Introduction
Ischemic cardiomyopathy is one of the main causes of
human mortality worldwide, accounting for ~30 million deaths
annually (1–3). In addition, myocardial
ischemia-reperfusion (I/R) injury (MIRI), which leads to the
aggravation of disordered metabolism of myocardial cells during
reperfusion following myocardial ischemia, thus causing myocardial
ultrastructural damage, is one of the main causes of death in
patients with ischemic heart disease (4,5).
Previous studies have shown that MIRI is associated with numerous
pathophysiological features, including the generation of oxygen
free radicals, calcium overload, endothelial dysfunction,
mitochondrial dysfunction, immune response, ferroptosis, myocardial
cell apoptosis and autophagy (6–8). At
present, ischemic preconditioning (IPC) and pharmacological
preconditioning (PPC) offer the main means to improve MIRI in
clinical practice (9); however,
due to the limited scope of their use, the identification of novel
therapeutic methods and targets is necessary.
In 1997, Sessler (10) proposed the concept of mild
perioperative hypothermia, highlighting that performing carotid
endarterectomy and neurosurgery at a temperature of 33–35°C could
protect against cerebral ischemia. This was later confirmed by a
large number of basic research studies and clinical trials
(11–13). In addition, further studies have
indicated that hypothermia can improve the ischemic injury of
organs through a variety of different mechanisms (14,15).
Notably, the PI3K/AKT signaling pathway has been shown to have an
important role in organ reperfusion injury. A previous study showed
that mild therapeutic hypothermia (MTH) combined with hydrogen
sulfide treatment could reduce damage caused to the hippocampal
neurons in rats via activating the PI3K/AKT signaling pathway,
thereby improving cerebral I/R injury in rats (11). In addition, MTH has been shown to
inhibit inflammatory reactions and reduce cell apoptosis through
activating the PI3K/AKT signaling pathway, thereby improving liver
I/R injury (16). MTH may also
ameliorate MIRI (17). In another
study, investigators revealed that MTH could effectively alleviate
MIRI in a rabbit model (18).
Mochizuki et al (19)
demonstrated that MTH could improve MIRI in rats by activating the
PI3K signaling pathway. In addition, certain studies have shown
that MTH can reduce inflammatory reactions by inhibiting NLR family
pyrin domain containing 3 inflammasomes, thereby inhibiting calcium
channels and improving MIRI (17,20).
However, the protective mechanism that accounts for the influence
of MTH on MIRI has yet to be fully elucidated.
Transient receptor potential cation channel
subfamily M member 7 (TRPM7) is a membrane protein with a dual
structure of ion channel and protein kinase that, as a member of
the TRP channel subfamily, is widely distributed in the heart,
brain, lung, kidney, and other organs and tissues (21,22).
TRPM7 is a non-selective cation channel with calcium-ion
permeability that serves an important role in the I/R injury of
various organs. Cerebral I/R injury has previously been studied in
a rat model with overexpression of TRPM7, and the high expression
of TRPM7 was shown to be inhibited by electroacupuncture through
the PI3K pathway (23).
Furthermore, inhibition of TRPM7 expression using small interfering
RNA has been shown to effectively suppress the calcium overload
induced by long-term oxygen-glucose deprivation, and to reduce the
generation of reactive oxygen species (ROS), thereby promoting the
survival of oxygen- and glucose-deprived neurons (24,25).
The expression of TRPM7 has also been reported to be upregulated
after renal ischemia (26). In
addition, microRNA-9-5p has been reported to decrease the
expression of TRPM7 by activating the PI3K/Akt pathway, thereby
promoting both the migration and invasion of endothelial progenitor
cells and angiogenesis (27).
Notably, a recent study revealed that inhibition of TRPM7
expression may have a protective effect on the reperfusion injury
of H9C2 cells, and that this could reduce the apoptotic rate of
H9C2 cells (28).
Ferroptosis is a cell death process that results
from the accumulation of iron-dependent lipid peroxides, which
differs from traditional apoptosis and necrosis (29). The occurrence of ferroptosis is
closely associated with various pathophysiological processes,
including the blockade of cystine transport, the accumulation of
reactive oxygen free radicals in cells, abnormal iron metabolism,
accumulation of iron ions and lipid peroxidation (29,30).
Of these processes, the accumulation of iron ions and lipid
peroxidation are biochemically the most important (31). Numerous studies have demonstrated
that ferroptosis has an important role in various types of
cardiovascular disease, including heart failure, myocardial
infarction and I/R injury (32–34).
Lapatinib has been shown to enhance Adriamycin-induced oxidative
stress and ferroptosis of cardiomyocytes through inhibiting the
PI3K/AKT signaling pathway (35).
With the knowledge that TRPM7 exerts a key role in
various cell death modalities (36), it was hypothesized that MTH could
regulate the expression of TRPM7 through its action on the PI3K/AKT
signaling pathway, thereby inhibiting ferroptosis induced by MIRI.
In order to further clarify the underlying protective mechanism of
MTH on MIRI, a rat model of MIRI was established, and the rats were
treated with various drugs to explore the protective mechanism of
MTH. Taken together, the findings of the present study offer
several insights that may inform future strategies to relieve MIRI
in animals or humans.
Materials and methods
Animals, chemical reagents and
kits
H9C2 cells, wortmannin (Wort) and erastin (Era) were
purchased from Beyotime Institute of Biotechnology. A total of 86
healthy adult male Sprague-Dawley rats (age, 6–8 weeks; weight,
250–350 g) were purchased from Hunan SJA Laboratory Animal Co.,
Ltd. 2-Aminoethoxydiphenyl borate (2-APB) was obtained from Selleck
Chemicals (cat. no. S6657). Antibodies against glutathione
peroxidase 4 (GPX4; cat. no. 52455; Cell Signaling Technology,
Inc.), acyl-CoA synthetase long chain family member 4 (ACSL4; cat.
no. 22401-1-AP; Proteintech Group, Inc.), ferroptosis suppressor
protein 1 (FSP1; cat. no. 20886-1-AP; Proteintech Group, Inc.),
phosphorylated (p)-PI3K (cat. no. 17366; Cell Signaling Technology,
Inc.), total (t)-PI3K (cat. no. 4249; Cell Signaling Technology,
Inc.), p-AKT (cat. no. 4060; Cell Signaling Technology, Inc.),
t-AKT (cat. no. 9272; Cell Signaling Technology, Inc.) and GAPDH
(cat. no. AF0006; Beyotime Institute of Biotechnology) were used in
the present study. ELISA kits for creatine kinase-MB (CK-MB; cat.
no. H197-1-1), lactate dehydrogenase (LDH; cat. no. A020-1-2),
cardiac troponin I (cTnI; cat. no. H149-2-1), malondialdehyde (MDA;
cat. no. A003-1-1), total superoxide dismutase (SOD; cat. no.
A001-1-1) and glutathione peroxidase (GSH-Px; cat. no. A005-1-2)
were supplied by Nanjing Jiancheng Bioengineering Institute. In
addition, the MTT cell viability assay kit was purchased from
APeXBIO Technology LLC, and the LDH cytotoxicity assay kit and the
cytoFLEX® Annexin V-FITC/PI apoptosis kit (cat. no.
KGA108) were purchased from Nanjing KeyGen Biotech Co., Ltd.
Cell culture and animal treatment
H9C2 cells were resuscitated and subcultured.
Subsequently, H9C2 cells in the exponential growth phase and in
good growth condition were prepared into a single cell suspension
in sugar-free and serum-free DMEM (Beyotime Institute of
Biotechnology), with the cell concentration adjusted to
2.5×105 cells/ml. Subsequently, the cells were evenly
inoculated into a 6-well plate (2 ml cell suspension/well), and the
plate was incubated overnight at 37°C in an atmosphere containing
5% CO2 with saturated humidity. Cells were randomly
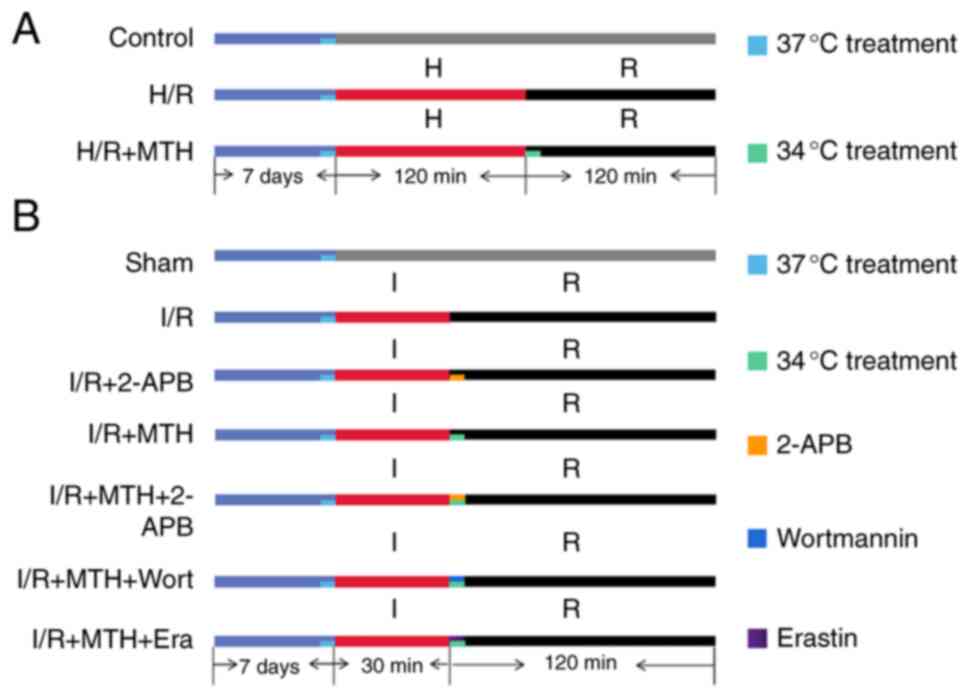
divided into three groups (Fig.
1A): The control group (no treatments); the H/R group
(subjected to hypoxia for 120 min, and then reoxygenation for 120
min at 37°C); and the H/R + MTH group [MTH (34°C) was induced
during the 120-min reperfusion period].
Regarding the animal experiments, the Animal
Experiment Ethics Committee of Nanchang University (Nanchang,
China) provided full approval for the present study (approval no.
NCULAE-20221121086). In addition, the present study followed the
Animal Research: Reporting In Vivo Experiments Guidelines
and the American Veterinary Medical Association Euthanasia
Guidelines 2020 (37). A total of
86 healthy adult male Sprague-Dawley rats (weight, 250–350 g) were
housed in cages, with each cage containing one rat, and were housed
under standard conditions (humidity, 45–55%; temperature, 23–25°C;
12-h light/dark cycle), with food and tap water given ad
libitum. The cages were cleaned every 3 days. All rats were
anesthetized prior to the experiment using 3% sodium pentobarbital
(50 mg/kg), which was injected intraperitoneally with heparin. All
rats were euthanized by an overdose of sodium pentobarbital (150
mg/kg), which was injected intraperitoneally at the end of the
experiment.
