Introduction
Toxic heavy metals including cadmium and nickel,
which are common contaminants in occupational and environmental
areas, may contribute to harmful effects on humans and other
organisms. Unsafe persistent exposure to such metals is mediated
through oral intake of contaminated water and food, inhalation of
polluted air, or dermal contact from manufacturing processes and
environmental contamination. Indeed, deleterious health effects
caused by cadmium and nickel are similar, but the underlying
mechanisms of toxicity and biological/toxicological signaling
pathways are individually different (1–3).
Cadmium is a natural constituent of ocean water and
the earth’s crust and is commonly associated with zinc, lead and
copper ores (4). Due to its
physicochemical properties, it is utilized in a variety of ways,
such as in batteries, pigment, coating, plating, plastic
stabilizers, non-ferrous alloys and photovoltaic devices. Ingestion
of contaminated food and water and inhalation by active and passive
smoking are considered the predominant routes for cadmium exposure
(5). Cadmium is classified as
carcinogenic to humans (group 1) by the World Health Organization’s
International Agency for Research on Cancer, as it acts as an
epigenetic or indirect genotoxic element. The molecular mechanisms
of cadmium carcinogenicity have been recently reviewed (6–8) and
range from interference with thiol-containing proteins and
consequent induction of oxidative stress to genomic expression
changes triggering cell cycle arrest, apoptosis, differentiation
and immortalization. Strong data from epidemiological studies on
occupational populations related to cadmium carcinogenicity showed
that lung cancer occurs by breathing the metal (9). Cadmium affects the transcription of
several genes; it induces protective genes, including those coding
for metallothioneins with a cadmium-chelating property (10,11),
heat shock proteins (HSPs) with roles in protein renaturation
(12,13) and heme-oxygenase I with an
anti-oxidative role (14). Notably,
cadmium may induce proto-oncogenes (15,16)
which can contribute to carcinogenesis. Its effects on the sex
hormone receptor genes (17) also
suggest endocrine-disrupting activity. However, these effects of
cadmium on gene expression are only a fraction of the effects, and
the unidentified effects on gene functioning in toxicity and
protection against cadmium must be clarified.
Nickel, a non-essential metal, occurs naturally in
soils and volcanic dust. It also has been categorized as a
carcinogenic heavy metal by the World Health Organization’s
International Agency for Research on Cancer. It combines with other
metals to form alloys that are widely used to produce coins,
jewelry and stainless steel. Nickel compounds are also applied to
refining, electroplating, welding, color pigmentation in ceramics,
the production of batteries, medical devices and carbon particles
(18). Although several animal
epidemiological and cell culture studies have documented that
nickel compounds are carcinogenic; the molecular mechanisms of
nickel carcinogenicity have not been investigated (19–22).
The mutagenic activity of nickel compounds to mammalian cells in
vitro in the Salmonella mutation assay was found to be
low, indicating that nickel-mediated mutagenic activity is not the
definite mechanism underlying nickel carcinogenicity (23,24). A
number of studies have implicated that aberrant modification of
structural chromatin and epigenetic alterations might be the
predominant contributable factors to nickel carcinogenesis.
Both cadmium and nickel have adverse effects on
human health by inducing oxidative stress, inhibiting DNA repair
proteins, and dysregulating signal transduction, thereby leading to
aberrant cell proliferation and differentiation (3). Furthermore, they have also been
associated with an increased risk for diseases, including cancer,
following chronic low-dose exposure (25,26).
However, activation of these pathways still remains to be
elucidated. In the present study, we comprehensively explored
molecular candidates and the mechanisms of toxicity, both common
and distinct, for nickel and cadmium using integrative
toxicogenomic and bioinformatic tools. We compared the effects of
cadmium to those of nickel using comparative genomic hybridization
(CGH) array, gene expression microarray and functional
proteomics-based technologies in p53-proficient human colon
carcinoma RKO cells. The cells were subchronically exposed to low
levels of cadmium chloride or nickel acetate, based on a survival
rate of at least 80% after a 24-h treatment. We also identified
both common and distinct mechanisms of toxicity underlying the
individual metals. Similarities and differences in gene expression
profiles, respective pathways and biological processes following
low-level exposure to each metal are also discussed.
Materials and methods
Cell culture and treatment
Human RKO colon carcinoma cells (ATCC no. CRL-2577)
were cultured in RPMI-1640 (Gibco, Grand Island, NY, USA)
supplemented with 10% fetal bovine serum (Gibco) and 1% antibiotics
(Gibco) at 37°C and 5% CO2. Cadmium chloride and nickel
(II) acetate were purchased from Sigma Co. (St. Louis, MO, USA).
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
assay and fluorescence-activated cell sorting analysis were
conducted to select the sublethal doses of cadmium and nickel for
our experimental design. The RKO cells were exposed to 50 μM
cadmium and 20 μM nickel for 24 h prior to subsequent testing.
Array comparative genomic
hybridization
Genomic DNA was extracted from RKO cells treated
with either metal and subjected to copy number variation (CNV)
analyses using Agilent SurePrint G3 microarrays with one
million-format slides (Agilent Technologies, Palo Alto, CA, USA),
in accordance with the manufacturer’s protocol. Briefly, DNA was
labeled with the exo-Klenow fragment using random primers and
cyanine 5 and cyanine 3 fluorescent-labeled nucleotides. Next,
hybridization with the hybridization master mix, washing and drying
were carried out. Subsequently, the microarray slides were scanned
at 2-μm resolution using a G2539A microarray scanner (Agilent
Technologies). Features were extracted from the scanned images
using Feature Extraction software (Agilent Technologies). The
extracted features were analyzed using Nexus Copy Number software
(BioDiscovery, El Segundo, CA, USA). Thresholds were set at a
minimum of three probes and a 0.4 average log ratio.
Gene expression microarray
Total RNA was extracted with the RNeasy Mini kit
(Qiagen, Hilden, Germany) according to the manufacturer’s
recommendations. RNA quality was assessed with an Agilent
Bioanalyzer Nano Chip 2100 (Agilent Technologies). The RNA samples
were then labeled using a Low Input Quick Amp Labeling kit (Agilent
Technologies), in accordance with the manufacturer’s protocol.
Hybridization was conducted using a Gene Expression Hybridization
kit (Agilent Technologies). A gene expression microarray chip was
designed to perceive pathways involving DNA damage and repair,
apoptosis, oxidative stress and the cell cycle. The arrays were
scanned on an Agilent scanner and analyzed using Feature Extraction
software. Subio platform version 1.6 was used for the
transcriptomic data analysis.
