Introduction
Lung cancer is the leading cause of cancer-related
deaths in Japan and also worldwide in most developed countries.
Every year, ~60,000 individuals succumb to lung cancer in Japan,
and the number is increasing rapidly. Even in early-stage lung
cancer, ~40% of patients with stage I and II non-small cell lung
cancer (NSCLC) die from recurrent disease within 5 years despite
complete resection (1,2). The precise diagnosis and
classification of cancers are critical for the selection of
appropriate therapies. However, since no reliable clinical or
molecular predictors are currently available, it is difficult to
select high-risk patients who require more aggressive therapies
such as adjuvant chemotherapy.
Genetic abnormalities that exist in a certain
population of early-stage lung cancer patients possibly induce
aggressive phenotypes that demonstrate rapid tumor growth,
persistent invasiveness and a high potential for distant
metastasis. The expression of a number of genes is altered in
cancer cells due to mutations, deletions, amplifications, and
either the upregulation or downregulation of mRNA transcription.
Comprehensive DNA microarray analysis of gene expression patterns
is a powerful tool that permits the simultaneous evaluation of a
large number of genes in cancer cells (3,4).
Microarray gene expression profiling has recently been used to
define prognostic signatures in patients with NSCLC (5–11).
However, information concerning gene expression profiling and
molecular pathways relating to the outcomes of patients with
early-stage lung cancer has yet to be well characterized.
Adenocarcinoma is currently the predominant
histological subtype of NSCLC. The results of several expression
profiling studies have demonstrated that the expression profiles
are distinctive and recapitulate the known histological subtypes
(5–7). As a significant proportion of patients
relapse within 2 years, identification of early-stage patients with
a poor prognosis could delineate the appropriate candidates for
adjuvant therapy. The present study aimed to identify a novel
prognostic signature in early-stage lung adenocarcinoma using cDNA
microarray and bioinformatics analysis.
Materials and methods
Patient samples
Intraoperatively, immediately upon removal of a lung
lobe in which a primary lung carcinoma was located, a 500-mg sample
of tumor tissue was cut and immediately immersed in liquid nitrogen
and stored at −80°C until use, as previously reported (12). We studied frozen specimens of lung
cancer tissue from 64 randomly selected patients who underwent
complete resection of stage I or II NSCLC lesions at Tokyo Medical
University, Tokyo, Japan from May 2003 to December 2006. Tumor
tissues were processed by the Human Tissue Bank section at our
department according to standard operating procedures and
protocols. Briefly, frozen tissue samples at −80°C were pulverized,
and total cellular RNA was collected from each flash-frozen sample
using TRIzol RNA isolation reagent (Invitrogen). Total RNA was
processed with an RNeasy Mini kit (Qiagen). In vitro
transcription-based RNA amplification was then performed on at
least 8 μg of total RNA from each sample. The RNA quality was
assessed using a bioanalyzer (model 2100, Agilent). According to
the results from the RNA quality assay, 24 lung adenocarcinoma
samples were selected as our dataset.
Microarray analysis
Complementary DNA was synthesized using the
T7-(dT)24 primer:
59-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG-(dT)24-39. The cDNA was
processed using phase-lock gel phenol/chloroform extraction
(#E0032005101, Fisher). Next, in vitro transcriptional
labeling with biotin was performed using the Enzo BioArray kit
(#900182, Affymetrix). The resulting cRNA was processed again using
the RNeasy Mini kit. Labeled cRNA was hybridized to an Affymetrix
GeneChip (Human Genome-133 Plus 2.0 Array) according to the
manufacturer's instructions. The raw fluorescence intensity data
within the CEL files were preprocessed with the robust multichip
average algorithm, as implemented with the R packages from
Bioconductor. This algorithm analyzes the microarray data in three
steps: a background adjustment, quantile normalization, and finally
summation of the probe intensities for each probe set using a log
scale linear additive model for the log transform of (background
corrected, normalized) PM intensities.
Data analysis
Affymetrix Human Genome-U133 Plus 2.0 GeneChip data,
quantified with MAS5, were imported into the Subio Platform (Subio
Inc., Tokyo, Japan). Signals <1 were replaced with 1, log2
transformed, and then mean-subtracted by each probe set to obtain
the log ratio against the average of the expression patterns. No
normalization was applied.
