Introduction
Pancreatic cancer is a disease with high mortality
due to the lack of early detection and resistance to gemcitabine
(GEM)-based chemotherapy. More than 90% of pancreatic cancer cases
are found to be metastatic at diagnosis. In addition, over 80% of
patients newly diagnosed with this disease are not eligible for
surgical resection (1). These
factors lead to an extremely low 5-year survival rate of patients
with pancreatic cancer, which is only 7.2% (2). Drug intervention is the general
treatment for pancreatic cancer. GEM has been used as the standard
chemotherapeutic agent for pancreatic cancer since 1997. However,
the clinical efficacy of GEM monotherapy is very limited mainly due
to drug resistance. Therefore, developing new rational combination
treatments with GEM is a promising strategy to overcome GEM
resistance in pancreatic cancer (3–5).
Once inside the cell, GEM is phosphorylated to its
monophosphate (dFdCMP), diphosphate (dFdCDP) and triphosphate
(dFdCTP) form consecutively (4).
dFdCTP incorporates into replicating DNA strands, resulting in
termination of DNA synthesis, induction of DNA replication stress
and DNA damage, and activation of the DNA damage response (DDR),
leading to GEM resistance (6). In
addition, dFdCDP also inhibits ribonucleotide reductase (RR), and
consequently decreased NTP pools (4). RR is composed of the regulatory
subunit (RRM1) and catalytic subunit (RRM2). Both RRM1 and RRM2
overexpression is associated with GEM resistance (7,8). Thus,
combination of GEM with a drug which targets the DDR and RR may
enhance its antitumor activity against pancreatic cancer.
Ataxia telangiectasia mutated and Rad3-related
(ATR), a key regulator of the DDR, has important functions in
sensing and repairing DNA damage, regulating the cell cycle,
stabilizing replication forks and restraining replication origin
firing (9–11). Furthermore, a recent study suggests
that ATR coordinates RRM2 accumulation to suppress replication
catastrophe (12). Thus, targeting
ATR could potentially overcome GEM resistance by inhibiting DNA
repair, abrogating GEM-induced activation of the cell cycle
checkpoints, and suppressing RR expression.
Cancer cells frequently harbor deficiencies in one
or more DDR pathways. These DDR deficiencies not only contribute to
tumorigenesis, but also render the cancer cells more dependent on
the remaining functional DDR pathways. Thus, targeting ATR to treat
pancreatic cancer may not only enhance GEM sensitivity, but also
show tumor selectivity (13–15).
Thus, ATR has been regarded as a promising target for cancer
treatment. Growing evidence shows that ATR inhibition increases the
antitumor activity of chemotherapeutic agents including GEM and
radiation in a variety of cancers including pancreatic cancer
(16,17). Inhibition of ATR sensitizes
pancreatic cancer cells to radiation accompanied by inhibition of
homologous recombination repair, decrease of checkpoint activation
and induction of more DNA damage (18,19).
However, the underlying molecular mechanisms are not fully
understood.
In the present study, we chose a novel ATR-selective
inhibitor, AZ20, which has shown potent anti-colorectal tumor
activity in both in vitro and in vivo preclinical
models (20), to investigate the
antitumor effect and the underlying molecular mechanism of ATR
inhibition either alone or in combination with GEM in pancreatic
cancer cell lines. Our results showed that although AZ20 treatment
had limited effect on cell death, it significantly enhanced that
induced by GEM. Notably, AZ20 enhanced GEM-induced cell death
mainly through enhancement of GEM-induced DNA damage rather than
abrogation of cell cycle checkpoints. It has been hypothesized that
p53-deficient cancer cells (resulting in loss of G1 checkpoint)
rely on the ATR/CHK1 pathway for DNA damage repair and are more
sensitive to ATR inhibition (21–24).
Our data suggests that the antitumor activity of the combination of
AZ20 and GEM was independent of the p53 status. These findings
suggest that targeting ATR may represent a promising strategy to
overcome GEM resistance in pancreatic cancer.
Materials and methods
Drugs
AZ20, GEM and roscovitine were purchased from
Selleck Chemicals (Houston, TX, USA).
Cell lines and treatments
The AsPC-1, BxPC-3, CFPAC-1, HPAC and MIAPaCa-2
human pancreatic cancer cell lines were purchased from the American
Type Culture Collection (ATCC; Manassas, VA, USA), and were
authenticated by the University of Arizona Genetics Core Facility
(Tucson, AZ, USA). The cells were cultured in Dulbecco's modified
Eagles medium (DMEM; Invitrogen, Carlsbad, CA, USA; HPAC and
MIAPaCa-2), RPMI-1640 medium (Invitrogen; AsPC-1 and BxPC-3) or
Iscove's modified Dulbecco's medium (IMDM; Invitrogen; CFPAC-1)
with 10% heat-inactivated fetal bovine serum (FBS; HyClone
Laboratories, Logan, UT, USA) plus 100 U/ml penicillin and 100
µg/ml streptomycin in a 37°C humidified atmosphere containing 5%
CO2/95% air. The cell lines were tested for the presence
of mycoplasma on a monthly basis.