Functional proteomics
Cells were collected by scrapping and were washed in
PBS (pH 7.4) for immunoprecipitation. After centrifugation, the
cells were resuspended and homogenized in RIPA buffer [50 mM
Tris-HCl pH 7.8, 150 mM NaCl, 0.5 mM EDTA, 0.1% sodium dodecyl
sulfate (SDS), 1% Triton X-100, 1 mM DTT and protease inhibitor
cocktail (Roche, Mannheim, Germany)]. The homogenate was incubated
on ice for 30 min and then centrifuged at 13,000 rpm and 4°C for 30
min to collect the supernatant. The samples were incubated with 4
μg rabbit anti-Gadd45a antibody (Santa Cruz Biotechnology, Santa
Cruz, CA, USA) for 10 h and consecutively with 100 μl of ExactaCruz
TMC to precipitate the Gadd45a-interacting proteins (Santa Cruz
Biotechnology) for 11 h. After a series of washes, the
immunoprecipitated samples were separated by SDS-polyacrylamide gel
electrophoresis and visualized by Coomassie Blue staining. Protein
bands with an altered expression pattern in response to the heavy
metals were selected and subjected to protein identification via
mass spectrometry analysis.
Pathway analysis
Molecular pathways among differentially expressed
genes identified by the microarray were dissected using Pathway
Studio 8.0 software (Ariadne Genomics, Rockville, MD, USA). This
program integrates relevant information among imported genes,
consequently allowing identification of biological pathways, gene
regulation networks and protein interaction maps.
Results
Identification of molecular candidates
for subchronic low-dose exposure to cadmium and nickel
The RKO cells were subchronically exposed to either
cadmium chloride or nickel acetate at concentrations of 50 or 20
μM, respectively, for different periods of time depending on the
target molecule of interest. These concentrations of the metal
compounds were determined as sublethal doses and resulted in
<20% cytotoxicity (27,28). Most of the genes differentially
expressed by cells exposed to cadmium or nickel were HSPs,
including HSP90AA1, HSP90AB1, HSPA1A, HSP90B1, HSPA2, HSPA5, HSPA8
and HSPA9 (Table I). Twenty genes
were significantly altered in the cadmium-exposed cells (Table II). Interestingly, most of them
overlapped with the commonly altered genes between the cadmium-and
nickel-exposed cells. However, there were unique molecular networks
involving the response to cadmium alone that were different from
the common networks, as represented in Figs. 1 and 2, respectively. Twenty-six genes were
differentially expressed in the nickel-exposed cells (Table III). Among them, several genes,
including FASN, HSP90AB1, HSP90B1, HSPA5, HSPA8, KRT18 and P4HB
were dysregulated both at the transcription and protein levels
following nickel exposure.
 | Table IKey genes among the commonly
expressed genes in cells subchronically exposed to low-level
cadmium and nickel after integrative toxicogenomic analysis. |
Table I
Key genes among the commonly
expressed genes in cells subchronically exposed to low-level
cadmium and nickel after integrative toxicogenomic analysis.
| Gene symbol | Gene ID | Gene name |
|---|
| ACTB | 60 | Actin, β |
| AKR1C2 | 1646 | Aldo-ketoreductase
family 1, member C2 (dihydrodiol dehydrogenase 2; bile acid binding
protein; 3-α hydroxysteroid dehydrogenase, type III) |
| DCD | 117159 | Dermcidin |
| HSP90AA1 | 3320 | Heat shock protein
90 kDa α (cytosolic), class A member 1 |
| HSP90AB1 | 3326 | Heat shock protein
90 kDa α (cytosolic), class B member 1 |
| HSPA1A
(HSPA1B) | 3303 | Heat shock 70 kDa
protein 1A |
| HSP90B1 | 7184 | Heat shock protein
90 kDa β (Grp94), member 1 |
| HSPA2 | 3306 | Heat shock 70 kDa
protein 2 |
| HSPA5 | 3309 | Heat shock 70 kDa
protein 5 (glucose-regulated protein, 78 kDa) |
| HSPA8 | 3312 | Heat shock 70 kDa
protein 8 |
| HSPA9 | 3313 | Heat shock 70 kDa
protein 9 (mortalin) |
| IDH1 | 3417 | Isocitrate
dehydrogenase 1 (NADP+), soluble |
| KRT18 | 3875 | Keratin 18 |
| MYH7 | 4625 | Myosin, heavy chain
7, cardiac muscle, β |
| NUDC | 10726 | Nuclear
distribution C homolog (A. nidulans) |
| P4HB | 5043 | Prolyl
4-hydroxylase, β polypeptide |
| PGK1 | 5230 | Phosphoglycerate
kinase 1 |
| PPP1R13B | 23368 | Protein phosphatase
1, regulatory subunit 13B |
| RNF128 | 79589 | Ring finger protein
128, E3 ubiquitin protein ligase |
| RPH3AL | 9501 | Rabphilin 3A-like
(without C2 domains) |
| TWIST1 | 7291 | Twist homolog 1
(Drosophila) |
| UTS2 | 10911 | Urotensin 2 |
| Table IIKey genes among the differentially
expressed genes in cells subchronically exposed to low-level
cadmium only after integrative toxicogenomic analysis. |
Table II
Key genes among the differentially
expressed genes in cells subchronically exposed to low-level
cadmium only after integrative toxicogenomic analysis.
| Gene symbol | Gene ID | Gene name |
|---|
| ACTB | 60 | Actin, β |
| AKR1C2 | 1646 | Aldo-ketoreductase
family 1, member C2 (dihydrodiol dehydrogenase 2; bile acid binding
protein; 3-α hydroxysteroid dehydrogenase, type III) |
| DCD | 117159 | Dermcidin |
| HSP90AA1 | 3320 | Heat shock protein
90 kDa α (cytosolic), class A member 1 |
| HSP90AB1 | 3326 | Heat shock protein
90 kDa α (cytosolic), class B member 1 |
| HSP90B1 | 7184 | Heat shock protein
90 kDa β (Grp94), member 1 |
| HSPA5 | 3309 | Heat shock 70 kDa
protein 5 (glucose-regulated protein, 78 kDa) |
| HSPA8 | 3312 | Heat shock 70 kDa
protein 8 |
| HSPA9 | 3313 | Heat shock 70 kDa
protein 9 (mortalin) |
| IDH1 | 3417 | Isocitrate
dehydrogenase 1 (NADP+), soluble |
| KRT18 | 3875 | Keratin 18 |
| MYH7 | 4625 | Myosin, heavy chain
7, cardiac muscle, β |
| NUDC | 10726 | Nuclear
distribution C homolog (A. nidulans) |
| P4HB | 5034 | Prolyl
4-hydroxylase, β polypeptide |
| PGK1 | 5230 | Phosphoglycerate
kinase 1 |
| PPP1R13B | 23368 | Protein phosphatase
1, regulatory subunit 13B |
| RNF128 | 79589 | Ring finger protein
128, E3 ubiquitin protein ligase |
| RPH3AL | 9501 | Rabphilin 3A-like
(without C2 domains) |
| TWIST1 | 7291 | Twist homolog 1
(Drosophila) |
| UTS2 | 10911 | Urotensin 2 |
| Table IIIKey genes among the differentially
expressed genes in cells subchronically exposed to low-level nickel
only after integrative toxicogenomic analysis. |
Table III
Key genes among the differentially
expressed genes in cells subchronically exposed to low-level nickel
only after integrative toxicogenomic analysis.
