Introduction
Gastric cancer (GC) is the fourth most frequent
malignancy and the second leading cause of cancer mortality
worldwide (1). In China, most of
the patients are identified at the advanced stage which leads to
dismal prognosis. Radical surgery is the only curative therapy and
chemotherapy serves as common strategy for late stage patients.
Patients with GC, however, are not particularly sensitive to
conventional chemotherapeutic drugs. Thus, it is of great interest
to find new approaches, and targeted manipulation of apoptosis is a
research hot spot (2).
TRAIL, a member of tumor necrosis factor (TNF) super
family, selectively triggers cell death in transformed cells while
causing virtually no toxic towards normal cells (3). TRAIL initiates apoptotic signals via
binding to two cell surface death receptors (DRs), DR4 (also known
as TRAIL-R1) and DR5 (also known as TRAIL-R2), leading to receptor
aggregation and recruitment of Fas-associated death domain (FADD)
followed by procaspase-8 (4). The
apoptotic process then follows two signaling pathways. In type I
cells, activated caspase-8 enables the autocatalytic cleavage of
caspase cascade triggers apoptosis. While in type II cells,
caspase-8 additionally triggers mitochondrial apoptosis pathway by
activating cleavage of Bid, which induces the release of cytochrome
c from mitochondria and in turn activates caspase-9 to
finally execute cell death (5).
Drugs targeting TRAIL pathway, including recombinant TRAIL and
agonistic antibodies have been demonstrated with robust anticancer
activity in a number of preclinical studies (6–8).
Despite the attractive tumoricidal potential,
TRAIL-based therapy is greatly hampered by its resistance, a major
obstacle to clinical application (8–10). The
most basic cause is dysfunction of death receptors due to
hyper-methylation (11), mutation
(12) and loss of cell surface
expression (13). The defects in
caspase protein (14) and
overexpression of pro-survival proteins, such as IAP family and
anti-apoptotic Bcl-2 family members (15–18),
are also linked to TRAIL sensitivity. In addition, more recent
findings suggested that cell survival signals, including
mitogen-activated protein kinase (MAPK) pathway,
phosphatidylinositol 3-kinase/Akt pathway, and transcription factor
NF-κB played pivotal roles in regulation of TRAIL signaling
(19–22). Previous studies demonstrated that
TRAIL resistance could be alleviated or even reversed by
combination therapy (23,24). PTX, one of the cytoskeletal drugs
targeting tubulin, is commonly used in advanced GC treatment. It
inhibits tumor cell proliferation through stabilizing microtubule
network and inhibiting microtubule dynamics (25). PTX has been explored as sensitizing
agent towards TRAIL in some cancer types (26–28),
yet, the molecular mechanisms were not fully elucidated.
In the present study, we demonstrate that combined
treatment of PTX and TRAIL significantly boosted apoptosis of
TRAIL-resistant GC cells both in vitro and in vivo.
The molecular mechanisms underlying the synergism involved enhanced
activation of caspases, upregulation of DRs and downregulation of
anti-apoptotic proteins. Moreover, we present herein the inhibitory
effect of MAPKs in TRAIL-induced apoptosis in GC and reveal for the
first time that TRAIL-augmenting effect of PTX was partly due to
suppression of the MAPK pathway.
Materials and methods
Cell culture and reagents
GC cell lines AGS, NCI-N87, SNU-1 and SNU-16 were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). MKN28 and NUGC3 were purchased from Health
Science Research Resources Bank (Tokyo, Japan), and SGC-7901,
BGC-823 and MGC-803 were obtained from Cell Research Institute
(Shanghai, China). All cells were maintained in Dulbecco's modified
Eagle's medium (DMEM; Gibco-BRL, Carlsbad, CA, USA) containing 10%
fetal bovine serum (FBS) and penicillin/streptomycin at 37°C with
5% CO2. Recombinant human TRAIL was purchased from
PeproTech EC Ltd. (London, UK). The small molecule inhibitors U0126
(#S1102), SP600125 (#S1460) and SB202190 (#S1077) were obtained
from Selleck Chemicals (Houston, TX, USA). The chemotherapeutic
drug PTX was from Beijing Union Pharmaceutical Factory (Beijing,
China).
Cell viability assay
Cell viability was assessed using Cell Counting
kit-8 assay (CCK-8) (Dojindo Molecular Technologies, Kumamoto,
Japan). Briefly, cells were seeded onto 96-well plates at a density
of 3–5×103 cells/well and allowed to attach for 24 h.
The cells were incubated with indicated concentrations of TRAIL
and/or PTX for 24 h. After incubation, CCK-8 solution (10
µl) was added to each well and absorbance was measured at
450 nm using multiscan spectrum.
Flow cytometry for apoptosis analysis and
expression of cell surface DRs
For cell apoptosis assay, cells (3×105)
were seeded onto 35-mm2 culture dishes and treated with
TRAIL and/or PTX for 24 h as indicated in the figure legends. Then,
the cells were stained using an Annexin V/PI double staining kit
(Dojindo Molecular Technologies) according to the manufacturer's
protocol.
For cell surface expression analysis, cells
(1×106) treated with PTX or vehicle for 24 h were
incubated with allophycocyanin (APC)-conjugated anti-DR4 and
anti-DR5 antibody, or isotype control (BioLegend, Inc., San Diego,
CA, USA) for 30 min at 4°C, and analyzed by flow cytometery (BD
Accuri C6; BD Biosciences, San Jose, CA, USA). Positive cells were
identified and measured by subtracting relative isotype control
values.
Western blot assay
Total proteins were extracted using RIPA lysis
buffer (Beijing Solarbio Science and Technology Co., Ltd., Beijing,
China) with protease inhibitors and phosphatase inhibitors (Beijing
Solarbio Science and Technology). Protein concentration was
quantified using the Bradford method (Pierce, Rockford, IL, USA).
Equivalent amount of protein (20 µg) was separated by 10%
SDS-PAGE gels and transferred onto PVDF membranes (Millipore,
Billerica, MA, USA). The membranes were blocked in 5% non-fat milk
in TBS-T for 1 h and incubated with primary antibodies overnight at
4°C, followed by incubation with HRP-conjugated secondary
antibodies. Signals were detected using chemiluminescent agents
(Pierce). Primary antibodies of DR4 and GAPDH were from Abcam
(Cambridge, MA, USA), and DR5 was from Sigma-Aldrich (St. Louis,
MO, USA). Other primary antibodies purchased from Cell Signaling
Technology (Danvers, MA, USA) were as follows: caspase-3 (#9662),
cleaved-caspase-3 (#9664), caspase-7 (#9492), cleaved-caspase-7
(#8438), caspase-8 (#9746), caspase-9 (#9502), cleaved-caspase-9
(#7237), PARP (#9542), cleaved-PARP (#5625), Bid (#2002), C-IAP1
(#7065), C-IAP2 (#3130), XIAP (#2045), Livin (#5471), Bcl-xL
(#2764), Bcl-2 (#2870), Mcl-1 (#5453), ERK (#4695),
phosphorylated-ERK (#4370), JNK (#9258), phosphorylated-JNK
(#4668), p38 (#8690), phosphorylated-p38 (#4511).
siRNA transfection
C-IAP1 siRNA and scrambled siRNA were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cells
(1×105) in 6-well plates were incubated for 24 h and
then transfected with siRNA (100 nmol/l) and Lipofectamine 2000
reagent (Invitrogen, Carlsbad, CA, USA) in Opti-MEM without serum,
according to the manufacturer's specifications. After 6 h, the
medium was replaced with fresh medium. Forty-eight hours after
transfection, C-IAP1 knockdown effect was verified by western
blotting.
