Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most
lethal human malignancy worldwide and has a very low
5-year-survival rate (approximately 6%). In the US alone,
approximately 40,000 patients succumbed to the disease in 2011
(1). The majority of PDAC patients
are diagnosed at advanced stages, since the disease does not cause
specific symptoms during the early stages. To date, surgery is the
primary treatment for pancreatic cancer. By contrast, chemotherapy
is only used for patients who are not suitable for surgical
resection with curative intent. A frequently used chemotherapeutic
drug is gemcitabine, which can effectively improve quality of life
and increase patient survival. Nevertheless, the high rate of
resistance to gemcitabine contributes to the poor prognosis of
pancreatic cancer (2,3). Thus, the development of novel
therapeutic strategies is mandatory.
The development of pancreatic cancer, like most
other cancer types of cancer, involves the activation of oncogenes
and the inactivation of tumor suppressor genes, dysregulating
signaling proteins critical to cell growth. Thus, the effective
control of pancreatic cancer cell growth or the induction of
apoptosis should target multiple gene pathways. Otherwise, drug
resistance will eventually develop. The molecular mechanisms
responsible for the resistance of PDAC patients to gemcitabine may
be due to: i) An altered tumor microenvironment, such as a dense
desmoplastic stroma, preventing gemcitabine infusion to the tumor
parenchyma. A previous study demonstrated that tumors from
gemcitabine-resistant patients had increased stimulation of
stroma-related gene pathways (4).
ii) The key molecular pathway responsible for gemcitabine
metabolism is altered; thus, the activation of gemcitabine is
anomalously inhibited or the excretion of gemcitabine is
accelerated. A previous study demonstrated that the inactivation
and downregulation of deoxycytidine kinase, a key enzyme in
gemcitabine activation, plays a significant role in acquiring
gemcitabine resistance (5).
Moreover, transmembrane xenobiotic transporters, such as
ATP-binding cassette sub-family G member 2 (ABCG-2), play a role in
gemcitabine resistance (6). iii)
The activation of aberrant proliferative and apoptotic pathways may
occur; thus, drug-resistant PDAC cells have a survival advantage
against chemotherapy (7,8).
In order to overcome drug resistance, research is
focusing on microRNAs (miRNAs or miRs). miRNAs are a family of
naturally occurring, non-coding RNA molecules, 19 to 25 nucleotides
in length. miRNAs post-transcriptionally regulate the expression of
target genes (9,10). Thus, miRNAs participate in a wide
range of biological processes, such as embryonic development, organ
formation and cell proliferation and apoptosis (11). miRNAs also play an important role in
tumorigenesis and chemosensitivity (12–14).
miR-181b has been found to be downregulated in glioblastoma and has
been widely studied in a variety of human cancers. For example, in
urothelial carcinoma (15), thyroid
papillary carcinoma (16), acute
lymphocytic leukemia (17), chronic
lymphocytic leukemia (CLL) (18),
colorectal cancer (19), breast
cancer (20), prostate cancer
(21), retinoblastoma (22) and PDAC (23), miRNA-181b is upregulated. Moreover,
its expression is reduced in glioblastoma (24) and gastric cancer (25). The loss of miRNA-181b expression may
contribute to the resistance of leukemia cells to chemotherapy
(18). miRNA-181b is expressed
during the stable stages of CLL; however, a reduction in miRNA-181b
expression has been associated with disease progression and drug
refraction (18).
Other studies have shown that miRNA-181b
significantly enhances drug sensitivity and its underexpression has
been associated with a shorter treatment-free survival in CLL cells
(26). However, another study
reported the downregulation of miRNA-181b in
chemotherapy-responsive acute promyelocytic leukemia (27). miRNA-181b expression is increased in
chemoresistant hepatocellular carcinoma (28), colorectal cancer (29) and breast cancer (30), as well as in drug-refractory gastric
and lung cancers (31). By
targeting different genes, miRNA-181b may play a complicated role
in chemotherapy-resistance, depending on the tumor type and
anti-neoplastic agent (12). In
this study, we investigated whether miR-181b is associated with the
sensitivity of PDAC cells to gemcitabine in vitro and in
nude mouse xenografts.
Materials and methods
Cell lines and culture
The PDAC SW1990 and CFPAC-1 cell lines were obtained
from Shanghai Cell Bank (Shanghai, China). These cell lines were
propagated and cultured in Dulbecco's modified Eagle's medium
(DMEM, Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal
bovine serum (FBS; Sigma, St. Louis, MO, USA), 100 μg/ml penicillin
and streptomycin, in a humidified chamber at 37°C with 5%
CO2.
Gemcitabine-resistant PDAC SW1990/GR and CFPAC-1/GR
cell sublines were acquired by culturing the parental cells with
gradually increasing concentrations of gemcitabine (Lilly,
Neuilly-sur-Seine Cedex, France) for approximately 6 months as
documented in a previous study (32). In brief, the cells were cultured in
gemcitabine-conditioned medium for 3 days at the concentration of 3
μM, followed by a recovery step, and agent-free medium culturing
until the cells recovered exponential growth. MTT assays were
performed to evaluate the IC50 of the
gemcitabine-treated cells. The IC50 dose was used for
the gemcitabine-conditioned medium to treat the cells. By
increasing the dosage of gemcitabine in the culture medium
intermittently for approximately 6 months (24 weeks for SW1990 and
21 weeks for CFPAC-1 cells), stable gemcitabine-resistant cell
sublines (SW1990/GR and CFPAC-1/GR) were acquired. The
IC50 in the SW1990/GR and CFPAC-1/GR cells was 232.2 and
314.4 μM, respectively.
miRNA mimics and inhibitor, gene
transfection, miRNA-overexpressed lentivirus and virus
infection
miR-181b mimics, inhibitor and negative control were
designed and synthesized by GenePharma Co., Ltd. (Shanghai, China).