| Gene symbol | Gene ID | Gene name |
|---|
| ACTB | 60 | Actin, β |
| AFAP1 | 60312 | Actin filament
associated protein 1 |
| AKR1C2 | 1646 | Aldo-ketoreductase
family 1, member C2 (dihydrodiol dehydrogenase 2; bile acid binding
protein; 3-α hydroxysteroid dehydrogenase, type III) |
| DCD | 117159 | Dermcidin |
| EIF3F | 8665 | Eukaryotic
translation initiation factor 3, subunit F |
| FASN | 2194 | Fatty acid
synthase |
| HSP90AA1 | 3320 | Heat shock protein
90 kDa α (cytosolic), class A member 1 |
| HSP90AB1 | 3326 | Heat shock protein
90 kDa α (cytosolic), class B member 1 |
| HSP90B1 | 7184 | Heat shock protein
90 kDa β (Grp94), member 1 |
| HSPA1A | 3303 | Heat shock 70 kDa
protein 1A |
| HSPA5 | 3309 | Heat shock 70 kDa
protein 5 (glucose-regulated protein, 78 kDa) |
| HSPA8 | 3312 | Heat shock 70 kDa
protein 8 |
| HSPA9 | 3313 | Heat shock 70 kDa
protein 9 (mortalin) |
| IDH1 | 3417 | Isocitrate
dehydrogenase 1 (NADP+), soluble |
| JUN | 3725 | Jun
proto-oncogene |
| KRT18 | 3875 | Keratin 18 |
| MYH7 | 4625 | Myosin, heavy chain
7, cardiac muscle, β |
| NUDC | 10726 | Nuclear
distribution C homolog (A. nidulans) |
| P4HB | 5034 | Prolyl
4-hydroxylase, β polypeptide |
| PGK1 | 5230 | Phosphoglycerate
kinase 1 |
| PPP1R13B | 23368 | Protein phosphatase
1, regulatory subunit 13B |
| RNF128 | 79589 | Ring finger protein
128, E3 ubiquitin protein ligase |
| RPH3AL | 9501 | Rabphilin 3A-like
(without C2 domains) |
