (MDA)
increased and GSH decreased in brain tissues of the treatment
group, closely resembling the biochemical characteristics of
ferroptosis (25,26). It is therefore hypothesized that
ferroptosis may contribute to the pathogenesis of neuronal
dysfunction induced by neonatal hemolytic hyperbilirubinemia
(8). However, the contribution of
ferroptosis to hemolytic HHIBD remains unclear. The present study,
utilizing both in vitro and in vivo assays, assessed
the role of ferroptosis in HHIBD.
Materials and methods
Modeling neonatal hemolytic
hyperbilirubinemia in rats
A total of 60 male Sprague-Dawley (SD) rats (aged
10–12 days; 24–28 g) were purchased from the Laboratory Animal
Center, Fujian Medical University (Fuzhou, China). Rat pups were
housed in the same standard laboratory polycarbonate cage with
their mothers at a suitable temperature (21±2°C) and humidity
(50±10%) with 12/12-h light/dark cycles to maintain their circadian
rhythm. Maternal milk was used to feed the rat pups, while adult
female rats had free access to standardized granular food (Beijing
Keao Xieli Feed Co., Ltd.) and tap water. The 10–12 day old rat
pups were randomly divided into the control, phenylhydrazine (PHZ)
and deferoxamine (DFO) + PHZ groups, with 12 rats in each
group.
To establish bilirubin encephalopathy secondary to
hemolysis, 75 mg/kg PHZ (Sigma-Aldrich; Merck KGaA) was injected
intraperitoneally once daily for 2 days, based on previous reports
(26,27). Rats in the DFO + PHZ group were
injected intraperitoneally with DFO (100 mg/kg; cat. no. D9533;
Sigma-Aldrich; Merck KGaA) 1 h prior to administration of PHZ
(28,29) whilst rats in the PHZ group received
the same volume of saline. Control rats received the same volume of
saline in parallel. Subsequently, 24 h after the last dosing, all
animals were anesthetized intraperitoneally with 100 mg/kg
pentobarbital sodium and then euthanized by decapitation to collect
trunk blood and brain tissue. A total of 0.5 ml blood from each rat
was collected and centrifuged at 1,500 × g at 4°C for 10 min and
then preserved at −80°C for later biochemical analysis. Brain
tissue was immediately collected, placed in liquid nitrogen for 5
min, and then stored at −80°C for future analysis. All treatments
were performed with care and rat suffering was minimized. During
the experiment, none of the rats reached any of the following
humane endpoints: Inability to drink the maternal milk, labored
breathing, inability to stand or no response to external stimuli.
Death was verified by monitoring the cessation of breathing and
heartbeat and pupil dilation. No animals were found dead before the
end of the experiment.
Cell culture and drug treatment
The highly differentiated PC12 cell line, derived
from a pheochromocytoma of the rat adrenal medulla, resembles
neurons and is widely used in the study of neurological diseases
in vitro (30,31). PC12 cells (Wuhan Boster Biological
Technology, Ltd.) were cultured in Dulbecco's Modified Eagle Medium
(cat. no. C11995500BT; Gibco; Thermo Fisher Scientific, Inc.),
supplemented with 10% fetal bovine serum (cat. no. SV30207.03;
HyClone; Cytiva) and 100 U/ml penicillin-streptomycin (cat. no.
V900929; Sigma-Aldrich; Merck KGaA). Cells were cultured at 37°C as
monolayers in a humidified atmosphere containing 5% CO2.
Based on MTT cell viability assay results (Fig. S1) and the level of superoxide
dismutase (SOD), MDA and reactive oxygen species (ROS) in the PC12
cells exposed to 0, 200 and 400 µM ferric ammonium citrate (FAC;
cat. no. F5879; Sigma-Aldrich; Merck KGaA) for 24 h (Fig. S2), 400 µM FAC treatment for 24 h
was selected for subsequent experiments, to assess the mechanism of
iron overload-induced neurotoxicity in vitro. PC12 cells
were pretreated with 10 µM ferrostatin-1 (Fer-1; cat. no. SML0583;
Sigma-Aldrich; Merck KGaA) or 100 µM DFO for 1 h at 37°C and
exposed to 400 µM FAC.
Measurement of serum hemoglobin,
hematocrit and bilirubin levels
Serum hemoglobin and hematocrit levels were assessed
using an automated analyzer (Coulter LH 780; Beckman Coulter,
Inc.). Another automated analyzer (Architect ci16200 Integrated
System; Abbott Pharmaceutical Co. Ltd.) was used to measure the
serum levels of total bilirubin, indirect bilirubin and direct
bilirubin using a diazo reagent-based method (32). All procedures strictly followed the
manufacturers' instructions.
Measurement of S100 calcium-binding
protein B (S100B) and serum neuron-specific enolase (NSE)
levels
Serum levels of S100B were assessed using the Rat
S100B ELISA kit (cat. no. E-EL-R0868; Elabscience Biotechnology,
Inc.), and those of serum NSE were assessed using the Rat NSE ELISA
Kit (cat. no. E-EL-R0058c; Elabscience Biotechnology, Inc.)
according to the manufacturer's instructions.
Measurement of SOD activity, MDA and
reduced GSH/oxidized glutathione disulfide (GSSG) levels
A total of 15 mg brain tissue from six rats in each
group was placed into 200 µl ice-cold 10% radioimmunoprecipitation
assay lysis buffer (RIPA; cat, no. P0013E; Beyotime Institute of
Biotechnology). Brain samples were homogenized using a
tissue homogenizer (cat. no. KZ-III-F; Wuhan Servicebio Technology
Co., Ltd.) and centrifuged at 1,000 × g for 10 min at 4°C. The
supernatant was then transferred into microcentrifuge tubes. For
cell extracts, PC12 cells were seeded in the 6-well plate at a
density of 50–60% for 24 h, and then exposed to FAC. A total of 200
µl RIPA lysis buffer were added to plate to homogenize the sample
and centrifuged at 1,000 × g for 10 min at 4°C, waiting for next
measurement.
SOD activity and total GSH/GSSG detection kits (cat.
nos. A001-3-1 and A061-1-1; Nanjing Jiancheng Bioengineering
Institute) were used to assess SOD and GSH/GSSG levels,
respectively. Tissue iron detection kits (cat. no. A039-2-1;
Nanjing Jiancheng Bioengineering Institute) were used to assess
tissue iron levels. An MDA detection kit (cat. no. S0131S; Beyotime
Institute of Biotechnology) was used to assess MDA levels. All
assays were conducted in triplicate according to the manufacturers'
protocols.
Kyoto Encyclopedia of Genes and
Genomes (KEGG) enrichment analysis
Proteomic analysis of separate triplicate brain
tissue samples from the control and PHZ groups was performed by
tandem mass tag (TMT) labeling (Cat. no. A44520; Thermo Fisher)
(33) and liquid
chromatography-tandem mass spectrometry (Q Exactive™ HF-X; Thermo
Fisher) (34). The detection was
conducted by multiple reaction monitoring (MRM) in negative
ionization mode. The electrospray voltage was 2.1 kV. The m/z scan
range was 350–1600 for the full scan, and intact peptides at a
resolution of 120,000 were detected in the Orbitrap. We set the
automatic gain control (AGC) at 3E6, and the fixed first mass was
100 m/z. The nitrogen gas temperature was at 300°C with a flow rate
of 5 l/min and nebulizer pressure was 40 psi. The differentially
abundant protein (DAP) screening parameters were adjusted to
P<0.05 and log2 fold change=0, KEGG enrichment
analysis was performed using ClueGO (v2.5.6; http://apps.cytoscape.org/apps/cluego) (35). The databases used were
KEGG_20.05.2019 (genome.jp/kegg/pathway.html) and Gene Ontology
(GO) (https://www.geneontology.org/)_Biological
Process-EBI-Uniprot-GO Annotation_20.05.2019 (https://david.ncifcrf.gov/home.jsp). The GO term
‘fusion mode’ was selected according to the P-value <0.05.
Protein-protein interaction network
analysis
The number of differential protein sequences in the
protein database (Maxquant: v1.6.15.0; http://www.maxquant.org/) screened according to a 1.2
expression difference multiplesin PHZ group vs. Control group, and
then compared with those in the protein network interaction
database of the Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING; v. 11.0; http://cn.string-db.org/), with a confidence score of
>0.7 (high confidence). The differential protein interaction was
analyzed, and the R package (Dev-version: 0.4;
christophergandrud.github.io/networkD3/) ‘networkD3’ function was
used to visually display the differential protein interaction
network.
Cell viability assay
In the FAC model, PC12 cells were seeded in 96-well
plates for 24 h at a density of 1×104 cells/well. Cells
were then treated with different concentrations of FAC (0, 100,
200, 400 and 800 µM, 1 and 5 mM) for 3, 6, 12 and 24 h. The
viability of PC12 cells was assessed using MTT (cat. no. F5655;
Sigma-Aldrich; Merck KGaA). Briefly, after the aforementioned
treatments, MTT (5 mg/ml) was added to the 96-well plate. The plate
was incubated at 37°C for 4 h to allow the formazan crystals to
form. The medium was then discarded and 100 µl dimethyl sulfoxide
was added to dissolve the formazan. Absorbance was measured at a
detection wavelength of 570 nm using a plate reader (Thermo Fisher
Scientific, Inc.).