The primer sequences are shown in Table
I. For gene transfection, the cells were cultured in 6-well
plates to 40% confluence. miR-181b mimics, inhibitor and negative
control were mixed with Lipofectamine 2000 (Invitrogen), and then
added to the cell culture medium according to the manufacturer's
instructions. After 24 h of transfection, total RNA and protein
were prepared from the cells and subjected to qRT-PCR and western
blot analyses, respectively.
 | Table ISequences of miR-181b mimics,
inhibitor and negative control. |
Table I
Sequences of miR-181b mimics,
inhibitor and negative control.
| Mimic (5′-3′) | Inhibitor
(5′-3′) |
|---|
| miRNA-181b |
AACAUUCAUUGCUGUCGGUGGGU |
ACCCACCGACAGCAAUGAAUGUU |
| Negative
control |
UUCUCCGAACGUGUCACGUTT | |
A miRNA-181b lentivirus overexpression system was
obtained from GenePharma. Briefly, the miRNA-181b lentiviral
expression vectors, LV3-pGLV-H1-miRNA-181b-GFP-Puro and
LV3-pGLV-H1-Null-GFP-Puro (null control) were constructed. The
vectors were then transfected into 293-T cells. The supernatant
titer was determined to be 1×108 TU/ml. Subsequently,
the lentivirus was used to infect PDAC cells following the
manufacturer's instructions.
qRT-PCR
Total cellular RNA from the cultured cells was
isolated using TRIzol reagent (Invitrogen). RNA samples (500 ng
each) were then reverse-transcribed into cDNA with miRNA-181b
reverse transcriptase primers (Applied Biosystems, Foster City, CA,
USA) using a TaqMan MicroRNA Reverse Transcription kit (Applied
Biosystems). Levels of miRNA-181b and U6 expression were determined
by qPCR with TaqMan MicroRNA Assays (Applied Biosystems) and an ABI
7500 machine (Applied Biosystems). The levels of mature miRNA-181b
expression were then normalized to U6 and calculated as the inverse
log of the ΔΔCT. All procedures were performed following the
manufacturer's instructions.
Protein extraction and western blot
analysis
Total cell protein lysates were prepared using an
RIPA buffer supplemented with 1% phenylmethylsulfonyl fluoride
(PMSF). Protein concentration was determined by a BCA kit (Keygen,
Nanjing, China). Subsequently, 30 μg of protein sample each was
separated by SDS-PAGE (10% gels) and transferred onto a 0.45 μm
polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, USA)
using a mini trans-blot system (Bio-Rad Laboratories, Hercules, CA,
USA). Primary antibodies and corresponding secondary antibodies
were then added followed by incubation. A rabbit anti-human BCL-2
antibody was purchased from Cell Signaling Technology (Danvers, MA,
USA), and a mouse anti-human GAPDH antibody and goat anti-rabbit or
anti-mouse secondary antibody were obtained from Beyotime (Nantong,
China). To quantify the protein expression, protein expression was
normalized to GAPDH levels (Chemilmager 5500; Alpha Innotech Corp.,
San Leandro, CA, USA).
Enzyme-linked immunosorbent assay
(ELISA)
To detect caspase-3 activity, we first seeded the
cells at a density of 1.5×106 cells per well in 6-well
plates. The cells were then transfected with miRNA-181b mimics,
inhibitor or negative controls for 48 h. Subsequently, caspase-3
activity was analyzed using a CaspACE Assay System (Promega,
Madison, Wisconsin, USA) following the manufacturer's
instructions.
Cell viability MTT assay
To detect cell viability, we first seeded
2×103 cells in 96-well plates, and then transfected them
with miR-181b mimics, inhibitor or negative controls for 72 h.
Subsequently, 20 μl per well of
3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
solution (5 mg/ml, Sigma) was added to the cells followed by
incubation at 37°C for 4 h. Then, 150 μl of dimethyl-sulphoxide
(DMSO) was substituted for the supernatant, followed by oscillation
for 10 min. Absorbance at 490 nm was detected using a microplate
reader (Multiskan MK3; Thermo Labsystems, Franklin, MA, USA). All
the results were normalized to the corresponding controls and the
percentage of the control was calculated.
Apoptosis Annexin V/flow cytometric
assay
Cells were incubated with culture medium containing
gemcitabine at a final concentration of 0.1 μM for 48 h. Cell
pellets were then collected, washed with phosphate-buffered saline
(PBS), resuspended in 100 μl of 1X binding buffer and stained with
5 μl phycoerythrin-Annexin V and 5 μl of 7-AAD (Becton-Dickinson,
Franklin Lakes, NJ, USA) at room temperature for 15 min in the
dark. A flow cytometer (Becton-Dickinson) was utilized to evaluate
the apoptotic levels in each sample.
Dual-luciferase reporter assay
Luciferase reporter constructs carrying 60-bp-long
synthetic oligonucleotides (Invitrogen, Shanghai, China) and
containing wild-type putative miRNA binding sites from the human
BCL-2 3′-UTR or their mutant versions (www.targetscan.org; Table
II) were inserted in XbaI-FseI sites of
pGL3-control vectors (Promega). The construct was confirmed by DNA
sequencing. Aliquots of 1.5×105 cells were seeded into
24-well plates. After 24 h, 200 ng of each independent luciferase
reporter plasmid plus 80 ng of pRL-TK (Promega) plasmid as the
control were co-transfected with 60 pmol of the miRNA-181b mimics,
inhibitor or control. Luciferase activity was then measured 48 h
after transfection using the Dual-Luciferase Reporter Assay System
with a GloMax Luminometer (Promega). Firefly luciferase activity
was normalized to Renilla luciferase activity for each transfected
cell sample.
| Table IITarget sites of BCL-2 3′-UTR and
their mutants or mismatch sequences. |
Table II
Target sites of BCL-2 3′-UTR and
their mutants or mismatch sequences.
