Introduction
Hepatocellular carcinoma (HCC) is one of the most
malignant tumors worldwide and Hepatitis B virus (HBV) has been
identified as the most important risk factor for developing HCC
(1–3). Of the four proteins translated by HBV,
the X-gene product (HBx) has been most closely associated with the
HCC pathogenesis (4). The
correlation between HBx and HCC development has been extensively
studied and the oncogenic roles of HBx include the following:
activation of a variety of transcription factors such as nuclear
factor-κB (NF-κB) (5), activator
protein 1 (AP-1) (6),
cAMP-responsive element binding protein/activating transcription
factor 2 (CREB/ATF-2) (7);
interaction with cellular oncogenes, such as Ras (8) and Src (9); regulation of cell apoptosis and the
cell cycle by interacting with caspases, CDK, CKI, and survivin
(10,11); and stimulation of cell signaling
pathways, such as the Wnt (12),
the Ras/MAPK (13), and the
PI3K-Akt/PKB pathway (14).
Notch signaling is a highly evolutionarily conserved
pathway which plays a pivotal role in regulating the development of
organs and tissues by affecting cell proliferation,
differentiation, apoptosis and stem cell maintenance (15). In mammals, the Notch family consists
of four transmembrane receptors (Notch-1-Notch-4) and five ligands
(Jagged-1, Jagged-2, Dll-1, Dll-3 and Dll-4) (16). Upon receptor-ligand binding, the
Notch receptor is proteolytically cleaved by a γ-secretase complex
(17), which results in releasing
of the Notch intracellular domain (NICD) (18). Then NICD translocates into the
nucleus and bands to the transcriptional factors known as CSL
(CBF-1/suppressor of hairless/Lag-1), leading to the
transcriptional activation of Notch target genes, including basic
helix-loop-helix (bHLH) proteins such as HES-1 and HES-5 (19,20).
Mounting evidence shows that perturbation of Notch signaling often
leads to tumorigenesis (21–24).
Some studies have shown the potential roles of Notch signaling in
the development of HCC (25,26).
However, up to now, only a few reports about the
role of Notch signaling in HBx-related HCC. In this study, we
investigated the relationship of the Notch pathway with HBx in
HepG2 cells, and found that HBx can enhance the progression of HCC
via the activation of Notch pathway, which may provide some new
clues for the potential role of Notch signaling in HBx-associated
liver cancer.
Materials and methods
Cell culture
The human hepatoma cell line HepG2 were obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). The HepG2/HBx and HepG2/pcDNA3.1 cell lines, were derived
from HepG2 cells by transfecting with HBx expression plasmid or an
empty plasmid (pcDNA3.1(+)/V5-HisB), respectively. Both cell lines
have been successfully established (27). All cell lines were cultured in DMEM
(Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum
(FBS; Gibco, Grand Island, NY, USA) and maintained in humidified
incubator at 37°C in a 5% CO2 atmosphere.
DAPT treatment
The γ-secretase inhibitor
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl
ester (DAPT) was purchased from the Sigma-Aldrich Company (St.
Louis, MO, USA). DAPT was dissolved in 100% dimethylsulphoxide
(DMSO, Sigma) to make a stock solution of 10 mM, which was then
diluted in culture medium to obtain the desired concentrations of
1, 5, 10 and 20 μM. DMSO diluted in culture medium at the final
concentration of 0.05% without DAPT was designated as 0 μM.
Untreated cells were those incubated in the culture medium without
any additives. Cells treated with or without DAPT were cultured for
48 h, after which the total RNA or protein was extracted and flow
cytometry was carried out.
Cell proliferation and viability
assays
Cell proliferation assays were performed by using a
Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) according to the
manufacturer’s instructions. Briefly, 1×104 cells/well
were plated in 96-well plates and cultured with growth medium. At
the indicated time points, the medium was aspirated. Then 100 μl
serum-free DMEM and 10 μl WST-8
[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H
tetrazolium, monosodium salt] were added to each well followed by
incubation at 37°C for 1.5 h. Absorbance was measured at 450 nm
with a reference wavelength of 630 nm on a spectrophotometer
(Molecular Devices, Sunnyvale, CA). To evaluate the viability of
HepG2/HBx cells, 1×104 cells/well were plated in 96-well
plates followed 12 h later by the addition of DAPT at
concentrations of 0, 1, 5, 10 or 20 μM. The cell viability was
assessed as the percent of viable cells relative to the untreated
control cells, which was determined for each concentration using
the following equation: % viability =
ODexperiment/ODcontrol × 100%. Control cells
were considered as 100% viable. All experiments were repeated five
times.
Cell cycle and apoptosis analysis by flow
cytometry
After treatment with or without DAPT for 48 h, cells
were harvested, immediately fixed in 75% ethanol at 4°C overnight,
treated with 50 mg/l RNAse A (Sigma) for 30 min at 37°C, and
stained with 50 mg/l PI (Sigma) for 10 min. Samples were then
analyzed for their DNA content by a FACSAria Cell Cytometer (BD
Biosciences, San Jose, CA, USA). The data were analyzed with the
CellQuest software (BD Biosciences). Apoptosis analysis was
performed by using a Annexin-V-FITC kit (Bender MedSystems,
Burlingame, CA, USA) according to the manufacturer’s instructions.