For these experiments, the rats were randomly
divided into seven groups (n=12/group) (Fig. 1B): The sham group (no treatments);
the I/R group, wherein the rats were subjected to ischemia for 30
min, followed by reperfusion for 120 min at 37°C; the I/R + 2-APB
group, in which the TRPM7 inhibitor 2-APB (5 µM) was added during
the reperfusion period; the I/R + MTH group, wherein MTH (at 34°C)
was induced during the 120-min reperfusion period; the I/R + MTH +
2-APB group, wherein 2-APB was administered combined with MTH
(34°C) during the reperfusion period; the I/R + MTH + Wort group,
wherein the PI3K inhibitor Wort (1.5 µM) was added and MTH (34°C)
was implemented during the reperfusion period; and the I/R + MTH +
Era group, wherein the ferroptosis promoter Era (10 µM) was added
and MTH (34°C) was implemented during the reperfusion period. All
intervention drugs were added to modified Krebs-Henseleit (MKH)
buffer during the first 15 min of reperfusion.
Isolated heart model preparation and
cell model establishment
Firstly, the Langendorff model for I/R injury was
constructed. Rats were first anesthetized using 3% sodium
pentobarbital (50 mg/kg), and then heparin (1,000 IU/kg; n=12 rats)
was injected intraperitoneally. After performing midline
thoracotomy, the heart was excised within 1 min, and the aorta was
cannulated for retrograde perfusion at a constant pressure of 80
mmHg using a Langendorff device with MKH buffer (120.0 mmol/l NaCl,
4.8 mmol/l KCl, 2.4 mmol/l CaCl2, 25.0 mmol/l
NaHCO3, 1.21 mmol/l KH2PO4, 1.2
mmol/l MgSO4 and 11.0 mmol/l glucose; pH, 7.40±0.05).
The buffer was gassed continuously with 95% O2 + 5%
CO2, and maintained at 37±0.2°C or 34±0.2°C with
circulating water. The perfusate MKH buffer during the reperfusion
phase needed to reach a temperature of 34°C within 5 min in the MTH
groups. The initial left ventricle end-diastolic pressure (LVEDP)
was set to 10 mmHg using a latex balloon filled with bubble-free
saline, and Med Lab 6.0 software (ZhongShiDiChuang Science and
Technology Development Co., Ltd.) was used to record hemodynamic
changes, including heart rate (HR), left ventricular systolic
pressure (LVSP), LVEDP and ± dP/dt(max). Rats with frequent
arrhythmia, refractory ventricular fibrillation, HR <180
beats/min or LVSP <75 mmHg were excluded from the study; a total
of two rats excluded.
Secondly, the H/R cell model was constructed. The
H9C2 cells were cultured in a low volume of substrate-free medium
(serum-free and glucose-free) in an anaerobic Plexiglas chamber
(Billups-Rothenberg, Inc.) containing 0.2% O2, saturated
with 95% N2 and 5% CO2 at 37°C for 2 h. The
volume of hypoxic medium used was the minimum volume required to
coat the cellular monolayer for the prevention of cellular
dehydration during the ischemic period. Simulated ischemia was
followed by a simulated reperfusion period, during which the cells
were exposed to normoxic culture medium at 37°C for 2 h (Fig. 1A). For the hypothermia treatment,
the ischemic and hypoxic cells were maintained in an incubator at
34°C for 2 h in the reoxygenation stage. The control cells were
cultured in growth medium at 37°C in an atmosphere containing 5%
CO2 and 95% humidified air.
Determination of myocardial infarct
size
After anesthetizing the rats, open chest surgery was
performed and the rats were sacrificed by cervical dislocation
before removing the heart. Subsequently, the hearts were
immediately removed and a Langendorff model was established. After
reperfusion for 2 h, the hearts were transferred to PBS solution
(pH 7.4) maintained at 4°C, and then frozen at −20°C for 80 min.
Each heart was cut into five cross-sections using a blade, and the
sections were incubated in 1% 2,3,5-triphenyltetrazolium chloride
(TTC) (prepared by dissolving 1 g TTC into 100 ml PBS; cat. no. TTC
0765; Ameresco, LLC) at 37°C for 20 min. The hearts were fixed in
4% paraformaldehyde overnight at 37°C. The myocardial infarct area
changed to white, whereas the area at risk remained red. An Epson
scanner (Seiko Epson Corporation) was used to scan each slice, and
the myocardial infarct area, was calculated using Image Pro Plus
software 6.0 (Media Cybernetics, Inc.).
Hematoxylin and eosin (H&E)
staining
The myocardial tissue obtained from rats was fixed
in 10% paraformaldehyde overnight at room temperature, followed by
embedding in paraffin and cutting into 4-µm sections. These
sections were subsequently dewaxed with xylene twice (5 min each)
and hydrated with 95% ethanol. Subsequently, 0.5% hematoxylin
solution was added for 10 min, and the sections were rinsed with
tap water, differentiated with 1% HCl in ethanol for 10 sec, and
finally stained with 5% eosin for 1–3 min; all steps were at room
temperature. The stained sections were dehydrated with ethanol for
25 min, dewaxed with xylene, sealed and dried with neutral resin,
and finally observed under a fluorescence microscope
(magnification, ×200; DMi8 DFC7000 T; Leica Microsystems,
Inc.).
Detection of cell viability and
cytotoxicity
Cell viability was measured using an MTT cell
viability assay kit and cytotoxi-city was measured using a LDH
cytotoxicity assay kit. All steps were performed in strict
accordance with the manufacturers' protocols.
Cell apoptosis detection
Cells were digested with 0.25% trypsin without EDTA.
After the digestion was terminated, the cells were collected and
centrifuged at 400 × g for 5 min at room temperature, after which
the supernatant was removed and the cells were washed with PBS. The
cells were then rinsed twice with PBS at 400 × g for 5 min at room
temperature, and a cytoFLEX Annexin V-FITC/PI cell apoptosis
detection kit was employed to assess the level of cell apoptosis.
All of the steps were performed in strict accordance with the
manufacturer's instructions. The samples were analyzed using a flow
cytometer (BD Accuri C6 Plus; BD Biosciences) with FlowJo software
(v10.6.2; FlowJo, LLC) according to the manufacturer's
instructions.
Determination of cardiac enzyme and
oxidative stress indicator levels
At the end of reperfusion in the Langendorff model,
coronary effluent was collected for 3 min. Subsequently, the levels
of cardiac muscle enzymes (CK-MB, LDH and c-TnI) were measured by
ELISA. All steps were performed in strict accordance with the
manufacturers' protocols. Furthermore, myocardial tissue was
homogenized in RIPA buffer (cat. no. P0013C; Beyotime Institute of
Biotechnology) containing benzamidine and benzoyl fluoride. The
homogenate was then centrifuged at 15,000 × g for 25 min at 4°C,
and the supernatant was separated and stored at −80°C. Oxidative
stress markers (GSH-Px, MDA and SOD) in the supernatant were then
measured by ELISA. All steps were performed strictly in accordance
with the manufacturer's instructions.
Western blot analysis
Total proteins were extracted from cell samples or
homogenized hearts using a kit (Total Protein Extraction Kit; cat.
no. P1250; Applygen Technologies, Inc.) according to the
manufacturer's instructions. The clarified supernatant was
separated from the sample by centrifugation at 12,000 × g for 5 min
using an MGL-16M desktop high-speed freezing centrifuge at 4°C.
Subsequently, a BCA reagent kit (cat. no. 23227; Thermo Fisher
Scientific, Inc.) was used to quantify the protein concentration,
according to the manufacturer's instructions. Proteins (20 µg/lane)
were then separated by SDS-PAGE on 8% gels and were transferred to
polyvinylidene fluoride (PVDF) membranes. Subsequently, the PVDF
membranes were blocked in 10% non-fat milk for 2 h at room
temperature, prior to incubation with primary antibodies against
TRPM7 (1:500; cat. no. ACC-047; Alomone Labs), t-PI3K (1:1,000),
p-PI3K (1:1,000), p-AKT (1:1,000), t-AKT (1:1,000), GPX4 (1:1,000),
ASCL4 (1:2,000) and FSP1 (1:1,000) at 4°C overnight, followed by
incubation with horseradish peroxidase-conjugated anti-rabbit IgG
(cat. no. 7074S; Cell Signaling Technology, Inc.) or anti-mouse IgG
(cat. no. 7076S; Cell Signaling Technology, Inc.) secondary
antibodies (1:700, diluted in 5% non-fat milk) at room temperature
for 2 h. The same membrane was probed with anti-GAPDH as a control
for normalization of the levels of the proteins of interest.
Protein bands were detected by chemiluminescence (cat. no. P90720;
MilliporeSigma) and the images were semi-quantified using ImageJ
version 1.51 software (National Institutes of Health).
Statistical analysis
All measurements were performed by two researchers
in a double-blinded manner. Data are shown as the mean ± standard
deviation, based on a minimum of three independent replicates.
One-way analysis of variance was employed, followed by Tukey's post
hoc test for multiple comparisons to assess the differences across
multiple groups and to evaluate the impact of treatments on data
derived from animal and cell studies. Statistical analysis was
performed using GraphPad Prism software (version 9.5; Dotmatics).
P<0.05 was considered to indicate a statistically significant
difference.