| Sequence
(5′-3′) |
|---|
| WT2896 |
CTTATTGTTAAAAACATGTTAGAAGCAATGAATGTATATAAAAGCCTCAACTAGTCATTT |
| MT2896 |
CTTATTGTTAAAAACATGTTAGAAGCAATTCCTACATATAAAAGCCTCAACTAGTCATTT |
| WT1752 |
ATACCATTTATCTGTATTAACTTTGGAATGTACTCTGTTCAATGTTTAATGCTGTGGTTG |
| MT1752 |
ATACCATTTATCTGTATTAACTTTGGCAGAGACTCTGTTCAATGTTTAATGCTGTGGTTG |
In vivo chemosensitivity assay
For in vivo chemosensitivity analyses,
2×106 cells from the SW1990 and SW1990/GR cell lines
transfected with LV3-pGLV-H1-miRNA-181b-GFP-Puro,
LV3-pGLV-H1-Null-GFP-Puro and mock control vectors were resuspended
in 25 μl of DMEM, and then injected into the pancreatic
undercapsule in 4-week-old BALB/c female nude mice (6 mice in each
group). BALB/c female nude mice were purchased from The Model
Animal Research Center of Nanjing University, Nanjing, China. Our
animal studies were approved by the Ethics Committee of Nanjing
Medical University. Two weeks after cell injection, gemcitabine
(150 mg/kg) was injected intraperitoneally twice weekly for 28
days. At the end of the experiments, the mice were euthanized and
the tumor lesions were excised. Tumor volume was determined as V =
(L × W2)/2, where L represents the length of the tumor
and W represents the width of the tumor.
Statistical analysis
All in vitro experiments were performed in
triplicate and repeated at least once. The Student's t-test was
performed to assess statistical differences between 2 groups and
the F-test was used for comparisons among 3 or more groups. The Q
test was used for multiple comparisons. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
Establishment of gemcitabine-resistant
pancreatic cancer cell sublines
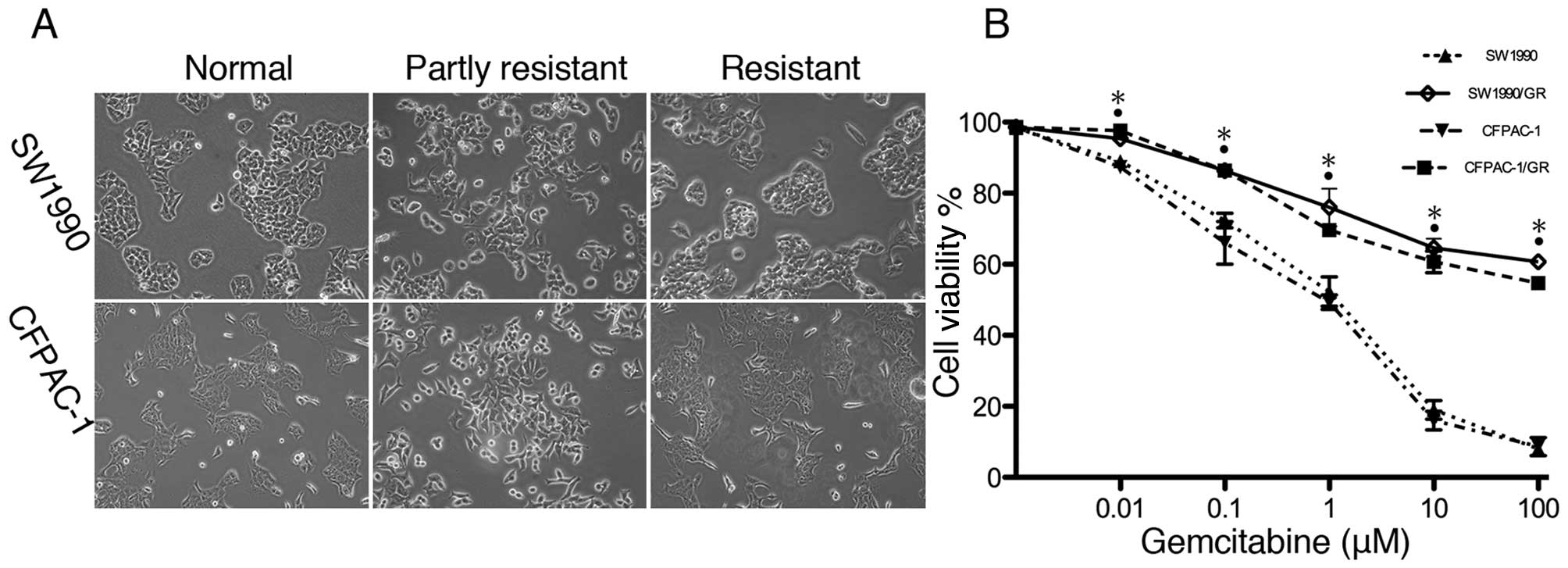
In this study, we first established
gemcitabine-resistant pancreatic cancer cell sublines by
cultivating SW1990 and CFPAC-1 cells with gradually increasing
concentrations of gemcitabine for 6 months. At first, SW1990 and
CFPAC-1 cells had partially increased viability with morphological
changes, followed by permanent gemcitabine resistance with
phenotypic recovery (Fig. 1A). The
cell viability MTT assay confirmed the increased resistance
(Fig. 1B).