The percentage of cells that were Annexin-V positive but PI
negative was compared among the different treatment groups.
Immunofluorescence assays
Immunofluorescence assays were performed as
previously described (28).
HepG2/HBx cells were cultured on glass coverslips for 24 h and
fixed with 4% paraformaldehyde. The fixed cells were incubated with
anti-HBx (1:200, Santa Cruz Biotechnology, Santa Cruz, CA) and
anti-NICD (1:200, Santa Cruz Biotechnology) for 12 h at 4°C, then
incubated with Cy3-conjugated goat anti-mouse IgG (1:100, Boster,
China) and FITC-conjugated goat anti-rabbit IgG (1:100, Boster) for
1 h. Nuclei were stained with diamidinophenyl indole (DAPI)
(Boster). The stained cells were observed using a Fluoview FV1000
laser scanning confocal microscope (Olympus, Japan).
Co-immunoprecipitation assays
Co-immunoprecipitation assays were performed as
previously described (28).
HepG2/HBx cells were lysed, and the lysates were pretreated with
protein G-agarose (Santa Cruz Biotechnology) to remove
non-specifically bound proteins. After centrifugation, one third of
the supernatants were stored at −80°C as a positive control. The
remaining supernatant samples were incubated for 2 h at 4°C with 1
μg of non-immune mouse IgG or mouse anti-HBx (Santa Cruz
Biotechnology), for the negative control and the experimental
group, respectively. Then, the mixture was incubated for 1 h to
overnight at 4°C with 20 μl protein G-agarose (Santa Cruz
Biotechnology). The immunocomplexes were extensively washed with
PBS, samples were boiled in electrophoresis sample buffer and
assayed by Western blot analysis.
Real-time PCR analysis
Total RNA was isolated from cultured cells using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and cDNA was
synthesized from 2 μg of total RNA using Moloney murine leukemia
virus reverse transcriptase (MMLV) (Promega, Madison, WI, USA).
Real-time quantitative PCR (qRT-PCR) using SYBR Premix (DRR041A,
Takara, Japan) was performed as previously described (29). Amplifications were performed in a
LightCycler machine (Roche Diagnostics, Basel, Switzerland)
following the manufacturer’s instructions. The HepG2 cDNA was used
as standard cDNA. A standard curve for each gene was generated from
serially diluted standards, and values for unknown samples were
extrapolated. β-actin was used as an internal control to normalize
samples. All standards and samples were assayed in triplicate. The
primer sequences used to amplify specific target genes are listed
in Table I.
 | Table IPrimer sequences for real-time
polymerase chain reaction analysis. |
Table I
Primer sequences for real-time
polymerase chain reaction analysis.
| Gene | Primer sequence | PCR product size
(bp) | GenBank accession
no. |
|---|
| Jagged-1 | F:
5′-CAACACGGTCCCCATCAAG-3′ | | |
| R:
5′-TACTTCAGAATTGTGTGTCCTTATTTTAGA-3′ | 76 | NM_000214.2 |
| Notch-1 | F:
5′-CCGCAGTTGTGCTCCTGAA-3′ | | |
| R:
5′-ACCTTGGCGGTCTCGTAGCT-3′ | 109 | NM_017617.3 |
| Hes-1 | F:
5′-GCTAAGGTGTTTGGAGGCT-3′ | | |
| R:
5′-CCGCTGTTGCTGGTGTA-3′ | 122 | NM_005524.2 |
| β-actin | F:
5′-GTTGCGTTACACCCTTTCTTG-3′ | | |
| R:
5′-GACTGCTGTCACCTTCACCGT-3′ | 157 | NM_001101.3 |
Western blot analysis
Cells were lysed as previously described (29) and the lysates were subjected to
electrophoresis on SDS-PAGE and transferred to PVDF membranes
(Millipore, Billerica, MA). The blotted membranes were blocked and
subsequently incubated with rabbit anti-Jagged-1 (1:600), rabbit
anti-Notch-1 (1:1,000), rabbit anti-Notch-1-IC (1:1,000), rabbit
anti-Hes-1 (1:600), and rabbit anti-actin (1:1,000). All antibodies
were from Santa Cruz Biotechnology except for anti-Notch-1-IC which
was obtained from Cell Signaling Technology (Danvers, MA). After
incubation with horseradish peroxidase-labeled secondary antibody
(1:5,000–10,000; Santa Cruz Biotechnology), visualization was
performed by an enhanced chemiluminescence kit (Pierce, Rockford,
IL) and exposure to X-ray film (Kodak, Rochester, NY).
Immunoblotting with the anti-actin antibody was used as an internal
control to confirm equivalent protein loading. Each experiment was
performed at least 3 times. The relative intensity of each protein
band was scanned by Quantity One software (Bio-Rad Laboratories,
Hercules, CA, USA).