Samples were classified into two groups,
recurrence-positive and recurrence-negative. Probe sets in both
groups whose detection values were absent in half of the samples
were removed. At this point, 28674 out of 54682 probe sets
remained. Finally, unvarying probe sets, whose log ratios were
between −1 and +1 in all samples, were filtered out to obtain the
final quality controlled probe sets (24420).
Principal component analysis (PCA) was applied to
the log ratio data of quality controlled genes. We recognized that
the samples in the recurrence-positive group might be
distinguishable as PC1 score negative (A) and positive (B)
subgroups.
We extracted the differentially expressed genes
(DEGs) for both A and B subgroups. We defined DEGs for A as being
>4-fold upregulated or downregulated compared with the average
of the recurrence-negative group, and having Mann-Whitney U-test
P-values of <0.05 between the recurrence-negative group and the
recurrence-positive A subgroup. A total of 721 probe sets were
selected as DEG for A. Similarly, we obtained 274 probe sets as
DEGs for B, which showed a >2-fold change and P-values of
<0.05 by the Mann-Whitney U-test, as compared with the
recurrence-negative group.
Biological analysis of the DEG lists
We searched 171 and 33 enriched GO terms for DEGs
determined for the A and B group, respectively, with the annotation
analysis plug-in of the Subio platform (data not shown). We further
analyzed these lists with the DAVID functional annotation web tool
(http://david.abcc.ncifcrf.gov) and
obtained the lists of enriched KEGG pathways (Tables I and II).
 | Table IPatient information regarding the 24
adenocarcinoma samples. |
Table I
Patient information regarding the 24
adenocarcinoma samples.
| | Early recurrence | |
|---|
| |
| |
|---|
| Variables | Total samples | Positive (n=8) | Negative (n=16) | P-value |
|---|
| Median age
(range) | 65.3 (42–76) | 67.0 (55–76) | 64.5 (42–76) | 0.551 |
| Gender | | | | 0.143 |
| Male | 14 | 3 | 11 | |
| Female | 10 | 5 | 5 | |
| Histological
differentiation | | | | 0.026a |
| Well/moderate | 14 | 2 | 11 | |
| Poor | 10 | 6 | 4 | |
| p-Stage | | | | 0.061 |
| IA | 14 | 2 | 12 | |
| IB | 8 | 5 | 3 | |
| IIA | 2 | 1 | 1 | |
| Table IIEnriched pathways in group A. |
Table II
Enriched pathways in group A.
| Category | Term | Count | % | P-value | Benjamini |
|---|
| KEGG_PATHWAY | Asthma | 8 | 1.5 | 0.000019 | 0.0024 |
| KEGG_PATHWAY | Allograft
rejection | 8 | 1.5 | 0.000085 | 0.0053 |
| KEGG_PATHWAY | Graft-versus-host
disease | 8 | 1.5 | 0.00014 | 0.0061 |
| KEGG_PATHWAY | Cell adhesion
molecules (CAMs) | 14 | 2.5 | 0.00017 | 0.0052 |
| KEGG_PATHWAY | Type I diabetes
mellitus | 8 | 1.5 | 0.00023 | 0.0059 |
| KEGG_PATHWAY | Intestinal immune
network for IgA production | 8 | 1.5 | 0.00062 | 0.013 |
| KEGG_PATHWAY | Autoimmune thyroid
disease | 8 | 1.5 | 0.0008 | 0.014 |
| KEGG_PATHWAY | Hematopoietic cell
lineage | 10 | 1.8 | 0.0011 | 0.017 |
| KEGG_PATHWAY | Systemic lupus
erythematosus | 10 | 1.8 | 0.003 | 0.041 |
| KEGG_PATHWAY | Antigen processing
and presentation | 8 | 1.5 | 0.013 | 0.15 |
| KEGG_PATHWAY | PPAR signaling
pathway | 7 | 1.3 | 0.018 | 0.19 |
| KEGG_PATHWAY | Oocyte meiosis | 9 | 1.6 | 0.018 | 0.18 |
| KEGG_PATHWAY | Viral
myocarditis | 7 | 1.3 | 0.02 | 0.18 |
| KEGG_PATHWAY | Arachidonic acid
metabolism | 6 | 1.1 | 0.027 | 0.22 |
| KEGG_PATHWAY | Cell cycle | 9 | 1.6 | 0.036 | 0.27 |
| KEGG_PATHWAY | Drug metabolism | 6 | 1.1 | 0.04 | 0.27 |
Ethical considerations
Written informed consent was obtained from the
patients for tissue procurement prior to surgery and their medical
records were maintained according to protocols approved by the
Institutional Review Board of Tokyo Medical University (no.