In vitro cytotoxicity assays
In vitro cytotoxicity of AZ20 and GEM, alone
or in combination, was determined using
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT)
(Sigma-Aldrich, St. Louis, MO, USA) reagent, as previously
described (25). IC50
values were calculated as drug concentrations necessary to inhibit
50% cell growth compared to vehicle control-treated cells using
GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA,
USA). The extent and direction of antitumor interactions between
AZ20 and GEM were determined by calculating combination index (CI)
values. CI < 0.9 indicates synergistic, 0.9 < CI <1.1
indicates additive, and CI > 1.1 indicates antagonistic
antitumor interactions, respectively.
Cell death and cell cycle
progression
Pancreatic cancer cell lines were treated with the
indicated drugs for up to 48 h. Cells were fixed with ice-cold 80%
(v/v) ethanol for 24 h at 4°C. The cells were pelleted, then were
washed with phosphate-buffered saline (PBS) (pH 7.4) and
resuspended in PBS containing propidium iodide (PI; 50 µg/ml),
Triton X-100 (0.1%, v/v), and DNase-free RNase (1 µg/ml). DNA
content was determined by flow cytometric analysis using a
FACSCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA),
as previously described (25). Cell
death events were expressed as the percentage of cells with sub-G1
DNA content. Cell cycle analysis and histogram generation were
carried out using FlowJo v7.6.5 (Tree Star, Ashland, OR, USA).
Western blot analysis
Soluble proteins were extracted in the presence of
protease and phosphatase inhibitors (Roche Applied Sciences China
Inc., Shanghai, China) and subjected to SDS-polyacrylamide gel
electrophoresis. Separated proteins were electrophoretically
transferred onto polyvinylidene difluoride (PVDF) membranes (Thermo
Fisher Inc., Rockford, IL, USA) and immunoblotted with anti-PARP-1
(9542), -pCDK1 (Y15) (9111), -CDK2 (2546), -γH2AX (2577), -GAPDH
(2118; Cell Signaling Technology, Danvers, MA, USA), -CHK1 (sc8408,
Santa Cruz Biotechnology, Santa Cruz, CA, USA), -RRM1 (ab137114),
-RRM2 (ab172476), -pCHK1 (S345) (ab47318), -pCDC25C (S216)
(ab32051), -pCDK2 (Y15) (ab76146) or -CDK1 (ab32094; Abcam,
Cambridge, MA, USA) antibodies, as previously described (25). Primary antibodies were diluted
1:1,000 in Odyssey Blocking Buffer (Li-Cor, Lincoln, NE, USA).
Immunoreactive proteins were visualized using the Odyssey Infrared
Imaging System (Li-Cor). Western blot analyses were repeated 3
times and one representative blot is shown.
Alkaline comet assay
BxPC-3 or HPAC cells were treated with the indicated
drugs for 8 h, and then subjected to alkaline comet assay as
previously described (26). Slides
were stained with SYBR Gold (Invitrogen), and then imaged on an
Olympus IX73 microscope equipped with a DP80 digital camera for
microscope and cellSens EN-V1 software (Olympus China Inc.,
Beijing, China). At least 100 comets/gel were scored by CometScore
(TriTek Corp., Sumerduck, VA, USA). The median percentage of DNA in
the tail was calculated and expressed as mean ± SEM.
Statistical analysis
Differences were compared using the pair-wise
two-sample t-test. Statistical analyses were performed using
GraphPad Prism 5.0. Error bars represent ± SEM. The level of
significance was set at p<0.05.