Statistics
SPSS version 17.0 software (SPSS for Windows, Inc.,
Chicago, IL, USA) was used for all statistical analyses. All
results are expressed as mean ± SEM. Statistical analysis of the
data was performed using a standard one-way ANOVA or one-way ANOVA
for repeated measures, followed by the least significant difference
(LSD) post-hoc test. Bonferroni’s correction was used to adjust for
multiple comparisons. A 2-tailed Student’s paired t-test was also
used to compare the differences in the values between two groups. A
P-value <0.05 was considered to be statistically
significant.
Results
The Notch signaling pathway is activated
in HepG2/HBx cells
Previously, we observed that the HBx protein
stimulated the proliferation as well as the cell cycle, inhibited
the apoptosis of HepG2 cells significantly, and promoted tumour
growth in nude mice (27). To
investigate the potential involvement of Notch signaling in the
development of HCC, we performed western blot analyses in HepG2,
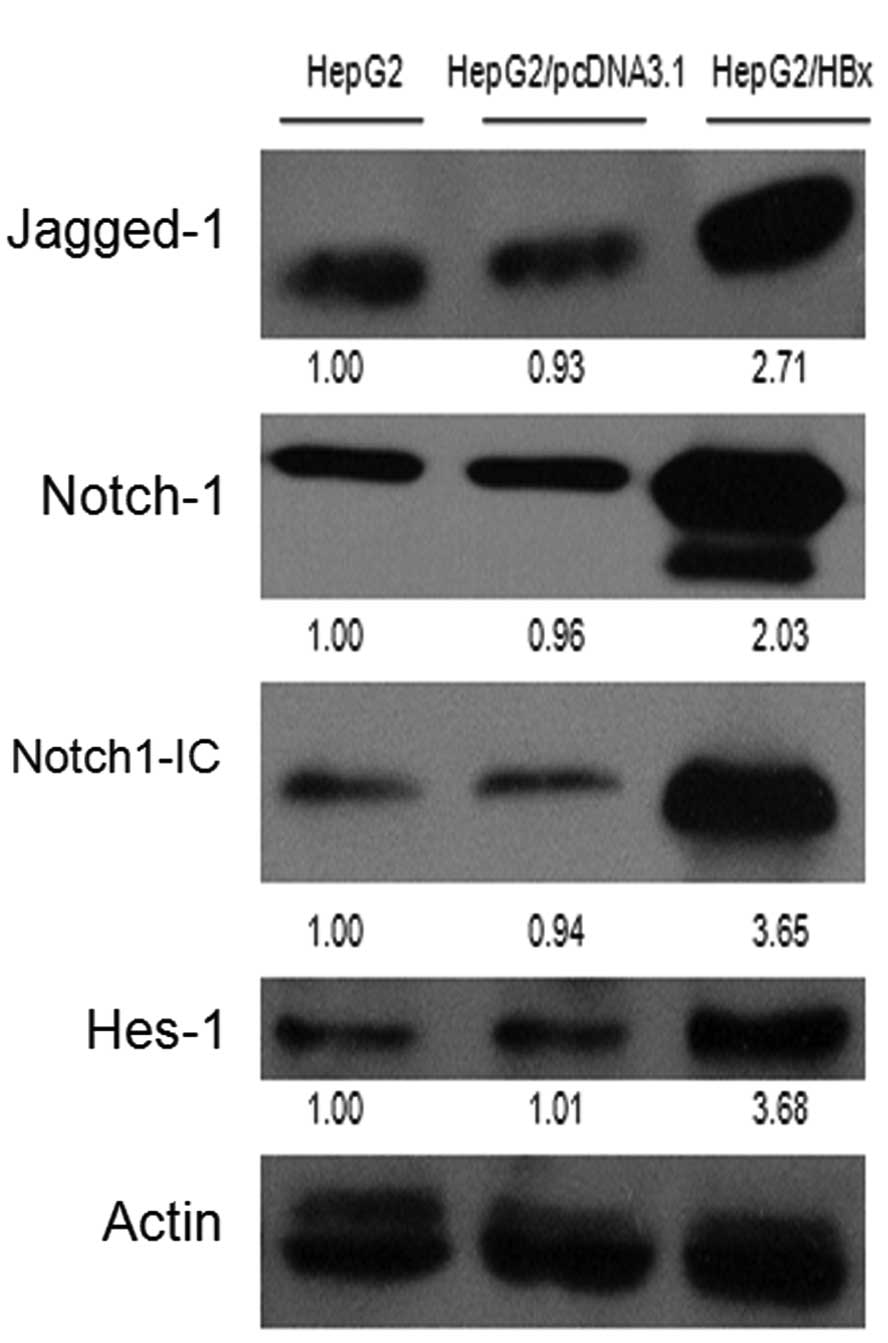
HepG2/pcDNA3.1 and HepG2/HBx cells. As shown in Fig. 1A, the protein levels of Jagged-1,
Notch-1, Notch-1C and Hes-1 in HepG2/HBx cells were elevated
relative to the controls. Then, we analyzed the mRNA levels of
Jagged-1, Notch-1 and Hes-1 using quantitative real-time RT-PCR
(qRT-PCR) and found that their mRNA levels were significantly
increased in HepG2/HBx cells as compared with the controls
(Fig. 1B).