965).
Results
Patient information
As shown in Table I,
there were 14 male and 10 female patients enrolled in this study.
The mean age was 65.3 years (range, 42–76). The histological
classifications were all adenocarcinoma; 14 were well/moderately
differentiated and 10 were poorly differentiated. The distribution
of clinical staging demonstrated that most of the patients were
early-stage IAB cases. Histological differentiation was
significantly correlated with early recurrence (P=0.026), whereas
no significant correlations were found among pathological stages
IA, IB and IIA (P=0.061).
Correlation of patient outcome with
putative adenocarcinoma classes
We aimed to ascertain whether lung cancer patient
outcome correlates with the subclasses of lung adenocarcinomas
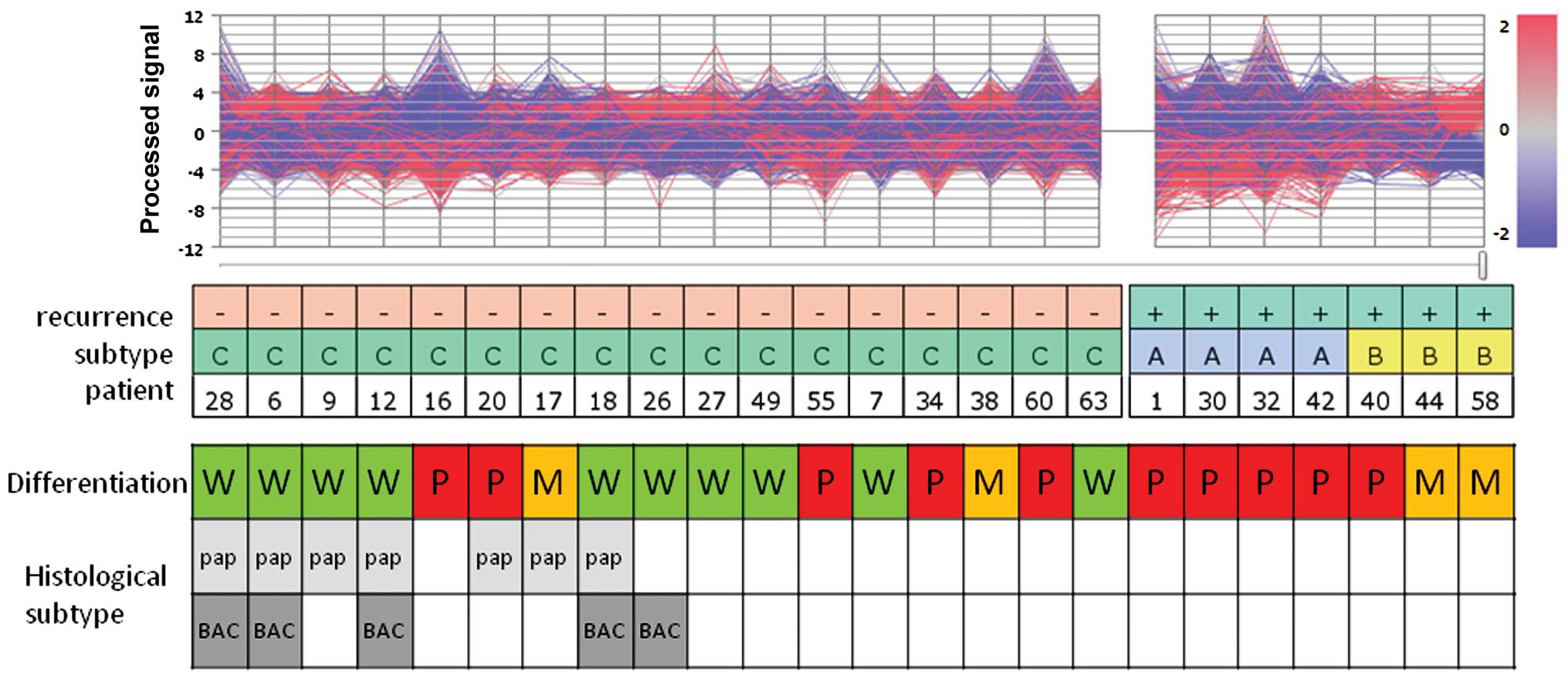
defined herein. Based on the results of PCA of this series, two
adenocarcinoma subgroups were identified within the early-relapse
group of early-stage adenocarcinoma cases, which differentially
expressed a broad range of gene patterns (Fig. 1).