Results
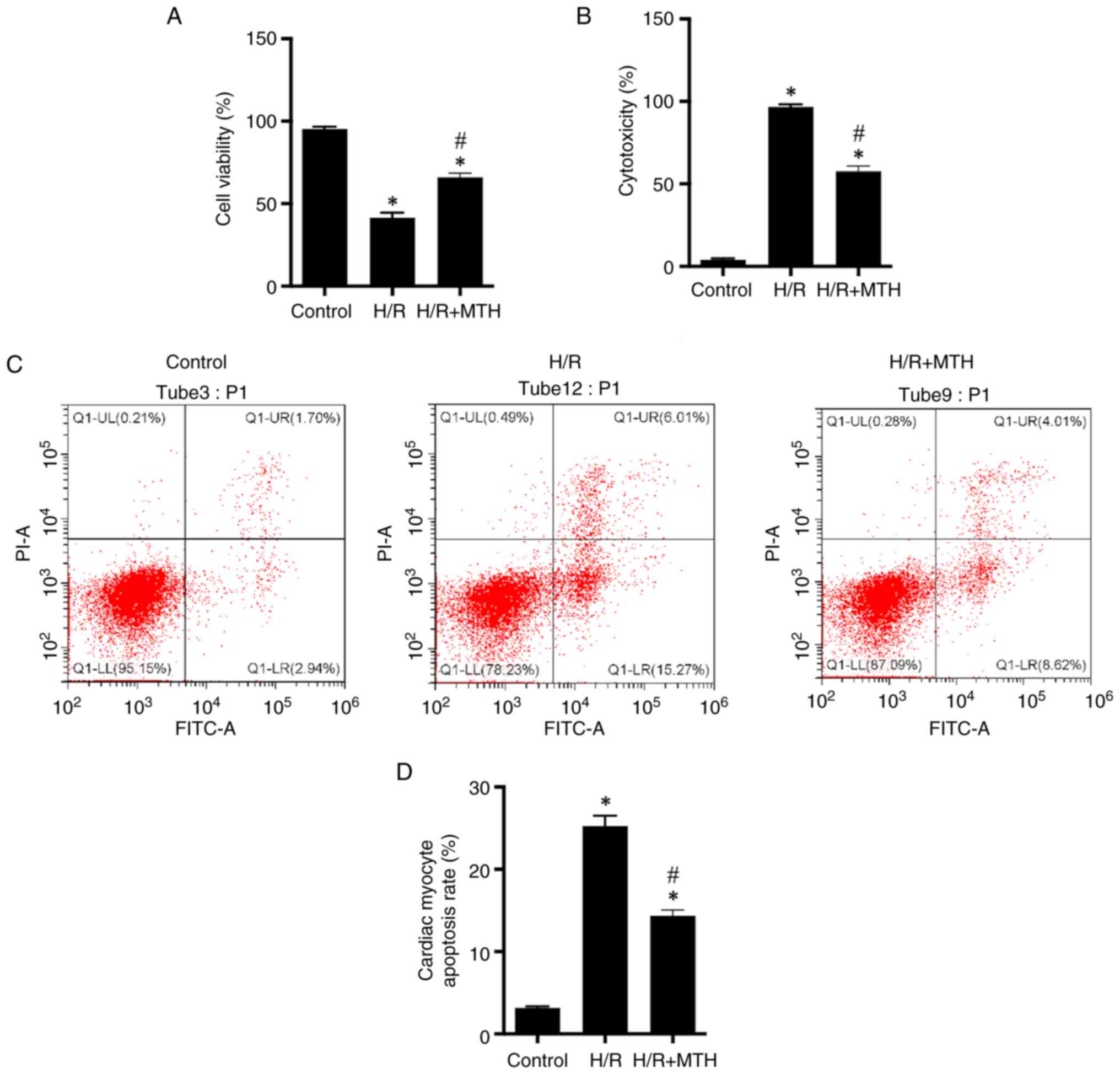
MTH improves cell viability, and
inhibits the release of LDH and cardiomyocyte apoptosis induced by
H/R
In our previous study (19), it was revealed that MTH (set at
34°C) was able to effectively improve MIRI. Therefore, based on
this previous research, the present study aimed to further
investigate the specific underlying protective mechanism. In the
present study, it was revealed that cell viability was reduced,
whereas the release of LDH and the level of apoptosis were
increased in the H/R group compared with those in the control group
(Fig. 2). However, implementing
conditions of MTH reversed this process through increasing cell
viability, and reducing the release of LDH and cardiac myocyte
apoptosis (Fig. 2), thus
suggesting that MTH nay exert a certain myocardial protective
effect.
MTH exerts its protective effect on
myocardial cells by inhibiting H/R-mediated ferroptosis through the
PI3K/AKT/TRPM7 signaling axis
In order to further clarify the myocardial
protective mechanism of MTH, a hypothesis was proposed based on
previous research (8), and western
blot analysis was performed to detect the corresponding indicators.
First, the expression levels of p-PI3K, p-AKT and TRPM7 were
measured in each group of cells, which revealed that the
phosphorylation levels of PI3K and AKT in the H/R group were
between one-half and one-third the levels of those in the control
group, and the expression levels of TRPM7 were 3–4 times higher
compared with those in the control group (Fig. 3A, D, F and H). By contrast, the
phosphorylation levels of PI3K and AKT in the H/R + MTH group were
higher compared with those in the H/R group, whereas the expression
levels of TRPM7 were reduced.
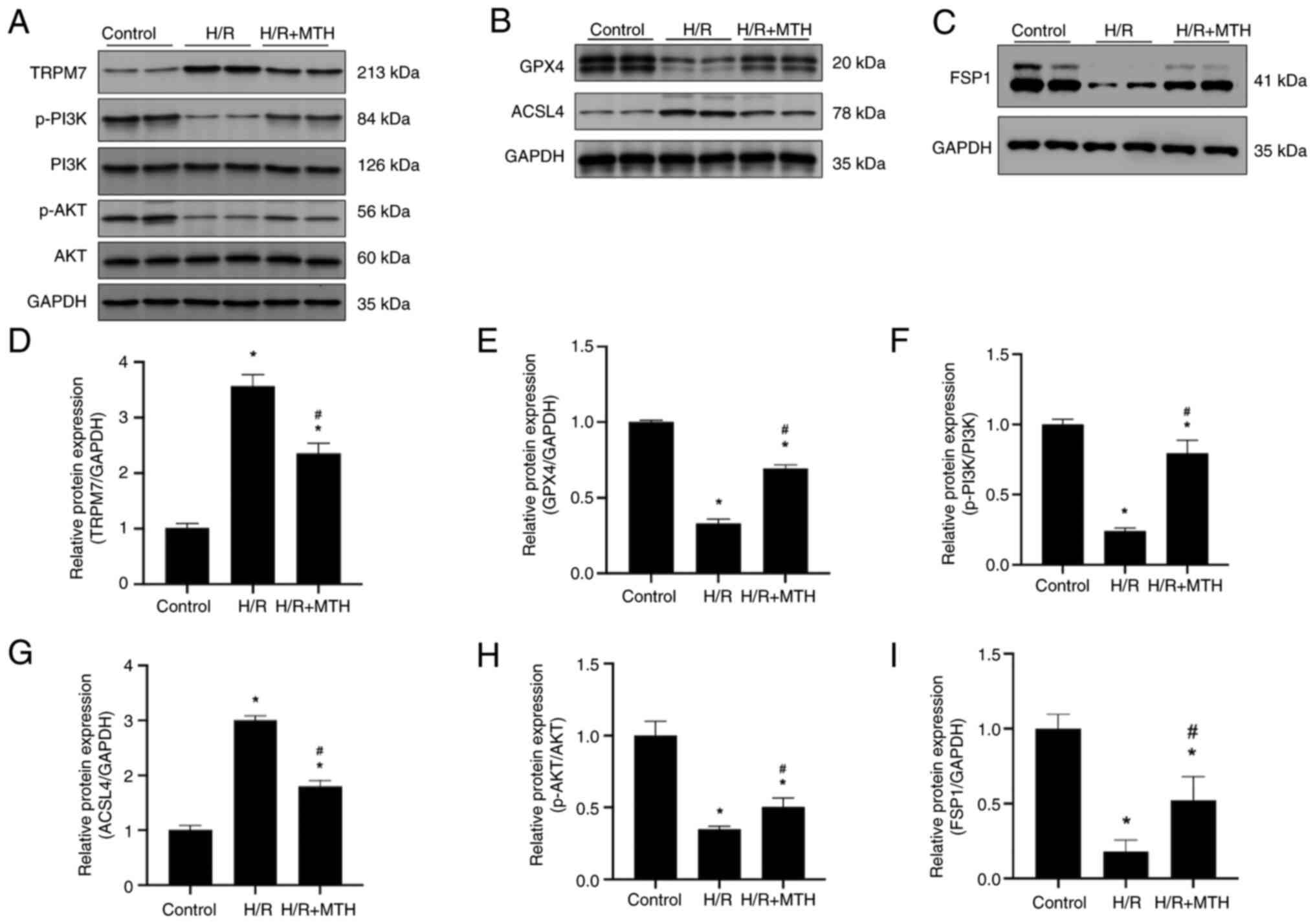 | Figure 3.PI3K/AKT phosphorylation, TRPM7
expression levels and the expression level of
ferroptosis-associated proteins in cells. Western blot analyses of
(A) TRPM7, p-PI3K, PI3K, p-AKT and AKT; (B) GPX4 and ACSL4; and (C)
FSP1. The expression levels of (D) TRPM7, (E) GPX4, (F) p-PI3K, (G)
ACSL4, (H) p-AKT and (I) FSP1 are shown. *P<0.05 vs. control
group; #P<0.05 vs. H/R group. ACSL4, acyl-CoA
synthetase long chain family member 4; FSP1, ferroptosis suppressor
protein 1; GPX4, glutathione peroxidase 4; H/R,
hypoxia-reperfusion; MTH, mild therapeutic hypothermia; p-,
phosphorylated; TRPM7, transient receptor potential cation channel
subfamily M member 7. |
Subsequently, the expression levels of GPX4, ACSL4
and FSP1 in the H9C2 cells were examined in each group (Fig. 3B, C, E, G and I). The expression
levels of GPX4 were decreased and the expression levels of ACSL4
were increased in the H/R group compared with those in the control
group. By contrast, MTH treatment reversed this process; ACSL4
expression levels were significantly decreased, whereas GPX4
expression levels were significantly increased compared with those
in the H/R group. Furthermore, the expression levels of FSP1 were
significantly decreased in the H/R group, whereas they were
significantly higher in the MTH group compared with those in the
H/R group. Taken together, these data suggested that MTH may reduce
H/R-mediated ferroptosis through the PI3K/AKT/TRPM7 signaling
axis.