| TRIP10 | 9322 | Thyroid hormone
receptor interactor 10 |
| TWIST1 | 7291 | Twist homolog 1
(Drosophila) |
| UTS2 | 10911 | Urotensin 2 |
Dissection of networks related to
subchronic low-dose exposure to cadmium or nickel, together or
alone
We also attempted to identify interactive networks
among cadmium-and/or nickel-responsive genes using Pathway Studio
software version 8.0. Based on a number of reliable studies,
relevant components in the putative signaling pathways were chosen
and incorporated into the established networks. As a result, common
biological processes disturbed by both cadmium and nickel were cell
proliferation and apoptotic cell death, which can lead to neoplasms
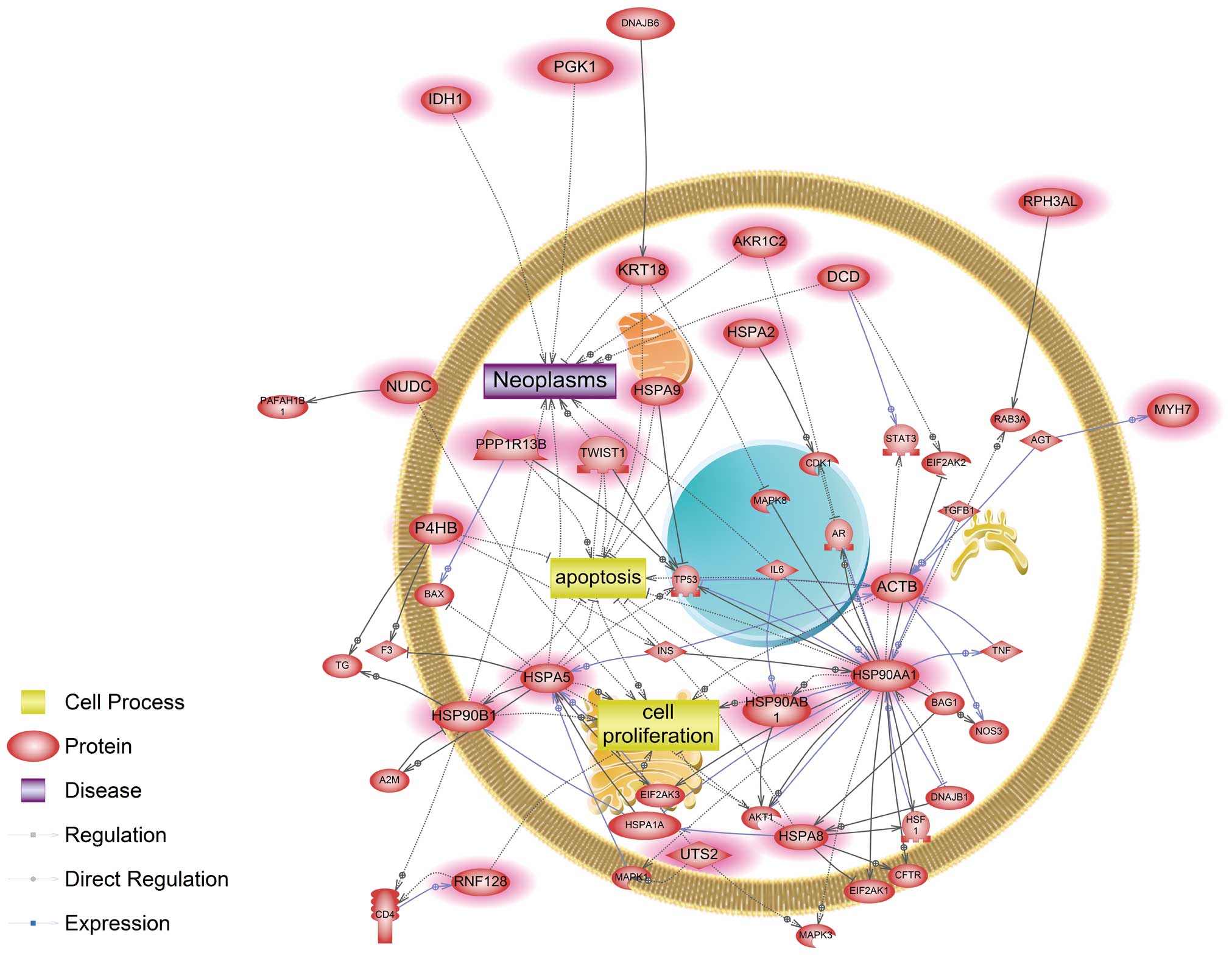
(Fig. 1). Four genes, ACTB,
HSP90AA1, HSPA5 and HSPA8, were identified as main modulators in a
common signaling pathway affected by cadmium and nickel. In
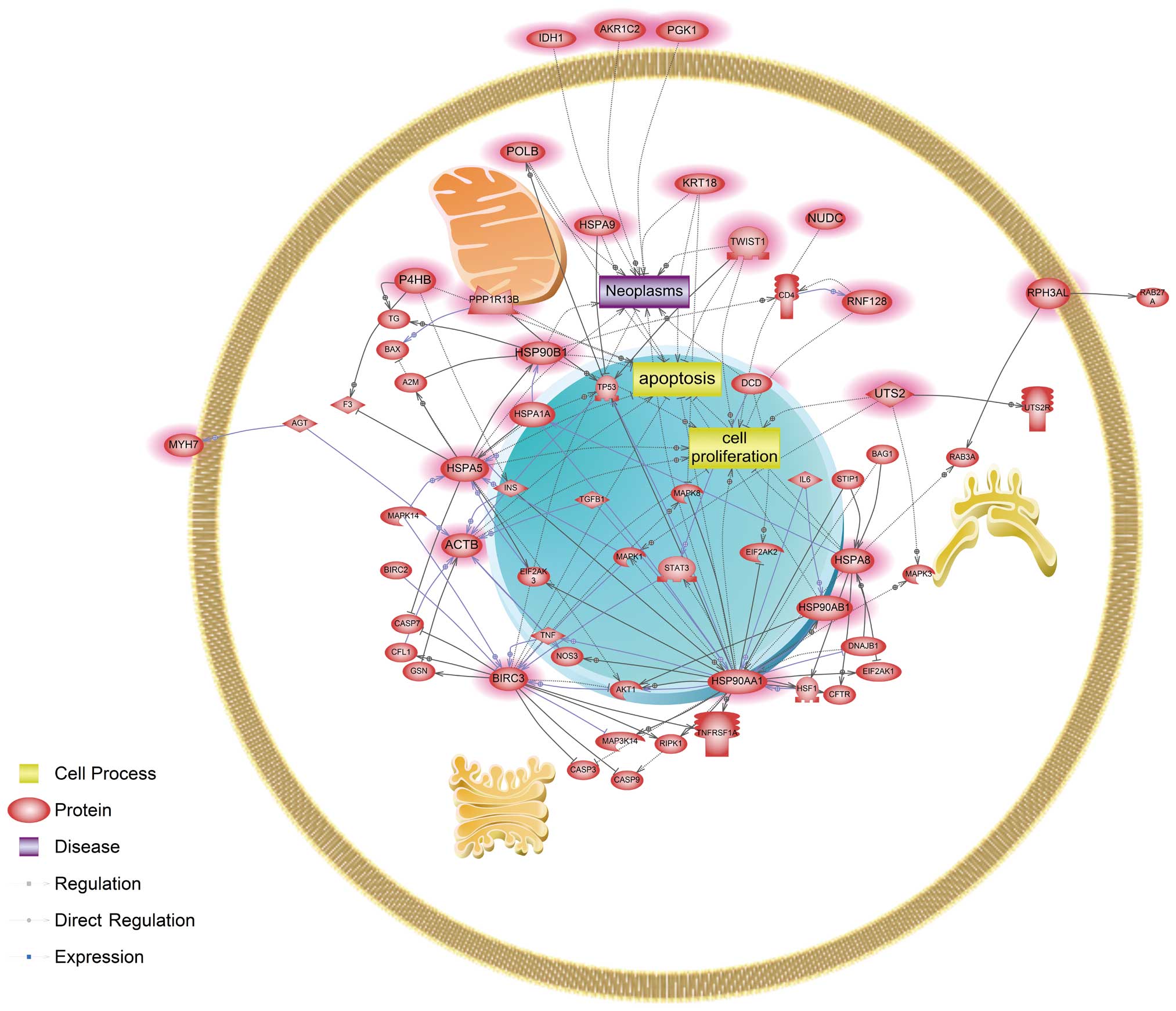
cadmium-exposed cells, signaling networks associated with
apoptosis, cell proliferation and neoplasm were affected. Several
genes, BIRC3, HSPA5, HSPA8 and HSP90AA1, were revealed to be key
modulators in cadmium only exposed cells (Fig. 2). Cells exposed only to nickel were
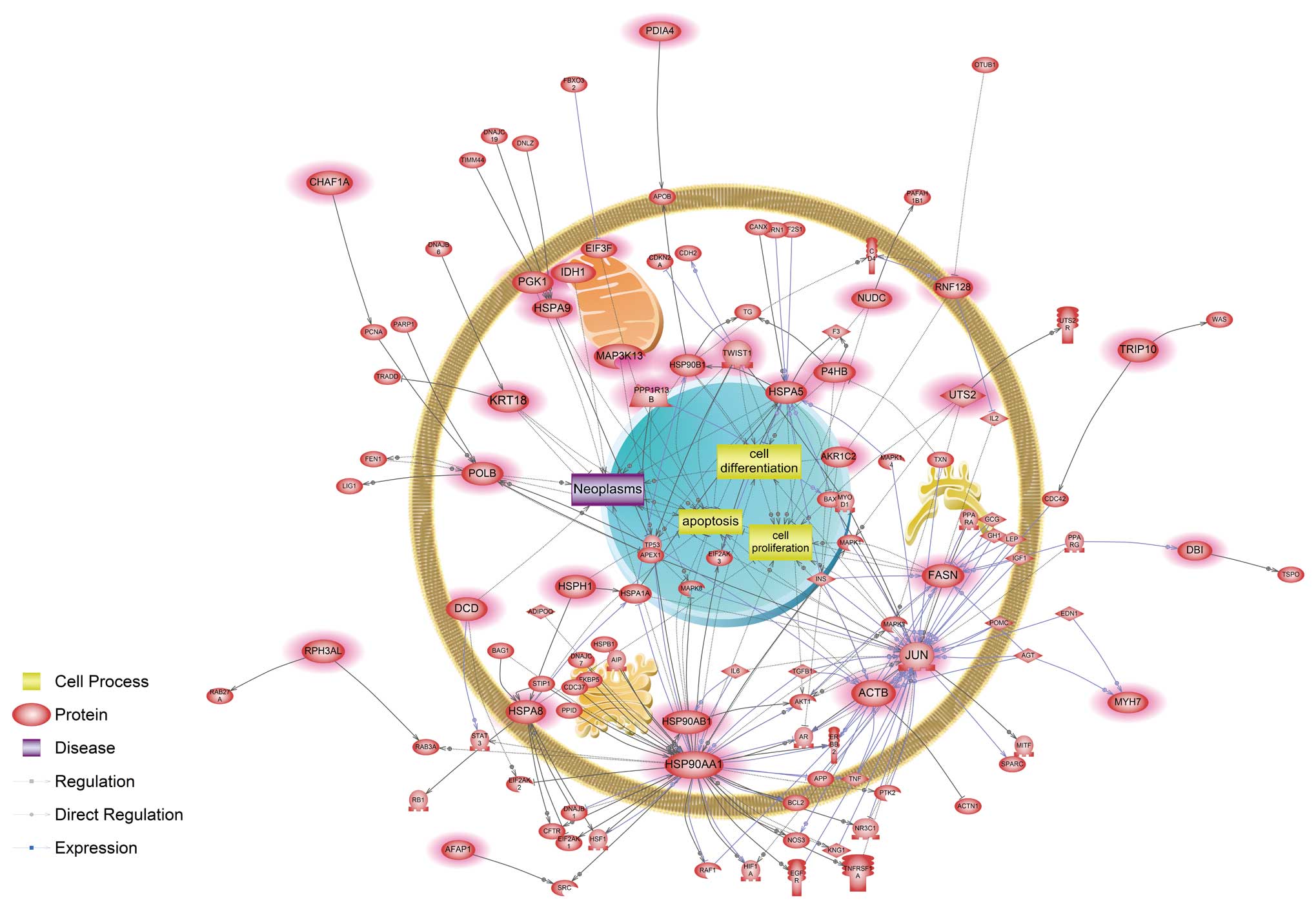
associated with a unique signaling pathway related to apoptosis,
cell differentiation, cell proliferation and neoplasm interacting
with FASN, HSP90AA1, HSP90AB1 and JUN genes (Fig. 3).