Statistical analysis of the microarray data, when
compared with the non-early-relapse group C, revealed 723 genes
with significant differences in expression in the samples of group
A, whereas 274 genes showed significant differences in expression
in samples of group B. We searched 171 and 33 altered GO terms for
DEGs in the A and B lists, respectively, with the annotation
analysis plug-in of the Subio platform (data not shown).
The histological classification of all samples of
group A was poorly differentiated, whereas only one out of three
cases in group B was classified as poorly differentiated. In this
series of early-stage IA-IIA adenocarcinomas, no papillary or
bronchio-alveolar carcinoma subtypes were associated with
recurrence within 2 years after complete resection.
Biological function analysis
Tables II and
III document the 16 and 17
enriched pathways in groups A and B, respectively. Clusters of
genes related to oncological or immunological functional signaling
were found enriched in group A as were pathways such as cell
adhesion molecules (CAMs), cell cycle, and antigen processing and
presentation. In group B, the pathways included CAMs, T cell
receptor signaling, cytokine-cytokine receptor interaction,
toll-like receptor signaling, chemokine signaling pathway, primary
immunodeficiency and natural killer cell mediated cytotoxicity. The
CAM pathway was found to be enriched in both groups A and B.
| Table IIIEnriched pathways in group B. |
Table III
Enriched pathways in group B.
| Category | Term | Count | % | P-value | Benjamini |
|---|
| KEGG_PATHWAY | T cell receptor
signaling pathway | 11 | 4.8 | 0.0000046 | 0.00048 |
| KEGG_PATHWAY | Cytokine-cytokine
receptor interaction | 16 | 7 | 0.0000075 | 0.00039 |
| KEGG_PATHWAY | Toll-like receptor
signaling pathway | 9 | 3.9 | 0.00014 | 0.0047 |
| KEGG_PATHWAY | Cell adhesion
molecules (CAMs) | 10 | 4.3 | 0.00016 | 0.0042 |
| KEGG_PATHWAY | Leukocyte
transendothelial migration | 8 | 3.5 | 0.0021 | 0.041 |
| KEGG_PATHWAY | Chemokine signaling
pathway | 10 | 4.3 | 0.0021 | 0.035 |
| KEGG_PATHWAY | Intestinal immune
network for IgA production | 5 | 2.2 | 0.0064 | 0.091 |
| KEGG_PATHWAY | Autoimmune thyroid
disease | 5 | 2.2 | 0.0074 | 0.091 |
| KEGG_PATHWAY | Primary
immunodeficiency | 4 | 1.7 | 0.016 | 0.17 |
| KEGG_PATHWAY | Type I diabetes
mellitus | 4 | 1.7 | 0.026 | 0.24 |
| KEGG_PATHWAY | Axon guidance | 6 | 2.6 | 0.047 | 0.36 |
| KEGG_PATHWAY | Cytosolic
DNA-sensing pathway | 4 | 1.7 | 0.052 | 0.37 |
| KEGG_PATHWAY | Natural killer cell
mediated cytotoxicity | 6 | 2.6 | 0.053 | 0.35 |
| KEGG_PATHWAY | NOD-like receptor
signaling pathway | 4 | 1.7 | 0.069 | 0.41 |
| KEGG_PATHWAY | Focal adhesion | 7 | 3 | 0.087 | 0.46 |
| KEGG_PATHWAY | RIG-I-like receptor
signaling pathway | 4 | 1.7 | 0.095 | 0.47 |
| KEGG_PATHWAY | Prion diseases | 3 | 1.3 | 0.1 | 0.47 |
Discussion
The development of microarray technologies has made
it possible to quantitate the expression of many thousands of genes
simultaneously in a given sample (3,4).
Comprehensive analysis of gene expression patterns in individual
tumors should, therefore, provide detailed molecular portraits that
can facilitate tumor classification. Several expression profiling
studies concluded that expression profiles are distinctive and
recapitulate known histological subtypes (5–7).