In vivo xenograft study
Animal studies were carried out in strict adherence
with institutional guidelines. SGC-7901 cells (2×106/200
µl per mouse) were subcutaneously injected into the right
hind legs of 6–8 week-old female nude mice. When tumors volume
reached 50–100 mm3, the mice were randomized to 4 groups
and dosing was initiated. They were: i) control (vehicle only); ii)
TRAIL (100 µg/kg intratumoral injection); iii) PTX (10 mg/kg
i.p.); and iv) the sequential administration of PTX (10 mg/kg i.p.)
followed by TRAIL (100 µg/kg intratumoral injection) after
24 h. All groups were treated once every 3 days for 18 days. The
tumor size and weight were monitored three times a week. Tumor
volume (V) was calculated as V = 0.5 × length × width2.
Tumor growth inhibition (TGI) was assessed in accordance with the
formula [1− (T−T0)/(C−C0)] × 100, where T and
T0 were the mean tumor volumes at the end of the drug
administration and day 0, respectively for treated group, and
C−C0 were those for vehicle control group.
Statistical analysis
All the values are presented as mean ± SD of three
independent experiments. The statistical significance among
experimental groups was assessed by two-sided Student's t-test.
P<0.05 were considered to be significant. All statistical
analyses were performed using GraphPad Prism 5.0 software.
Results
Response of GC cells to TRAIL and
expression of TRAIL receptors
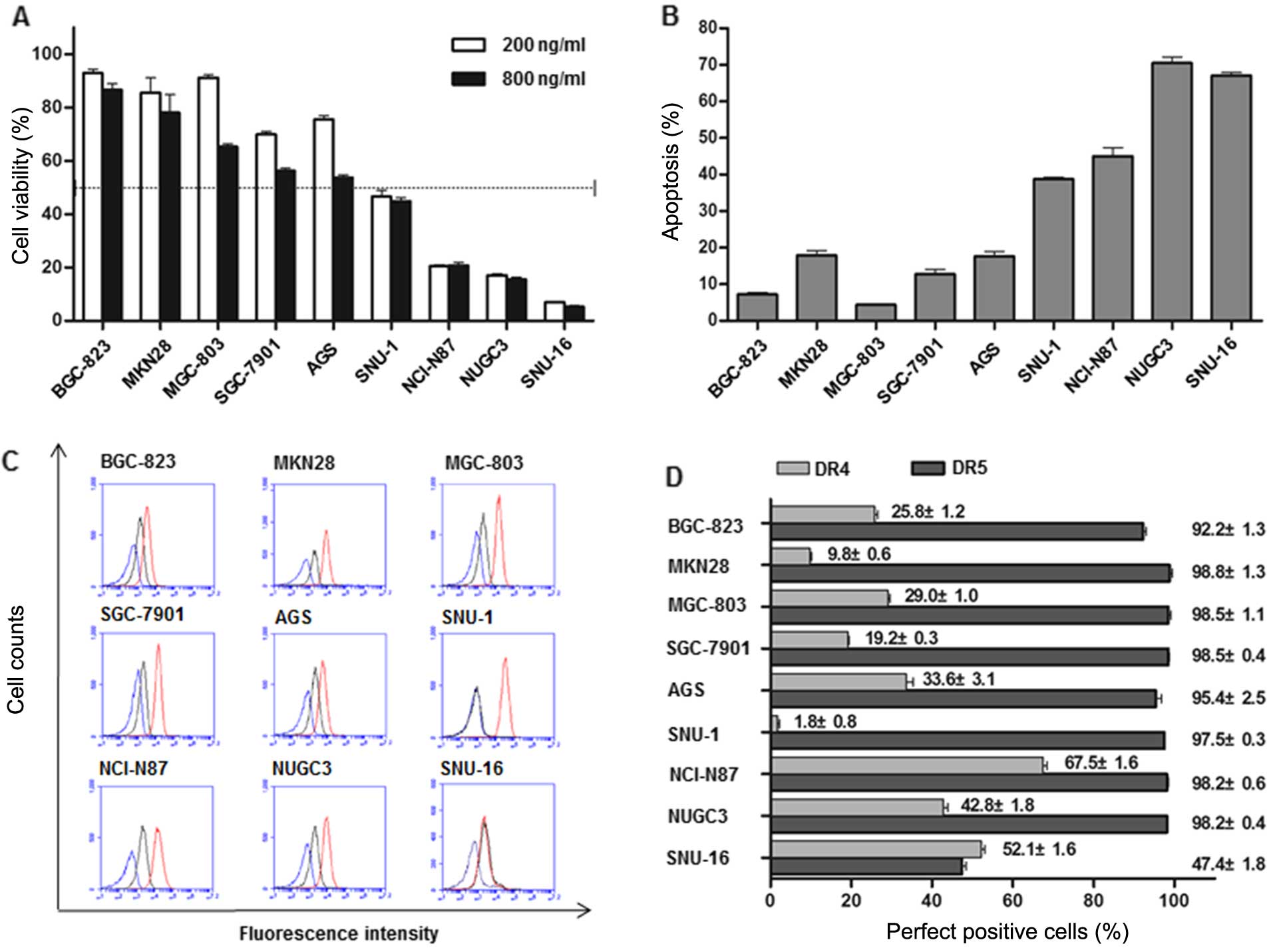
First, we analyzed the effects of TRAIL on cell
viability in 9 GC cells using CCK-8 assay. As shown in Fig. 1A, GC cells had varying degree of
sensitivity to TRAIL. SNU-16, NUGC3, NCI-N87 and SNU-1 were defined
as sensitive cells, in which >50% growth inhibition was achieved
when treated with TRAIL (200 ng/ml) for 24 h. By contrast, the
growth inhibition rates of TRAIL-resistant cells (BGC-823, MKN28,
MGC-803, SGC-7901 and AGS) were <50% even under condition of
elevated concentration or prolonged exposure time indicating an
intrinsic resistance mechanisms in these cells. TRAIL-induced
apoptosis was also confirmed by Annexin V/PI double staining in the
same cell lines (Fig. 1B). To
determine whether TRAIL pathway was intact, we measured cell
surface expression of DR4 and DR5 (Fig.
1C and D). Flow cytometric analysis revealed a predominant
expression of DR5 in all these cells except SNU-16. DR4, however,
exhibited diverse levels in 9 cell lines and the top three
sensitive cells (SNU-16, NUGC3 and NCI-N87) expressed the highest
levels.