Notch-1 colocalizes and interacts with
HBx in HepG2/HBx cells
To study the possible relationship between Notch-1
and HBx, we performed immunofluorescence and co-immunoprecipitation
assays. As shown in Fig. 1C, the
nuclei were stained blue (i), HBx was stained red (ii) whereas the
NICD was stained green (iii) in HepG2/HBx cells. Yellow staining in
the dual-labeling experiments indicates overlapping areas of red
and green fluorescent labels (iv), suggesting co-localization of
NICD with HBx in the HepG2/HBx cells. The compounds
immunoprecipitated with anti-HBx or non-immune mouse IgG from
HepG2/HBx cells were subjected to western blot analysis with
anti-NICD and anti-Jagged-1, respectively. As shown in Fig. 1D, NICD was co-immunoprecipitated
with HBx. No specific interaction was found between Jagged-1 and
HBx or the protein immunoprecipitated by non-immune IgG, indicating
the specificity of the NICD-HBx interaction.
Inhibition of Notch signaling attenuates
the growth of HepG2/HBx cells
In order to further establish that active Notch
signaling is important for HBx to function as an oncoprotein, we
treated HepG2/HBx cells with various concentrations of DAPT for 12
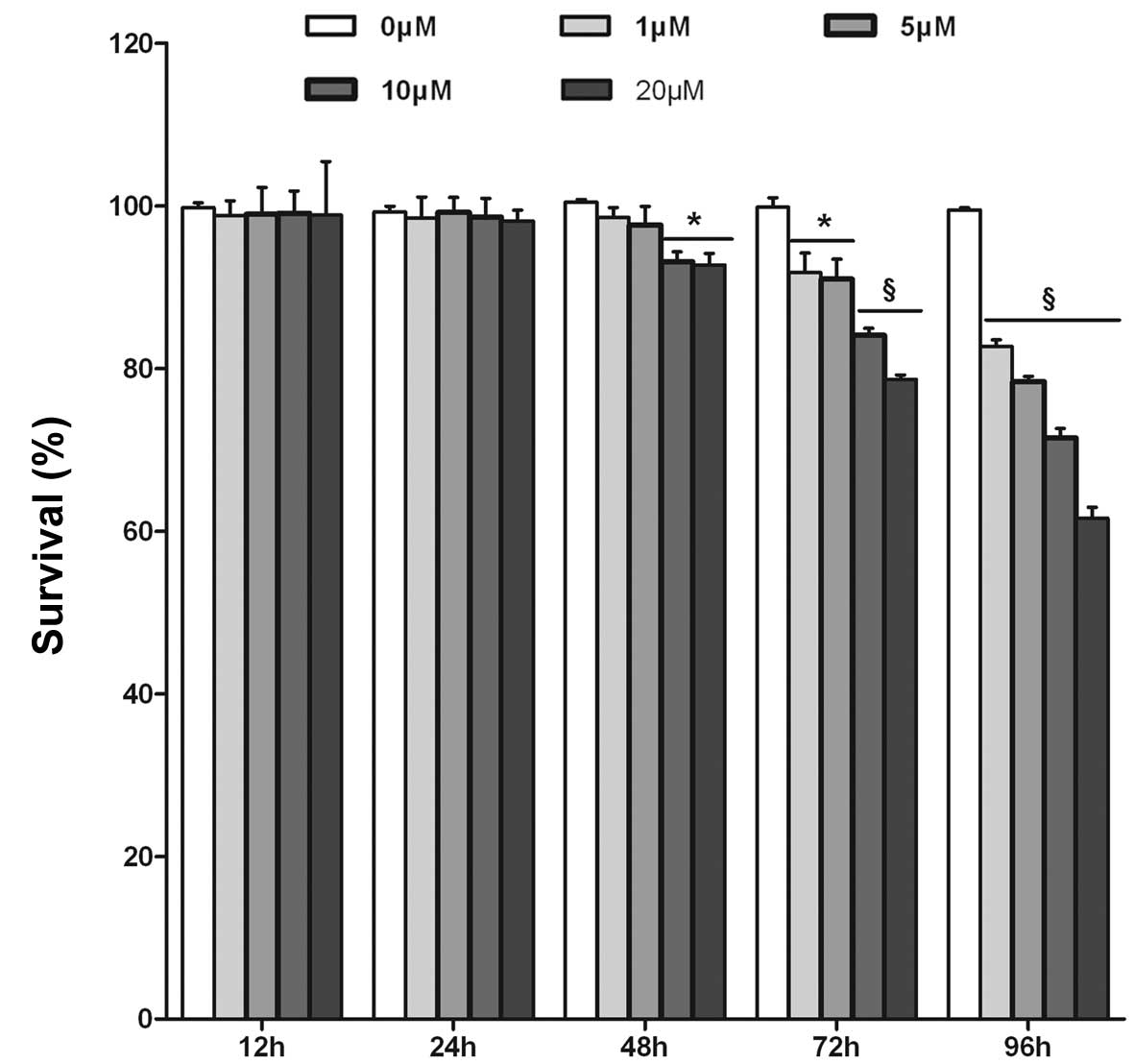
to 96 h and assessed cell proliferation by the WST-8 assay. As
shown in Fig. 2A, in HepG2/HBx
cells, increasing concentrations and durations of treatment with
DAPT resulted in a decrease of cell viability. In contrast,
treatment with various concentrations of DAPT for 12 or 24 h did
not produce any significant reduction in cell viability. However,
treatment of DAPT for >48 h resulted in significant dose- and
time-dependent reduction in cell viability of HepG2/HBx cells. A
significant reduction in cell viability by DAPT treatment was
observed at concentrations of 10 and 20 μM after 48 h, with
inhibition rates of 7.30 and 7.73%, respectively (P<0.05,
Fig. 2A). We, therefore, selected
the DAPT treatment time point of 48 h for further studies.