Discussion
The mechanisms of toxicity for low-dose exposure to
cadmium and nickel are unclear. Therefore, we comprehensively
explored molecular candidates and the mechanisms of toxicity, both
common and distinct, for both metals using integrative
toxicogenomic and bioinformatic tools. Similarities and differences
in gene subsets, pathways and biological processes in response to
each metal are further discussed.
Using a high-resolution CGH array, we found no
significant difference in recurrent CNV following continuous
low-level exposure to both metals tested when comparing the tenth
passage of the culture to the first. Less than 700 gene entities
with altered expression at the mRNA and protein levels were
identified by gene expression microarray and functional proteomic
analysis after a 24-h exposure to either metal individually. This
small subset of genes exhibiting a very slight to mild extent of
metal-induced profiling change in terms of gene structure might be
attributable to the low-level metal exposure of the cells. A number
of studies have reported an association between DNA copy number
variation and predisposition to several genetic diseases, including
cancers, neurodegenerative disorders and obesity (29,30).
These findings suggest that the significant contribution of a
particular CNV to pathogenesis is involved in an altered dose of
genes within the chromosomal region affected. Furthermore, cadmium
and nickel induce carcinogenesis in diverse experimental models
in vitro and in vivo. However, we found no
significant alteration in recurrent CNV following low-level
exposure to the metals after ten passages, suggesting that such a
metal treatment has an undetectable impact on DNA structural
variations based on aberrant copy number. However, exposure
duration will be prolonged with such low doses in future research,
as ten passages may have been insufficient for CNV to occur in
p53-proficient carcinoma cells.
Alterations in gene and protein expression levels
were analyzed in RKO cells following a 24-h low-dose exposure to
either metals using gene expression microarray and functional
proteomics, respectively. Nineteen transcripts and 69 protein
entities were altered in the cadmium-exposed cells, whereas 629
transcripts and 69 protein entities were differentially expressed
in nickel-exposed cells. Among them, 22 protein entities, including
several members of the heat shock protein (HSP) family, were
commonly altered in the cells treated with either cadmium or
nickel. These proteins might be considered valuable biomarkers for
cadmium and nickel exposure even at the low levels used. These
entities are classified according to their functions, such as
ligands (i.e., UTS2), stress proteins (i.e., ACTB, AKR1C2, DCD,
HSPA1A, HSPA2, HSP90AA1, HSP90B1, KRT18 and MYH7), kinases and
phosphatases (i.e., PGK1 and PPP1R13B, respectively), transcription
factors (i.e., TWIST1), and other enzymes (i.e., AKR1C2, IDH1, P4HB
and RNF128). Our data suggest that the molecular candidates
identified here, including many regulatory factors and stress
proteins, are potential metal carcinogenicity markers, which were
consistently observed in previous studies (3). HSPs are stress-inducible proteins that
fold and unfold other proteins and represent a suite of highly
conserved and broadly distributed proteins in nature (31). HSP expression is regulated by
temperature and other stressors (32). Recent studies have shown that
diverse HSPs are activated by heavy metals including cadmium,
copper, lead and zinc in cell lines, shrimp and the sea star
(31,33,34).
Although our data showed downregulation of HSPs, they can be
suggested as potential carcinogenesis-related genes in response to
low-dose cadmium or nickel. Six genes, including AFAP1, EIF3F,
FASN, HSPA1A, JUN and TRIP10 were distinct in nickel-exposed cells.
Interestingly, EIF3F increased significantly in response to
low-level nickel exposure. EIF3F is a protein complex with at least
13 non-identical subunits (35).
The functions of the individual subunits have not been clarified;
however, a recent study demonstrated that EIF3F is involved in
apoptotic signaling (36). The
EIF3F gene is also differentially expressed by volatile organic
compounds (37); however, no
reports have found a nickel-modulated signaling network involving
EIF3F. Thus, these data are the first results indicating that the
EIF3F gene is dysregulated by subchronic exposure to low-dose
nickel and also suggest that this gene might be involved in
apoptotic signaling. The FASN gene product is a key enzyme for
palmitate biosynthesis into long-chain saturated fatty acids in the
presence of the reduced form of nicotinamide adenine dinucleotide
phosphate (38,39). Additionally, this protein fuses with
estrogen receptor α in some cancer cell lines. Another study
demonstrated alterations in rat liver lipid metabolism induced by
nickel deficiency (40). Thus,
nickel has detrimental impacts on lipid metabolism involving FASN.
JUN, a c-jun oncoprotein, is an essential component of the
activator protein (AP) transcription complex. Upon
heterodimerization with other Jun/Fos members, JUN forms an active
AP-1 complex that regulates expression of many target genes
harboring AP-1-binding DNA elements within their promoters. These
AP-1 target genes are involved in various crucial cellular
functions such as cell cycle progression, migration, proliferation
and apoptosis. JUN is activated upon phosphorylation by
c-jun-N-terminal kinase, which responds to diverse biological
stress signals (41).
Interestingly, JUN both induces and inhibits cellular apoptosis
(42–44).
A number of reports have demonstrated that oxidative
stress, interference with cell proliferation, dysregulation of
oncogenes or tumor-suppressor genes, and an impaired DNA repair
system are predominant mechanisms driven by carcinogenic metal
compounds (3,45,46).