PTX and TRAIL act synergistically to
inhibit GC cell growth
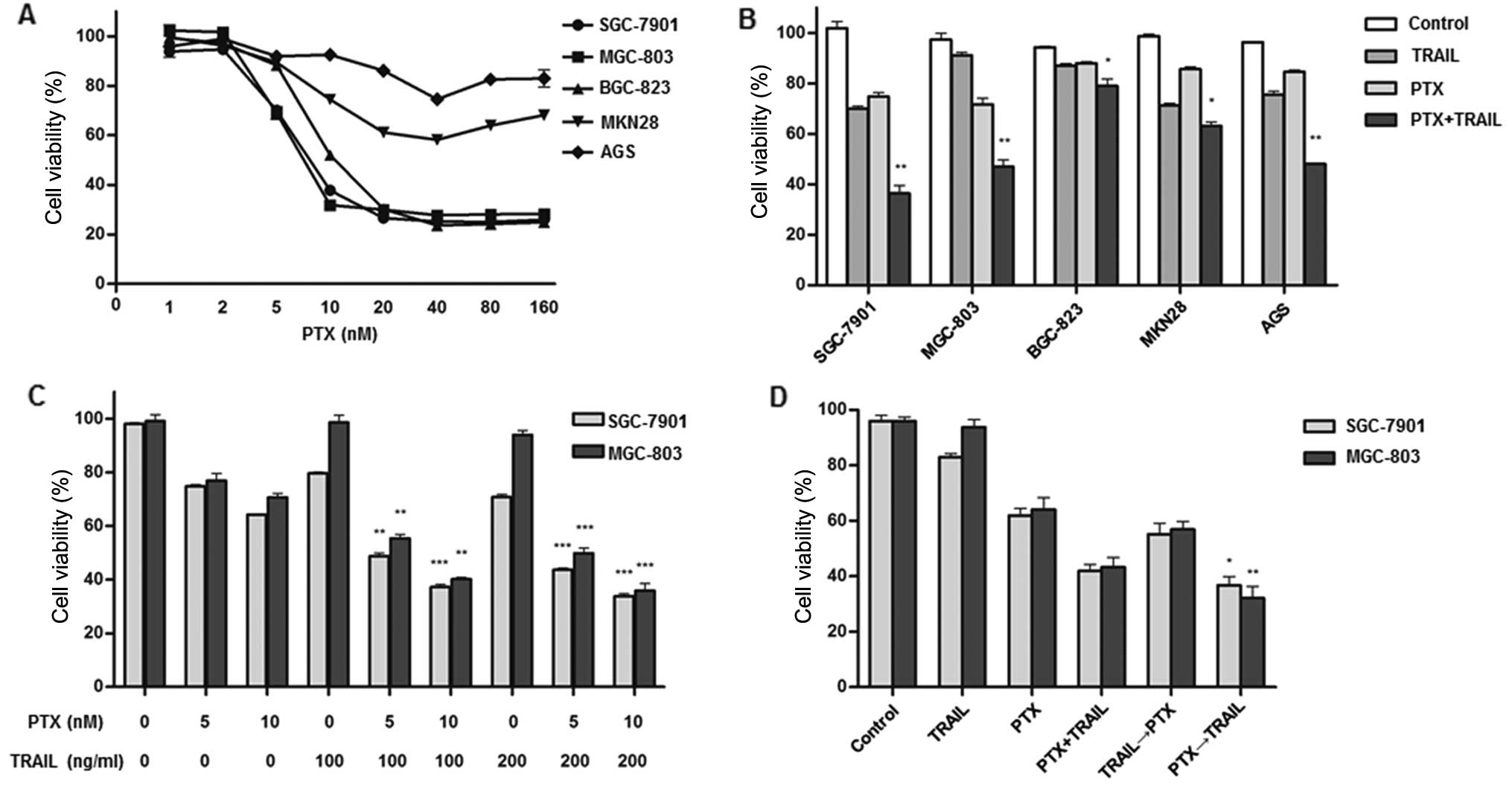
PTX exhibited growth inhibiting effect in
TRAIL-resistant cell lines (Fig.
2A). We attempted to investigate whether PTX augments
TRAIL-inducing apoptosis and found a significant decrease on cell
viability in combination group compared with TRAIL or PTX alone in
all 5 resistant cell lines (P<0.05) (Fig. 2B). To confirm this synergism,
SGC-7901 and MGC-803 cells were cultured with different
combinations of TRAIL (100 and 200 ng/ml) and PTX (5 and 10 nM).
The results indicated that PTX promoted TRAIL-mediated cytotoxic
effect in a dose-dependent manner (Fig.
2C). Considering changes in the order of drug exposure could
enhance cell death (29) and
sequential application of PTX followed by other anticancer
chemicals is common in GC treatment (30), we tested the possibility of
sequential administration of PTX and TRAIL. Cells were
pre-incubated with PTX alone for 12 h and subsequently treated with
TRAIL for an additional 12 h. Reduction of cell viability was more
distinct in the sequential exposure group compared with
simultaneous administration group (Fig.
2D), indicating that pre-treatment of PTX rendered cancer cell
more vulnerable to TRAIL.