Confirmation of the inhibition of Notch
signaling in HepG2/HBx cells
HepG2/HBx cells treated with various concentrations
of DAPT for 48 h and assessed for the DAPT inhibitory effects on
Notch-1 signaling by western blot analysis of Jagged-1, Notch-1,
Notch-1-IC, Hes-1 protein levels, and by qRT-PCR of Hes-1
transcripts. As shown in Fig. 2B and
C, DAPT treatment greatly reduced the amount of Notch-1-IC and
Hes-1 protein in a dose-dependent manner, while there was no
significant effect on Jagged-1 and Notch-1 levels (Fig. 2B). A significant reduction of
Notch-1-IC and Hes-1 protein levels were observed at 10 and 20 μM
of DAPT treatment. qRT-PCR experiments showed that the
downregulation of Hes-1 transcripts in HepG2/HBx cells occurred
after treatment with DAPT at 10 (P<0.05) and 20 μM (P<0.05)
(Fig. 2C). These data were
consistent with the previous results that treatment with 10 and 20
μM DAPT for 48 h significantly inhibited the proliferation of
HepG2/HBx cells.
Inhibition of Notch signaling arrests
G0/G1 phase, shortens the S phase and induces apoptosis in
HepG2/HBx cells
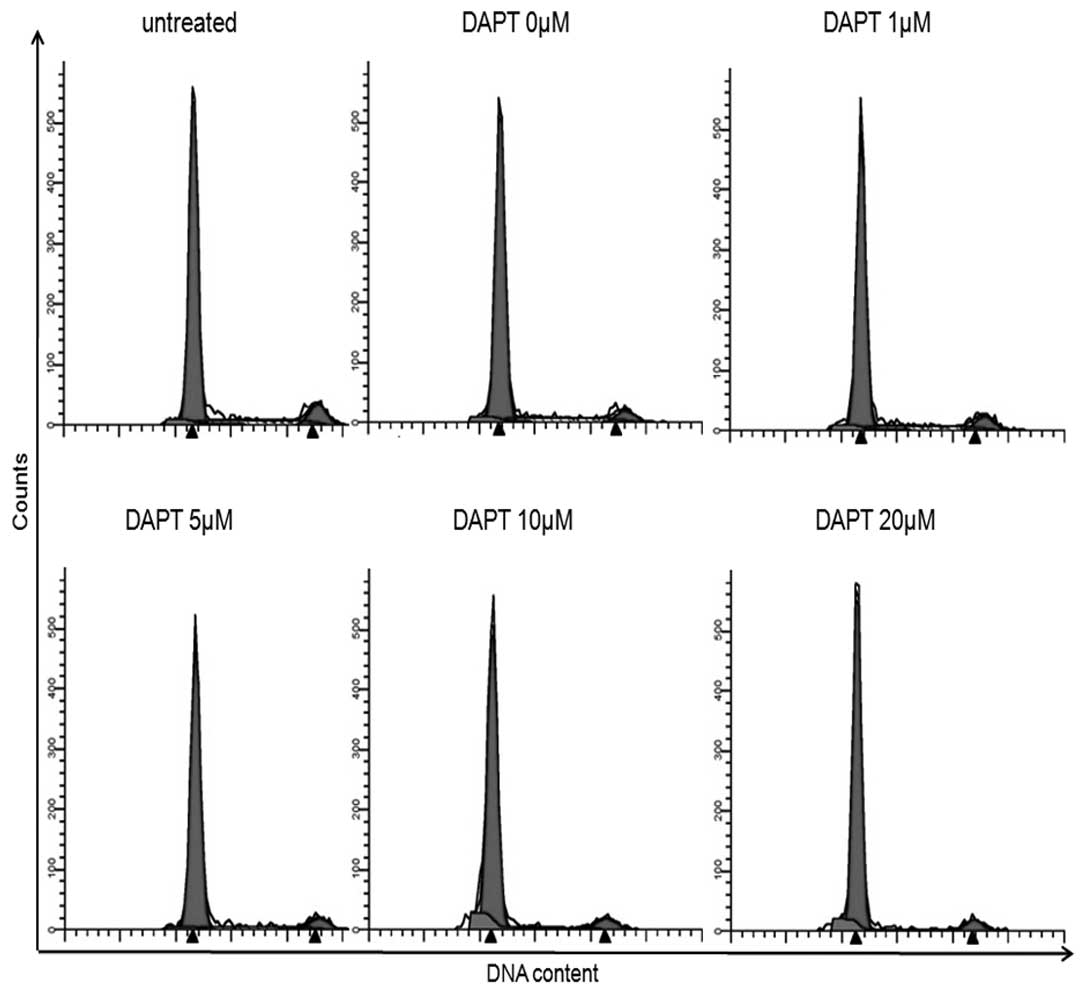
We performed PI staining and flow cytometry to
define the cell-cycle distribution of DAPT-treated HepG2/HBx cells.