miRNA-181b regulates gemcitabine
resistance in PDAC cells
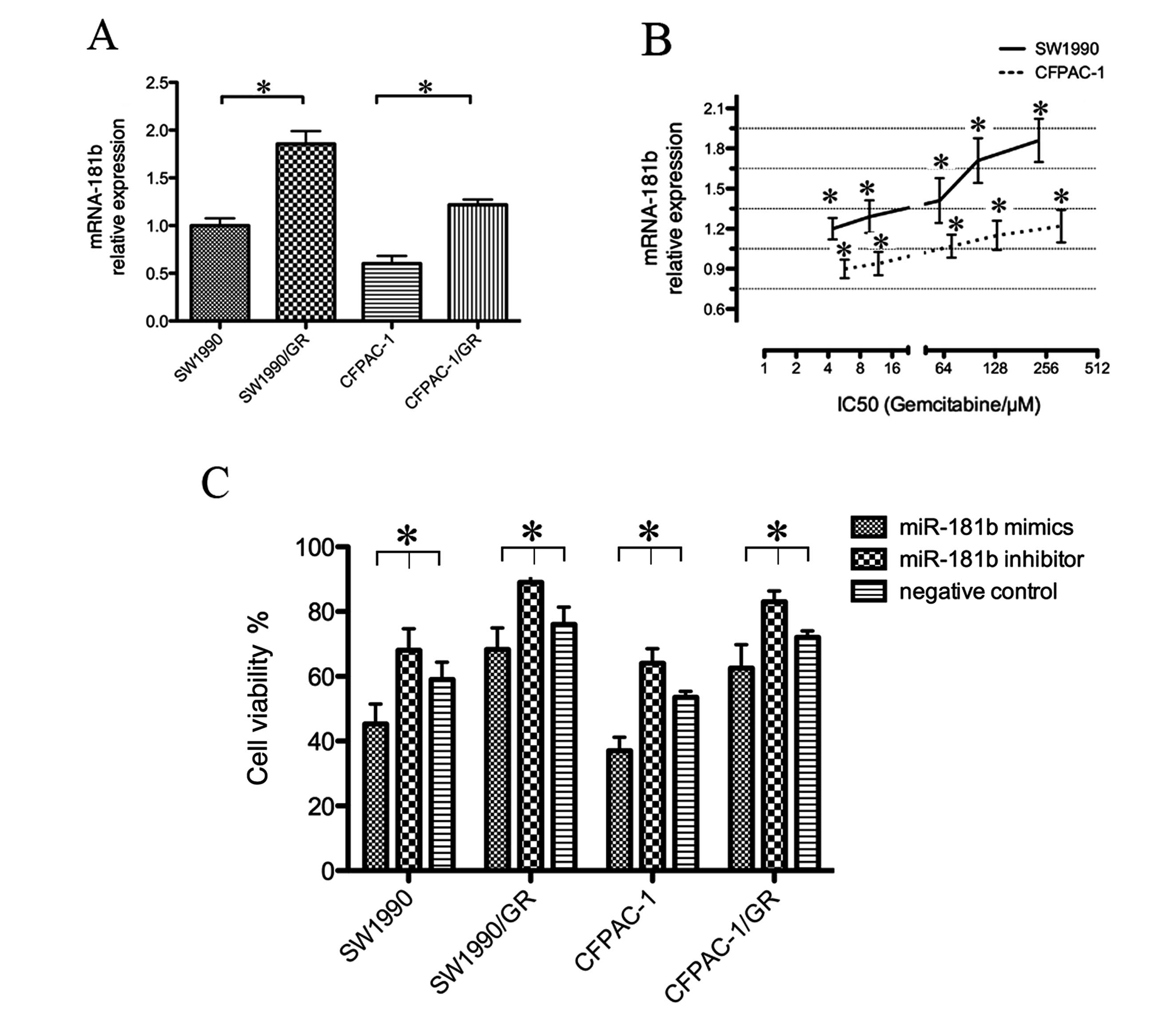
To assess the role of miR-181b in PDAC cells, we
first determined the miRNA-181b expression in normal and resistant
cell lines. miR-181b indeed was differentially expressed between
the parental and resistant cell lines, suggesting that miR-181b
affects gemcitabine sensitivity in PDAC cells. In particular,
miRNA-181b was expressed in the SW1990 and CFPAC-1 cells, as
detected by qRT-PCR (Fig. 2A).
However, the SW1990/GR and CFPAC-1/GR cells expressed significantly
higher levels of miRNA-181b (Fig.
2B). SW1990, CFPAC-1, SW1990/GR and CFPAC-1/GR cells were
transiently transfected with miRNA-181b mimics, inhibitor, or
control, and then treated with a gemcitabine-conditioned medium (1
μM) for 72 h. The MTT assay showed that miRNA-181b mimics increased
the gemcitabine sensitivity of these 4 cell lines (Table III and Fig. 2C).
| Table IIIEffects of miR-181b on PDAC cell
viability following treatment with gemcitabine (1 μM) for 72 h (%
of control). |
Table III
Effects of miR-181b on PDAC cell
viability following treatment with gemcitabine (1 μM) for 72 h (%
of control).
| Cell line |
|---|
|
|
|---|