PTX sensitizes GC cells to TRAIL-induced
apoptosis by activating caspase-dependent mitochondrial apoptotic
pathway
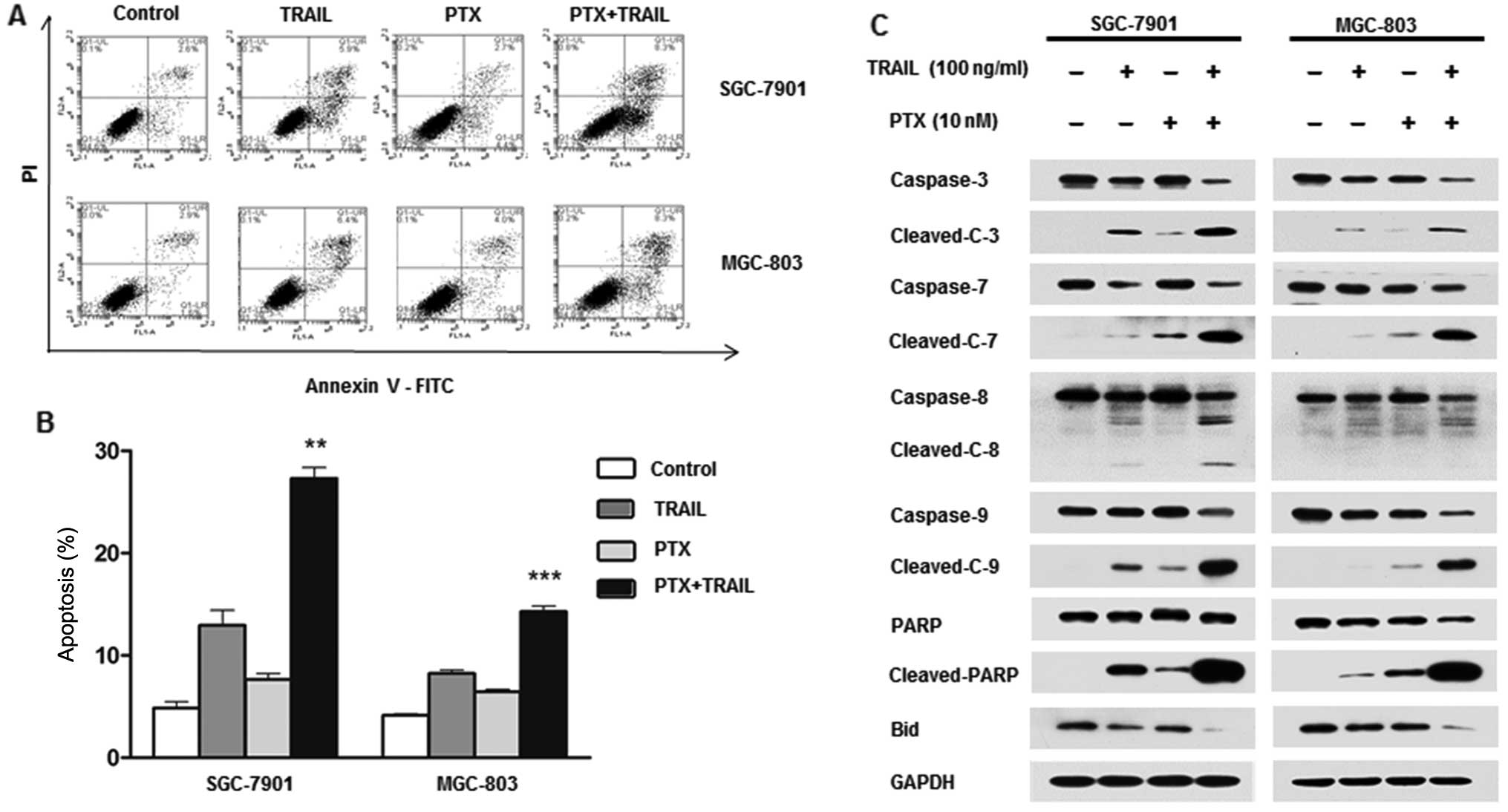
To ascertan whether cell growth inhibition was due
to apoptosis, Annexin V/PI double staining was performed. A
significant increase in apoptosis was observed in the combination
treatment group compared with either agent alone (Fig. 3A and B). Taking SGC-7901 cell line
as an example, the mean apoptotic rate was 2.8 and 8% treated with
PTX and TRAIL alone, respectively, but as much as 22.4% after
combination treatment. The expression of full length and cleaved
caspase-3, -7, -8 and -9 and PARP was measured by western blotting
(Fig. 3C). Combination of TRAIL and
PTX markedly enhanced cleavage of all the caspase proteins compared
with single agent alone. Noteworthy, cleavage of caspase-9, but not
caspase-8, was induced by PTX, indicating the effect of PTX on
activation of mitochondrial pathway. We further tested expression
of Bid, another key component in mitochondrial pathway, which was
significantly decreased in combination group.
PTX upregulated TRAIL receptors and
downregulated anti-apoptotic proteins
Given the therapeutic potential of combined regimen,
we focused the following studies on unraveling the molecular
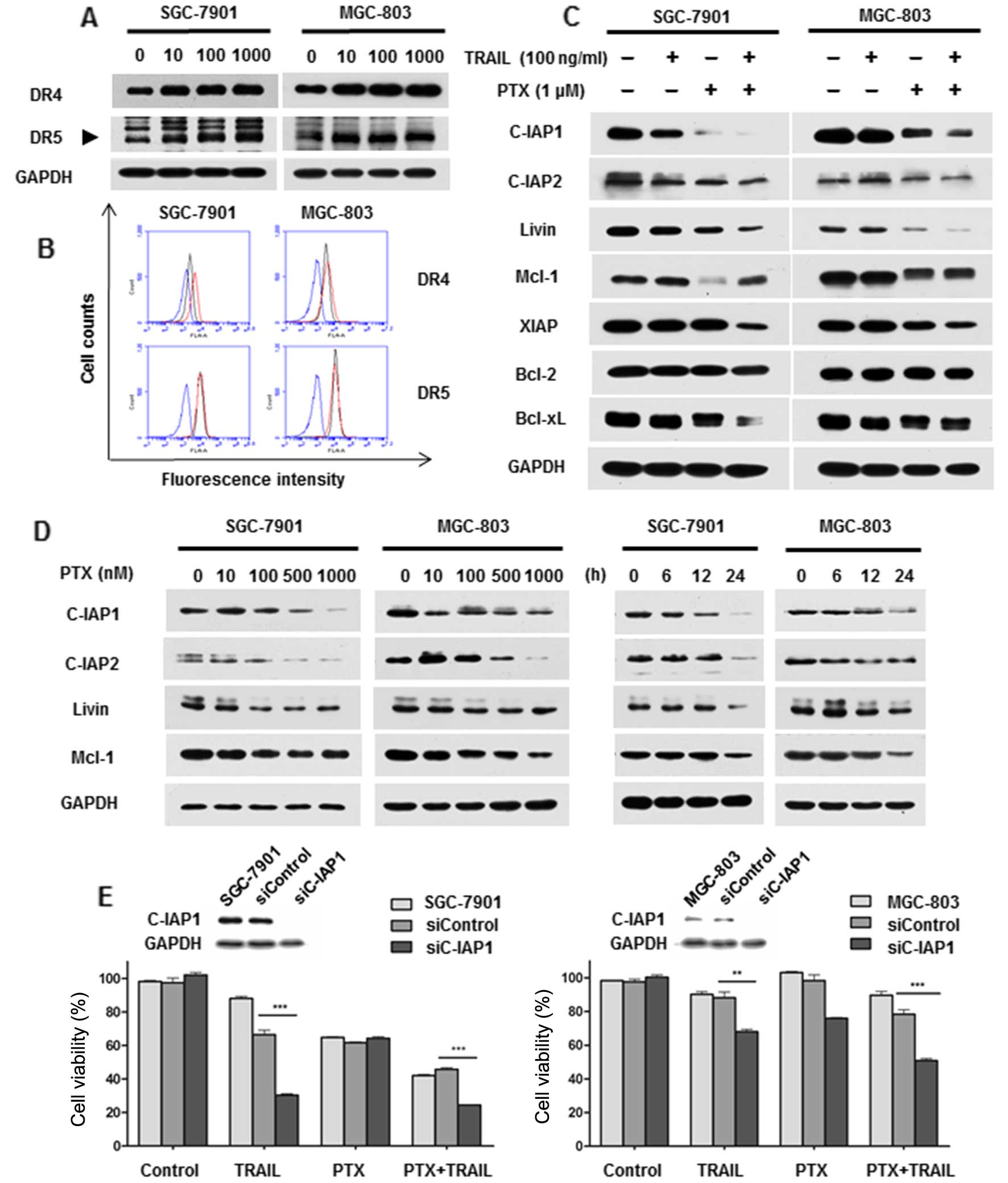
mechanism of the synergism. As shown in Fig. 4A, PTX induced DR4 and DR5 expression
in both SGC-7901 and MGC-803 cells, albeit the trend was not
distinct in MGC-803 cells. Additionally, enhanced cell surface
expression of DR4, not DR5, was also examined after PTX treatment
by flow cytometric analysis (Fig.