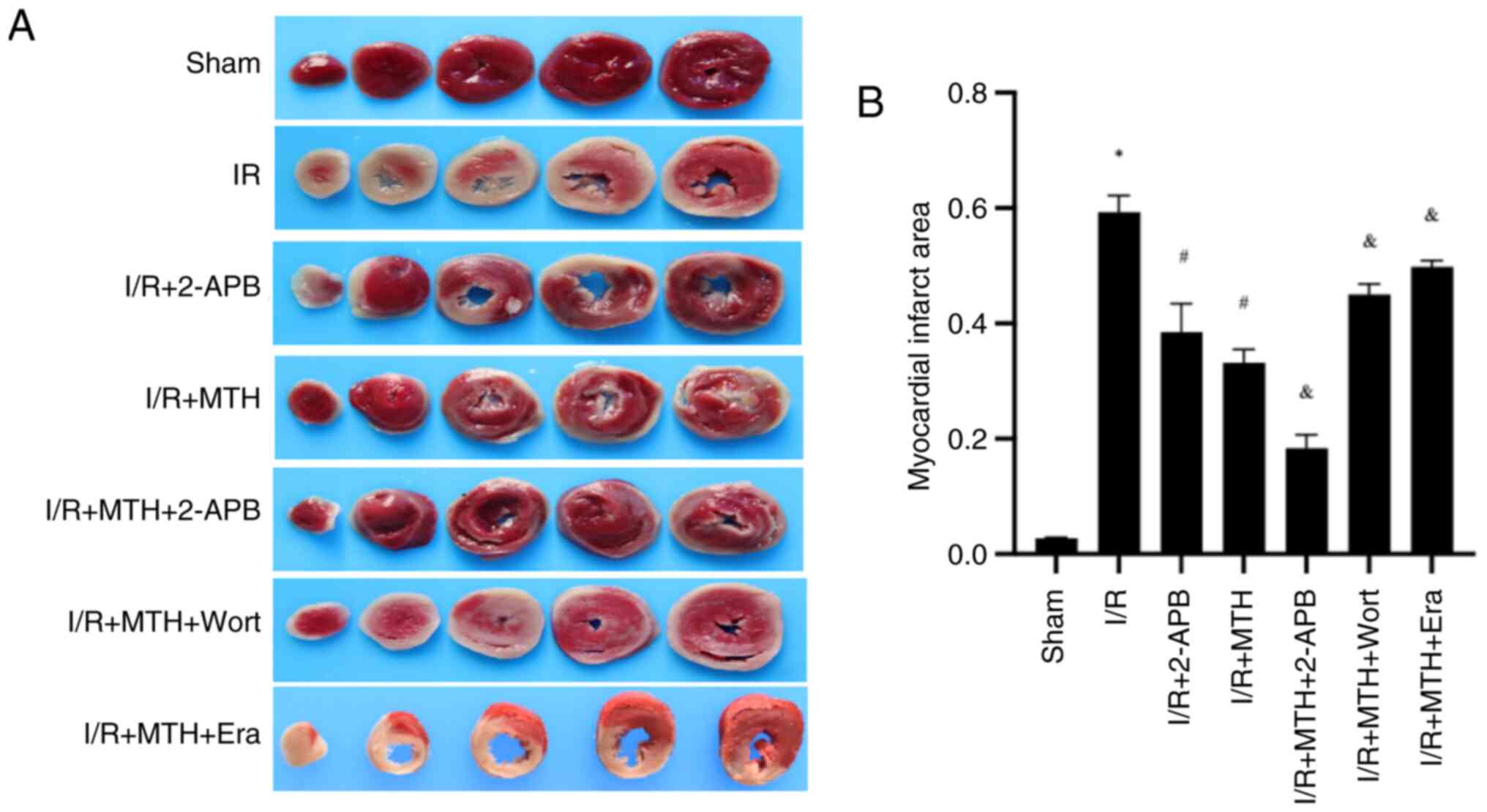
MTH reduces the myocardial infarct
area and improves hemodynamic performance following I/R injury in
rats
To determine whether MTH exerts a protective effect
on MIRI, analysis of the myocardial infarct area and the
hemodynamics of rats was performed for each group. Compared with in
the I/R group, the myocardial infarct area was significantly
reduced in all rats with I/R injury subjected to MTH, except in the
MTH + Wort/Era groups (Fig. 4),
which confirmed that MTH could protect against myocardial injury
caused by I/R. In addition, it was revealed that the ferroptosis
promoter Era, the PI3K inhibitor Wort and the TRPM7 inhibitor 2-APB
could affect the protective effect of MTH on MIRI. Compared with in
the I/R + MTH group, the myocardial infarct area of the I/R + MTH +
2-APB group was significantly reduced, whereas the area was
significantly increased in the I/R + MTH + Wort and I/R + MTH + Era
groups, suggesting that I/R could induce ferroptosis of myocardial
cells. Moreover, the inhibitory effect of MTH on myocardial cell
ferroptosis induced by I/R may be associated with the expression
levels of p-PI3K and TRPM7.
In addition, the hemodynamics of all groups of rats
were recorded to further elucidate the effects of MTH and various
drugs on myocardial infarction (Table
I). The results showed no significant differences in the HR or
heart rate pressure product values among all of the groups at
baseline. However, compared with in the sham group, significant
changes in HR, LVEDP, LVSP and ± dP/dt(max) were observed in other
groups at 30, 60, 80 and 120 min. As expected, MTH treatment led to
significant reductions in the HR, LVSP and ± dP/dt(max) values,
whereas the LVEDP values were increased. The PI3K inhibitor Wort
and the ferroptosis promoter Era were able to inhibit the effects
of MTH, whereas treatment with the TRPM7 inhibitor 2-APB was found
to enhance the effect of MTH, suggesting that MTH exerts an
influence on the expression levels of PI3K and TRPM7, and on the
occurrence of ferroptosis.
 | Table I.Detection of hemodynamic
parameters. |
Table I.
Detection of hemodynamic
parameters.
|
| Reperfusion |
|---|
| Hemodynamic
assessment |
|
|---|
| Baseline (T0) | 30 min (T1) | 60 min (T2) | 90 min (T3) | 120 min (T4) |
|---|
| HR, bpm |
|
|
|
|
|
| Sham
group | 302.66±18.20 | 287.10±5.00 | 286.59±8.26 | 281.30±11.43 | 280.26±8.41 |
| I/R
group | 288.64±6.02 |
218.45±22.52a |
205.43±5.67a |
180.41±5.16a |
150.04±4.75a |
| I/R +
2-APB group | 299.98±4.75 |
255.04±4.96a,b |
219.27±15.23a,b |
225.37±15.39a,b |
213.42±8.19a,b |
| I/R +
MTH group | 297.76±5.69 |
250.07±7.05a,b |
277.42±14.40a,b |
209.66±6.93a,b |
196.74±9.57a,b |
| I/R +
MTH + 2-APB group | 300.47±6.65 | 273.91±3.19 |
255.71±14.69a |
251.91±8.83a |
242.83±5.71a |
| I/R +
MTH + Wort group | 293.66±8.27 |
208.10±5.99a,c |
210.96±11.88a,c |
186.37±8.40a |
167.74±3.87a,c |
| I/R +
MTH + Era group | 296.66±6.51 |
204.67±4.16a,c |
208.04±8.15a,c |
189.74±9.75a |
165.69±2.31a,c |
| LVEDP, mmHg |
|
|
|
|
|
| Sham
group | 7.18±0.39 | 6.89±0.59 | 6.06±0.29 | 6.11±0.52 | 5.67±0.46 |
| I/R
group | 6.85±0.59 |
33.79±2.60a |
39.85±1.10a |
45.12±1.63a |
48.91±2.44a |
| I/R +
2-APB group | 7.29±0.42 |
19.12±0.33a |
19.68±0.83a,b |
22.03±0.24a,b |
27.10±0.25a,b |
| I/R +
MTH group | 7.27±0.68 |
19.87±0.36a |
21.57±0.86a,b |
25.14±0.60a,b |
29.98±0.11a,b |
| I/R +
MTH + 2-APB group | 6.94±0.86 |
10.14±0.69a |
11.12±0.59a |
14.06±0.55a |
17.01±0.29a |
| I/R +
MTH + Wort group | 6.65±0.45 |
29.09±1.46a |
33.56±0.41a,c |
41.87±1.75a,c |
47.10±0.52a,c |
| I/R +
MTH + Era group | 7.33±0.48 |
29.39±1.10a |
34.40±0.67a,c |
43.58±1.29a,c |
46.52±0.98a,c |
| LVSP, mmHg |
|
|
|
|
|
| Sham
group | 103.81±7.09 | 95.15±8.98 | 91.16±9.93 |
93.76±11.92a |
88.05±3.35a |
| I/R
group | 103.15±4.98 |
62.02±8.12a |
50.08±4.72a |
41.08±3.82a |
28.63±5.42a |
| I/R +
2-APB group | 96.37±7.12 |
84.30±1.23a |
69.61±4.89a |
62.52±3.42a |
67.51±2.34a |
| I/R +
MTH group | 103.27±8.58 |
81.33±2.73a |
66.85±2.44a |
57.34±1.61a |
56.03±4.07a |
| I/R +
MTH + 2-APB group | 99.73±2.61 | 88.92±1.41 |
79.74±5.37a |
76.63±4.89a |
69.57±2.87a |
| I/R +
MTH + Wort group | 63.83±1.00 |
63.83±1.00a |
57.30±1.67a |
46.18±2.76a |
32.26±8.13a |
| I/R +
MTH + Era group | 64.63±0.88 |
64.63±0.88a |
56.88±1.08a |
45.87±2.83a |
31.23±9.06a |
|
+dP/dt(max), mmHg/sec |
|
|
|
|
|
| Sham
group |
2,703.57±166.22 |
2,605.05±128.14 |
2,660.96±160.54 |
2,609.83±176.35 |
2,515.96±168.90 |
| I/R
group |
2,891.98±101.97 |
1,724.36±179.88a |
1,538.13±106.34a,b |
1,205.16±246.80a,b |
851.32±60.81a,b |
| I/R +
2-APB group | 2,862.30±81.92 |
2,518.11±162.52b |
2,150.19±131.84a,b |
2,033.59±108.93a,b |
1,896.02±189.31a,b |
| I/R +
MTH group |
2,773.18±156.28 |
2,393.79±78.29b |
2,023.00±66.09a |
1,900.44±86.25a |
1,830.33±181.71a |
| I/R +
MTH + 2-APB group |
2,918.62±118.30 | 2,573.60±80.10 |
2,354.58±78.47a | 2,367.53±83.92 |
2,518.84±122.80a |
| I/R +
MTH + Wort group |
2,759.03±253.44 |
1,810.25±89.80a,c |
1,475.58±83.63a,c |
1,192.34±86.02a,c |
979.77±98.81a,c |
| I/R +
MTH + Era group |
2,714.76±150.42 |
1,657.27±341.90a,c |
1,481.72±92.60a,c |
1,210.00±120.82a,c |
932.13±112.38a,c |
|
-dP/dt(max), mmHg/sec |
|
|
|
|
|
| Sham
group |
−2,785.34±133.79 |
−2,528.84±187.12 |
−2,589.51±96.94 |
−2,514.40±292.89 |
−2,696.33±181.73 |
| I/R
group |
−2,750.15±86.02 |
−1,758.31±114.46a |
−1,514.12±77.14a |
−1,227.65±84.21a |
−830.36±64.71a |
| I/R +
2-APB group |
−2,867.99±163.77 |
−2,566.42±86.04b |
−2,066.89±54.34a,b |
−2,043.36±107.16a,b |
−1,905.83±170.75a,b |
| I/R +
MTH group |
−2,757.58±173.15 |
−2,439.87±152.32b |
−1,927.35±161.08a,b |
−1,908.28±133.05a,b |
−1,863.66±68.43a,b |
| I/R +
MTH + 2-APB group |
−2,882.22±112.48 |
−2,598.29±65.88 |
−2,413.16±96.44 |
−2,322.91±84.80 |
−2,138.46±45.25a |
| I/R +
MTH + Wort group |
−2,733.51±253.68 |
−1,863.77±79.23a,c |
−1,581.44±75.12a,c |
−1,184.09±62.19a,c |
−954.36±112.69a,c |
| I/R +
MTH + Era group |
−2,660.97±245.14 |
−1,669.67±156.29a,c |
−1,496.65±168.57a,c |
−1,196.06±111.58a,c |
−949.58±104.47a,c |
MTH alleviates MIRI in rats
To further verify the protective effect of MTH on
MIRI, the myocardial tissue of rats was stained with H&E, and
the results obtained showed that, compared with in the control
group, the myocardial tissue of rats in the I/R group exhibited
clear pathological changes. Specifically, the myocardial cells were
disordered; some myocardial cells were seen to be broken and
dissolved; edema and some degeneration were observed; and the cell
gap was notably widened (Fig. 5A).