| SW1990 | SW1990/GR | CFPAC-1 | CFPAC-1/GR |
|---|
| miRNA-181b
mimics | 76.41% | 89.64% | 69.34% | 86.43% |
| miRNA-181b
inhibitor | 114.15% | 117.71% | 120.94% | 116.62% |
miRNA-181b promotes the apoptosis of PDAC
cells
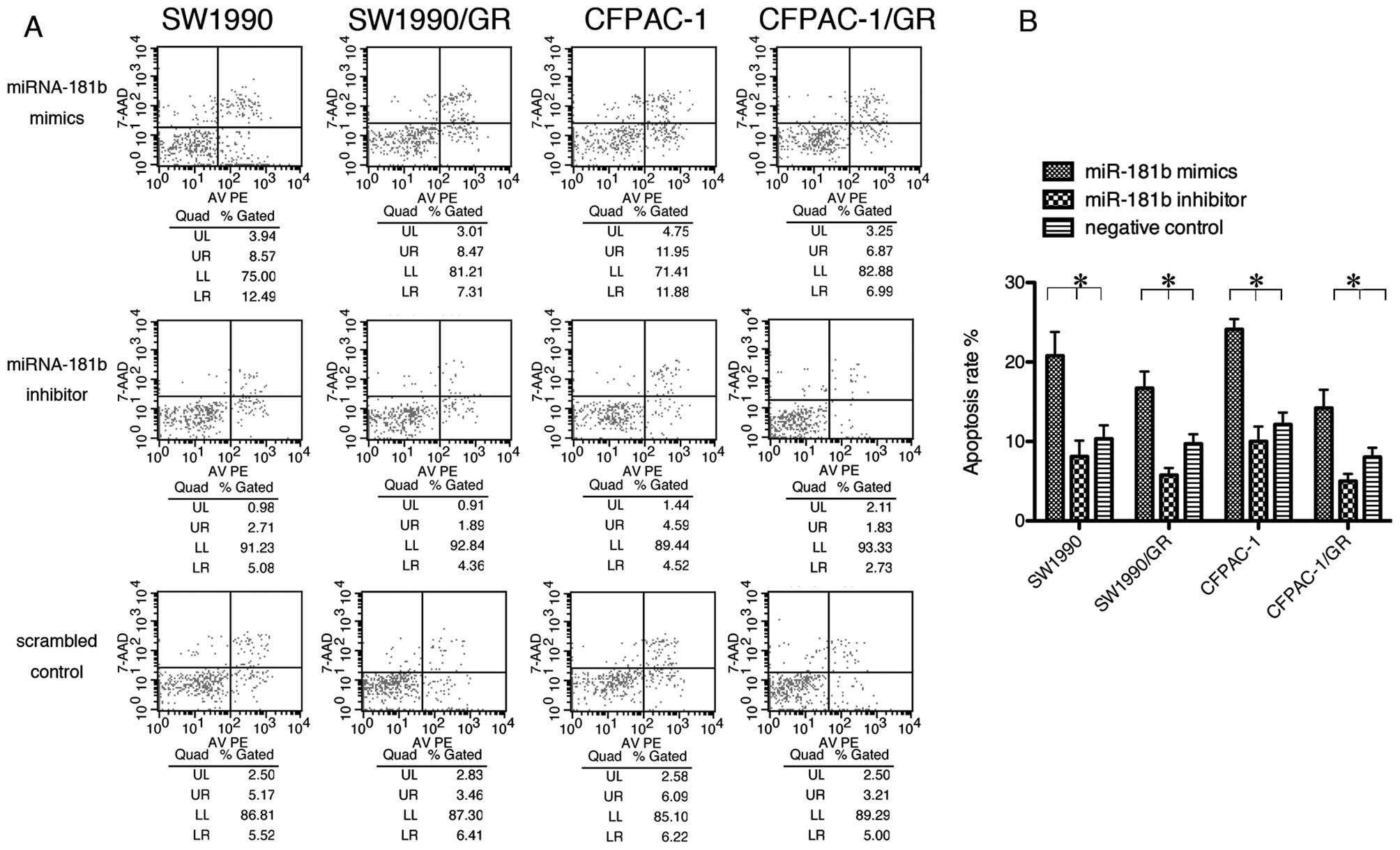
To further elucidate gemcitabine sensitivity in PDAC
cells, we performed a flow cytometric assay. Tumor cells were
transiently transfected with miRNA-181b mimics, inhibitor or
control, and then treated with gemcitabine (0.1 μM). The data
showed that miRNA-181b caused the PDAC cells to undergo apoptosis
(Fig. 3).
miRNA-181b promotes gemcitabine
sensitivity in PDAC cells in vivo
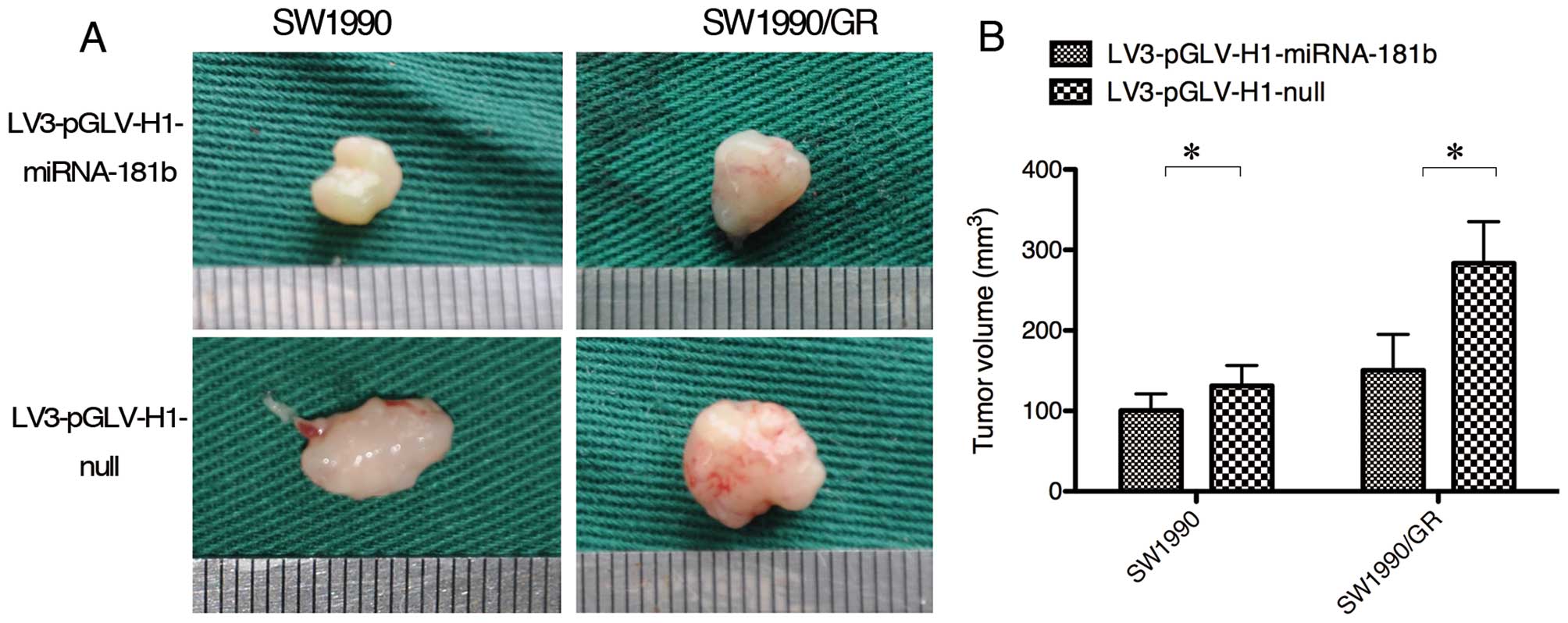
To confirm our in vitro data, we performed
nude mouse xenograft assays. PDAC SW1990 and SW1990/GR cells were
transfected with a lentivirus carrying miR-181b mimics or negative
control sequences in vitro that were then transplanted into
the pancreata of nude mice. Two weeks after tumor cell
transplantation, gemcitabine was administered to the mice at 150
mg/kg twice weekly for 4 weeks. The tumor size in the mice in the
miRNA-181b overexpression group was smaller than that in the mice
of the null control group (Fig.