Measurement of lipid oxidation using
confocal laser scanning microscopy
After PC12 cells were seeded in the 35 mm confocal
dish (cat. no. FCFC016; Beyotime Institute of Biotechnology; China)
at a density of 50 %, the cells were treated with FAC as
aforementioned for 24 h, and then were rinsed with
phosphate-buffered saline (PBS) three times. To visualize neuronal
lipid oxidation, 10 µM BODIPY 581/591 C11 (cat. no. D3861; Thermo
Fisher Scientific, Inc.) was added to each well and incubated at
37°C for 0.5 h and stained with 1 µg/ml DAPI for 5 min at 37°C.
Cells were rinsed with PBS three times and imaged using a confocal
laser scanning microscope (SP5; Leica Microsystems GmbH) (36). The argon laser (excitation, 488 nm;
emission, 500–535 nm) was used to detect oxidized lipids, whereas
the white light laser (excitation, 561 nm; emission, 573–613 nm)
was used to detect non-oxidized lipids.
Measurement of ROS levels
PC12 cells were seeded in the 6-well plates for 24 h
at a density of 2×105 cells/well and then were treated
with FAC for 24 h as aforementioned. Subsequently, cells were
stained with the ROS indicator 2′,7′-dichlorodihydrofluorescein
diacetate (cat. no. S0033S; Beyotime Institute of Biotechnology)
for 30 min at 37°C. Subsequently, the cells were washed with PBS
three times and digested with 0.25 % trypsin (cat. no. C0201S;
Beyotime Institute of Biotechnology) at 37°C for 2 min. Finally,
labelled cells were detected with a flow cytometer (LSRFortessa™
X-20; BD Biosciences) and analyzed by Flow v10.9 (BD Biosciences)
(37).
Detection of intracellular free iron
using fluorescence microscopy
FerroOrange (cat. no. F374; Dojindo Laboratories,
Inc.) was used to detect intracellular Fe2+. PC12 cells
were seeded in the 12-well plates for 24 h at a density of
2×105 cells/well and then were treated with 400 µM FAC
as aforementioned., They were rinsed with PBS and stained with 1 µM
FerroOrange at 37°C for 0.5 h. The cells were rinsed with PBS three
times and imaged under a fluorescence microscope (38).
Transmission electron microscopy
(TEM)
PC12 cells were seeded in a 10 cm dish for 24 h at a
density of 1×106 cells/well and then exposed to FAC as
aforementioned. Subsequently, cells were fixed in 2.5%
glutaraldehyde (diluted in 0.1 µM PBS; pH 7.4) at 25°C for 24 h and
then post-fixed in 1% osmium tetroxide (dissolved in 0.1 µM PBS; pH
7.4) at 25°C for 60 min. After dehydration with ethanol (50, 70,
80, 90 and 100%), samples were embedded by resin (cat. no. 45347;
Merck) with different condition (37°C for 12 h; 45°C for 12 h; 60°C
for 12 h) and ultrathin sectioning (50 nm). Then, the samples were
stained with uranyl acetate at room temperature for 30 min and
stained with lead citrate at room temperature for 8 min. Digital
images were captured using TEM (FEI Tecnai G2 F30; Thermo Fisher
Scientific, Inc.) (39).
Western blotting
Proteins were extracted from brain tissue using RIPA
lysis buffer (cat. no. P0013E; Beyotime Institute of Biotechnology)
supplemented with phenylmethylsulfonyl fluoride and then denatured
at 95°C in 5X protein loading buffer (cat. no. P0286; Beyotime
Institute of Biotechnology) for 10 min. The concentration of
protein in sample were measured by Enhanced BCA Protein Assay Kit
(cat. no. P0009; Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. An equal amount of protein samples
(20 µg/lane) were added to per lane and then were separated on 10%
gels by SDS-PAGE. Proteins were then transferred to 0.2 µm PVDF
membranes and blocked with 5% non-fat milk for 60 min at room
temperature. PVDF membranes were incubated overnight at 4°C with
primary antibodies against the following: Rabbit acyl-coenzyme A
synthetase long-chain family member 4 (ACSL4; 1:1,000; cat. no.
A16848; ABclonal Biotech Co., Ltd.); ferritin heavy chain (FTH;
1:1,000; cat. no. A19544; ABclonal Biotech Co., Ltd.); transferrin
receptor 1 (TFRC; 1:1,000; cat. no. A5865; ABclonal Biotech Co.,
Ltd.); ferroportin 1 (FPN1; 1:1,000; cat. no. A14884; ABclonal
Biotech Co., Ltd.); divalent metal transporter 1 (DMT1; 1:1,000;
cat. no. A10231; ABclonal Biotech Co., Ltd.); iron regulatory
protein 2 (IRP2; 1:1,000; cat. no. A6382; ABclonal Biotech Co.,
Ltd.); GAPDH (1:5,000; cat. no. A19056; ABclonal Biotech Co.,
Ltd.); xCT (1:1,000; cat. no. ab175186; Abcam); GSH peroxidase 4
(GPX4; 1:1,000; cat. no. ab125066; Abcam) and ferroptosis
suppressor protein 1 (FSP1; 1:1,000; cat. no. AVARP09054_P050;
Aviva Systems Biology, Corp). After washing by TBS buffer with 0.1%
Tween-20 (cat. no. ST1727; Beyotime Institute of Biotechnology),
PVDF membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (1:8,000; cat. no.
31460; Thermo Fisher) at 37°C for 60 min. Enhanced
chemiluminescence detection was performed (cat. no. 1705061;
BIO-RAD) (40) and ImageJ software
(version 1.53; National Institutes of Health) was used to
semi-quantify the protein bands.
Statistical analysis
Data are presented as mean ± standard error of the
mean. All analyses were conducted using GraphPad Prism 8.0
(GraphPad Software; Dotmatics). Significance of differences between
two groups was analyzed using two-way independent-sample t-test.
For three or more groups, one-way analysis of variance followed by
Tukey's post-hoc test was used to determine significance. P<0.05
was considered to indicate a statistically significant
difference.
Results
Successful establishment of the
hemolytic hyperbilirubinemia model
To establish a hemolytic hyperbilirubinemia model of
neonatal SD rats, PHZ was used to increase erythrocyte turnover via
hemolysis (Fig. S3A). In the PHZ
group, the appearance of the neonatal rats and their brains was
more jaundiced than that of the control group (Fig. S3B and C). PHZ group had
significantly decreased hematocrit and hemoglobin levels compared
with the control group (Fig. S3D and
E). Moreover, the serum levels of three types of bilirubin
(total, direct and indirect bilirubin) significantly increased in
the PHZ group compared with that of the control group (Fig. S3F-H), indicating that the
hemolytic hyperbilirubinemia model was established
successfully.
Subsequently, changes in the brain injury markers
NSE and S100B in the PHZ group were assessed. Compared with those
in the control group, serum S100B and NSE levels were significantly
increased in the PHZ group (Fig. 1A
and B). Simultaneously, the concentration of iron significantly
increased in the brain tissue of the PHZ group compared with that
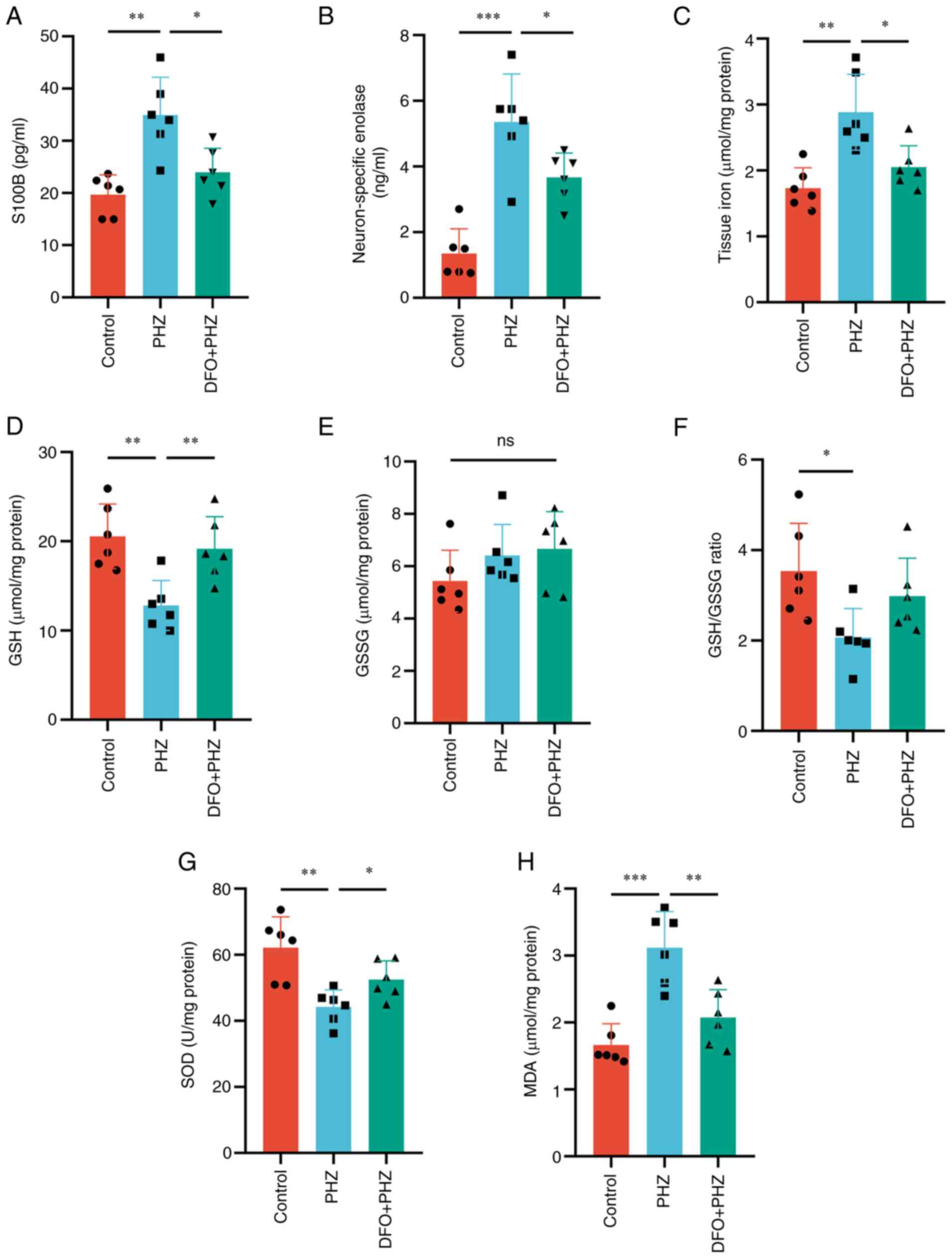
of the control group (Fig. 1C).