However, the negative pathological changes observed in the
myocardial tissue of I/R model rats were found to be improved
following treatment with MTH or 2-APB, and the myocardial cells
were arranged neatly. Notably, compared with in the I/R + MTH
group, the pathological changes observed in the myocardial tissue
of the I/R model rats treated with MTH + 2-APB were further
improved; myocardial cells and clear morphological structures were
both found to be well-arranged, and myocardial cell edema and cell
degeneration were suppressed, thus suggesting that 2-APB may
enhance the effect of MTH through inhibiting the expression of
TRPM7. However, the I/R model rats in the MTH + Wort and MTH + Era
treatment groups exhibited obvious pathological changes, which were
not notably different from those observed in the I/R group.
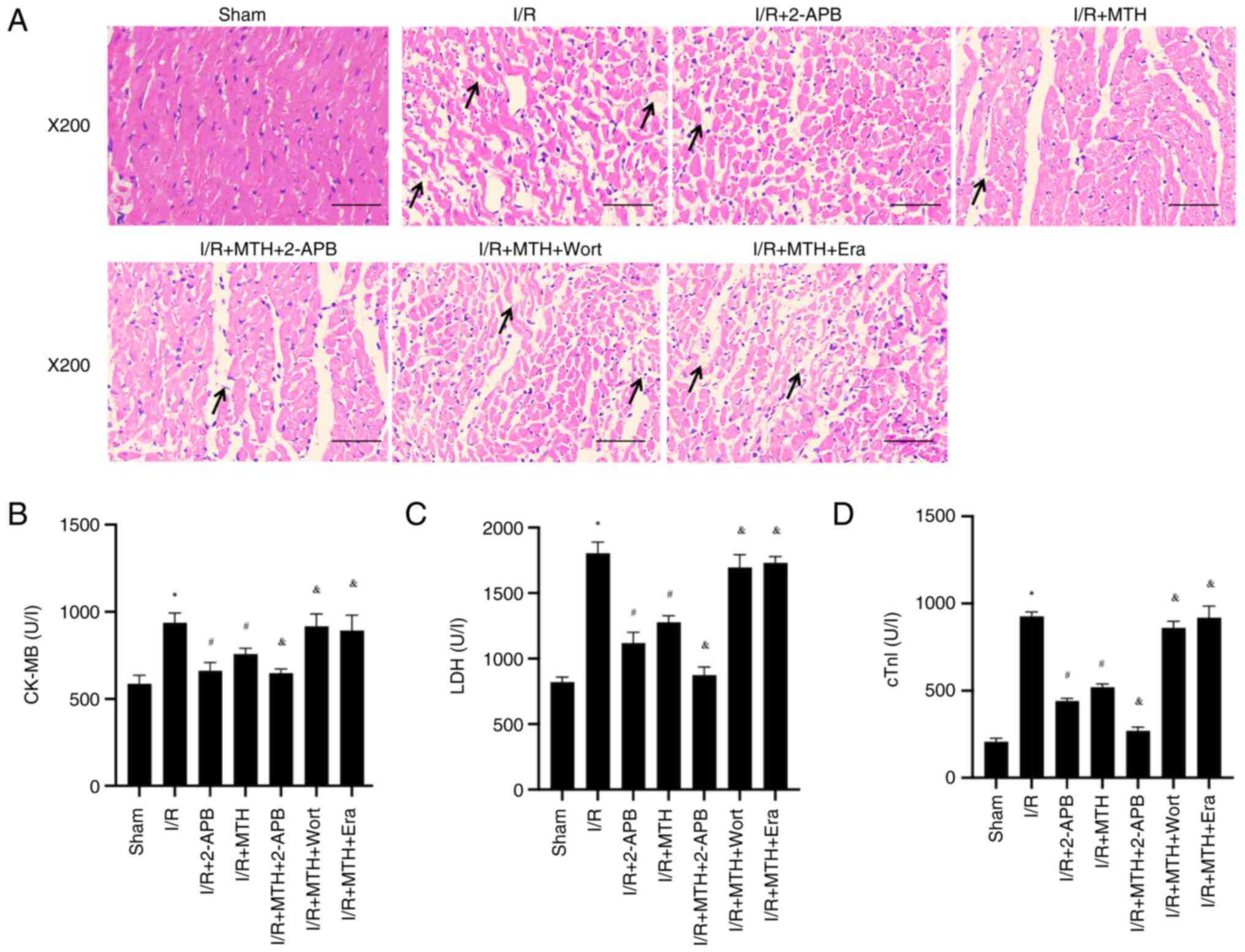 | Figure 5.Pathological changes of myocardial
tissue and levels of myocardial enzymes. (A) Hematoxylin and eosin
staining, arrows indicate the location of myocardial fiber rupture
or dissolution. Levels of (B) CK-MB, (C) LDH and (D) cTnI.
*P<0.05 vs. sham group; #P<0.05 vs. I/R group;
&P<0.05 vs. I/R + MTH group. 2-APB,
2-aminoethoxydiphenyl borate; CK-MB, creatine kinase-MB; cTnI,
cardiac troponin I; Era, erastin; I/R, ischemia-reperfusion; LDH,
lactate dehydrogenase; MTH, mild therapeutic hypothermia; Wort,
wortmannin. |
In addition, the concentrations of CK-MB, LDH and
cTnI were measured by ELISA. Compared with those in the sham group,
the levels of CK-MB, LDH and cTnI in the I/R group rats were
significantly increased, whereas the levels of CK-MB, LDH and cTnI
in the rats treated with MTH or 2-APB were decreased (Fig. 5B-D). In addition, compared with in
the I/R + MTH group, the levels of CK-MB, LDH and cTnI were
significantly decreased in the I/R + MTH + 2-APB group, whereas
they were increased in the I/R + MTH + Wort and I/R + MTH + Era
groups. Taken together, these results suggested that MTH could
improve MIRI, and that the protective effect of MTH was affected by
the expression levels of PI3K and TRPM7, and by whether ferroptosis
occurred.
MTH is able to reduce MIRI in rats
through activating the PI3K/AKT signaling pathway
Subsequently, the activation levels of PI3K and AKT
were detected in the rats (Fig. 6A, C
and D). Compared with in the sham group, the phosphorylation
levels of PI3K and AKT in the I/R group rats were significantly
decreased; however, a higher phosphorylation level of PI3K and AKT
was observed in the I/R + MTH group compared with that in the I/R
group. Furthermore, the phosphorylation levels of PI3K and AKT were
significantly decreased following treatment with Wort, compared
with those in the I/R + MTH group. Notably, no significant
differences in the phosphorylation levels of PI3K and AKT were
observed between the I/R and I/R + 2-APB groups, between the I/R +
MTH and I/R + MTH + 2-APB groups, or between the I/R + MTH and I/R
+ MTH + Era groups. Collectively, these data revealed that neither
Era nor 2-APB affected the phosphorylation levels of PI3K and AKT,
suggesting that MTH is able to regulate the expression of TRPM7 and
the occurrence of ferroptosis through activating the PI3K/AKT
signaling pathway, thereby protecting MIRI.
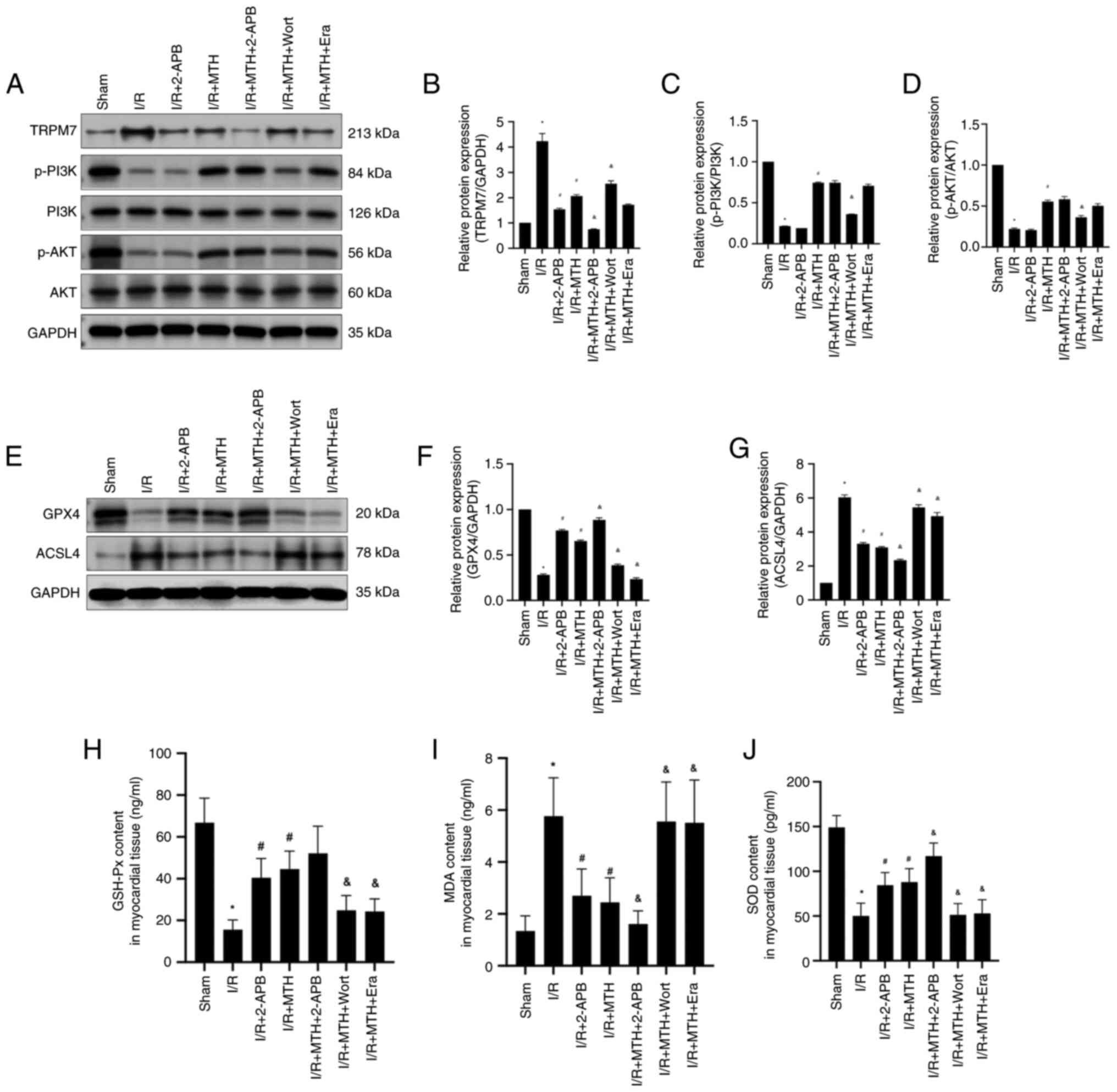 | Figure 6.PI3K/AKT phosphorylation, TRPM7
expression levels and the expression level of
ferroptosis-associated proteins. (A) Western blot analysis of
TRPM7, p-PI3K, PI3K, p-AKT and AKT. Expression levels of (B) TRPM7,
(C) p-PI3K and (D) p-AKT. (E) Western blot analysis of GPX4 and
ACSL4. Expression levels of (F) GPX4 and (G) ACSL4. Levels of the
oxidative stress indicators (H) GSH-Px, (I) MDA and (J) SOD.