Genomic methods offer promise for the classification
of human lung carcinomas. In one previous study, it is important to
note that the performance of the adenocarcinoma classifier showed a
better predictive accuracy than the squamous cell lung carcinoma
(SCC) classifier (adenocarcinoma AUC = 0.83, SCC AUC = 0.68). This
could have been due to the heterogeneity of the SCC samples as
indicated by the two distinct subgroups showing differing clinical
outcomes in this tumor type (9).
Multiple independent studies of mRNA expression profiles in lung
adenocarcinoma have proven highly reproducible. Analyses of the
relationship between expression profiles and tumor development and
differentiation, the presence or absence of specific pathogenic
mutations, patient prognosis and survival after surgical treatment,
and specific histopathology all appear to be promising (13).
Adenocarcinoma is currently the predominant
histological subtype of NSCLC. NSCLC composes the majority of
bronchogenic carcinoma cases with a lesser fraction being
small-cell lung carcinomas. The three main subtypes of NSCLC are
adenocarcinoma (60%), SCC (25%) and large-cell cancer (5%).
Adenocarcinoma has replaced SCC as the most frequent histological
subtype over the last 25 years (1,2,14).
Therefore, we focused on adenocarcinoma of the lung, and
particularly whether we could identify a novel prognostic signature
of early recurrence in early-stage lung adenocarcinoma using cDNA
microarray techniques.
The data indicated that patterns of gene expression
obtained from cDNA microarray studies of crudely dissected lung
tumors can be used to detect tumor subtypes that correlate with
biological and clinical phenotypes. Specifically, patterns of gene
expression were found that corresponded to the major morphological
classes of lung tumors. In addition, we were able to define two
subgroups of early recurrence in the adenocarcinoma cases that
differed not only in gene expression patterns, but also in clinical
and pathological properties, including histological differentiation
and subtype. In the statistical analysis of microarray data, when
compared with the non-early-recurrence group C, we revealed 723
genes with significant differences in expression in the samples of
group A, whereas 274 genes showed significant differences in
expression in group B. The differentially expressed genes were
classified according to biological processes. We searched 171 and
33 enriched GO terms for DEGs for the A and B lists, respectively,
with the annotation analysis plug-in of the Subio platform (data
not shown).
Gene annotation enrichment analysis is a functional
analysis technique that has gained widespread attention and for
which many tools have been developed. The differentially expressed
genes were classified according to biological processes and
molecular functions using the functional annotation clustering tool
of the DAVID bioinformatics resources. The DAVID functional
clustering analysis revealed 16 significantly altered biological
pathways in group A that included 3 distinct functionally related
metastatic categories, specifically CAMs, cell cycle, and antigen
processing and presentation. In group B, there were 17
significantly altered biological pathways, including 7 distinct
functionally related metastatic categories. Notably, the CAM
pathway was the most interrelated in both groups. In addition, the
T cell receptor signaling pathway, cytokine-cytokine receptor
interaction, toll-like receptor signaling pathway, chemokine
signaling pathway, primary immunodeficiency and natural killer cell
mediated cytotoxicity were also altered (Tables II and III). These results suggest that the
possibility of metastasis of early-stage lung adenocarcinoma was
closely related to the CAM pathway. Interestingly, considering the
relationship between group A or group B and histological
differentiation as poor or well/moderate, respectively, the
metastatic possibility of poorly differentiated early
adenocarcinoma appeared to be correlated with tumor development
factors, such as the cell cycle, whereas that of well/moderately
differentiated early-stage adenocarcinoma appeared to be correlated
with host immunological factors, such as the T cell receptor
signaling pathway, cytokine-cytokine receptor interaction, the
toll-like receptor signaling pathway, the chemokine signaling
pathway, primary immunodeficiency and natural killer cell mediated
cytotoxicity.
Our results suggest that the particular genes that
define the clusters and molecular pathways, or that are associated
with early recurrence, likely reflect the characteristics of the
particular tumors included in the analysis. Current therapy for
patients with early-stage disease usually consists of surgical
resection without adjuvant treatment. Clearly, the identification
of a high-risk group among early-stage patients would lead to
consideration of additional therapeutic interventions, possibly
leading to improved survival of these patients.