4).
miRNA-181b affects the gemcitabine
resistance of PDAC cells by the downregulation of BCL-2
expression
To explore the underlying molecular events, we
performed western blot analysis and found that miRNA-181b reduced
the expression of BCL-2 protein in vitro and in tumor
xenografts (Fig. 5B). We also
performed ELISA to detect caspase-3 activity and found that
caspase-3 activity increased with miRNA-181b overexpression, but
was reduced following treatment with a miRNA-181b inhibitor
(Fig. 5C).
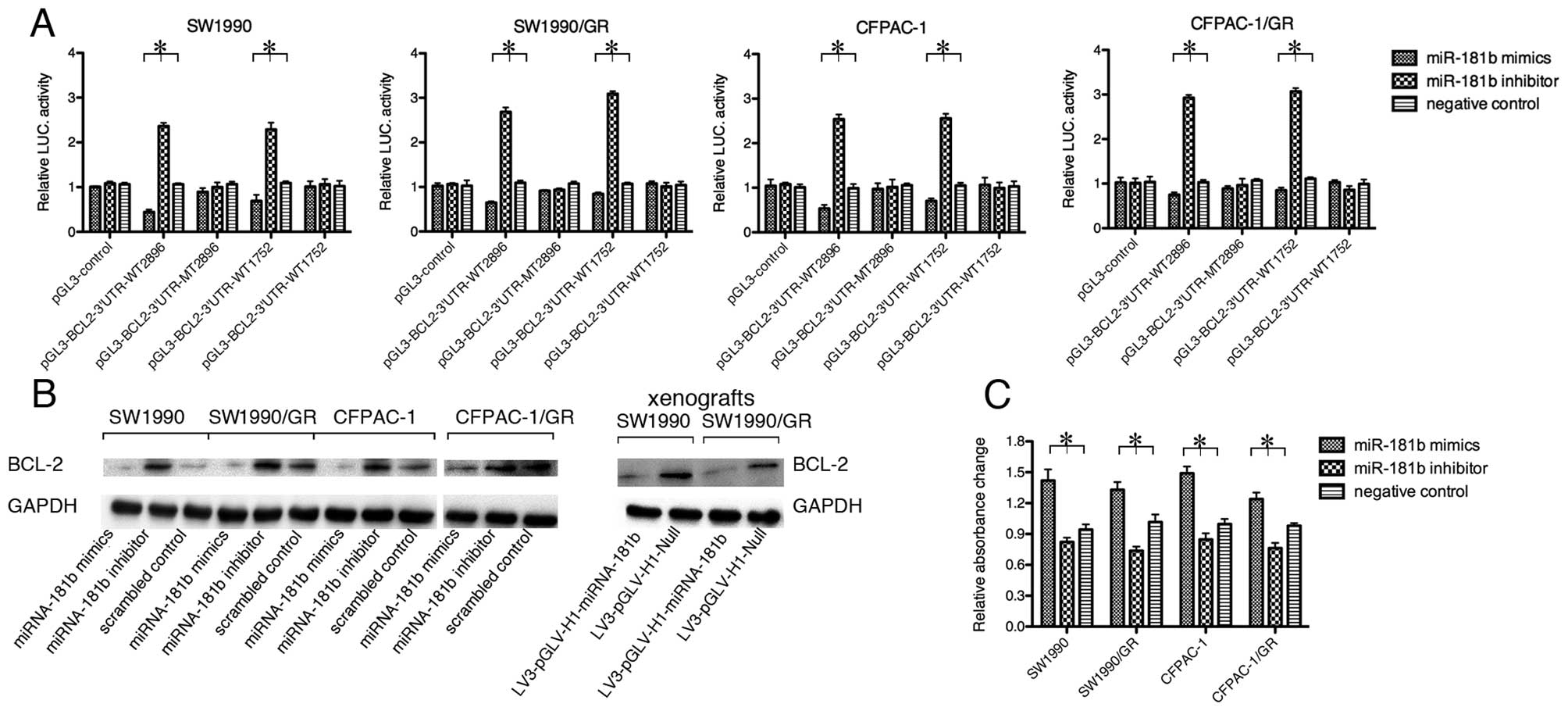 | Figure 5miR-181b targets BCL-2. (A)
Dual-luciferase reporter assay. BCL-2 3′UTR sequences or mutated
sequences were designed as MT2896, WT2896, MT1752 and WT1752, and
then inserted into a pGL3 vector. Subsequently, they were
co-transfected with miRNA-181b mimics, inhibitor, or a negative
control into pancreatic ductal adenocarcinoma (PDAC) cells. The
Renilla luciferase expression plasmid, pRL-TK, and the original
pGL3 control were utilized as an internal and external standard,
respectively. Luciferase activity was downregulated by miRNA-181b
mimics, but elevated by a miRNA-181b inhibitor in the normal and
resistant cell lines. Luciferase activity with the mutant sequences
showed no significant changes among cells transfected with
miRNA-181b mimics, inhibitor or negative control
(*p<0.05). (B) Western blot analysis. PDAC cells were
transiently transfected with miRNA-181b mimics, inhibitor, or
control for 48 h. Total cellular protein was extracted and blotted
using a BCL-2 antibody. The data revealed that miRNA-181b
overexpression reduced BCL-2 expression, whereas a miRNA-181b
inhibitor increased BCL-2 levels. (C) ELISA. Caspase-3 activity was
significantly increased by the exogenous expression of miRNA-181b
and decreased by a miRNA-181b inhibitor in the parental and
drug-resistant cells (*p<0.05). |
We also performed bioinformatics analyses and found
there are 2 potential miR-181b target sites in the BCL-2 3′UTR. We
performed a dual-luciferase reporter assay to assess whether BCL-2
is indeed a direct downstream target of miRNA-181b. The luciferase
activity was significantly restrained by miRNA-181b mimics, while
the miRNA-181b inhibitor led to an enhancement of luciferase
activity in normal and resistant cell lines (Fig. 5A).