*P<0.05 vs. sham group; #P<0.05 vs. I/R group;
&P<0.05 vs. I/R + MTH group. 2-APB,
2-aminoethoxydiphenyl borate; ACSL4, acyl-CoA synthetase long chain
family member 4; Era, erastin; GPX4, glutathione peroxidase 4;
GSH-Px, glutathione peroxidase; I/R, ischemia-reperfusion; MDA,
malondialdehyde; MTH, mild therapeutic hypothermia; p-,
phosphorylated; SOD, superoxide dismutase; TRPM7, transient
receptor potential cation channel subfamily M member 7; Wort,
wortmannin. |
MTH improves MIRI by activating the
PI3K/AKT signaling pathway to inhibit the expression of TRPM7
To further verify the hypothesis, the expression
levels of TRPM7 were determined (Fig.
6A and B). Compared with in the sham group, the expression
levels of TRPM7 in the I/R group were significantly increased,
whereas the expression levels of TRPM7 were inhibited in the I/R +
MTH group, suggesting that MTH treatment could downregulate the
expression of TRPM7. Moreover, compared with in the I/R + MTH
group, the expression levels of TRPM7 in the I/R + MTH + 2-APB
group were significantly decreased, and a higher expression of
TRPM7 was detected in the I/R + MTH + Wort group. These findings
suggested that, under MTH conditions, the expression of TRPM7 may
be inhibited through activating the PI3K/AKT signaling pathway, and
that 2-APB may enhance the inhibitory effect of MTH. No significant
difference in the expression levels of TRPM7 was observed between
the I/R + MTH and I/R + MTH + Era groups, revealing that Era did
not affect the expression levels of TRPM7.
MTH is able to inhibit ferroptosis
induced by MIRI through the PI3K/AKT signaling pathway
To further clarify the association between
ferroptosis and the PI3K/AKT signaling pathway, the expression
levels of GPX4 and ACSL4 were examined in the rats in each group
(Fig. 6E-G). The expression levels
of GPX4 in the I/R group were significantly decreased compared with
those in the sham group. Following MTH or 2-APB treatment, the
expression levels of GPX4 were increased, suggesting that MTH and
2-APB could inhibit the occurrence of ferroptosis. Moreover,
compared with in the I/R + MTH group, the expression levels of GPX4
in the I/R + MTH + 2-APB group were significantly increased, and
the expression levels of GPX4 in the I/R + MTH + Wort group were
decreased by 50%. These findings suggested that 2-APB may enhance
the protective effect of MTH through inhibiting TRPM7, whereas Wort
may damage the protective effect of MTH through inhibiting the
PI3K/AKT signaling pathway. These findings were consistent with the
aforementioned experimental results obtained from investigating the
myocardial infarct area and improvements in hemodynamic performance
following I/R injury in rats.
The expression changes of ACSL4 in each group were
found to be the opposite of those reported for GPX4. Compared with
in the sham group, the expression levels of ACSL4 in the I/R group
were significantly increased. Following treatment with MTH or
2-APB, the expression levels of ACSL4 were decreased. Moreover, a
greater decrease in the expression levels of ACSL4 was observed in
the I/R + MTH + 2-APB group when compared with the I/R + MTH group,
whereas the change in expression of ACSL4 showed an opposite trend
in the I/R + MTH + Wort and I/R + MTH + Era groups. These findings
suggested that MTH may be able to inhibit the expression of TRPM7
through activating the PI3K/AKT signaling pathway, which
subsequently inhibits the occurrence of ferroptosis, thereby
eventually producing a protective effect on MIRI. In addition, the
levels of the oxidative stress markers (GSH-Px, SOD and MDA) were
further tested in the myocardial tissue of each group of rats
(Fig. 6H-J). These experiments
revealed that the levels of GSH-Px and SOD in the MTH group were
significantly higher compared with those in the I/R group, whereas
the levels of MDA were significantly lower compared with those in
the I/R group, further confirming that MTH may alleviate
ferroptosis induced by MIRI by inhibiting oxidative stress.
Discussion
MIRI, which may arise after myocardial ischemia,
cardiac surgery or circulatory arrest, contributes to adverse
cardiovascular outcomes. During this process, the lack of blood
flow to the heart leads to an imbalance between oxygen demand and
supply and, in turn, damage to the cardiac tissue (37). To date, a number of studies have
identified several mechanisms involved in MIRI, including
ferroptosis, calcium overload, inflammation and oxidative stress
(38–40). Myocardial ischemia leads to the
abnormal functioning of ion channels in cardiomyocytes, increasing
intracellular calcium concentration. Following blood reperfusion,
the calpain system is rapidly activated, thereby promoting
cardiomyocyte apoptosis (38,39).
At the same time, following myocardial infarction and reperfusion,
MIRI promotes the increase of iron ions in the myocardium and
induces ferroptosis (40). IPC and
PPC represent the main treatments for MIRI; however, their
therapeutic effects remain unsatisfactory.
Numerous studies have suggested that MTH is able to
effectively improve MIRI (41–43),
and the results of the present study were consistent with these
previous findings. In the present study, it was observed that MTH
improved cell viability, inhibited LDH release and reduced the rate
of cardiac myocyte apoptosis. In addition, the results revealed
that the myocardial infarct area in all rats with I/R injury under
MTH conditions was significantly reduced compared with that in the
I/R group; however, Era and Wort could effectively abolish the
cardioprotective effect of MTH. Furthermore, subjecting the rats to
MTH conditions led to a significant reduction in the HR, LVSP and ±
dP/dt(max) values, and an increase in the LVEDP of rats with I/R
injury.
Several studies have indicated that MTH exerts a
protective effect on various types of cardiovascular injury. For
example, MTH has been shown to inhibit both inflammatory reactions
and cardiomyocyte apoptosis, thereby effectively improving MIRI in
septic rats (44). Moreover,
clinical trials showed that MTH effectively led to an improvement
in the prognosis of patients with out-of-hospital cardiac arrest
(45–47). Furthermore, MTH may exert a
protective effect on acute myocardial infarction. Notably, MTH (at
34°C) has been shown to effectively improve MIRI (17) and the findings of the present study
lend support to this previous study. In the present study, clear
pathological changes were identified in the myocardial tissue of
I/R rats, with disorganized myocardial cell arrangement, partial
myocardial cell fracture and lysis, myocardial cell edema and
degeneration, and a notable widening of cell gaps. However, the
disruptive pathological changes observed in the myocardial tissue
of I/R model rats were markedly ameliorated following treatment
with MTH or 2-APB, and the myocardial cells were shown to be neatly
arranged. Compared with those in the I/R group, the levels of
CK-MB, LDH and cTnI also exhibited significant decreases in the
rats treated with MTH or 2-APB. Similarly, it was confirmed that
implementing MTH conditions led to improvements in the H/R-induced
decrease in cell viability, a reduction in the rate of LDH release
and a reduction in the apoptotic rate of cardiac myocytes in
vitro.
PI3K/AKT is an important signaling pathway in
organisms that comprises PI3K and the downstream molecule, AKT
(48). The PI3K/AKT pathway is
activated via the receptor tyrosine kinase. Tyrosine residues are
subsequently phosphorylated, providing binding sites for PI3K
translocation to the cell membrane, thereby transducing signals to
various extracellular matrices and cytokines (48). The pathway serves important roles
in numerous cellular processes, including adhesion, proliferation,
migration, metabolism, invasion and survival (49). In addition, numerous studies have
shown that the PI3K/AKT signaling pathway participates in the
occurrence and development of various types of cardiovascular
disease (50). Notably,
resveratrol has been shown to reduce myocardial cell apoptosis and
mitochondrial oxidative damage caused by MIRI by activating
PI3K/AKT (51,52), suggesting that activation of the
PI3K/AKT signaling pathway may protect against MIRI. Furthermore,
MTH has been shown to activate the PI3K signaling pathway to
protect the myocardium from I/R injury (19). In addition, as an inhibitor of
PI3K, a previous study demonstrated that 1 µM Wort could
effectively inhibit the protective effect of MTH on MIRI (17). Therefore, the same concentration of
Wort was chosen for the experiments in the present study. Notably,
the PI3K signaling pathway is able to improve I/R injury through
regulating the expression levels of TRPM7 (21). The present study revealed that MTH
affected the physiological levels of PI3K and AKT in cells in
vitro. Subsequently, the animal intervention experiments showed
that, compared with in the I/R group, the phosphorylation levels of
PI3K and AKT were significantly increased in the I/R + MTH group;
however, no significant differences in the PI3K or AKT
phosphorylation levels were observed between the I/R + MTH and the
I/R + MTH + 2-APB groups. These results provided further evidence
to indicate that the expression levels of TRPM7 are regulated by
the PI3K/AKT signaling pathway, thereby providing further evidence
for our hypothesis.
TRPM7 is a protein with a dual structure consisting
of a cationic channel and a serine/threonine protein kinase, which
has been reported to be temperature-sensitive (49); 2-APB is as an inhibitor of TRPM7. A
previous study showed that 5 µM 2-APB was able to effectively
inhibit MIRI (53). Therefore,
2-APB at the same dose was used in the relevant experiments in the
present study. The results demonstrated that MTH was able to
inhibit the expression of TRPM7 and the occurrence of H/R-induced
ferroptosis. In addition, the expression levels of TRPM7 were found
to be closely associated with ferroptosis. Compared with in the I/R
+ MTH group, the expression levels of GPX4 in the I/R + MTH + 2-APB
group were significantly increased, whereas the expression levels
of ACSL4 were decreased. Notably, a previous study reported that
lipopolysaccharide (LPS) induced upregulation of TRPM7 expression
in cardiomyocytes, and knocking down TRPM7 could inhibit
LPS-induced ferroptosis in cardiomyocytes, which was consistent
with the present research findings (54).
TRPM7 serves a key role in ischemic cardiomyopathy.
Inhibiting TRPM7 can exert a protective effect on the reperfusion
injury of H9C2 cells and reduce the apoptotic rate of H9C2 cells
(54). Under neuronal hypoxic
conditions or in oxygen-glucose deprivation models, the TRPM7
channels are induced to open, causing calcium overload and inducing
cells to produce large amounts of ROS, which in turn can promote
the opening of TRPM7, leading to neuronal damage (28).
Ferroptosis is a type of programmed cell death
characterized by iron-dependent lipid peroxidation (29). The accumulation of lipid ROS is one
of the characteristics of ferroptosis. When ferroptosis occurs,
large amounts of ROS are produced, which, in turn, promotes cell
ferroptosis (29). GPX4 and ACSL4
have key roles in the occurrence and development of ferroptosis,
and may be used as indicators to monitor ferroptosis. GPX4 has been
shown to inhibit lipid peroxidation, reduce ROS production and
inhibit the occurrence of cell ferroptosis (55). By contrast, ACSL4 can increase the
content of phosphatidylethanolamines in cells, thereby leading to
cell ferroptosis (56). Notably, a
previously published study suggested that FSP1 is one of the key
targets for inhibiting ferroptosis, and it is considered that
FSP1-mediated ferroptosis may be independent of the GPX4-mediated
ferroptosis signaling pathway (57). Therefore, GPX4 and ACSL4 were
selected as the monitoring indicators of ferroptosis in the present
study. As an activator of ferroptosis, our previous study (58) confirmed that 10 µM Era could
effectively activate the occurrence of ferroptosis. Therefore, the
same concentration of Era was chosen for the experiments performed
in the present study. The expression levels of GPX4 in the I/R +
MTH + 2-APB group were significantly increased compared with those
in the I/R + MTH group, whereas the expression of GPX4 in the I/R +
MTH + Wort group was decreased. Moreover, the expression of ACSL4
exhibited the opposite trend. In addition, several studies have
indicated that ferroptosis serves a key role in cardiovascular
injury caused by various diseases, including heart failure,
myocardial injury and I/R injury (30–32).