4B). To further uncover the role of PTX on modulating cell
death, apoptosis-related proteins were investigated. Notably,
combination treatment significantly inhibited expression of
anti-apoptotic regulators including C-IAP1, C-IAP2, Livin and Mcl-1
(Fig. 4C). Consistent with these
findings, decreased expression of these proteins was also observed
in SGC-7901 and MGC-803 cells treated with PTX alone in a dose- and
time-dependent manner (Fig. 4D).
Moreover, a representative protein C-IAP1 was chosen for functional
verification. Silencing of C-IAP1 by siRNA enhanced TRAIL-mediated
cytotoxicity and amplified the TRAIL-sensitizing effect of PTX
(Fig. 4E). All these results
suggested that PTX sensitized TRAIL-induced apoptosis via
upregulating DRs and downregulating anti-apoptotic proteins.
PTX induced inactivation of the MAPK
pathway
Considering MAPKs have been implicated in regulation
of TRAIL-resistance (19,31) in conjunction with the reports that
PTX could modulate activation of MAPKs (32), we hypothesized that PTX-mediated
MAPK activity may contribute to TRAIL sensitivity in GC cells. To
this end, we first evaluated the effect of PTX on MAPK activity.
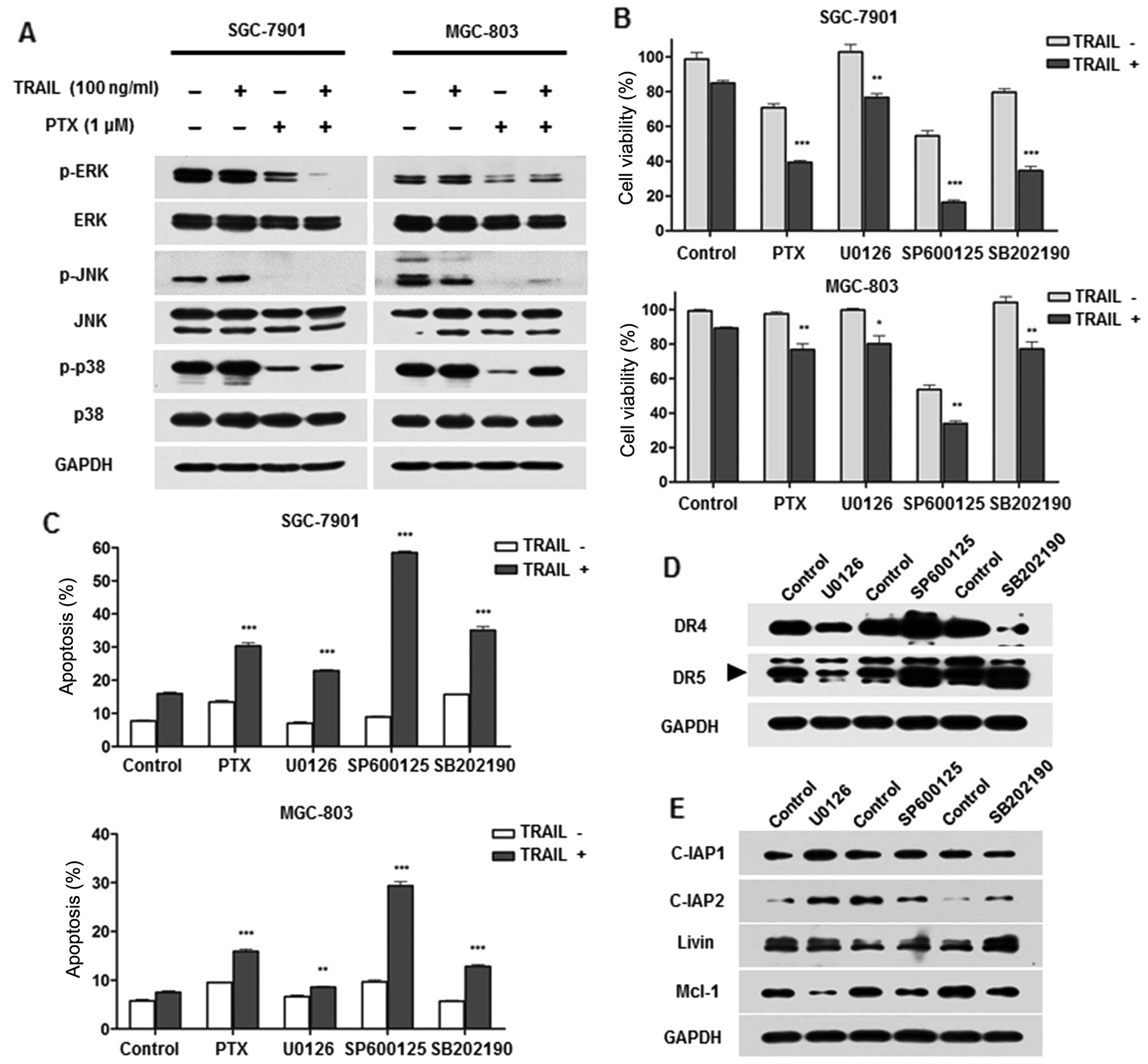
Results depicted in Fig. 5A
demonstrated ERK, JNK and p38 were strongly suppressed after
treatment of PTX alone as well as in combination with TRAIL. Next,
we investigated the possible role of MAPK deprivation on the
sensitivity to TRAIL. Treatment of U0126 (ERK inhibitor), SP600125
(JNK inhibitor) and SB202190 (p38 inhibitor) respectively,
significantly enhanced TRAIL-induced apoptosis and cytotoxicity, as
did PTX (Fig. 5B and C). Among the
three inhibitors, SP600125 showed the strongest sensitization
effect. To further assess the effect of MAPKs on TRAIL signalling,
expression of DRs and anti-apoptotic proteins were evaluated after
pretreatment with specific inhibitors. Results showed SP600125
increased DR4 and DR5 expression while U0126 suppressed them
indicating MAPKs exhibited opposite effect on DRs (Fig. 5D). Moreover, C-IAP2 was suppressed
by SP600125 and Mcl-1 was decreased by all three inhibitors
(Fig. 5E). Collectively, these
results suggested that PTX-mediated inhibition of MAPKs might
contribute to facilitate TRAIL potential in GC cells.