Discussion
In the present study, we investigated the effects of
miRNA-181b on PDAC cell sensitivity to gemcitabine and the
potential underlying molecular mechanisms. We found that although
gemcitabine-resistant PDAC sublines expressed higher levels of
miR-181b than the parental cell lines, miRNA-181b mimics
significantly induced PDAC cell sensitivity to gemcitabine in both
parental and drug-resistant PDAC cells in vitro and in nude
mouse xenografts. By contrast, a miRNA-181b inhibitor led
gemcitabine resistance in PDAC cells. miR-181b mimics also induced
significantly higher levels of tumor cell apoptosis than the
controls. Molecularly, miR-181b suppressed BCL-2 expression and
induced caspase-3 activity in PDAC cells. Therefore, it can be
concluded that miRNA-181b may be useful in the clinical treatment
of gemcitabine-resistant PDAC.
In this study, we first established
gemcitabine-resistant PDAC sublines and unexpectedly discovered
that miR-181b expression was higher in drug-resistant cells when
compared to parental cells, for an unknown reason. These data
indicate that during the establishment of gemcitabine resistance,
miR-181b itself is not sufficient to antagonize gemcitabine. By
contrast, the transient transfection of a miR-181b mimics
sensitizes PDAC cells to gemcitabine. This was further confirmed in
parental cell lines, as well as in in vivo experiments using
nude mice. In clinical practice, gemcitabine is frequently used as
the first-line chemotherapy for PDAC. A previous study showed
however, that gemcitabine had a response rate of <20% (33). Thus, increasing drug sensitivity by
miR-181b may be a novel tool for PDAC chemotherapy and improving
patient survival. In this study, miR-181b increased the sensitivity
of PDAC cells to gemcitabine and induced PDAC cell apoptosis. The
evasion of apoptosis has long been acknowledged as one of the
hallmarks of cancer (34). The
induction of apoptosis could effectively control PDAC
progression.
Recently, miRNA has been recognized to play a
critical role in the regulation of cell apoptosis and
chemosensitivity (13,14). Functionally, miRNA can
complementarily target the mRNAs of target genes, and thereby
degrade or inhibit them from translating into proteins. Thus, the
altered expression of miRNAs contributes to a variety of human
diseases, such as inherited diseases, heart disease and cancer
(35,36). miRNA-181b has been reported to be
involved in certain types of human malignancies. Tissue inhibitor
of metalloprotease 3 (TIMP3) and BCL-2 mRNAs are major targets of
miRNA-181b. Thus, the overexpression of miR-181b may lead to the
downregulation of these genes and alter tumor progression and
chemoresistance. Previous studies have shown that miRNA-181b
suppresses TIMP3 expression in breast cancer and hepatocellular
carcinoma cells, and facilitates tumor progression and
chemoresistance (28,30). By contrast, miRNA-181b expression
has been shown to enhance the sensitivity of B-cell CLL, as well as
that of gastric and lung cancer cells to chemotherapy through the
downregulation of BCL-2 expression (26,31).
Moreover, in pancreatic cancer, miRNA-181b is considered a
prospective biomarker for pre-operative diagnosis (23,37,38).
However, the mechanisms of miRNA-181b action in PDAC carcinogenesis
and chemoresistance remain unclear.
A number of gene pathways, including the Akt, EGFR,
SHH, Notch, MAPK and NFκB pathways, have been associated with
chemoresistance (39). BCL-2 plays
an important role in apoptosis and gemcitabine resistance (13,40).
BCL-2 protein maintains the integrity of the mitochondrial membrane
by preventing caspase activation (e.g., caspase-3), resulting in
cell survival (41).
Transcriptionally, various kinases and transcription factors can
induce BCL-2 expression (13,42).
As a target gene of miRNA-181b, BCL-2 facilitates cell survival
against chemotherapy via the blockage of Bax/Bak-induced apoptosis
(18,26,31). A
previous study showed that BCL-2 also participates in gemcitabine
resistance in PDAC (43). Thus, our
current data support the positive role of miR-181b in sensitizing
PDAC cells to gemcitabine.
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li W, Ma Q, Liu J, et al: Hyperglycemia as
a mechanism of pancreatic cancer metastasis. Front Biosci.
17:1761–1774. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garrido-Laguna I, Uson M, Rajeshkumar NV,
et al: Tumor engraftment in nude mice and enrichment in
stroma-related gene pathways predict poor survival and resistance
to gemcitabine in patients with pancreatic cancer. Clin Cancer Res.
17:5793–5800. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saiki Y, Yoshino Y, Fujimura H, et al: DCK
is frequently inactivated in acquired gemcitabine-resistant human
cancer cells. Biochem Biophys Res Commun. 421:98–104. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Wolf C, Jansen R, Yamaguchi H, et al:
Contribution of the drug transporter ABCG2 (breast cancer
resistance protein) to resistance against anticancer nucleosides.
Mol Cancer Ther. 7:3092–3102. 2008.PubMed/NCBI
|
|
7
|
Schniewind B, Christgen M, Kurdow R, et
al: Resistance of pancreatic cancer to gemcitabine treatment is
dependent on mitochondria-mediated apoptosis. Int J Cancer.
109:182–188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kagawa S, Takano S, Yoshitomi H, et al:
Akt/mTOR signaling pathway is crucial for gemcitabine resistance
induced by Annexin II in pancreatic cancer cells. J Surg Res.
178:758–767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu H, Buchan RJ and Cook SA: MicroRNA-223
regulates Glut4 expression and cardiomyocyte glucose metabolism.
Cardiovasc Res. 86:410–420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma F, Liu X, Li D, et al: MicroRNA-466l
upregulates IL-10 expression in TLR-triggered macrophages by
antagonizing RNA-binding protein tristetraprolin-mediated IL-10
mRNA degradation. J Immunol. 184:6053–6059. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ambros V: MicroRNA pathways in flies and
worms: growth, death, fat, stress, and timing. Cell. 113:673–676.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hummel R, Hussey DJ and Haier J:
MicroRNAs: predictors and modifiers of chemo- and radiotherapy in
different tumour types. Eur J Cancer. 46:298–311. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lima RT, Busacca S, Almeida GM, Gaudino G,
Fennell DA and Vasconcelos MH: MicroRNA regulation of core
apoptosis pathways in cancer. Eur J Cancer. 47:163–174. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schoof CR, Botelho EL, Izzotti A and dos
Vasques LR: MicroRNAs in cancer treatment and prognosis. Am J
Cancer Res. 2:414–433. 2012.PubMed/NCBI
|
|
15
|
Ratert N, Meyer HA, Jung M, et al:
Reference miRNAs for miRNAome analysis of urothelial carcinomas.