Rehmannioside A has been shown to inhibit the occurrence of
ferroptosis induced by I/R through activating the PI3K/AKT
signaling pathway, suggesting that the PI3K/AKT signaling pathway
is of crucial importance in regulating ferroptosis that is induced
by I/R (33), consistent with the
present experimental results. As aforementioned, according to the
results from the isolated rat model and in cell experiments, the
present study revealed that MTH could inhibit the expression of
TRPM7 through the PI3K/AKT signaling pathway, thereby inhibiting
the occurrence of ferroptosis induced by I/R and H/R, ultimately
exerting a myocardial protective effect.
The present study, however, had some limitations.
First, specific inhibitors were only used in the isolated rat model
to study the mechanism of action of MTH. The present study did not
use the C11-BODIPY method to detect lipid peroxidation levels in
myocardial tissue and myocardial cells, nor was the iron ion
content in myocardial tissues and cells measured. In addition, the
current study did not directly manipulate the expression levels of
TRPM7 using transgenic, plasmid transfection or lentiviral
interference techniques, which may have confirmed that MTH could
exert cardioprotective effects by manipulating the expression of
TRPM7. Furthermore, the exploration of downstream sites affected by
TRPM7 in ferroptosis will be an important focus of subsequent
experiments. We aim to construct transgenic animal models of TRPM7,
and knockdown and overexpression cell models, and explore the
effects of TRPM7 on mitochondrial function and ferroptosis through
molecular biology methods. Whether this mechanism can be further
validated in future clinical studies, and whether it can enhance
the therapeutic effect of MTH by regulating TRPM7 to inhibit
ferroptosis, deserves further investigation.
The present study indicated that when MIRI occurs,
the expression of TRPM7 on the myocardial cell membrane may be
increased. As a non-selective cation channel, TRPM7 is permeable to
calcium and magnesium ions in the plasma. As its expression level
is increased, this may lead to calcium ion influx, causing calcium
overload of myocardial cells and, finally, myocardial cell damage.
In addition, calcium overload is positively associated with a large
amount of ROS accumulation produced by myocardial cells, further
causing myocardial damage. It is worth noting that the accumulation
of ROS can induce ferroptosis, and further aggravate myocardial
injury. Therefore, it may be suggested that MTH therapy inhibits
the expression of TRPM7 through activating the PI3K/AKT signaling
pathway, thereby inhibiting the occurrence of ferroptosis,
ultimately participating in myocardial protection (Fig. 7). In conclusion, MTH protection
activates the PI3K/AKT signaling pathway to inhibit TRPM7 and
suppress ferroptosis induced by MIRI. However, further studies are
required to investigate the clinical application of MTH.
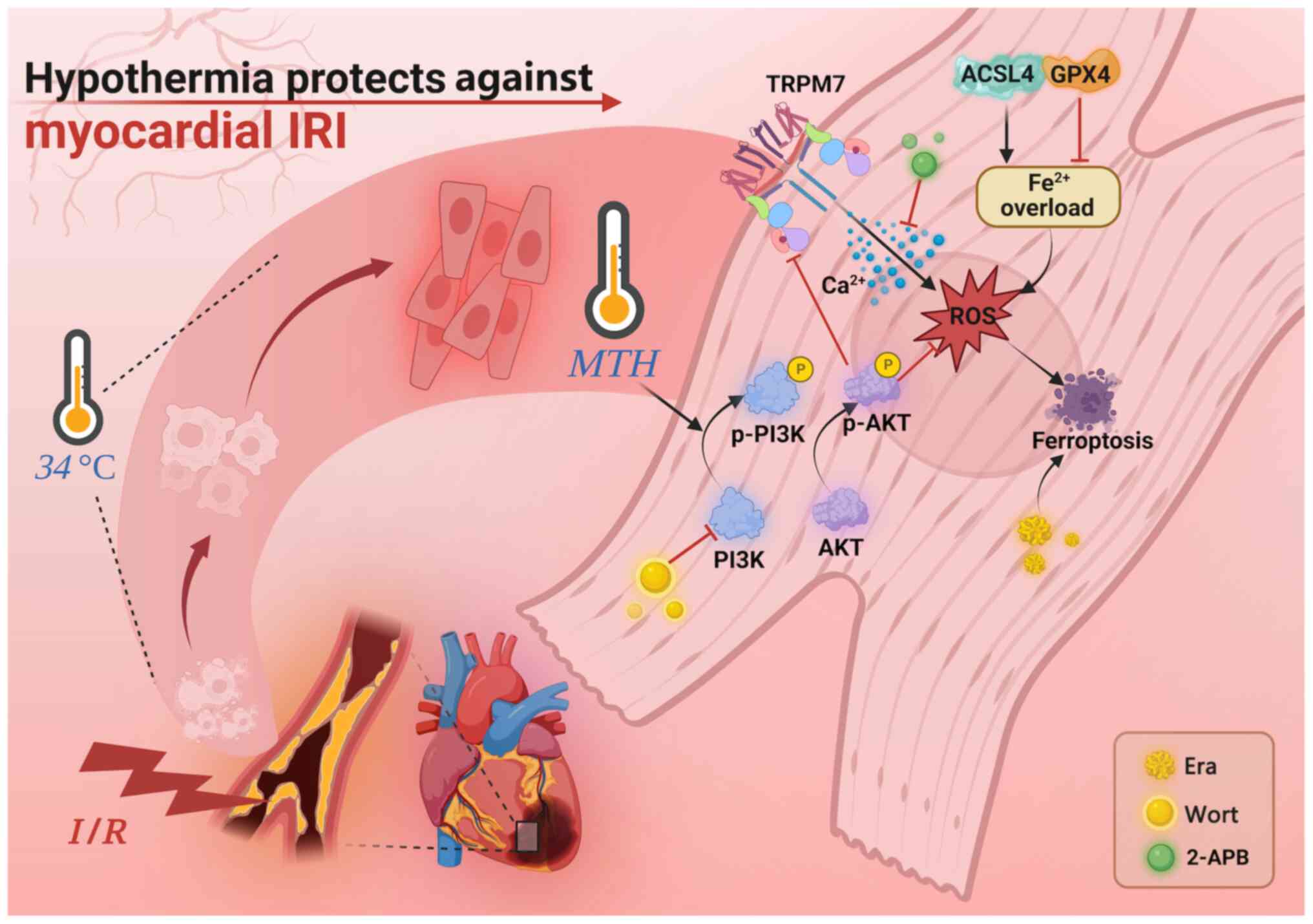 | Figure 7.Protective mechanism of MTH on
myocardial IRI. MTH can reduce the ferroptosis induced by
myocardial I/R via inhibiting the expression of TRPM7, thereby
alleviating myocardial IRI. 2-APB, 2-aminoethoxydiphenyl borate;
ACSL4, acyl-CoA synthetase long chain family member 4; Era,
erastin; GPX4, glutathione peroxidase 4; I/R, ischemia-reperfusion;
IRI, I/R injury; MTH, mild therapeutic hypothermia; p-,
phosphorylated; ROS, reactive oxygen species; TRPM7, transient
receptor potential cation channel subfamily M member 7; Wort,
wortmannin. |
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 82160371 to JZ, grant no. 81760338
to SY); the Key Projects of Jiangxi Provincial Department of
Education Science and Technology Plan (grant no. GJJ210134 to SY);
the Natural Science Foundation in Jiangxi Province grant (grant no.
20224ACB216009 to JZ); the Science and Technology Plan of Jiangxi
Provincial Administration of Traditional Chinese Medicine (grant
no. 2022B1038 to YQL); and the Science and Technology Plan of
Jiangxi Provincial Health Commission (grant no. 202310106 to
YQL).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YQL, YXC, PY, DJZ, XYT, ZCZ, FX, WD, YL and ZYT
performed nearly all of the experiments. YQL, YXC, PY and JZ
analyzed the data. YQL, YXC, PY, JZ and SY designed the study and
all of the experiments were performed under their guidance. YXC, PY
and DJZ were the major contributors in writing the manuscript. YQL
and SY confirm the authenticity of all the raw data. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The Animal Experiment Ethics Committee of Nanchang
University provided full approval for this research (no.
NCULAE-20221121086). In addition, this study also followed the
Animal Research: Reporting In Vivo Experiments Guidelines
and the American Veterinary Medical Association Euthanasia
Guidelines 2020.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Petrie MC, Verma S, Docherty KF, Inzucchi
SE, Anand I, Belohlávek J, Böhm M, Chiang CE, Chopra VK, de Boer
RA, et al: Effect of dapagliflozin on worsening heart failure and
cardiovascular death in patients with heart failure with and
without diabetes. JAMA. 323:1353–1368. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Raphael CE, Roger VL, Sandoval Y, Singh M,
Bell M, Lerman A, Rihal CS, Gersh BJ, Lewis B, Lennon RJ, et al:
Incidence, trends, and outcomes of type 2 myocardial infarction in
a community cohort. Circulation. 141:454–463. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang X, Toan S, Li R and Zhou H:
Therapeutic strategies in ischemic cardiomyopathy: Focus on
mitochondrial quality surveillance. EBioMedicine. 84:1042602022.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang K, Li Y, Qiang T, Chen J and Wang X:
Role of epigenetic regulation in myocardial ischemia/reperfusion
injury. Pharmacol Res. 170:1057432021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Algoet M, Janssens S, Himmelreich U, Gsell
W, Pusovnik M, Van den Eynde J and Oosterlinck W: Myocardial
ischemia-reperfusion injury and the influence of inflammation.
Trends Cardiovasc Med. 33:357–366. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao WK, Zhou Y, Xu TT and Wu Q:
Ferroptosis: Opportunities and challenges in myocardial
ischemia-reperfusion injury. Oxid Med Cell Longev.
2021:99296872021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen CL, Zhang L, Jin Z, Kasumov T and
Chen YR: Mitochondrial redox regulation and myocardial
ischemia-reperfusion injury. Am J Physiol Cell Physiol.
322:C12–C23. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mokhtari-Zaer A, Marefati N, Atkin SL,
Butler AE and Sahebkar A: The protective role of curcumin in
myocardial ischemia-reperfusion injury. J Cell Physiol.