Disturbances in oxidative balance caused by hemolytic
hyperbilirubinemia were then assessed. SOD, which serves an
important function in the first line of antioxidant defense,
significantly decreased in the brains of the PHZ group compared
with that of the control group (Fig.
1G). Compared with the control group, the GSH and GSH/GSSG
levels significantly decreased in the PHZ group. However, there was
no difference in the level of GSSG between the two groups (Fig. 1D-F). Similarly, the level of lipid
peroxidation, as assessed by detecting the levels of MDA,
significantly increased in the PHZ group compared with that of the
control group (Fig. 1H). Overall,
these results suggested that hemolytic hyperbilirubinemia could
disrupt the redox balance in rat brains, resulting in brain injury.
However, the specific mechanism of action remained unclear.
Therefore, mass spectrometry analysis was performed to evaluate the
potential molecular mechanisms of hemolytic hyperbilirubinemia.
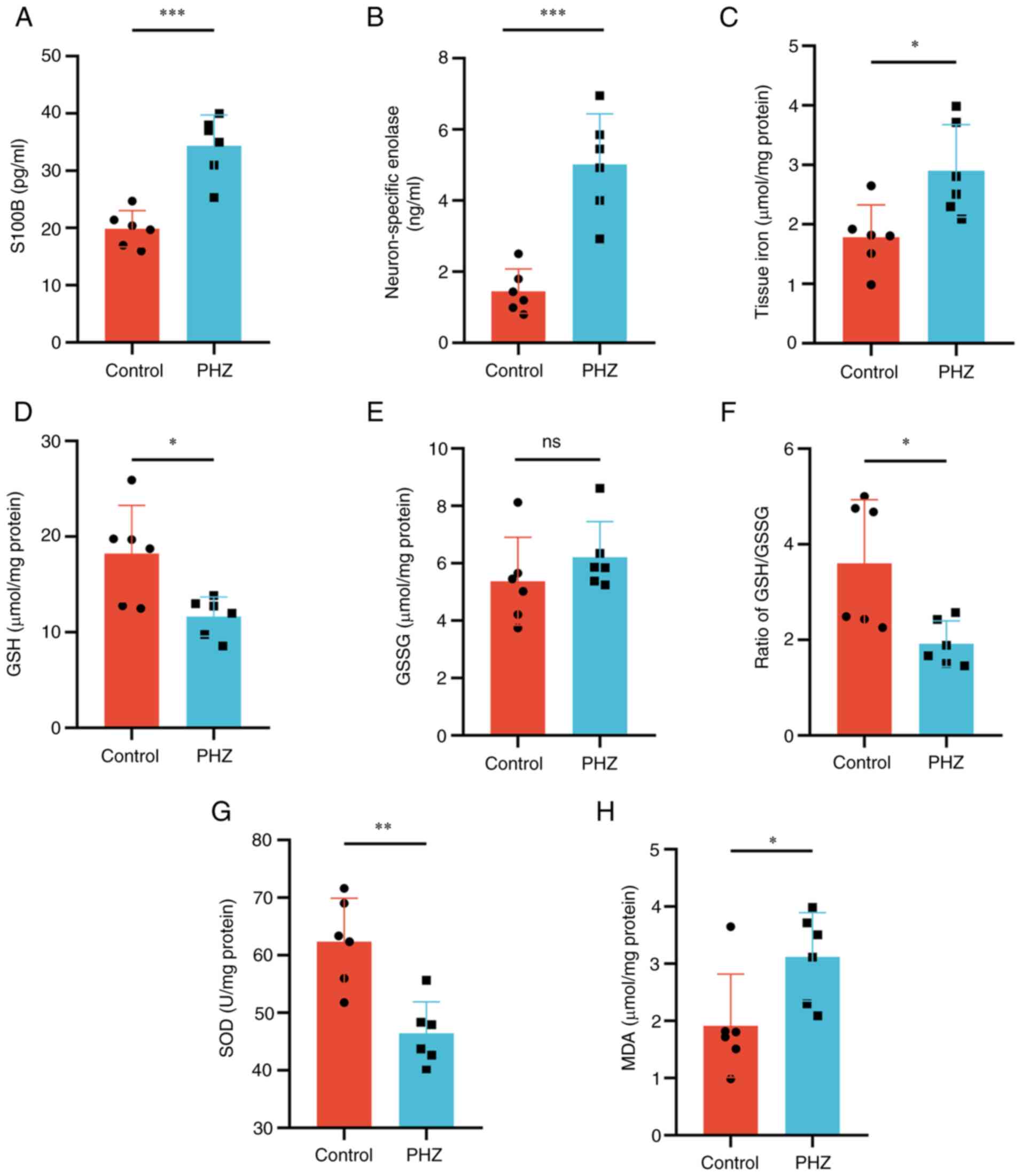 | Figure 1.Hemolytic hyperbilirubinemia-induced
brain damage in neonatal Sprague-Dawley rats. Serum levels of (A)
S100B and (B) neuron-specific enolase. (C) Tissue iron levels in
the brain. Redox imbalances assessed by measuring levels of (D)
GSH, (E) GSSG, (F) GSH/GSSG, (G) SOD and (H) MDA. n=6. *P<0.05;
**P<0.01; ***P<0.001. S100B, S100 calcium-binding protein B;
GSH, glutathione; GSSG, glutathione disulfide; SOD, superoxide
dismutase; MDA, malondialdehyde; PHZ, phenylhydrazine; ns, not
significant. |
Ferroptosis participates in brain
damage induced by hemolytic hyperbilirubinemia
Proteomic analysis of brain tissue samples from the
control and PHZ groups were performed in triplicate using TMT
labelling to evaluate whether ferroptosis is involved in HHIBD.
KEGG analysis using ClueGO software demonstrated the enrichment of
ferroptosis-related proteins in the PHZ group compared with those
in the control group. When the DAPs screening parameters were
adjusted to P<0.05 and log2 fold change=0, compared
with the control, a total of 26 ferroptosis-associated DAPs were
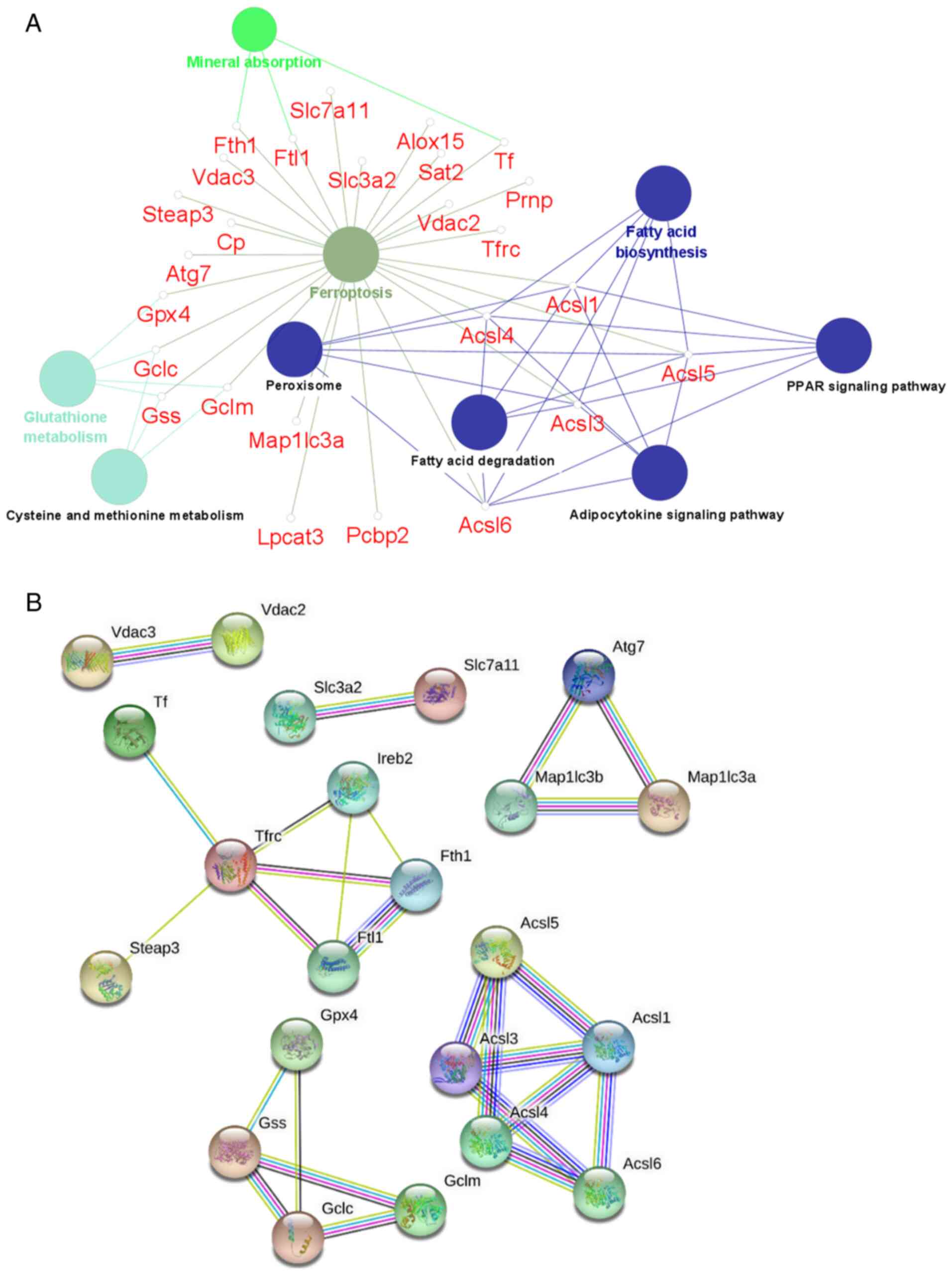
demonstrated to be enriched in the PHZ group (Fig. 2A). These DAPs were also involved in
other metabolic pathways, including mineral absorption, glutathione
metabolism, and fatty acid biosynthesis. The STRING database was
then used to determine the protein-protein interaction network
(Fig. 2B; confidence score
>0.7) among the screened ferroptosis-associated DAPs.