Treatment of HepG2/HBx cells with 0–20 M of DAPT for 48 h resulted
in arrest in the G0/G1 phase and shortening of the S phase. The
proportion of HepG2/HBx cells in the G0/G1 phase was significantly
increased at concentrations of 10 (88.99%, P<0.05) and 20 μM
(90.95%, P<0.05), compared to that of 80.23% in untreated
control cells (Fig. 3A and B). The
proportion of cells in S phase was significantly decreased at
concentrations of 10 (4.60%, P<0.05) and 20 μM (3.93%,
P<0.05), relative to that of 12.55% in untreated control cells
(Fig. 3A and B). No significant
difference was observed between the G2/M phases. As shown in
Fig. 3C and D, the apoptotic rates
were increased from 5.45% in non-DAPT-treated cells to 13.28% at a
concentration of 10 μM (P<0.01, Fig.
3C and D) and to (20.22%) at 20 μM (P<0.001, Fig. 3C and D) after treatment with DAPT
for 48 h. Taken together, these results indicate that the
inhibition of Notch signaling was associated with decreased DNA
synthesis (S phase) and increased apoptosis, which contributed to
the impaired growth of HepG2/HBx cells treated with DAPT.
HepG2 cells are not significantly
affected by Notch inhibition
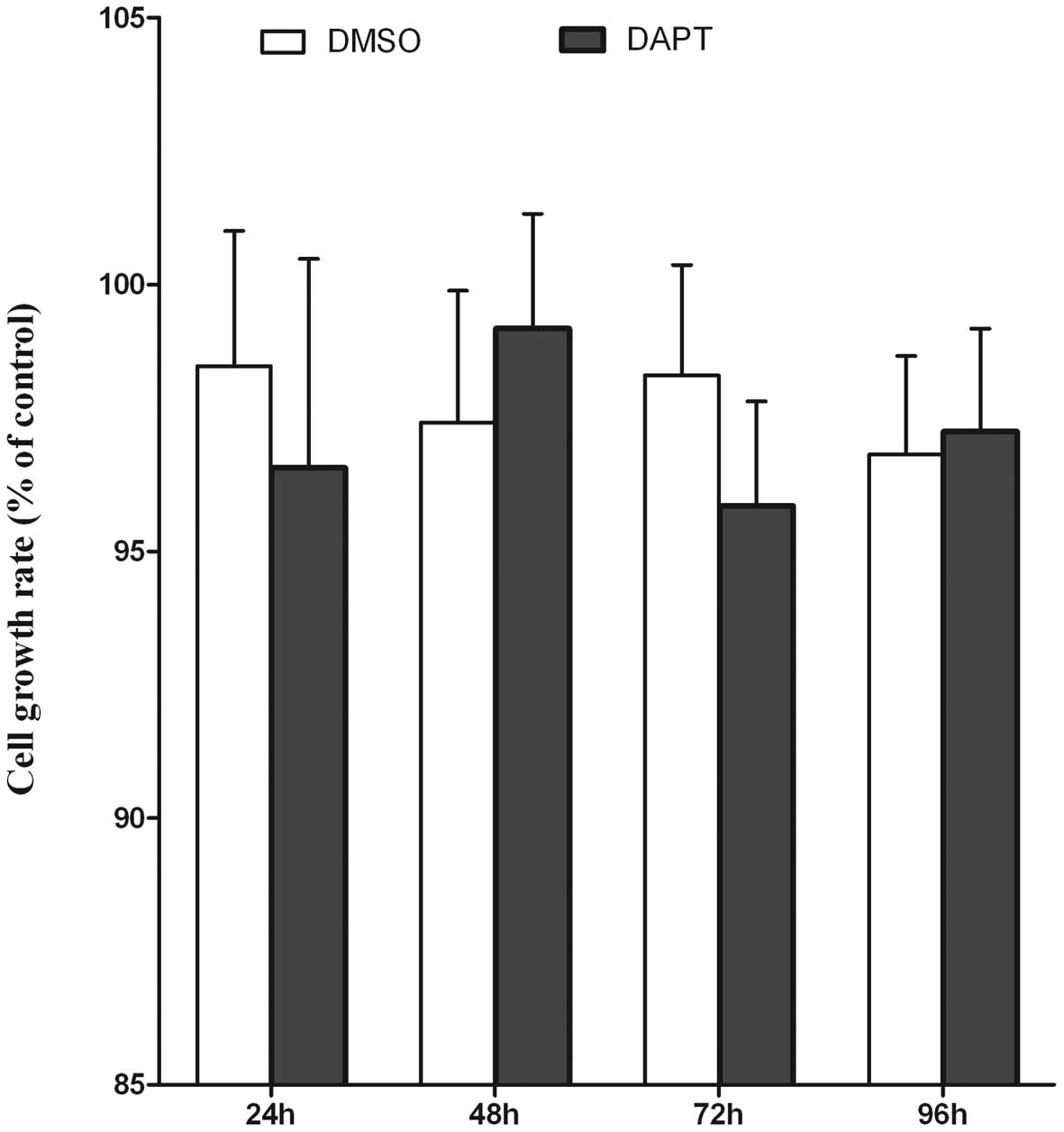
As a control, Notch signaling was blocked in HepG2
cells using DAPT as well. HepG2 cells were treated with DAPT at the
final concentration of 10 μM or with 0.05% DMSO. The proliferation
rate of HepG2 cells was measured after DAPT treatment using WST-8
assay. No effect on cell proliferation could be observed in HepG2
cells upon ablation of Notch signaling (Fig. 4A). The protein expression level of
Hes-1 was monitored using western blot analysis. As shown in
Fig. 4B, treatment of HepG2 cells
with DAPT led to a distinct Hes-1 downregulation after 48 h. These
results demonstrated that Notch signaling was much less important
for the growth of HepG2 cells than for that of HepG2/HBx cells.
Discussion
We previously reported that HBx protein stimulated
the proliferation as well as cell cycle, inhibited the apoptosis of
HepG2 cells significantly, and promoted tumor growth in nude mice
(27). However, little is known
about the mechanism of HBx action. A large number of studies have
demonstrated that Notch signaling plays a crucial role in various
malignant tumors (21–24). Expression and localization of Notch
receptors and their ligands have been observed in the normal human
liver tissue and the deregulated Notch signaling had been found in
malignant liver tumors (25,26).
To examine whether Notch signaling was involved in
the HBx-related HCC, we investigated the relationship between HBx
and Notch signaling in HepG2 cells after being transfected with the