We found that ACTB, HSP90AA1, HSPA5 and HSPA8 acted as key
components of pathways associated with proliferation, apoptosis and
neoplastic processes in response to cadmium and nickel (Fig. 1). The metals also interacted with
important components in multiple signaling pathways crucial for
cell growth, proliferation, survival, cell cycle, drug resistance,
the stress response and carcinogenesis, including AKT1, BAG1, BAX,
CDK1, CFTR, HSF1, TP53, MAPK3, MAPK8, STAT3 and TNF (Fig. 1). Herein, we showed that both
cadmium and nickel influenced key components in the AKT1-mediated
pathway, similar to a recent study showing that cadmium-induced
reactive oxygen species activate the Akt/mTOR pathway by activating
the positive regulator PI3K, resulting in neuronal apoptosis
(47). These findings suggest that
particular stress-inducible proteins, protein kinases, or
transcription factors may be prominent biomarkers of subchronic
exposure to cadmium or nickel and may be used to develop preventive
approaches for associated disorders and/or diseases; however, the
underlying mechanisms of toxicity must be identified. Furthermore,
targeting regulatory component crosstalk among the
mitogen-activated protein kinase pathways representing a cascade of
sequential phosphorylation events might offer new opportunities to
develop novel anticancer agents designed to be target-specific
chemotherapeutic drugs. In addition to common responses, we also
examined the mechanisms of toxicity that were unique to each metal
compound. Our pathway analysis results of cadmium-treated cells
found that the caspase (CASP)-associated pathway was a mechanism
unique to cadmium involving CASP3, CASP7 and CASP9 (Fig. 2). Gene expression changes in
nickel-exposed cells were associated with a hypoxic response. Our
data showed that HSP90AA1, one of the main modulators, directly
interacted with hypoxia inducible factor-1α (HIF-1α) (Fig. 3). HIF-1α is a transcription factor
that induces the transcription of genes involved in glycolysis,
glucose transport, apoptosis and other cellular processes as a
result of a change in intracellular oxygen concentration (45). PGK1, encoding phosphoglycerate
kinase 1, was found to be one of the HIF-1α targets to be
dysregulated in nickel-exposed cells, suggesting that nickel
induces a hypoxic-like response through HIF-1α, which was
consistent with the results of a comparative genomic expression
analysis in rat liver-derived cells exposed to nickel, cadmium, or
chromium (46). Dysregulation of
HIF-1α interferes with cellular energy metabolism, such as
glycolysis, causing cells to shift toward non-oxidative forms of
ATP production and altering production of glycolytic enzymes and
glucose transporters (48).
HSP90AA1 directly interacted with the androgen receptor (AR) based
on the nickel-responsive pathway analysis. An abundance of evidence
has revealed that the AR forms a heterodimer complex with Hsp90,
resulting in a stable, unbound AR (49). Growth of a normal and neoplastic
prostate is mediated by the AR, a ligand-dependent transcription
factor activated by high affinity androgen binding. The AR is
highly expressed in recurrent prostate cancer cells. Here, HIF-1α
interacted with the AR in response to subchronic nickel exposure
(Fig. 3). Interestingly, we also
found that HSP90AA1 was involved in the BCL2-related apoptotic
pathway uniquely in response to nickel exposure (Fig. 3), whereas it interacted with several
CASP genes functioning in CASP-associated apoptotic signaling
unique to cadmium exposure alone (Fig.
2). These data suggest that interference with the apoptotic
process was likely common to both metals, but that the responsive
interaction network with molecular targets was distinct in response
to each metal. These findings support that HSP90 contributes to the
pathogenesis of chronic diseases, including cancers, rheumatoid and
arthritis, particularly through apoptosis (50,51).
Targeting HSP90 has emerged as a potential avenue for therapeutic
intervention. HSP90 is a molecular chaperone required for the
post-translational stability and function of numerous key signal
transduction proteins, termed ‘client’ proteins (52,53). A
number of these client proteins have been causally implicated in
the pathogenesis of prostate cancer, including AR, HER2, AKT and
RAF1 (54–56). The interaction of these proteins
with HSP90 regulates their half-life. We revealed from our pathway
analysis that HSP90AA1 directly interacted with Src encoding Src
protein-tyrosine kinase. Src is a member of a large family of
structurally related kinases, many of which are expressed in highly
differentiated cell types (57,58).
Src and its family members play a key regulatory role transducing
signals from cell surface receptors and coupling receptors with the
cytoplasmic signaling machinery involved in several cellular
processes, such as cell growth, differentiation, migration,
proliferation, survival and specialized cell signaling.
Additionally, JUN and FASN were shown here to be main modulators in
unique nickel-responsive signaling pathways (Fig. 3) and their products have been
implicated in tumor initiation and development of various tissues,
as evidenced from cellular and/or epidemiological studies (59–61).
JUN has a regulatory role in HIF-1α function through these
interactions. Stabilizing HIF-1α is dependent on c-Jun domains for
DNA binding and heterodimerization (62). Indeed, HIF-1α is activated by
interacting with c-Jun, allowing increased expression of vascular
endothelial growth factor, which is a signal protein involved in
stimulating vasculogenesis and angiogenesis (63). JUN was regulated by interaction with
the AR. Intriguingly, JUN and FASN were also governed by
interacting with AKT1, INS and LEP in nickel-responsive signaling
pathways. We also found that JUN was controlled by an interaction
with APEX1, a redox-sensitive repair protein that plays an
essential role in the base excision repair (BER) system. Apart from
APEX1, we found that many other proteins were related to the DNA
damage response and DNA repair particularly in the BER pathway;
FEN1, PARP1, PCNA and POLB were uniquely responsible for the
nickel-associated network. JUN interacted with MITF, whose product
is called microphthalmia-associated transcription factor, which has
a key role in the development, survival and function of particular
cell types, such as melanocytes, retinal pigment epithelial cells
and osteoclasts (64). FASN was
found to regulate ERBB2 or HER2 expression and vice versa. Previous
studies have consistently reported FASN-mediated regulation of
ERBB2 expression and a suppressive effect on ERBB2 overexpression
by inhibiting FASN-encoded fatty acid synthase in cancer cells
(65). Other evidence supports that
ERBB2, previously identified in preneoplastic breast lesions,
upregulates FASN expression (66).
Furthermore, FASN overexpression increases ERBB1 or epidermal
growth factor receptor (EGFR) and ERBB2 protein expression levels
as well as tyrosine phosphorylation (67). Treatment of EGFR and ERBB2 with
kinase inhibitors suggests that both EGFR and ERBB2 are activated
by regulating EGF-mediated FASN expression (68). Interestingly, nickel seemed to
uniquely perturb the cell differentiation process associated with
expression and function of both common (HSP90AA1, HSP90B1, HSPA5,
NUDC and TWIST1) and distinct (JUN) components (Fig. 3). This result suggests that although
a similar subset of genes was altered upon exposure to cadmium and
nickel individually, they might contribute to unique network
interactions and cellular processes (Figs. 1–3).
This might be attributable to the combined effects of the
expression patterns and regulation of the components involved in
different signaling pathways. Taken together, our results
identified valuable biomarkers and distinctive signaling networks
in response to subchronic low-dose exposure of carcinogenic metals
cadmium and nickel.