To evaluate the hypothesis that ferroptosis has a
role in HHIBD, ferroptosis-associated proteins were assessed using
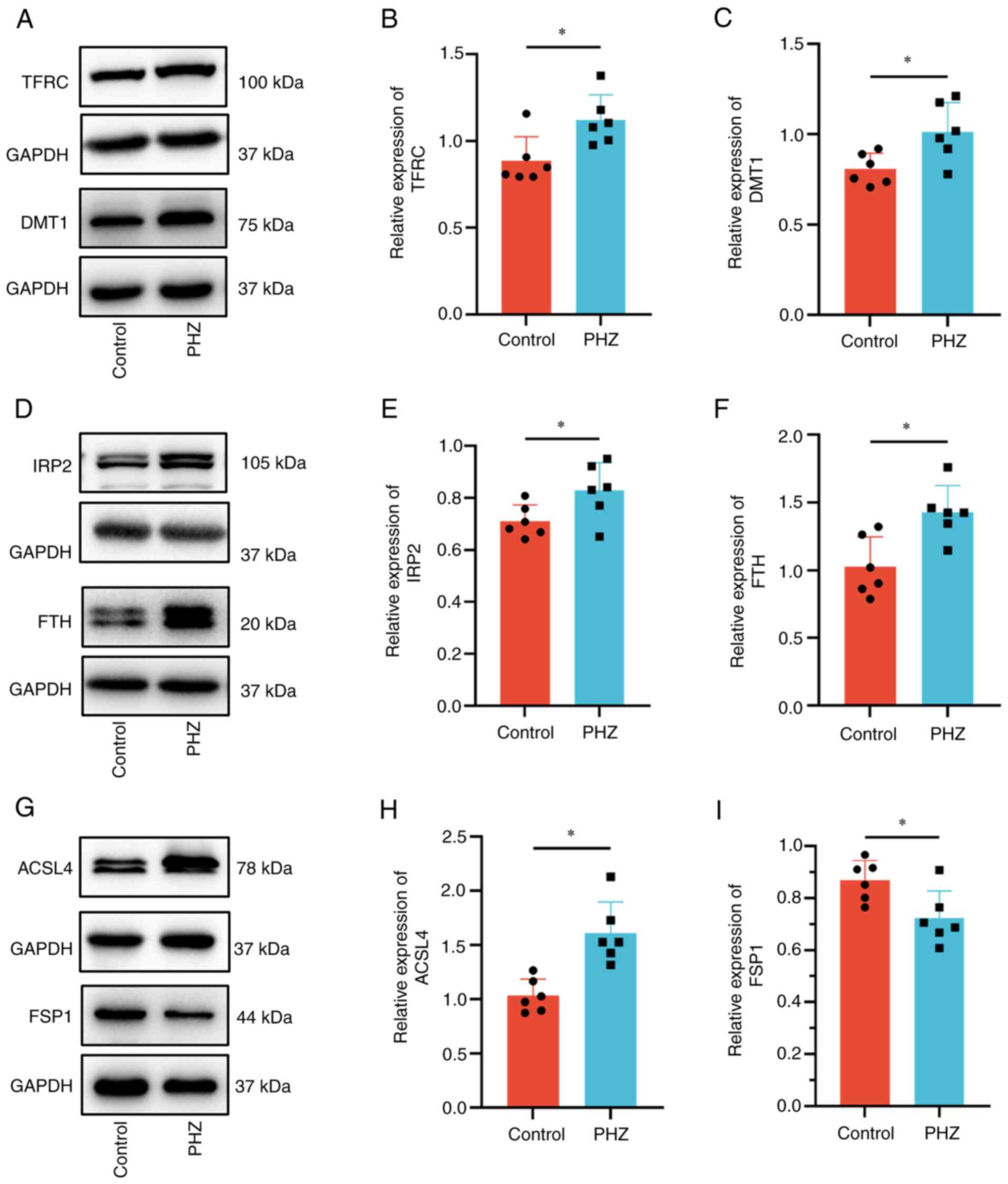
western blotting. As it was previously demonstrated that the iron
concentration in the brain tissue of the PHZ group significantly
increased compared with that of the control group (Fig. 1C), protein-controlled iron import
and export were assessed. The levels of the iron transporters TFRC
(Fig. 3A and B) and DMT1 (Fig. 3A and C), which represent important
components of ferroptosis that regulate iron levels, significantly
increased in the PHZ group compared with that of the control group,
whilst FPN1 levels did not significantly change (Fig. S4A and B). The results also
demonstrated that levels of the iron storage protein FTH
significantly increased in the PHZ group compared with that of the
control group (Fig. 3D and F).
Furthermore, the levels of IRP2, which is involved in the
regulation of iron metabolism, significantly increased in the brain
in the PHZ group compared with that of the control group (Fig. 3D and E). ACSL4 is a specific
biomarker and driver of ferroptosis (41), while FSP1 has been identified as
key molecules in independent pathways associated with ferroptosis
inhibition (42). ACSL4 levels
significantly increased and FSP1 levels significantly decreased in
the PHZ group compared with that of the control group (Fig. 3G-I). The level of GPX4, an
inhibitor of ferroptosis that converts lipid hydroperoxides into
non-toxic lipid alcohols, was also demonstrated to have
significantly increased after the onset of hemolytic
hyperbilirubinemia in the PHZ group compared with that of the
control group (Fig. S4A and C).
By contrast, the level of xCT, an important antioxidant defense
molecule, did not change significantly (Fig. S4A and D).
Effect of the neuroprotective
ferroptosis inhibitor DFO on HHIBD
DFO and its function as an iron chelator and
specific inhibitor of ferroptosis (28,29),
was used to further assess the function of ferroptosis in HHIBD.
Compared with that in the PHZ group, DFO did not significantly
influence the levels of hematocrit, hemoglobin and bilirubin in the
DFO + PHZ group (Fig. S5).
However, DFO pretreatment significantly reduced the levels of NSE
and S100B, compared with that of the PHZ only group, indicating
that brain injury was alleviated (Fig.
4A and B). DFO mitigated the imbalanced iron levels in the
brain (Fig. 4C), thereby partially
alleviating the redox imbalance compared with the PHZ group. The
levels of GSH and SOD were increased following treatment with DFO
(Fig. 4D-G), whereas MDA levels
were significantly reduced, compared with that of the PHZ only
group (Fig. 4H). There was no
difference in the level of GSSG between the DFO + PHZ groups and
the PHZ group (Fig. 4E).
 | Figure 4.DFO pretreatment alleviates hemolytic
hyperbilirubinemia-induced brain damage in neonatal Sprague-Dawley
rats. Serum levels of (A) S100B and (B) neuron-specific enolase.