To our knowledge, this is the first study utilizing
cDNA microarray techniques, followed by molecular functional
pathway analysis, concerning the early recurrence of early-stage
adenocarcinoma of the lung. However, there were some limitations to
this study. Firstly, this was a small data set analysis at a single
institute. A large cohort sample of patients from multiple
institutions is needed. Secondly, the potential interactions of the
many specific individual genes and their clusters in lung tumor
biology and clinical outcome exist. This may be due to the
different platforms used (different genes analyzed) and the
different algorithms for selecting functional categories. Thirdly,
hierarchical clustering methods and functional analysis offer a
powerful approach to class discovery, but provide no means of
determining validity for the classes discovered. This is still a
putative functional analysis. It is important to state that several
in vitro and in vivo studies are still needed to
demonstrate whether these mechanisms are effective in reality.
In conclusion, in the present study, we present a
comprehensive gene expression analysis and functional pathway
analysis of early-stage lung adenocarcinomas, wherein we identified
a distinct molecular pathway category, the CAMs, which correlated
with the early relapse of early-stage lung adenocarcinoma
subclasses. Further in vitro and in vivo studies,
which can demonstrate these mechanisms, are warranted.
Acknowledgements
We are indebted to Dr Clifford A. Kolba, to
Associate Professor Edward F. Barroga and to Professor J. Patrick
Barron, Chairman of the Department of International Medical
Communications of Tokyo Medical University, for their editorial
review of the English manuscript. This study was supported by
grants from the Ministry of Education, Culture, Sports, Science and
Technology (grant no. 21791332).
References
|
1
|
Sawabata N, Asamura H, Goya T, et al:
Japanese Lung Cancer Registry Study: first prospective enrollment
of a large number of surgical and nonsurgical cases in 2002. J
Thorac Oncol. 5:1369–1375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Asamura H, Goya T, Koshiishi Y, et al: A
Japanese Lung Cancer Registry study: prognosis of 13,010 resected
lung cancers. J Thorac Oncol. 3:46–52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schena M, Shalon D, Davis RW and Brown PO:
Quantitative monitoring of gene expression patterns with a
complementary DNA microarray. Science. 270:467–470. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chee M, Yang R, Hubbell E, et al:
Accessing genetic information with high-density DNA arrays.
Science. 274:610–614. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beer DG, Kardia SL, Huang CC, et al:
Gene-expression profiles predict survival of patients with lung
adenocarcinoma. Nat Med. 8:816–824. 2002.PubMed/NCBI
|
|
6
|
Bhattacharjee A, Richards WG, Staunton J,
et al: Classification of human lung carcinomas by mRNA expression
profiling reveals distinct adenocarcinoma subclasses. Proc Natl
Acad Sci USA. 98:13790–13795. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garber ME, Troyanskaya OG, Schluens K, et
al: Diversity of gene expression in adenocarcinoma of the lung.
Proc Natl Acad Sci USA. 98:13784–13789. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wigle DA, Jurisica I, Radulovich N, et al:
Molecular profiling of non-small cell lung cancer and correlation
with disease-free survival. Cancer Res. 62:3005–3008.
2002.PubMed/NCBI
|
|
9
|
Raponi M, Zhang Y, Yu J, et al: Gene
expression signatures for predicting prognosis of squamous cell and
adenocarcinomas of the lung. Cancer Res. 66:7466–7472. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu Y, Yao R, Yan Y, et al: A gene
expression signature that can predict green tea exposure and
chemopreventive efficacy of lung cancer in mice. Cancer Res.
66:1956–1963. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Potti A, Mukherjee S, Petersen R, et al: A
genomic strategy to refine prognosis in early-stage non-small-cell
lung cancer. N Engl J Med. 355:570–580. 2006. View Article : Google Scholar
|
|
12
|
Nakamura H, Saji H, Ogata A, et al: cDNA
microarray analysis of gene expression in pathologic stage IA
nonsmall cell lung carcinomas. Cancer. 97:2798–2805. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meyerson M and Carbone D: Genomic and
proteomic profiling of lung cancers: lung cancer classification in
the age of targeted therapy. J Clin Oncol. 23:3219–3226. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sawabata N, Miyaoka E, Asamura H, et al:
Japanese lung cancer registry study of 11,663 surgical cases in
2004: demographic and prognosis changes over decade. J Thorac
Oncol. 6:1229–1235. 2011. View Article : Google Scholar : PubMed/NCBI
|