HBx gene. It was found that HBx protein upregulated the expression
of Notch-1, Jagged-1 and Hes-1 at the transcriptional level, which
is related with the stimulated growth of HepG2 cells by HBx in
vitro and in vivo. However, when Notch signaling was
blocked with a γ-secretase inhibitor DAPT, the inhibited Notch
signaling decreased the proliferation, arrested the G0/G1 phase and
shortened the S phase of the cell cycle and induced apoptosis in
HepG2/HBx cells. It seemed that when Notch signaling was blocked,
the abnormality caused by HBx in HepG2 cells was partially
reversed. However, the inhibition of Notch signaling in
untransfected HepG2 cells had barely any effect on their growth.
Moreover, we found that HBx colocalized and interacted with NICD
(Notch intracellular domain) in HepG2/HBx cells. These findings
demonstrated that HBx can activate Notch signaling by binding to
NICD, which may contribute to the stimulated growth of HepG2 cells
both in vitro and in vivo. Considering that the HepG2
cell line was originated histologically from human hepatocellular
carcinoma, our studies suggest that HBx can promote the progression
of HCC via the activated Notch pathway.
In summary, our results demonstrate that HBx can act
as an oncogenic factor to promote the progression of HCC by binding
to NICD to activate the Notch signaling pathway, which may provide
a new clue for the potential role of Notch signaling in the
HBx-associated human liver carcinoma.
Acknowledgements
This study was supported by the National Science
Foundation of China, no. 30570821 and no. 30971352.
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
HBV
|
hepatitis B virus
|
|
HBx
|
HBV X protein
|
|
Notch-IC or NICD
|
Notch intracellular domain
|
|
CSL
|
mammalian CBF1, Drosophila Su(H),
C. elegans LAG-1
|
|
DAPT
|
γ-secretase inhibitor
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl
ester
|
|
qRT-PCR
|
quantitative real-time RT-PCR
|
References
|
1
|
El-Serag HB: Hepatocellular carcinoma:
recent trends in the United States. Gastroenterology. 127:S27–S34.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar
|
|
3
|
Shibuya K, Mathers CD, Boschi-Pinto C,
Lopez AD and Murray CJ: Global and regional estimates of cancer
mortality and incidence by site: II. Results for the global burden
of disease 2000. BMC Cancer. 2:372002. View Article : Google Scholar
|
|
4
|
Koike K: Hepatitis B virus HBx gene and
hepatocarcinogenesis. Intervirology. 38:134–142. 1995.PubMed/NCBI
|
|
5
|
Lucito R and Schneider RJ: Hepatitis B
virus X protein activates transcription factor NF-kappa B without a
requirement for protein kinase C. J Virol. 66:983–991.