Acknowledgements
This study was supported by Korea Ministry of
Enviroment as ‘The Ecoinnovation Project’ (412-112-011).
References
|
1
|
Andrew AS, Warren AJ, Barchowsky A, et al:
Genomic and proteomic profiling of responses to toxic metals in
human lung cells. Environ Health Perspect. 111:825–835.
2003.PubMed/NCBI
|
|
2
|
Arita A and Costa M: Epigenetics in metal
carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics.
1:222–228. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koedrith P and Seo YR: Advances in
carcinogenic metal toxicity and potential molecular markers. Int J
Mol Sci. 12:9576–9595. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fabbri M, Urani C, Sacco MG, Procaccianti
C and Gribaldo L: Whole genome analysis and microRNAs regulation in
HepG2 cells exposed to cadmium. ALTEX. 29:173–182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Talio MC, Luconi MO, Masi AN and Fernandez
LP: Cadmium monitoring in saliva and urine as indicator of smoking
addiction. Sci Total Environ. 408:3125–3132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martelli A, Rousselet E, Dycke C, Bouron A
and Moulis JM: Cadmium toxicity in animal cells by interference
with essential metals. Biochimie. 88:1807–1814. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bertin G and Averbeck D: Cadmium: cellular
effects, modifications of biomolecules, modulation of DNA repair
and genotoxic consequences (Review). Biochimie. 88:1549–1559. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hartwig A: Mechanisms in cadmium-induced
carcinogenicity: recent insights. Biometals. 23:951–960. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Waalkes MP: Cadmium carcinogenesis. Mutat
Res. 533:107–120. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Karin M, Haslinger A, Holtgreve H, et al:
Characterization of DNA sequences through which cadmium and
glucocorticoid hormones induce human metallothionein-IIA gene.
Nature. 308:513–519. 1984. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schmidt CJ, Jubier MF and Hamer DH:
Structure and expression of two human metallothionein-I isoform
genes and a related pseudogene. J Biol Chem. 260:7731–7737.
1985.PubMed/NCBI
|
|
12
|
Williams GT and Morimoto RI: Maximal
stress-induced transcription from the human HSP70 promoter requires
interactions with the basal promoter elements independent of
rotational alignment. Mol Cell Biol. 10:3125–3136. 1990.
|
|
13
|
Hiranuma K, Hirata K, Abe T, et al:
Induction of mitochondrial chaperonin, hsp60, by cadmium in human
hepatoma cells. Biochem Biophys Res Commun. 194:531–536. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takeda K, Ishizawa S, Sato M, Yoshida T
and Shibahara S: Identification of a cis-acting element that
is responsible for cadmium-mediated induction of the human heme
oxygenase gene. J Biol Chem. 269:22858–22867. 1994.PubMed/NCBI
|
|
15
|
Jin P and Ringertz NR: Cadmium induces
transcription of proto-oncogenes c-jun and c-myc in
rat L6 myoblasts. J Biol Chem. 265:14061–14064. 1990.PubMed/NCBI
|
|
16
|
Epner DE and Herschman HR: Heavy metals
induce expression of the TPA-inducible sequence (TIS) genes. J Cell
Physiol. 148:68–74. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Garcia-Morales P, Saceda M, Kenney N, et
al: Effect of cadmium on estrogen receptor levels and
estrogen-induced responses in human breast cancer cells. J Biol
Chem. 269:16896–16901. 1994.PubMed/NCBI
|
|
18
|
Salnikow K and Zhitkovich A: Genetic and
epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis:
nickel, arsenic, and chromium. Chem Res Toxicol. 21:28–44. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Doll R, Morgan LG and Speizer FE: Cancers
of the lung and nasal sinuses in nickel workers. Br J Cancer.
24:623–632. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kerckaert GA, LeBoeuf RA and Isfort RJ:
Use of the Syrian hamster embryo cell transformation assay for
determining the carcinogenic potential of heavy metal compounds.
Fundam Appl Toxicol. 34:67–72. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuper CF, Woutersen RA, Slootweg PJ and
Feron VJ: Carcinogenic response of the nasal cavity to inhaled
chemical mixtures. Mutat Res. 380:19–26. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miller AC, Mog S, McKinney L, et al:
Neoplastic transformation of human osteoblast cells to the
tumorigenic phenotype by heavy metal-tungsten alloy particles:
induction of genotoxic effects. Carcinogenesis. 22:115–125. 2001.
View Article : Google Scholar
|
|
23
|
Fletcher GG, Rossetto FE, Turnbull JD and
Nieboer E: Toxicity, uptake, and mutagenicity of particulate and
soluble nickel compounds. Environ Health Perspect. 102(Suppl 3):
69–79. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kargacin B, Klein CB and Costa M:
Mutagenic responses of nickel oxides and nickel sulfides in Chinese
hamster V79 cell lines at the xanthine-guanine phosphoribosyl
transferase locus. Mutat Res. 300:63–72. 1993. View Article : Google Scholar
|
|
25
|
Liao CM, Shen HH, Chen CL, et al: Risk
assessment of arsenic-induced internal cancer at long-term low dose
exposure. J Hazard Mater. 165:652–663. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Satarug S and Moore MR: Adverse health
effects of chronic exposure to low-level cadmium in foodstuffs and
cigarette smoke. Environ Health Perspect. 112:1099–1103. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park JYS and Seo YR: The protective role
of Nrf2 in cadmium-induced DNA damage. Mol Cell Tox. 7:61–66. 2011.
View Article : Google Scholar
|
|
28
|
Kim HL and Seo YR: Synergistic genotoxic
effect between gene and environmental pollutant: oxidative DNA
damage induced by thioredoxin reductase 1 silencing under nickel
treatment. Mol Cell Tox. 7:251–257. 2011. View Article : Google Scholar
|
|
29
|
Pollack JR, Sørlie T, Perou CM, et al:
Microarray analysis reveals a major direct role of DNA copy number
alteration in the transcriptional program of human breast tumors.
Proc Natl Acad Sci USA. 99:12963–12968. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ledet EM, Hu X, Sartor O, Rayford W, Li M
and Mandal D: Characterization of germline copy number variation in
high-risk African American families with prostate cancer. Prostate.
73:614–623. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qian Z, Liu X, Wang L, et al: Gene
expression profiles of four heat shock proteins in response to
different acute stresses in shrimp, Litopenaeus vannamei.
Comp Biochem Physiol C Toxicol Pharmacol. 156:211–220. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
De Maio A: Heat shock proteins: facts,
thoughts, and dreams. Shock. 11:1–12. 1999.
|
|
33
|
Leal RB, Posser T, Rigon AP, et al:
Cadmium stimulates MAPKs and Hsp27 phosphorylation in bovine
adrenal chromaffin cells. Toxicology. 234:34–43. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matranga V, Pinsino A, Randazzo D,
Giallongo A and Dubois P: Long-term environmental exposure to
metals (Cu, Cd, Pb, Zn) activates the immune cell stress response
in the common European sea star (Asterias rubens). Mar
Environ Res. 76:122–127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dong Z and Zhang JT: Initiation factor
eIF3 and regulation of mRNA translation, cell growth, and cancer.