(C) Levels of iron in the brain. Redox imbalances assessed by
measuring the levels of (D) GSH, (E) GSSG, (F) GSH/GSSG, (G) SOD
and (H) MDA. n=6. *P<0.05; **P<0.01; ***P<0.001. DFO,
deferoxamine; S100B, S100 calcium-binding protein B; GSH,
glutathione; GSSG, glutathione disulfide; SOD, superoxide
dismutase; MDA, malondialdehyde; PHZ, phenylhydrazine; ns, not
significant. |
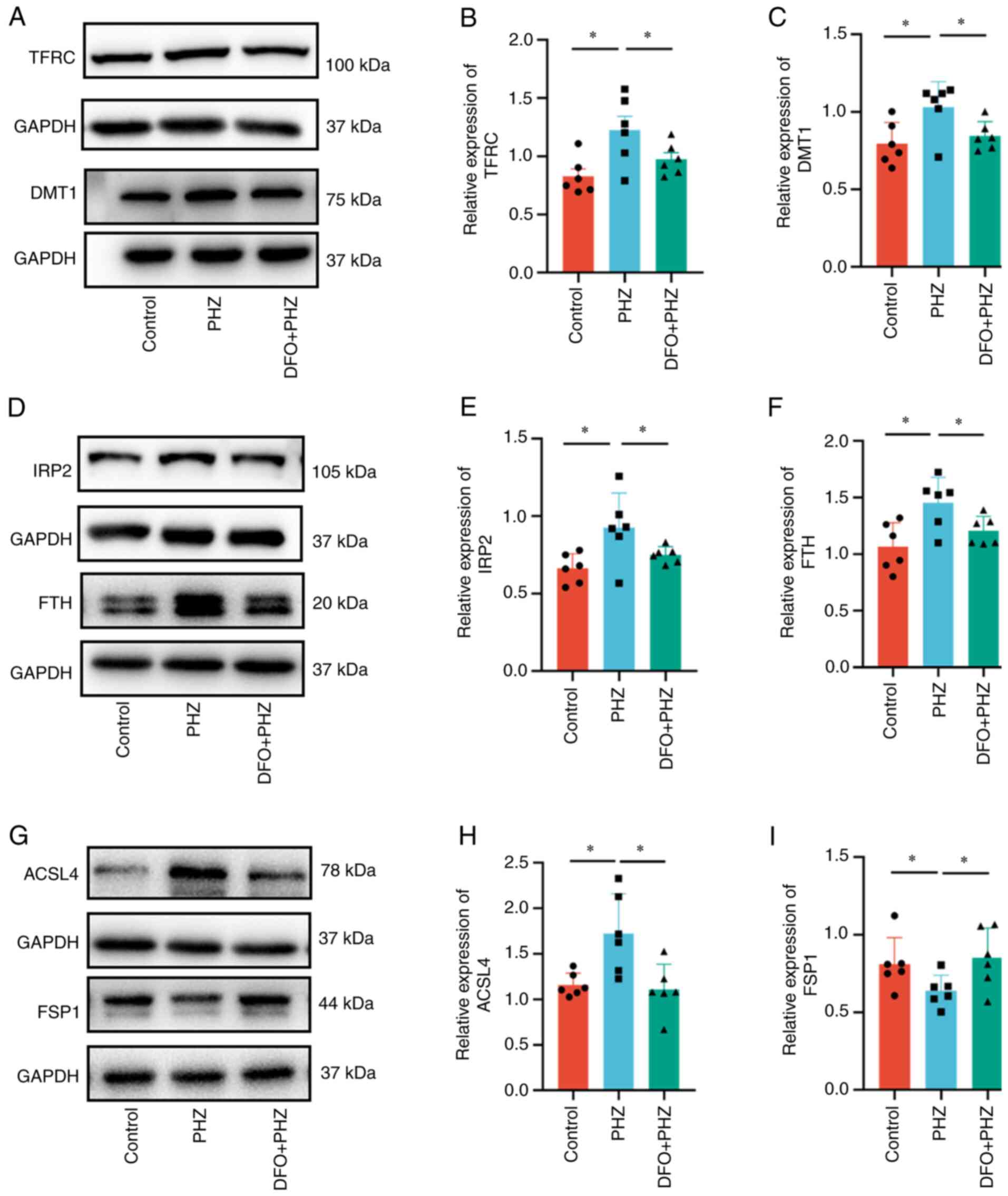
The effect of DFO on the proteins related to
ferroptosis was then evaluated. The protein levels of DMT1 and
TFRC, which regulate iron transport in and out of cells, were
decreased following DFO treatment compared with those of the PHZ
group (Fig. 5A-C), whereas the
FPN1 level was not significantly altered (Fig. S6A and B). Similarly, DFO restored
the levels of IRP2 and FTH proteins compared with those of the PHZ
group (Fig. 5D-F). Moreover, the
altered levels of the key proteins in the ferroptosis pathway,
ACSL4 and FSP1, were decreased following DFO treatment (Fig. 5G-I). However, the protein level of
xCT did not significantly change following DFO pretreatment
(Fig. S6A and D). Furthermore,
compared with the PHZ group, the GPX4 level decreased in the DFO +
PHZ group (Fig. S6A and C).
Iron overload promotes ferroptosis in
PC12 cells
Pretreatment with Fer-1 or DFO was demonstrated to
antagonize the reduction in cell proliferation induced by FAC
(Fig. 6A). The results also
demonstrated that FAC exposure significantly increased MDA and
reduced SOD activity levels and induced excessive generation of ROS
compared with those in control group (Fig. 6B-E). Furthermore, FAC treatment
also increased the Fe2+ content in PC12 cells, as
indicated by significantly higher FerroOrange signals compared with
those observed in control cells (Fig.
6F and G). Changes in the mitochondria were also demonstrated,
with FAC treatment resulting in a reduction in mitochondrial volume
and increased membrane density (Fig.
6H). Furthermore, the level of lipid peroxidation assessed
using confocal laser scanning microscopy demonstrated that FAC
significantly increased lipid peroxidation compared with that in
control cells (Fig. 6I and J).
However, pretreatment with 10 µM Fer-1 and/or 100 µM DFO
effectively attenuated the aforementioned damage induced by FAC
treatment.
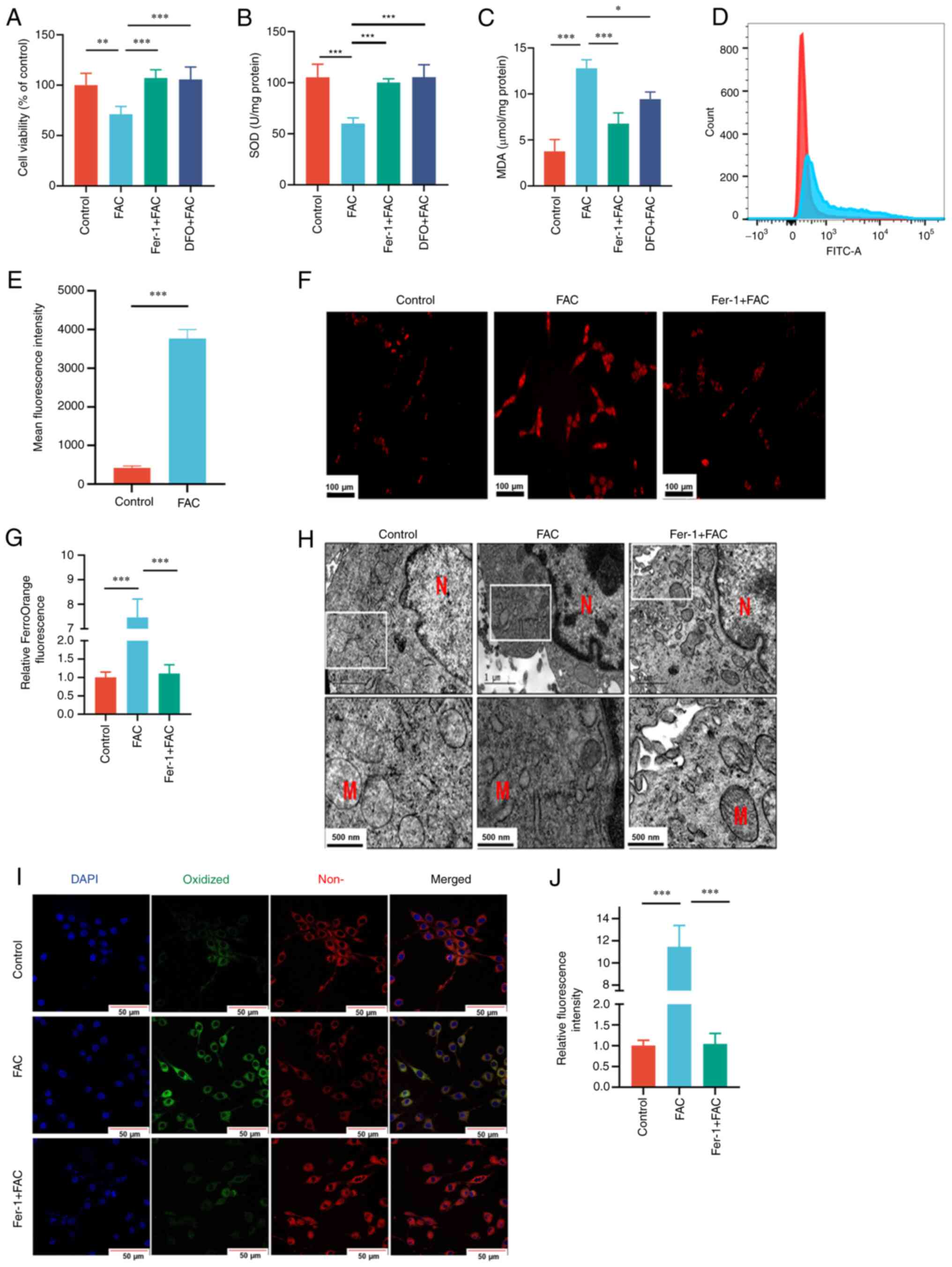 | Figure 6.PC12 cells were pretreated with 100
µM DFO or 10 µM Fer-1 for 1 h and then exposed to 400 µM FAC for 24
h. (A) Cell viability was assessed using an MTT assay. Level of (B)
SOD activity and (C) MDA in PC12 cells. (D) Level of reactive
oxygen species in PC12 cells assessed using
2′,7′-dichlorodihydrofluorescein diacetate via flow cytometry and
(E) quantified. (F) Concentrations of free iron assessed using
FerroOrange staining (scale bar, 100 µm). (G) Relative FerroOrange
fluorescence. (H) Mitochondrial morphology observed using
transmission electron microscopy (scale bar, 1 µm, upper images;
500 nm, lower images). (I) Confocal microscopy images of the
oxidized and non-oxidized variants of lipid peroxides stained using
a BODIPY C11 probe (scale bar, 50 µm). (J) Relative fluorescence
intensity of oxidized variants of lipid peroxides. n=3. *P<0.05;
**P<0.01; ***P<0.001. DFO, deferoxamine; Fer-1,
ferrostatin-1; FAC, ferric ammonium citrate; SOD, superoxide
dismutase; MDA, malondialdehyde; N, nucleus; M, mitochondria. |
Discussion
In the present study, the results demonstrated that
iron accumulation and ferroptosis occur in rats with HHIBD. DFO
treatment restored the normal expression of ferroptosis-associated
proteins (IRP2, FTH, TFRC, DMT1, ACSL4 and FSP1) and the levels of
ferroptosis-associated biochemical markers (MDA, GSH, SOD and
tissue iron).