1992.PubMed/NCBI
|
|
6
|
Kekule AS, Lauer U, Weiss L, Luber B and
Hofschneider PH: Hepatitis B virus transactivator HBx uses a tumour
promoter signalling pathway. Nature. 361:742–745. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maguire HF, Hoeffler JP and Siddiqui A:
HBV X protein alters the DNA binding specificity of CREB and ATF-2
by protein-protein interactions. Science. 252:842–844. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Doria M, Klein N, Lucito R and Schneider
RJ: The hepatitis B virus HBx protein is a dual specificity
cytoplasmic activator of Ras and nuclear activator of transcription
factors. EMBO J. 14:4747–4757. 1995.PubMed/NCBI
|
|
9
|
Klein NP and Schneider RJ: Activation of
Src family kinases by hepatitis B virus HBx protein and coupled
signaling to Ras. Mol Cell Biol. 17:6427–6436. 1997.PubMed/NCBI
|
|
10
|
Gottlob K, Fulco M, Levrero M and
Graessmann A: The hepatitis B virus HBx protein inhibits caspase 3
activity. J Biol Chem. 273:33347–33353. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Benn J and Schneider RJ: Hepatitis B virus
HBx protein deregulates cell cycle checkpoint controls. Proc Natl
Acad Sci USA. 92:11215–11219. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ding Q, Xia W, Liu JC, et al: Erk
associates with and primes GSK-3beta for its inactivation resulting
in upregulation of beta-catenin. Mol Cell. 19:159–170. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Benn J and Schneider RJ: Hepatitis B virus
HBx protein activates Ras-GTP complex formation and establishes a
Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci USA.
91:10350–10354. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee YI, Kang-Park S and Do SI: The
hepatitis B virus-X protein activates a phosphatidylinositol
3-kinase-dependent survival signaling cascade. J Biol Chem.
276:16969–16977. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miele L: Notch signaling. Clin Cancer Res.
12:1074–1079. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kopan R and Ilagan MX: Gamma-secretase:
proteasome of the membrane? Nat Rev Mol Cell Biol. 5:499–504. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maillard I, Adler SH and Pear WS: Notch
and the immune system. Immunity. 19:781–791. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jarriault S, Brou C, Logeat F, Schroeter
EH, Kopan R and Israel A: Signalling downstream of activated
mammalian Notch. Nature. 377:355–358. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ohtsuka T, Sakamoto M, Guillemot F and
Kageyama R: Roles of the basic helix-loop-helix genes Hes-1 and
Hes5 in expansion of neural stem cells of the developing brain. J
Biol Chem. 276:30467–30474. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miele L, Golde T and Osborne B: Notch
signaling in cancer. Curr Mol Med. 6:905–918. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yanagawa S, Lee JS, Kakimi K, Matsuda Y,
Honjo T and Ishimoto A: Identification of Notch-1 as a frequent
target for provirus insertional mutagenesis in T-cell lymphomas
induced by leukemogenic mutants of mouse mammary tumor virus. J
Virol. 74:9786–9791. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ellisen LW, Bird J, West DC, et al: TAN-1,
the human homolog of the Drosophila notch gene, is broken by
chromosomal translocations in T lymphoblastic neoplasms. Cell.
66:649–661. 1991.PubMed/NCBI
|
|
24
|
Sjolund J, Manetopoulos C, Stockhausen MT
and Axelson H: The Notch pathway in cancer: differentiation gone
awry. Eur J Cancer. 41:2620–2629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nijjar SS, Crosby HA, Wallace L, Hubscher
SG and Strain AJ: Notch receptor expression in adult human liver: a
possible role in bile duct formation and hepatic
neovascularization. Hepatology. 34:1184–1192. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nijjar SS, Wallace L, Crosby HA, Hubscher
SG and Strain AJ: Altered Notch ligand expression in human liver
disease: further evidence for a role of the Notch signaling pathway
in hepatic neovascularization and biliary ductular defects. Am J
Pathol. 160:1695–1703. 2002. View Article : Google Scholar
|
|
27
|
Cheng B, Guo X, Zheng Y, Wang Y, Liu C and
Li P: The effects of HBx gene on the expression of DNA repair
enzymes hOGG1 and hMYHalpha mRNA in HepG2 cells. J Huazhong Univ
Sci Technolog Med Sci. 29:187–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao J, Chen C, Hong L, et al: Expression
of Jagged-1 and its association with hepatitis B virus X protein in
hepatocellular carcinoma. Biochem Biophys Res Commun. 356:341–347.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Balint K, Xiao M, Pinnix CC, et al:
Activation of Notch-1 signaling is required for
beta-catenin-mediated human primary melanoma progression. J Clin
Invest. 115:3166–3176. 2005. View
Article : Google Scholar : PubMed/NCBI
|