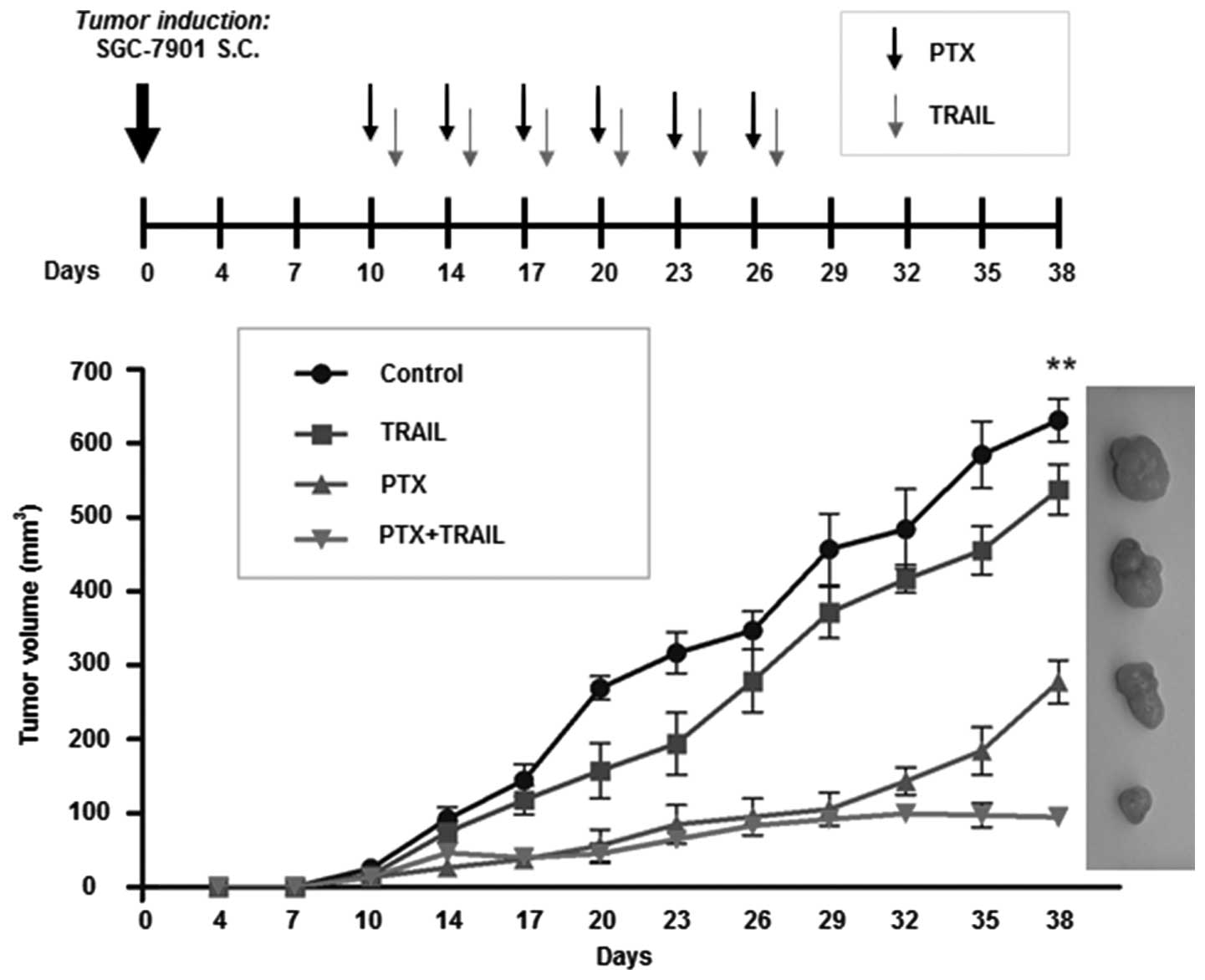
PTX enhances the antitumor effect of
TRAIL in a tumor xenograft model
To assess the therapeutic effect of PTX and TRAIL,
we established nude mouse models bearing SGC-7901 tumor xenografts.
The results revealed that TRAIL or PTX alone only slightly
suppressed growth of tumors (TGI was 14.8 and 56.0%, respectively),
whereas in the combination group, TGI was 85.1% at the end of drug
administration (Fig. 6). The data
further confirmed that PTX enhanced the tumor-suppressing capacity
of TRAIL in resistant GC cells.
Discussion
Identification of new drugs causing tumor specific
apoptosis has roused enormous interest. TRAIL legend and its
receptors are attractive targets for the selective eradication of
tumor cells. Recombinant TRAIL protein and TRAIL receptor agonistic
antibodies have been tested in clinical trials, displaying
encouraging antitumor activities with mild side-effects.
Nevertheless, resistance to TRAIL limits their clinical
application. In the present study, we found not all the cell lines
underwent significant apoptosis when treated with TRAIL. Numerous
chemotherapeutic agents can sensitize tumor cells to TRAIL-mediated
apoptosis (26–28). However, there are scarce data
elucidating the synergistic interaction between PTX and TRAIL in GC
cells. In the present study, we assessed the tumoricidal potential
of TRAIL combined with PTX both in vitro and in vivo,
and analyzed the mechanism by which PTX sensitized TRAIL-resistant
GC cells.
One possible mechanism is synergistic activation of
caspase. Apoptosis can be induced by two pathways. TRAIL and PTX
share two complementary characteristics for performing apoptosis:
one is high efficacy of cell death triggered by death
receptor-mediated pathway, and the other is activation of
mitochondria-controlled signaling. In the present study, we
observed that TRAIL alone only slightly activated caspase-3, -8 and
PARP in SGC-7901 and MGC-803 cells. In contrast, simultaneous
administration of PTX and TRAIL resulted in a dramatic increase in
cleavage of all caspase proteins. Notably, the combined
administration markedly activated caspase-9 and induced cleavage of
Bid, which are key events in mitochondrial apoptosis signaling,
suggesting that PTX facilitated sensitivity to TRAIL by reinforcing
mitochondria-mediated apoptosis. This observation is supported by
others reporting that PTX induces apoptosis via a death
receptor-independent, caspase-3/-8-driven mitochondrial
amplification loop (33).
Apoptosis signals are initiated when TRAIL binds to
DR4 and DR5. Upregulation of DRs can enhance the responsiveness of
cancer cells to TRAIL-mediated cell death (26,34).
According to our data, high DR5 expression was prevalent and almost
undifferentiated in GC cells. Intriguingly, GC cells shared diverse
surface expression of DR4 with the tendency that TRAIL-resistant
cell lines exhibited relatively low levels. These findings provided
novel evidence that susceptibility of GC cells to TRAIL might be
mainly ascribed to the surface expression of DR4. As we found PTX
could increase DR4 expression, it was plausible that induction of
DR4 would be an efficient way to potentiate tumoricidal potential
of TRAIL in GC. Another crucial point for increasing cell
susceptibility to TRAIL is inhibition of the anti-apoptotic
protein. Previous studies supported that TRAIL-resistant cells were
re-sensitized by Bcl-2 or IAP antagonist (17,18).
Here we revealed that PTX itself markedly inhibited expression of
C-IAP1, C-IAP2, Livin and Mcl-1 in both SGC-7901 and MGC-803 cells,
and combined application of PTX and TRAIL exhibited similar
effects. In addition, knockdown of C-IAP1 by siRNA augmented
cytotoxic potential of TRAIL indicating PTX-mediated suppression of
anti-apoptotic proteins contributed to the sensitizing effect.