Intraperitoneal injection of PHZ can induce
hemolysis to increase the concentration of unconjugated bilirubin
(25,26). Thus, compared with exogenous
administration of bilirubin and Gunn rat (43,44),
the PHZ-induced rat model provides a reproduction of the states of
bilirubin encephalopathy secondary to hemolytic disease. According
to previous data, the highest permeability of the blood-brain
barrier occurs on the 10th day of life of a newborn rat pup
(45). Thus, 10- to 12-day-old
rats were administered PHZ and it was demonstrated that the total
bilirubin level significantly increased, whereas the hemoglobin and
hematocrit levels significantly decreased compared with the control
group. Notably, the total bilirubin level exceeded the accepted
hyperbilirubinemia threshold (>51.3 µM) in rats (26). It was also confirmed that the
neonatal rats developed HHIBD by measuring the NSE and S100B
levels.
Extensive research has been conducted on the
potential mechanisms of bilirubin neurotoxicity in vitro and
in vivo, including studies assessing oxidative stress
(46), endoplasmic reticulum
stress (47,48), inflammation (49), autophagy (50), apoptosis (51,52)
and abnormal synthesis of both DNA and protein (53). However, the underlying mechanisms
of neurotoxicity induced by bilirubin remain unclear and require
further investigation. The contribution of other hemolysis products
in the development of neurotoxicity have also not been well
studied. Moreover, excess ferrous ion can accumulate in the basal
ganglia and cause damage, and the basal ganglia is also a target
site of bilirubin neurotoxicity (54).
Accumulation of peroxidized lipids is a typical
characteristic of ferroptosis (55). For example, in the present study,
the level of MDA was significantly increased in the PHZ group
compared with the control group. Furthermore, GSH has been
indicated to exhibit neuroprotective effects against
bilirubin-induced cytotoxicity (56). Brain tissue GSH is reported to be
significantly reduced following the induction of HHIBD in rats
(26,27). Moreover, in the current study, it
was demonstrated that HHIBD resulted in the downregulation of GSH
levels and SOD activity and the upregulation of MDA levels. DFO, an
iron-chelating agent and ferroptosis inhibitor, has been reported
to induce antioxidant activity and reduce oxidative damage
(57). Accordingly, pretreatment
with DFO reversed the aforementioned changes. In addition, compared
with the PHZ group, DFO pretreatment was demonstrated to
significantly reduce the levels of NSE and S100B in the DFO + PHZ
group, while the hemolysis related indicators such as hematocrit,
hemoglobin and bilirubin were not alleviated. Thus, DFO
pretreatment could alleviate HHIBD.
ACSL4 is considered to be a specific biomarker and
driver of ferroptosis. Low expression of ACSL4 mediates glioma cell
proliferation by inhibiting ferroptosis (58). In an animal model of acute brain
injury, ACSL4 levels increased significantly, and knockout of ACSL4
reduced brain injury by inhibiting ferroptosis (59,60).
In the present study, the expression of ACSL4 was significantly
upregulated in the rat HHIBD model, which further demonstrated that
ferroptosis may serve an important role in HHIBD and provides a
possible intervention target for further study. Additionally, FSP1
acts as an oxidoreductase, restraining the propagation of lipid
peroxides and preventing ferroptosis (61). Moreover, the expression of FSP1 was
decreased in the brain tissue of rats with hemolytic
hyperbilirubinemia in the present study, thereby suggesting excess
lipid peroxidation in the rat model.
Another biochemical characteristic of ferroptosis is
iron accumulation (62). Iron
overload and an increase in lipid peroxidation products have been
reported in infants with hemolytic diseases (63). These findings indicate that iron
toxicity may serve a vital role in the pathobiology of severe
hemolytic diseases. Results from the present study demonstrated
that the level of iron was significantly elevated in the brain
tissue of rats with HHIBD, which is consistent with the
aforementioned studies (8,25). Intracellular iron levels are
controlled by highly regulated mechanisms involving proteins, such
as the iron importer TFRC, iron storage protein ferritin and FPN1
(64,65). IRP2 is the main regulator of iron
metabolism and maintains intracellular iron homeostasis (66). When intracellular iron is
insufficient, IRP2 can promote iron transport into cells by
inhibiting the degradation of TFRC (67). TFRC is considered a positive
regulator of ferroptosis, as it promotes iron intake (68,69).
In the present study, it was demonstrated that iron overload caused
by hemolytic hyperbilirubinemia leads to ferroptosis and serves an
important role in HHIBD, based on the upregulated expression of
IRP2 and TFRC in combination with the increased total iron
content.
Ferritin consists of FTH and light chain subunits
and is considered to be the pivotal protein for storing and
dislodging excess iron to reduce cell damage and stress (64). Autophagic degradation of ferritin,
mediated by lysosomes, has been reported to increase intracellular
Fe2+ levels and promote ferroptosis (70). However, it has been reported that
ferroptosis induced by iron overload is accompanied by the
upregulation of FTH expression under certain pathological
conditions, such as traumatic brain injury and cerebral
ischemia/reperfusion injury (71).
Moreover, the present study demonstrated that the level of FTH
significantly increased in the PHZ group, whilst this change was
restored in the DFO + PHZ group. In summary, FTH may serve a key
role in ferroptosis in HHIBD, which requires further
exploration.
To further evaluate the role of ferroptosis in the
neurotoxicity of iron overload, a FAC model was established in
vitro. FAC, which has been widely used to induce iron overload,
has high solubility and the ability to convert into ferrous ion
(72). Typical mitochondrial
features of ferroptosis include shrinking, increased membrane
density and reduced or absent cristae (73,74).
In the present study, ferroptosis-specific mitochondrial
morphological changes in PC12 cells, induced by FAC, were
demonstrated using TEM. In the FAC-induced PC12 cell damage model,
Fer-1 and DFO pretreatment antagonized the increase in lipid
peroxides, demonstrated using confocal laser scanning microscopy.
These findings further demonstrated that iron overload could lead
to neuronal damage via ferroptosis.
The present study has certain limitations. Firstly,
histological analysis was not performed to visually detect the
tissue damage caused by HHIBD. Secondly, recent studies have
pointed out that activation of inflammation, including the
activation of multiple inflammation-related signaling pathways, can
lead to ferroptosis (75,76). However, plasma TNF-α, IFN-γ, IL-2
or IL-10 levels were not assessed in the animal model. In future
studies, the role of inflammatory reactions in
hyperbilirubinemia-induced brain damage and the interplay between
inflammation and ferroptosis should be evaluated. However, higher
levels of iron and MDA and lower levels of GSH and SOD activity
were measured in the brain tissues of the PHZ group compared with
those in the control group. In addition, HHIBD resulted in
increases in the expression of the ferroptosis-related proteins
ACSL4, FTH, TFRC and DMT1 and a reduction in the expression of
FSP1. KEGG pathway enrichment analysis demonstrated that the
differentially expressed rat brain proteins between the control and
PHZ groups were significantly enriched in ferroptosis, GSH
metabolism and fatty acid biosynthesis pathways. Pretreatment with
DFO alleviated redox imbalance and lipid peroxidation-mediated
HHIBD. PC12 cells treated with FAC showed shrinking mitochondria,
high mitochondrial membrane density, and increased lipid reactive
oxygen species and intracellular ferrous iron, which were
antagonized by pretreatment with ferrostatin-1 or DFO.
To the best of our knowledge, this is the first
study to elucidate the role of ferroptosis in HHIBD in vivo.
The present study demonstrated the vital role of ferroptosis in
HHIBD, and iron overload was demonstrated to trigger ferroptosis
potentially via the activation of lipid peroxidation in the brain.