PLoS One. 7:e393092012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pallante P, Visone R, Ferracin M, et al:
MicroRNA deregulation in human thyroid papillary carcinomas. Endocr
Relat Cancer. 13:497–508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zanette DL, Rivadavia F, Molfetta GA, et
al: miRNA expression profiles in chronic lymphocytic and acute
lymphocytic leukemia. Braz J Med Biol Res. 40:1435–1440. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Visone R, Veronese A, Rassenti LZ, et al:
miR-181b is a biomarker of disease progression in chronic
lymphocytic leukemia. Blood. 118:3072–3079. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xi Y, Formentini A, Chien M, et al:
Prognostic values of microRNAs in colorectal cancer. Biomark
Insights. 2:113–121. 2006.PubMed/NCBI
|
|
20
|
Yan LX, Huang XF, Shao Q, et al: MicroRNA
miR-21 overexpression in human breast cancer is associated with
advanced clinical stage, lymph node metastasis and patient poor
prognosis. RNA. 14:2348–2360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schaefer A, Jung M, Mollenkopf HJ, et al:
Diagnostic and prognostic implications of microRNA profiling in
prostate carcinoma. Int J Cancer. 126:1166–1176. 2010.PubMed/NCBI
|
|
22
|
Xu X, Jia R, Zhou Y, et al:
Microarray-based analysis: Identification of hypoxia-regulated
microRNAs in retinoblastoma cells. Int J Oncol. 38:1385–1393.
2011.PubMed/NCBI
|
|
23
|
Panarelli NC, Chen YT, Zhou XK,
Kitabayashi N and Yantiss RK: MicroRNA expression aids the
preoperative diagnosis of pancreatic ductal adenocarcinoma.
Pancreas. 41:685–690. 2012.PubMed/NCBI
|
|
24
|
Ciafre SA, Galardi S, Mangiola A, et al:
Extensive modulation of a set of microRNAs in primary glioblastoma.
Biochem Biophys Res Commun. 334:1351–1358. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen L, Yang Q, Kong WQ, et al:
MicroRNA-181b targets cAMP responsive element binding protein 1 in
gastric adenocarcinomas. IUBMB Life. 64:628–635. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dan Xia Z, Wei Z, Cheng F, et al:
miR-181a/b significantly enhances drug sensitivity in chronic
lymphocytic leukemia cells via targeting multiple anti-apoptosis
genes. Carcinogenesis. 33:1294–1301. 2012.PubMed/NCBI
|
|
27
|
Careccia S, Mainardi S, Pelosi A, et al: A
restricted signature of miRNAs distinguishes APL blasts from normal
promyelocytes. Oncogene. 28:4034–4040. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang B, Hsu SH, Majumder S, et al:
TGFbeta-mediated upregulation of hepatic miR-181b promotes
hepatocarcinogenesis by targeting TIMP3. Oncogene. 29:1787–1797.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakajima G, Hayashi K, Xi Y, et al:
Non-coding MicroRNAs hsa-let-7g and hsa-miR-181b are associated
with chemoresponse to S-1 in colon cancer. Cancer Genomics
Proteomics. 3:317–324. 2006.PubMed/NCBI
|
|
30
|
Lu Y, Roy S, Nuovo G, et al:
Anti-microRNA-222 (anti-miR-222) and -181B suppress growth of
tamoxifen-resistant xenografts in mouse by targeting TIMP3 protein
and modulating mitogenic signal. J Biol Chem. 286:42292–42302.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu W, Shan X, Wang T, Shu Y and Liu P:
miR-181b modulates multidrug resistance by targeting BCL2 in human
cancer cell lines. Int J Cancer. 127:2520–2529. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
An Y, Yao J, Wei JS, et al: Establish a
gemcitabine-resistant pancreatic cancer cell line SW1990/GZ and
research the relationship between SW1990/GZ and pancreatic cancer
stem cell. Zhonghua Wai Ke Za Zhi. 48:999–1003. 2010.(In
Chinese).
|
|
33
|
Friess H, Langrehr JM, Oettle H, et al: A
randomized multi-center phase II trial of the angiogenesis
inhibitor Cilengitide (EMD 121974) and gemcitabine compared with
gemcitabine alone in advanced unresectable pancreatic cancer. BMC
Cancer. 6:2852006. View Article : Google Scholar
|
|
34
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
35
|
Osman A: MicroRNAs in health and disease -
basic science and clinical applications. Clin Lab. 58:393–402.
2012.PubMed/NCBI
|
|
36
|
Fillat C and Altafaj X: Gene therapy for
Down syndrome. Prog Brain Res. 197:237–247. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ren Y, Gao J, Liu JQ, et al: Differential
signature of fecal microRNAs in patients with pancreatic cancer.
Mol Med Rep. 6:201–209. 2012.PubMed/NCBI
|
|
38
|
Liu J, Gao J, Du Y, et al: Combination of
plasma microRNAs with serum CA19-9 for early detection of
pancreatic cancer. Int J Cancer. 131:683–691. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hung SW, Mody HR and Govindarajan R:
Overcoming nucleoside analog chemoresistance of pancreatic cancer:
a therapeutic challenge. Cancer Lett. 320:138–149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cory S and Adams JM: The Bcl2 family:
regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
De Botton S, Sabri S, Daugas E, et al:
Platelet formation is the consequence of caspase activation within
megakaryocytes. Blood. 100:1310–1317. 2002.PubMed/NCBI
|
|
42
|
Zhao Y, Shen S, Guo J, et al:
Mitogen-activated protein kinases and chemoresistance in pancreatic
cancer cells. J Surg Res. 136:325–335. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dong J, Zhao YP, Zhou L, Zhang TP and Chen
G: Bcl-2 upregulation induced by miR-21 via a direct interaction is
associated with apoptosis and chemoresistance in MIA PaCa-2
pancreatic cancer cells. Arch Med Res. 42:8–14. 2011. View Article : Google Scholar
|