Noteworthy, we found MAPKs were involved in the
PTX-mediated sensitization to TRAIL. MAPK pathway, regulating cell
proliferation, differentiation, mitosis and apoptosis, mainly
consists of ERK, JNK1/2 and p38 MAPK members. They are frequently
activated in GC (35). In this
study, we found MAPKs (including ERK, JNK and p38) acted as
negative regulators in TRAIL-induced apoptosis. PTX, however,
suppressed MAPK activation, abolished the inhibition effect, and
therefore partly restored TRAIL sensitivity. We also revealed
suppression of JNK by the specific inhibitor SP600125, which showed
the most effective sensitization effect. Consistent with the
results of Mucha et al (19), JNK inhibition sensitised
hepatocellular carcinoma cells to TRAIL. Similarly, ERK abrogated
TRAIL-induced apoptosis by phosphorylating pro-caspase-8 and
inhibited the cleavage of Bid (36,37).
However, other groups reported that enforced activation of ERK and
p38 conferred sensitization of tumor cells to TRAIL by suppressing
expression of FLIP and increasing expression of DRs (34,38).
These inconsistent results indicate that MAPK signaling may play
various roles in the way the diverse cancer cells react to TRAIL.
Moreover, our results showed that MAPKs played opposing roles on
modulating DRs and only JNK inhibitor could induce both DR4 and DR5
expression. In agreement with our results, Kim et al
(39) have shown SP600125
upregulated DRs surface expression on hepatocellular cancer cells.
We also found MAPK inhibitors could suppress expression of
anti-apoptotic protein C-IAP2 and Mcl-1. All these results
suggested that inhibition of MAPKs was involved in
TRAIL-sensitizing effect of PTX.
In summary, the present study suggested that PTX
sensitized resistant GC cells to TRAIL-mediated tumoricidal effect
both in vitro and in vivo. Potentiation of
mitochondrial apoptotic signals, upregulation of death receptors,
downregulation of anti-apoptotic proteins and inactivation of MAPKs
were all involved in the synergistic interaction. Our findings
strongly suggested that the combination of PTX with TRAIL could
serve as a new therapeutic strategy for GC. Further study on the
potential application in this direction is warranted.
Acknowledgments
The present study was supported in part by the
National Nature Science Foundation of China (nos. 81402308 and
81301874), the Natural Science Foundation of Beijing (no. 7132051),
the 985 special project sponsored by the Peking University Health
Science Center (no. 2013-5-09), and the Ministry of Science and
Technology of the People's Republic of China (Nos. 2014AA020603,
2012AA02A203-B01, 2012AA02A504-B01, 2012AA020101 and
2014AA020603).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Call JA, Eckhardt SG and Camidge DR:
Targeted manipulation of apoptosis in cancer treatment. Lancet
Oncol. 9:1002–1011. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pitti RM, Marsters SA, Ruppert S, Donahue
CJ, Moore A and Ashkenazi A: Induction of apoptosis by Apo-2
ligand, a new member of the tumor necrosis factor cytokine family.
J Biol Chem. 271:12687–12690. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:611–620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Samraj AK, Keil E, Ueffing N,
Schulze-Osthoff K and Schmitz I: Loss of caspase-9 provides genetic
evidence for the type I/II concept of CD95-mediated apoptosis. J
Biol Chem. 281:29652–29659. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herbst RS, Eckhardt SG, Kurzrock R,
Ebbinghaus S, O'Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA,
Tohnya TM, Lum BL, et al: Phase I dose-escalation study of
recombinant human Apo2L/TRAIL, a dual proapoptotic receptor
agonist, in patients with advanced cancer. J Clin Oncol.
28:2839–2846. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Soria JC, Smit E, Khayat D, Besse B, Yang
X, Hsu CP, Reese D, Wiezorek J and Blackhall F: Phase 1b study of
dulanermin (recombinant human Apo2L/TRAIL) in combination with
paclitaxel, carboplatin, and bevacizumab in patients with advanced
non-squamous non-small-cell lung cancer. J Clin Oncol.
28:1527–1533. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wiezorek J, Holland P and Graves J: Death
receptor agonists as a targeted therapy for cancer. Clin Cancer
Res. 16:1701–1708. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 32:1341–1350. 2013. View Article : Google Scholar
|
|
10
|
Prasad S, Kim JH, Gupta SC and Aggarwal
BB: Targeting death receptors for TRAIL by agents designed by
Mother Nature. Trends Pharmacol Sci. 35:520–536. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Horak P, Pils D, Haller G, Pribill I,
Roessler M, Tomek S, Horvat R, Zeillinger R, Zielinski C and
Krainer M: Contribution of epigenetic silencing of tumor necrosis
factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL
resistance and ovarian cancer. Mol Cancer Res. 3:335–343. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bin L, Thorburn J, Thomas LR, Clark PE,
Humphreys R and Thorburn A: Tumor-derived mutations in the TRAIL
receptor DR5 inhibit TRAIL signaling through the DR4 receptor by
competing for ligand binding. J Biol Chem. 282:28189–28194. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y and Zhang B: TRAIL resistance of
breast cancer cells is associated with constitutive endocytosis of
death receptors 4 and 5. Mol Cancer Res. 6:1861–1871. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grotzer MA, Eggert A, Zuzak TJ, Janss AJ,
Marwaha S, Wiewrodt BR, Ikegaki N, Brodeur GM and Phillips PC:
Resistance to TRAIL-induced apoptosis in primitive neuroectodermal
brain tumor cells correlates with a loss of caspase-8 expression.
Oncogene. 19:4604–4610. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ricci MS, Kim SH, Ogi K, Plastaras JP,
Ling J, Wang W, Jin Z, Liu YY, Dicker DT, Chiao PJ, et al:
Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib
sensitizes resistant human cancer cells to TRAIL-induced death.
Cancer Cell. 12:66–80. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Munshi A, Pappas G, Honda T, McDonnell TJ,
Younes A, Li Y and Meyn RE: TRAIL (APO-2L) induces apoptosis in
human prostate cancer cells that is inhibitable by Bcl-2. Oncogene.
20:3757–3765. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Finlay D, Vamos M, González-López M,
Ardecky RJ, Ganji SR, Yuan H, Su Y, Cooley TR, Hauser CT, Welsh K,
et al: Small-molecule IAP antagonists sensitize cancer cells to
TRAIL-induced apoptosis: Roles of XIAP and cIAPs. Mol Cancer Ther.