These findings demonstrate that ferroptosis is involved in HHIBD
and shed light on the potential underlying molecular mechanisms of
HHIBD. Furthermore, the present study provides new insights into
candidate proteins that are potentially involved in ferroptosis in
the brain during hemolytic hyperbilirubinemia.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Junjin Lin
and Professor Linying Zhou (Public Technology Service Center,
Fujian Medical University, Fuzhou, China) and Dr Liping Zhu, Dr
Xudong Zhuang and Dr Xinrui Wang (NHC Key Laboratory of Technical
Evaluation of Fertility Regulation for Non-Human Primate, Fujian
Maternity and Child Health Hospital, Fuzhou, China) for their
technical assistance.
Funding
The present study was supported by the Natural Science
Foundation of Fujian Province (grant no. 2020J01327), the Fujian
Provincial Health Technology Project (grant no. 2020GGB017), the
Joint Funds for the Innovation of Science and Technology, Fujian
Province (grant no. 2020Y9143), the Open Project of the Key
Laboratory of Environment and Health of Fujian Medical University
(grant no. GWGXZD-202002) and Fujian Maternity and Child Health
Hospital (grant no. YCXM 20-08).
Availability of data and materials
Mass spectrometry proteomics data have been
deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via
the iProX partner repository with the dataset identifier PXD041604.
Other datasets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
HL and JZ confirm the authenticity of all the raw
data. HL and LX conceived and designed the study. JZ, XL, SL, GL,
JT, JL, CZ and SW performed the experiments. JZ and XL collected
and analyzed the experimental data. JZ and XL prepared the first
version of the manuscript. XL, SL, GL, JT, JL, CZ, SW and LX
provided critical comments on the revision of the manuscript. HL
and LX revised and finalized the manuscript. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Fujian Maternity and Child Health
Hospital (approval no. 2020KY093; Fuzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lauer BJ and Spector ND:
Hyperbilirubinemia in the Newborn. Pediatr Rev. 32:341–349. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Soto Conti CP: Bilirubin: The toxic
mechanisms of an antioxidant molecule. Arch Argent Pediatr.
119:e18–e25. 2021.(In English, Spanish). PubMed/NCBI
|
|
3
|
Christensen RD, Agarwal AM, George TI,
Bhutani VK and Yaish HM: Acute neonatal bilirubin encephalopathy in
the State of Utah 2009–2018. Blood Cells Mol Dis. 72:10–13. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wong RJ and Stevenson DK: Neonatal
hemolysis and risk of bilirubin-induced neurologic dysfunction.
Semin Fetal Neonatal Med. 20:26–30. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kumar V, Kumar P, Sundaram V, Munjal SK,
Malhi P and Panda NK: Childhood neurodevelopmental outcomes of
survivors of acute bilirubin encephalopathy: A retrospective cohort
study. Early Hum Dev. 158:1053802021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Subspecialty Group of Neonatology, Society
of Pediatrics, Chinese Medical Association and Chinese Multicenter
Study Coordination Group for Neonatal Bilirubin Encephalopathy, .
Clinical characteristics of bilirubin encephalopathy in Chinese
newborn infants-a national multicenter survey. Zhonghua Er Ke Za
Zhi. 50:331–335. 2012.(In Chinese). PubMed/NCBI
|
|
7
|
Par EJ, Hughes CA and DeRico P: Neonatal
Hyperbilirubinemia: Evaluation and treatment. Am Fam Physician.
107:525–534. 2023.PubMed/NCBI
|
|
8
|
Viktorinova A: Iron-mediated oxidative
cell death is a potential contributor to neuronal dysfunction
induced by neonatal hemolytic hyperbilirubinemia. Arch Biochem
Biophys. 654:185–193. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khdair-Ahmad F, Aladily T, Khdair-Ahmad O
and Badran EF: Chelation therapy for secondary neonatal iron over
load: Lessons learned from rhesus hemolytic disease. Turk J
Pediatr. 60:335–339. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aygun C, Tekinalp G and Gurgey A:
Increased Fetal iron load in rhesus hemolytic disease. Pediatr
Hematol Oncol. 21:329–333. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaplan M, Bromiker R and Hammerman C:
Hyperbilirubinemia, hemolysis, and increased bilirubin
neurotoxicity. Semin Perinatol. 38:429–437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nishiie-Yano R, Hirayama S, Tamura M,
Kanemochi T, Ueno T, Hirayama A, Hori A, Ai T, Hirose N and Miida
T: Hemolysis is responsible for elevation of serum iron
concentration after Regular exercises in judo athletes. Biol Trace
Elem Res. 197:63–69. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Comporti M, Signorini C, Buonocore G and
Ciccoli L: Iron release, oxidative stress and erythrocyte ageing.
Free Radic Biol Med. 32:568–576. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun S, Shen J, Jiang J, Wang F and Min J:
Targeting ferroptosis opens new avenues for the development of
novel therapeutics. Signal Transduct Target Ther. 8:3722023.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ashraf A, Jeandriens J, Parkes HG and So
PW: Iron dyshomeostasis, lipid peroxidation and perturbed
expression of cystine/glutamate antiporter in Alzheimer's disease:
Evidence of ferroptosis. Redox Biol. 32:1014942020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rui T, Wang H, Li Q, Cheng Y, Gao Y, Fang
X, Ma X, Chen G, Gao C, Gu Z, et al: Deletion of ferritin H in
neurons counteracts the protective effect of melatonin against
traumatic brain injury-induced ferroptosis. J Pineal Res.
70:e127042021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vitalakumar D, Sharma A and Flora SJS:
Ferroptosis: A potential therapeutic target for neurodegenerative
diseases. J Biochem Mol Toxicol. 35:e228302021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ding X, Gao L, Han Z, Eleuteri S, Shi W,
Shen Y, Song ZY, Su M, Yang Q, Qu Y, et al: Ferroptosis in
Parkinson's disease: Molecular mechanisms and therapeutic
potential. Ageing Res Rev. 91:1020772023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bu ZQ, Yu HY, Wang J, He X, Cui YR, Feng
JC and Feng J: Emerging Role of Ferroptosis in the Pathogenesis of
Ischemic Stroke: A new therapeutic target? ASN Neuro.
13:1759091421103752021. View Article : Google Scholar
|
|
21
|
Lu C, Tan C, Ouyang H, Chen Z, Yan Z and
Zhang M: Ferroptosis in Intracerebral hemorrhage: A panoramic
perspective of the metabolism, mechanism and theranostics. Aging
Dis. 13:13482022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li QS and Jia YJ: Ferroptosis: A critical
player and potential therapeutic target in traumatic brain injury
and spinal cord injury. Neural Regen Res. 18:5062023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ren S, Chen Y, Wang L and Wu G: Neuronal
ferroptosis after intracerebral hemorrhage. Front Mol Biosci.
9:9664782022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luykx LM, Berger HM, Geerdink J, Kanhai
HHH and Egberts J: Non-protein-bound iron and free radical damage
in fetuses with rhesus haemolytic disease: Influence of
intrauterine transfusions. BJOG. 111:303–310. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mejia GB, Sanz CR, Avila MM, Peraza AV,
Guzmán DC, Olguín HJ, Ramírez AM and Cruz EG: Experimental
hemolysis model to study bilirubin encephalopathy in rat brain. J
Neurosci Methods. 168:35–41. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pazar A, Kolgazi M, Memisoglu A, Bahadir
E, Sirvanci S, Yaman A, Yeğen BÇ and Ozek E: The neuroprotective
and anti-apoptotic effects of melatonin on hemolytic
hyperbilirubinemia-induced oxidative brain damage. J Pineal Res.
60:74–83. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luo Y, Peng M and Wei H: Melatonin
promotes brain-derived neurotrophic factor (BDNF) expression and
anti-apoptotic effects in neonatal hemolytic hyperbilirubinemia via
a phospholipase (PLC)-mediated mechanism. Med Sci Monit.
23:5951–5959. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX,
Sun C, Li B, Fan BY, Wang X, Li WX, et al: Deferoxamine promotes
recovery of traumatic spinal cord injury by inhibiting ferroptosis.
Neural Regen Res. 14:532–541. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin T, He Q, Cheng C, Li H, Liang L, Zhang
G, Su C, Xiao Y, Bradley J, Peberdy MA, et al: UAMC-3203 or/and
Deferoxamine improve Post-Resuscitation myocardial dysfunction
through suppressing ferroptosis in a rat model of cardiac arrest.
Shock. 57:344–350. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chian S, Jiang ZC, Jiang LX, Wang KT, Fan
YX, Liao T, Chen WS and Yao WX: Caffeine-induced neurotoxicity
mediated by Nrf2 pathway in PC12 cells and zebrafish larvae. J Appl
Toxicol. 42:629–637. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wiatrak B, Kubis-Kubiak A, Piwowar A and
Barg E: PC12 cell line: Cell types, coating of culture vessels,
differentiation and other culture conditions. Cells. 9:9582020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rand RN and Di Pasqua A: A new diazo
method for the determination of bilirubin. Clin Chem. 8:570–578.
1962. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zecha J, Satpathy S, Kanashova T,
Avanessian SC, Kane MH, Clauser KR, Mertins P, Carr SA and Kuster
B: TMT Labeling for the Masses: A Robust and Cost-efficient,
In-solution Labeling Approach. Mol Cell Proteomics. 18:1468–1478.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grebe SK and Singh RJ: LC-MS/MS in the
Clinical Laboratory-Where to from here? Clin Biochem Rev. 32:5–31.