234:214–222. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ibáñez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sessler DI: Mild perioperative
hypothermia. N Engl J Med. 336:1730–1737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi H, Su Z, Su H, Chen H, Zhang Y and
Cheng Y: Mild hypothermia improves brain injury in rats with
intracerebral hemorrhage by inhibiting IRAK2/NF-κB signaling
pathway. Brain Behav. 11:e019472021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iaizzo PA, Kehler CH, Carr RJ, Sessler DI
and Belani KG: Prior hypothermia attenuates malignant hyperthermia
in susceptible swine. Anesth Analg. 82:803–809. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wass CT, Lanier WL, Hofer RE, Scheithauer
BW and Andrews AG: Temperature changes of > or=1 degree C alter
functional neurologic outcome and histopathology in a canine model
of complete cerebral ischemia. Anesthesiology. 83:325–335. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schleef M, Gonnot F, Pillot B, Leon C,
Chanon S, Vieille-Marchiset A, Rabeyrin M, Bidaux G,
Guebre-Egziabher F, Juillard L, et al: Mild Therapeutic hypothermia
protects from acute and chronic renal ischemia-reperfusion injury
in mice by mitigated mitochondrial dysfunction and modulation of
local and systemic inflammation. Int J Mol Sci. 23:92292022.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu X, Wen S, Zhao S, Yan F, Zhao S, Wu D
and Ji X: Mild therapeutic hypothermia protects the brain from
ischemia/reperfusion injury through upregulation of iASPP. Aging
Dis. 9:401–411. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiao Q, Ye Q, Wang W, Xiao J, Fu B, Xia Z,
Zhang X, Liu Z and Zeng X: Mild hypothermia pretreatment protects
against liver ischemia reperfusion injury via the PI3K/AKT/FOXO3a
pathway. Mol Med Rep. 16:7520–7526. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tissier R, Chenoune M, Ghaleh B, Cohen MV,
Downey JM and Berdeaux A: The small chill: Mild hypothermia for
cardioprotection? Cardiovasc Res. 88:406–414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kanemoto S, Matsubara M, Noma M, Leshnower
BG, Parish LM, Jackson BM, Hinmon R, Hamamoto H, Gorman JH III and
Gorman RC: Mild hypothermia to limit myocardial
ischemia-reperfusion injury: Importance of timing. Ann Thorac Surg.
87:157–163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mochizuki T, Yu S, Katoh T, Aoki K and
Sato S: Cardioprotective effect of therapeutic hypothermia at 34°C
against ischaemia/reperfusion injury mediated by PI3K and nitric
oxide in a rat isolated heart model. Resuscitation. 83:238–242.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao R, Zhao H, Wang X, Tang B, Cai Y,
Zhang X, Zong H, Li Y and Wang Y: Mild hypothermia therapy lowers
the inflammatory level and apoptosis rate of myocardial cells of
rats with myocardial ischemia-reperfusion injury via the NLRP3
inflammasome pathway. Comput Math Methods Med. 2021:64152752021.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harteneck C, Plant TD and Schultz G: From
worm to man: Three subfamilies of TRP channels. Trends Neurosci.
23:159–166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sah R, Mesirca P, Mason X, Gibson W,
Bates-Withers C, Van den Boogert M, Chaudhuri D, Pu WT, Mangoni ME
and Clapham DE: Timing of myocardial trpm7 deletion during
cardiogenesis variably disrupts adult ventricular function,
conduction, and repolarization. Circulation. 128:101–114. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao L, Wang Y, Sun N, Liu X, Li L and Shi
J: Electroacupuncture regulates TRPM7 expression through the
trkA/PI3K pathway after cerebral ischemia-reperfusion in rats. Life
Sci. 81:1211–1222. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aarts M, Iihara K, Wei WL, Xiong ZG,
Arundine M, Cerwinski W, MacDonald JF and Tymianski M: A key role
for TRPM7 channels in anoxic neuronal death. Cell. 115:863–877.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fatima G, Sharma VP, Das SK and Mahdi AA:
Oxidative stress and antioxidative parameters in patients with
spinal cord injury: Implications in the pathogenesis of disease.
Spinal Cord. 53:3–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Meng Z, Wang X, Yang Z and Xiang F:
Expression of transient receptor potential melastatin 7
up-regulated in the early stage of renal ischemia-reperfusion.
Transplant Proc. 44:1206–1210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou DM, Sun LL, Zhu J, Chen B, Li XQ and
Li WD: MiR-9 promotes angiogenesis of endothelial progenitor cell
to facilitate thrombi recanalization via targeting TRPM7 through
PI3K/Akt/autophagy pathway. J Cell Mol Med. 24:4624–4632. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang J, Hu S, Huang L, Zhou J, Xiang H,
Yang H, Cheng H and Tang Y: Protective effect of inhibiting TRPM7
expression on hypoxia post-treatment H9C2 cardiomyocytes. Clin
Hemorheol Microcirc. 77:91–105. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C
and Li B: Ferroptosis, a new form of cell death: Opportunities and
challenges in cancer. J Hematol Oncol. 12:342019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su LJ, Zhang JH, Gomez H, Murugan R, Hong
X, Xu D, Jiang F and Peng ZY: Reactive oxygen species-induced lipid
peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med
Cell Longev. 2019:50808432019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu X, Li Y, Zhang S and Zhou X:
Ferroptosis as a novel therapeutic target for cardiovascular
disease. Theranostics. 11:3052–3059. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Z, Tang J, Song J, Xie M, Liu Y,
Dong Z, Liu X, Li X, Zhang M, Chen Y, et al: Elabela alleviates
ferroptosis, myocardial remodeling, fibrosis and heart dysfunction
in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling.
Free Radic Biol Med. 181:130–142. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Chen X, Zhou W, Men H, Bao T, Sun
Y, Wang Q, Tan Y, Keller BB, Tong Q, et al: Ferroptosis is
essential for diabetic cardiomyopathy and is prevented by
sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 12:708–722.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun L, Wang H, Xu D, Yu S, Zhang L and Li
X: Lapatinib induces mitochondrial dysfunction to enhance oxidative
stress and ferroptosis in doxorubicin-induced cardiomyocytes via
inhibition of PI3K/AKT signaling pathway. Bioengineered. 13:48–60.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shi R, Fu Y, Zhao D, Boczek T, Wang W and
Guo F: Cell death modulation by transient receptor potential
melastatin channels TRPM2 and TRPM7 and their underlying molecular
mechanisms. Biochem Pharmacol. 190:1146642021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hickman DL: Euthanasia of neonatal rats
and mice using carbon monoxide. J Am Assoc Lab Anim Sci.
62:274–278. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Montaigne D, Marechal X, Modine T, Coisne
A, Mouton S, Fayad G, Ninni S, Klein C, Ortmans S, Seunes C, et al:
Daytime variation of perioperative myocardial injury in cardiac
surgery and its prevention by Rev-Erbα antagonism: a single-centre
propensity-matched cohort study and a randomised study. Lancet.
391:59–69. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lian ZX, Wang F, Fu JH, Chen ZY, Xin H and
Yao RY: ATP-induced cardioprotection against myocardial
ischemia/reperfusion injury is mediated through the RISK pathway.
Exp Ther Med. 12:2063–2068. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ma X, Godar RJ, Liu H and Diwan A:
Enhancing lysosome biogenesis attenuates BNIP3-induced
cardiomyocyte death. Autophagy. 8:297–309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Moon BF, Iyer SK, Hwuang E, Solomon MP,
Hall AT, Kumar R, Josselyn NJ, Higbee-Dempsey EM, Tsourkas A, Imai
A, et al: Iron imaging in myocardial infarction reperfusion injury.
Nat Commun. 11:32732020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Götberg M, Olivecrona GK, Koul S, Carlsson
M, Engblom H, Ugander M, van der Pals J, Algotsson L, Arheden H and
Erlinge D: A pilot study of rapid cooling by cold saline and
endovascular cooling before reperfusion in patients with
ST-elevation myocardial infarction. Circ Cardiovasc Interv.
3:400–407. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hamamoto H, Leshnower BG, Parish LM,
Sakamoto H, Kanemoto S, Hinmon R, Miyamoto S, Gorman JH III and
Gorman RC: Regional heterogeneity of myocardial reperfusion injury:
Effect of mild hypothermia. Ann Thorac Surg. 87:164–171. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jung KT, Bapat A, Kim YK, Hucker WJ and
Lee K: Therapeutic hypothermia for acute myocardial infarction: A
narrative review of evidence from animal and clinical studies.
Korean J Anesthesiol. 75:216–230. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qin Z, Shen S, Qu K, Nie Y and Zhang H:
Mild hypothermia in rat with acute myocardial ischaemia-reperfusion
injury complicating severe sepsis. J Cell Mol Med. 25:6448–6454.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Maynard C, Longstreth WT Jr, Nichol G,
Hallstrom A, Kudenchuk PJ, Rea T, Copass MK, Carlbom D, Deem S,
Olsufka M, et al: Effect of prehospital induction of mild
hypothermia on 3-month neurological status and 1-year survival
among adults with cardiac arrest: Long-term follow-up of a
randomized, clinical trial. J Am Heart Assoc. 4:e0016932015.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fuernau G, Beck J, Desch S, Eitel I, Jung
C, Erbs S, Mangner N, Lurz P, Fengler K, Jobs A, et al: Mild
Hypothermia in Cardiogenic Shock Complicating Myocardial
Infarction. Circulation. 139:448–457. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sánchez-Alegría K, Flores-León M,
Avila-Muñoz E, Rodríguez-Corona N and Arias C: PI3K signaling in
neurons: A central node for the control of multiple functions. Int
J Mol Sci. 19:37252018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Di-Luoffo M, Ben-Meriem Z, Lefebvre P,
Delarue M and Guillermet-Guibert J: PI3K functions as a hub in
mechanotransduction. Trends Biochem Sci. 46:878–888. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ghigo A and Li M: Phosphoinositide
3-kinase: Friend and foe in cardiovascular disease. Front
Pharmacol. 6:1692015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yu D, Xiong J, Gao Y, Li J, Zhu D, Shen X,
Sun L and Wang X: Resveratrol activates PI3K/AKT to reduce
myocardial cell apoptosis and mitochondrial oxidative damage caused
by myocardial ischemia/reperfusion injury. Acta Histochemica.
123:1517392021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shen YC, Shen YJ, Lee WS, Chen MYC, Tu WC
and Yang KT: Two benzene rings with a boron atom comprise the core
structure of 2-APB responsible for the anti-oxidative and
protective effect on the ischemia/reperfusion-induced rat heart
injury. Antioxidants (Basel). 10:16672021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Deng W, Ren G, Luo J, Gao S, Huang W, Liu
W and Ye S: TRPM7 mediates endoplasmic reticulum stress and
ferroptosis in sepsis-induced myocardial injury. J Bioenerg
Biomembr. 55:207–217. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yu P, Zhang J, Ding Y, Chen D, Sun H, Yuan
F, Li S, Li X, Yang P, Fu L, et al: Dexmedetomidine
post-conditioning alleviates myocardial ischemia-reperfusion injury
in rats by ferroptosis inhibition via SLC7A11/GPX4 axis activation.
Hum Cell. 35:836–848. 2022. View Article : Google Scholar : PubMed/NCBI
|