13:5–15. 2014. View Article : Google Scholar :
|
|
18
|
Kocab AJ, Veloso A, Paulsen MT, Ljungman M
and Duckett CS: Effects of physiological and synthetic IAP
antagonism on c-IAP-dependent signaling. Oncogene. 34:5472–5481.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mucha SR, Rizzani A, Gerbes AL, Camaj P,
Thasler WE, Bruns CJ, Eichhorst ST, Gallmeier E, Kolligs FT, Göke
B, et al: JNK inhibition sensitises hepatocellular carcinoma cells
but not normal hepatocytes to the TNF-related apoptosis-inducing
ligand. Gut. 58:688–698. 2009. View Article : Google Scholar
|
|
20
|
Lee HH, Jeong JW, Lee JH, Kim GY, Cheong
J, Jeong YK, Yoo YH and Choi YH: Cordycepin increases sensitivity
of Hep3B human hepatocellular carcinoma cells to TRAIL-mediated
apoptosis by inactivating the JNK signaling pathway. Oncol Rep.
30:1257–1264. 2013.PubMed/NCBI
|
|
21
|
Liu J, Qu X, Xu L, Zhang Y, Qu J, Hou K
and Liu Y: Phosphoinositide 3-kinase/Akt and nuclear factor κB
pathways are involved in tumor necrosis factor-related
apoptosis-inducing ligand resistance in human gastric cancer cells.
Mol Med Rep. 3:491–496. 2010. View Article : Google Scholar
|
|
22
|
Wan Z, Pan H, Liu S, Zhu J, Qi W, Fu K,
Zhao T and Liang J: Downregulation of SNAIL sensitizes
hepatocellular carcinoma cells to TRAIL-induced apoptosis by
regulating the NF-κB pathway. Oncol Rep. 33:1560–1566.
2015.PubMed/NCBI
|
|
23
|
Nazim UM, Jeong JK, Seol JW, Hur J, Eo SK,
Lee JH and Park SY: Inhibition of the autophagy flux by gingerol
enhances TRAIL-induced tumor cell death. Oncol Rep. 33:2331–2336.
2015.PubMed/NCBI
|
|
24
|
Liu YJ, Lin YC, Lee JC, Kuo SC, Ho CT,
Huang LJ, Kuo DH and Way TD: CCT327 enhances TRAIL-induced
apoptosis through the induction of death receptors and
downregulation of cell survival proteins in TRAIL-resistant human
leukemia cells. Oncol Rep. 32:1257–1264. 2014.PubMed/NCBI
|
|
25
|
Pasquier E, Carré M, Pourroy B, Camoin L,
Rebaï O, Briand C and Braguer D: Antiangiogenic activity of
paclitaxel is associated with its cytostatic effect, mediated by
the initiation but not completion of a mitochondrial apoptotic
signaling pathway. Mol Cancer Ther. 3:1301–1310. 2004.PubMed/NCBI
|
|
26
|
Gong J, Yang D, Kohanim S, Humphreys R,
Broemeling L and Kurzrock R: Novel in vivo imaging shows
up-regulation of death receptors by paclitaxel and correlates with
enhanced antitumor effects of receptor agonist antibodies. Mol
Cancer Ther. 5:2991–3000. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hunter TB, Manimala NJ, Luddy KA, Catlin T
and Antonia SJ: Paclitaxel and TRAIL synergize to kill
paclitaxel-resistant small cell lung cancer cells through a
caspase-independent mechanism mediated through AIF. Anticancer Res.
31:3193–3204. 2011.PubMed/NCBI
|
|
28
|
Nimmanapalli R, Perkins CL, Orlando M,
O'Bryan E, Nguyen D and Bhalla KN: Pretreatment with paclitaxel
enhances apo-2 ligand/tumor necrosis factor-related
apoptosis-inducing ligand-induced apoptosis of prostate cancer
cells by inducing death receptors 4 and 5 protein levels. Cancer
Res. 61:759–763. 2001.PubMed/NCBI
|
|
29
|
Lee MJ, Ye AS, Gardino AK, Heijink AM,
Sorger PK, MacBeath G and Yaffe MB: Sequential application of
anticancer drugs enhances cell death by rewiring apoptotic
signaling networks. Cell. 149:780–794. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsuburaya A, Yoshida K, Kobayashi M,
Yoshino S, Takahashi M, Takiguchi N, Tanabe K, Takahashi N, Imamura
H, Tatsumoto N, et al: Sequential paclitaxel followed by tegafur
and uracil (UFT) or S-1 versus UFT or S-1 monotherapy as adjuvant
chemotherapy for T4a/b gastric cancer (SAMIT): A phase 3 factorial
randomised controlled trial. Lancet Oncol. 15:886–893. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Trivedi R, Maurya R and Mishra DP:
Medicarpin, a legume phytoalexin sensitizes myeloid leukemia cells
to TRAIL-induced apoptosis through the induction of DR5 and
activation of the ROS-JNK-CHOP pathway. Cell Death Dis.
5:e14652014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McDaid HM and Horwitz SB: Selective
potentiation of paclitaxel (taxol)-induced cell death by
mitogen-activated protein kinase kinase inhibition in human cancer
cell lines. Mol Pharmacol. 60:290–301. 2001.PubMed/NCBI
|
|
33
|
von Haefen C, Wieder T, Essmann F,
Schulze-Osthoff K, Dörken B and Daniel PT: Paclitaxel-induced
apoptosis in BJAB cells proceeds via a death receptor-independent,
caspases-3/-8-driven mitochondrial amplification loop. Oncogene.
22:2236–2247. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Do MT, Na M, Kim HG, Khanal T, Choi JH,
Jin SW, Oh SH, Hwang IH, Chung YC, Kim HS, et al: Ilimaquinone
induces death receptor expression and sensitizes human colon cancer
cells to TRAIL-induced apoptosis through activation of ROS-ERK/p38
MAPK-CHOP signaling pathways. Food Chem Toxicol. 71:51–59. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Paterson AL, Shannon NB, Lao-Sirieix P,
Ong CA, Peters CJ, O'Donovan M and Fitzgerald RC: A systematic
approach to therapeutic target selection in oesophago-gastric
cancer. Gut. 62:1415–1424. 2013. View Article : Google Scholar
|
|
36
|
Söderström TS, Poukkula M, Holmström TH,
Heiskanen KM and Eriksson JE: Mitogen-activated protein
kinase/extracellular signal-regulated kinase signaling in activated
T cells abrogates TRAIL-induced apoptosis upstream of the
mitochondrial amplification loop and caspase-8. J Immunol.
169:2851–2860. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mandal R, Raab M, Matthess Y, Becker S,
Knecht R and Strebhardt K: pERK 1/2 inhibit caspase-8 induced
apoptosis in cancer cells by phosphorylating it in a cell cycle
specific manner. Mol Oncol. 8:232–249. 2014. View Article : Google Scholar
|
|
38
|
Yerbes R, López-Rivas A, Reginato MJ and
Palacios C: Control of FLIP(L) expression and TRAIL resistance by
the extracellular signal-regulated kinase1/2 pathway in breast
epithelial cells. Cell Death Differ. 19:1908–1916. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim EY, Ryu JH and Kim AK: CAPE promotes
TRAIL-induced apoptosis through the upregulation of TRAIL receptors
via activation of p38 and suppression of JNK in SK-Hep1
hepatocellular carcinoma cells. Int J Oncol. 43:1291–1300.
2013.PubMed/NCBI
|