Crit Rev Oncol Hematol. 59:169–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shi J, Feng Y, Goulet AC, et al: The
p34cdc2-related cyclin-dependent kinase 11 interacts with the p47
subunit of eukaryotic initiation factor 3 during apoptosis. J Biol
Chem. 278:5062–5071. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yim WC, Lee MB and Kwon Y:
Cross-experimental analysis of microarray gene expression datasets
for in silico risk assessment of TiO2
nano-particles. Mol Cell Tox. 8:229–239. 2012. View Article : Google Scholar
|
|
38
|
Hillgartner FB, Salati LM and Goodridge
AG: Physiological and molecular mechanisms involved in nutritional
regulation of fatty acid synthesis. Physiol Rev. 75:47–76.
1995.PubMed/NCBI
|
|
39
|
Smith S, Witkowski A and Joshi AK:
Structural and functional organization of the animal fatty acid
synthase. Prog Lipid Res. 42:289–317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stangl GI and Kirchgessner M: Nickel
deficiency alters liver lipid metabolism in rats. J Nutr.
126:2466–2473. 1996.PubMed/NCBI
|
|
41
|
Kyriakis JM: Signaling by the germinal
center kinase family of protein kinases. J Biol Chem.
274:5259–5262. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ham J, Babij C, Whitfield J, et al: A
c-Jun dominant negative mutant protects sympathetic neurons against
programmed cell death. Neuron. 14:927–939. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Behrens A, Sibilia M and Wagner EF:
Amino-terminal phosphorylation of c-Jun regulates stress-induced
apoptosis and cellular proliferation. Nat Genet. 21:326–329. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Eferl R, Ricci R, Kenner L, et al: Liver
tumor development. c-Jun antagonizes the proapoptotic activity of
p53. Cell. 112:181–192. 2003.PubMed/NCBI
|
|
45
|
Beyersmann D and Hartwig A: Carcinogenic
metal compounds: recent insight into molecular and cellular
mechanisms. Arch Toxicol. 82:493–512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Permenter MG, Lewis JA and Jackson DA:
Exposure to nickel, chromium, or cadmium causes distinct changes in
the gene expression patterns of a rat liver derived cell line. PLoS
One. 6:e277302011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen L, Xu B, Liu L, et al: Cadmium
induction of reactive oxygen species activates the mTOR pathway,
leading to neuronal cell death. Free Radic Biol Med. 50:624–632.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gordan JD, Thompson CB and Simon MC: HIF
and c-Myc: sibling rivals for control of cancer cell metabolism and
proliferation. Cancer Cell. 12:108–113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
De Leon JT, Iwai A, Feau C, et al:
Targeting the regulation of androgen receptor signaling by the heat
shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc
Natl Acad Sci USA. 108:11878–11883. 2011.
|
|
50
|
Mahalingam D, Swords R, Carew JS, Nawrocki
ST, Bhalla K and Giles FJ: Targeting HSP90 for cancer therapy. Br J
Cancer. 100:1523–1529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hashiramoto A, Murata M, Kawazoe T, et al:
Heat shock protein 90 maintains the tumour-like character of
rheumatoid synovial cells by stabilizing integrin-linked kinase,
extracellular signal-regulated kinase and protein kinase B.
Rheumatology. 50:852–861. 2011. View Article : Google Scholar
|
|
52
|
Hahn JS: The Hsp90 chaperone machinery:
from structure to drug development. BMB Rep. 42:623–630. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Saporita AJ, Ai J and Wang Z: The Hsp90
inhibitor, 17-AAG, prevents the ligand-independent nuclear
localization of androgen receptor in refractory prostate cancer
cells. Prostate. 67:509–520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Trepel J, Mollapour M, Giaccone G and
Neckers L: Targeting the dynamic HSP90 complex in cancer. Nat Rev
Cancer. 10:537–549. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Vanaja DK, Mitchell SH, Toft DO and Young
CY: Effect of geldanamycin on androgen receptor function and
stability. Cell Stress Chaperones. 7:55–64. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Parsons SJ and Parsons JT: Src family
kinases, key regulators of signal transduction. Oncogene.
23:7906–7909. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Roskoski R Jr: Src kinase regulation by
phosphorylation and dephosphorylation. Biochem Biophys Res Commun.
331:1–14. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ishimura N, Amano Y, Sanchez-Siles AA, et
al: Fatty acid synthase expression in Barrett’s esophagus:
implications for carcinogenesis. J Clin Gastroenterol. 45:665–672.
2011.
|
|
60
|
Beyersmann D: Effects of carcinogenic
metals on gene expression. Toxicol Lett. 127:63–68. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wu HC, Yang CY, Hung DZ, et al: Nickel(II)
induced JNK activation-regulated mitochondria-dependent apoptotic
pathway leading to cultured rat pancreatic β-cell death.
Toxicology. 289:103–111. 2011.PubMed/NCBI
|
|
62
|
Yu B, Miao ZH, Jiang Y, et al: c-Jun
protects hypoxia-inducible factor-1α from degradation via its
oxygen-dependent degradation domain in a nontranscriptional manner.
Cancer Res. 69:7704–7712. 2009.
|
|
63
|
Duyndam MC, Hulscher ST, van der Wall E,
Pinedo HM and Boven E: Evidence for a role of p38 kinase in
hypoxia-inducible factor 1-independent induction of vascular
endothelial growth factor expression by sodium arsenite. J Biol
Chem. 278:6885–6895. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ogihara H, Morii E, Kim DK, Oboki K and
Kitamura Y: Inhibitory effect of the transcription factor encoded
by the mutant mi microphthalmia allele on transactivation of
mouse mast cell protease 7 gene. Blood. 97:645–651. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Menendez JA, Vellon L, Mehmi I, et al:
Inhibition of fatty acid synthase (FAS) suppresses HER2/neu
(erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad
Sci USA. 101:10715–10720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Vazquez-Martin A, Colomer R, Brunet J,
Lupu R and Menendez JA: Overexpression of fatty acid synthase gene
activates HER1/HER2 tyrosine kinase receptors in human breast
epithelial cells. Cell Prolif. 41:59–85. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lin VC, Chou CH, Lin YC, et al: Osthole
suppresses fatty acid synthase expression in HER2-overexpressing
breast cancer cells through modulating Akt/mTOR pathway. J Agric
Food Chem. 58:4786–4793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Alli PM, Pinn ML, Jaffee EM, McFadden JM
and Kuhajda FP: Fatty acid synthase inhibitors are chemopreventive
for mammary cancer in neu-N transgenic mice. Oncogene. 24:39–46.
2005. View Article : Google Scholar : PubMed/NCBI
|