2011.PubMed/NCBI
|
|
35
|
Men L, Li Y, Wang X, Li R, Zhang T, Meng
X, Liu S, Gong X and Gou M: Protein biomarkers associated with
frozen Japanese puffer fish (Takifugu rubripes) quality traits.
Food Chem. 327:1270022020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang Y, Zhao J, Li R, Liu Y, Zhou L, Wang
C, Lv C, Gao L and Cui D: CircLRFN5 inhibits the progression of
glioblastoma via PRRX2/GCH1 mediated ferroptosis. J Exp Clin Cancer
Res. 41:3072022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou Y, Li L, Mao C and Zhou D:
Astragaloside IV ameliorates spinal cord injury through controlling
ferroptosis in H2O2-damaged PC12 cells in vitro. Ann Transl Med.
10:11762022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zuo X, Zeng H, Wang B, Yang X, He D, Wang
L, Ouyang H and Yuan J: AKR1C1 Protects corneal epithelial cells
against oxidative stress-mediated ferroptosis in dry eye. Invest
Ophthalmol Vis Sci. 63:32022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang S, Cao B, Zhang J, Feng Y, Wang L,
Chen X, Su H, Liao S, Liu J, Yan J and Liang B: Induction of
ferroptosis in human nasopharyngeal cancer cells by cucurbitacin B:
Molecular mechanism and therapeutic potential. Cell Death Dis.
12:2372021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lorimier P, Lamarcq L, Labat-Moleur F,
Guillermet C, Bethier R and Stoebner P: Enhanced chemiluminescence:
A high-sensitivity detection system for in situ hybridization and
immunohistochemistry. J Histochem Cytochem. 41:1591–1597. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jia B, Li J, Song Y and Luo C:
ACSL4-Mediated ferroptosis and its potential role in central
nervous system diseases and injuries. Int J Mol Sci. 24:100212023.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hankø E, Hansen TWR, Almaas R, Lindstad J
and Rootwelt T: Bilirubin induces apoptosis and necrosis in human
NT2-N Neurons. Pediatr Res. 57:179–184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shapiro SM: Somatosensory and brainstem
auditory evoked potentials in the gunn rat model of acute bilirubin
neurotoxicity. Pediatr Res. 52:844–849. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Roger C, Koziel V, Vert P and Nehlig A:
Autoradiographic mapping of local cerebral permeability to
bilirubin in immature rats: Effects of hyperbilirubinemia. Pediatr
Res. 39:64–71. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qaisiya M, Coda Zabetta CD, Bellarosa C
and Tiribelli C: Bilirubin mediated oxidative stress involves
antioxidant response activation via Nrf2 pathway. Cell Signal.
26:512–520. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qaisiya M, Brischetto C, Jašprová J, Vitek
L, Tiribelli C and Bellarosa C: Bilirubin-induced ER stress
contributes to the inflammatory response and apoptosis in neuronal
cells. Arch Toxicol. 91:1847–1858. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schiavon E, Smalley JL, Newton S, Greig NH
and Forsythe ID: Neuroinflammation and ER-stress are key mechanisms
of acute bilirubin toxicity and hearing loss in a mouse model. PLoS
One. 13:e02010222018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Vodret S, Bortolussi G, Iaconcig A,
Martinelli E, Tiribelli C and Muro AF: Attenuation of
neuro-inflammation improves survival and neurodegeneration in a
mouse model of severe neonatal hyperbilirubinemia. Brain Behav
Immun. 70:166–178. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Qaisiya M, Mardešić P, Pastore B,
Tiribelli C and Bellarosa C: The activation of autophagy protects
neurons and astrocytes against bilirubin-induced cytotoxicity.
Neurosci Lett. 661:96–103. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shi HS, Lai K, Yin XL, Liang M, Ye HB, Shi
HB, Wang LY and Yin SK: Ca2+-dependent recruitment of voltage-gated
sodium channels underlies bilirubin-induced overexcitation and
neurotoxicity. Cell Death Dis. 10:7742019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ye H, Xing Y, Zhang L, Zhang J, Jiang H,
Ding D, Shi H and Yin S: Bilirubin-induced neurotoxic and ototoxic
effects in rat cochlear and vestibular organotypic cultures.
Neurotoxicology. 71:75–86. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rawat V, Bortolussi G, Gazzin S, Tiribelli
C and Muro AF: Bilirubin-induced oxidative stress leads to DNA
damage in the cerebellum of hyperbilirubinemic neonatal mice and
activates DNA Double-Strand break repair pathways in human cells.
Oxid Med Cell Longev. 2018:18012432018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Youdim MB, Ben-Shachar D, Yehuda S and
Riederer P: The role of iron in the basal ganglion. Adv Neurol.
53:155–162. 1990.PubMed/NCBI
|
|
55
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Al-Abdi S: Decreased glutathione
S-transferase level and neonatal hyperbilirubinemia associated with
Glucose-6-phosphate dehydrogenase deficiency: A perspective review.
Am J Perinatol. 34:305–314. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen GH, Song CC, Pantopoulos K, Wei XL,
Zheng H and Luo Z: Mitochondrial oxidative stress mediated
Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol
Med. 180:95–107. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cheng J, Fan Y, Liu B, Zhou H, Wang J and
Chen Q: ACSL4 suppresses glioma cells proliferation via activating
ferroptosis. Oncol Rep. 43:147–158. 2020.PubMed/NCBI
|
|
59
|
Chen J, Yang L, Geng L, He J, Chen L, Sun
Q, Zhao J and Wang X: Inhibition of Acyl-CoA synthetase long-chain
family member 4 facilitates neurological recovery after stroke by
regulation ferroptosis. Front Cell Neurosci. 15:6323542021.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cui Y, Zhang Y, Zhao X, Shao L, Liu G, Sun
C, Xu R and Zhang Z: ACSL4 exacerbates ischemic stroke by promoting
ferroptosis-induced brain injury and neuroinflammation. Brain Behav
Immun. 93:312–321. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Doll S, Freitas FP, Shah R, Aldrovandi M,
da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius
E, Scheel CH, et al: FSP1 is a glutathione-independent ferroptosis
suppressor. Nature. 575:693–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Doll S and Conrad M: Iron and ferroptosis:
A still ill-defined liaison. IUBMB Life. 69:423–434. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Rath MEA, Smits-Wintjens VEHJ, Oepkes D,
Walther FJ and Lopriore E: Iron status in infants with alloimmune
haemolytic disease in the first three months of life. Vox Sang.
105:328–333. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mleczko-Sanecka K and Silvestri L:
Cell-type-specific insights into iron regulatory processes. Am J
Hematol. 96:110–127. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ganz T: New regulators of systemic iron
homeostasis. Signal Transduct Target Ther. 6:2802021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gao G, Li J, Zhang Y and Chang YZ:
Cellular iron metabolism and regulation. Adv Exp Med Biol.
1173:21–32. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sfera A, Bullock K, Price A, Inderias L
and Osorio C: Ferrosenescence: The iron age of neurodegeneration?
Mech Ageing Dev. 174:63–75. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lu Y, Yang Q, Su Y, Ji Y, Li G, Yang X, Xu
L, Lu Z, Dong J, Wu Y, et al: MYCN mediates TFRC-dependent
ferroptosis and reveals vulnerabilities in neuroblastoma. Cell
Death Dis. 12:5112021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Xiong Q, Li X, Li W, Chen G, Xiao H, Li P
and Wu C: WDR45 Mutation impairs the autophagic degradation of
transferrin receptor and promotes ferroptosis. Front Mol Biosci.
8:6458312021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhang J, Chen X, Hong J, Tang A, Liu Y,
Xie N, Nie G, Yan X and Liang M: Biochemistry of mammalian
ferritins in the regulation of cellular iron homeostasis and
oxidative responses. Sci China Life Sci. 64:352–362. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wang P, Cui Y, Ren Q, Yan B, Zhao Y, Yu P,
Gao G, Shi H, Chang S and Chang YZ: Mitochondrial ferritin
attenuates cerebral ischaemia/reperfusion injury by inhibiting
ferroptosis. Cell Death Dis. 12:4472021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu Y, Bell BA, Song Y, Kim HJ, Sterling
JK, Kim BJ, Poli M, Guo M, Zhang K, Rao A, et al: Intraocular iron
injection induces oxidative stress followed by elements of
geographic atrophy and sympathetic ophthalmia. Aging Cell.
20:e134902021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yang J, Zhou Y, Xie S, Wang J, Li Z, Chen
L, Mao M, Chen C, Huang A, Chen Y, et al: Metformin induces
ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J
Exp Clin Cancer Res. 40:2062021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yu H, Yang C, Jian L, Guo S, Chen R, Li K,
Qu F, Tao K, Fu Y, Luo F and v Liu S: Sulfasalazine-induced
ferroptosis in breast cancer cells is reduced by the inhibitory
effect of estrogen receptor on the transferrin receptor. Oncol Rep.
42:826–838. 2019.PubMed/NCBI
|
|
75
|
Chen Y, Fang Z-M, Yi X, Wei X and Jiang
DS: The interaction between ferroptosis and inflammatory signaling
pathways. Cell Death Dis. 14:2052023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Dou J, Liu X, Yang L, Huang D and Tan X:
Ferroptosis interaction with inflammatory microenvironments:
Mechanism, biology, and treatment. Biomed Pharmacother.
155:1137112022. View Article : Google Scholar : PubMed/NCBI
|