Results
AZ20 treatment causes growth arrest,
but limited cell death in pancreatic cancer cells
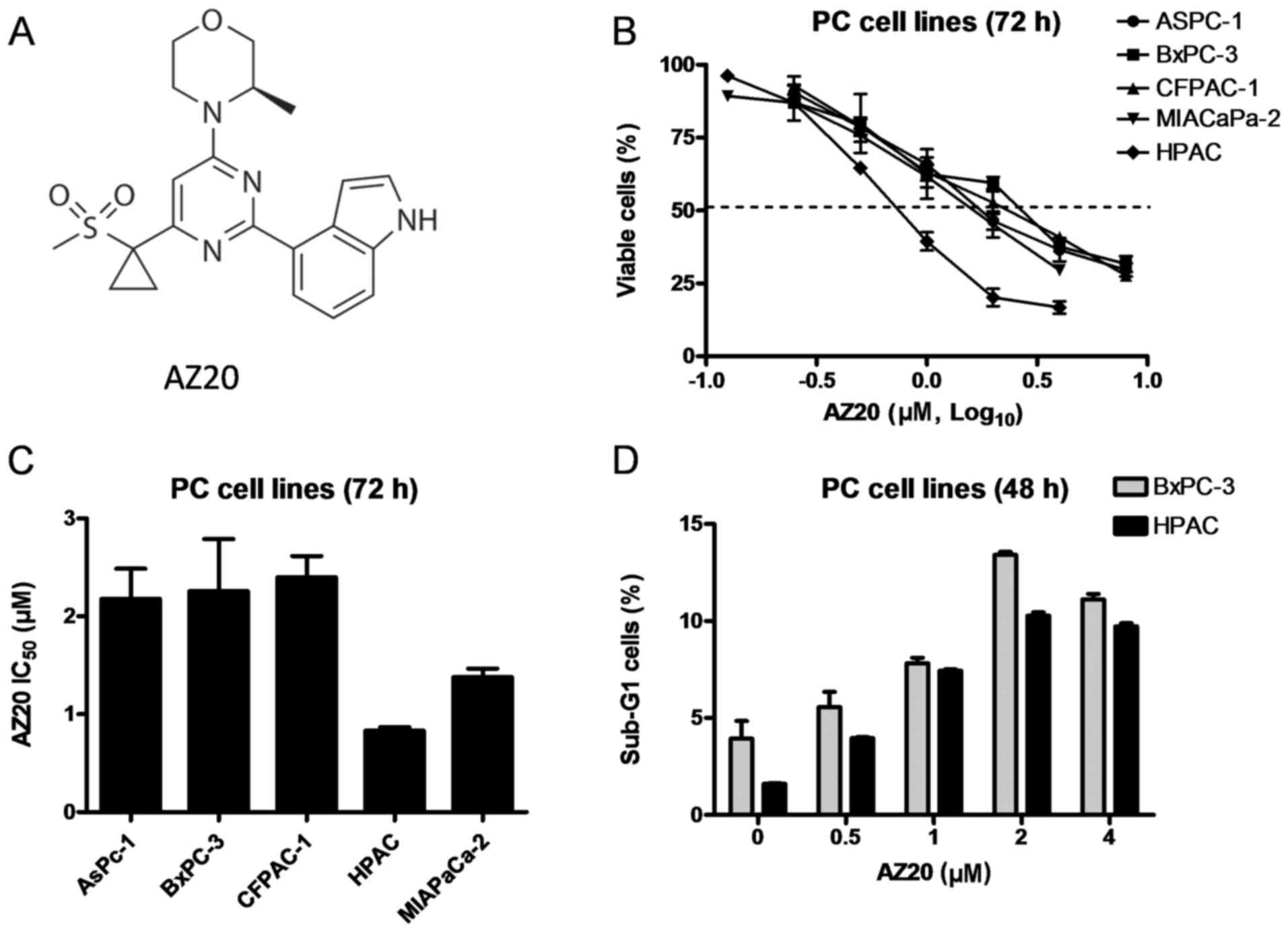
To begin our investigation, the sensitivity of AZ20
was determined in a panel of pancreatic cancer cell lines using MTT
assays after 72 h of drug treatment. As shown in Fig. 1B and C, AZ20 treatment caused growth
inhibition in a concentration-dependent manner with IC50
values ranging from 0.8 µM in HPAC cells to 2.4 µM in CFPAC-1
cells. In contrast to a previous hypothesis that p53-deficient
cancer cells are more sensitive to ATR inhibition, our data showed
that p53-wild-type HPAC cells responded better to AZ20 treatment
compared to the p53-mutant pancreatic cancer cell lines (Fig. 1C). Since HPAC and BxPC-3 cell lines
are widely used in preclinical studies, we chose these 2 cell lines
for the rest of our studies.
To determine whether AZ20 treatment causes cell
death, BxPC-3 and HPAC cells were treated with variable
concentrations of AZ20 for 48 h and then subjected to flow
cytometric analysis to detect sub-G1 cells. As compared to the no
drug treatment control, AZ20 treatment induced statistically
significant but biologically limited cell death (<14%). This was
accompanied by low levels of PARP-1 cleavage in both cell lines,
indicating that the AZ20 treatment-induced cell death was through
apoptosis (Figs. 1D, and 2C and D). These results demonstrated that
AZ20 treatment causes growth inhibition and limited cell death in
the pancreatic cancer cells.
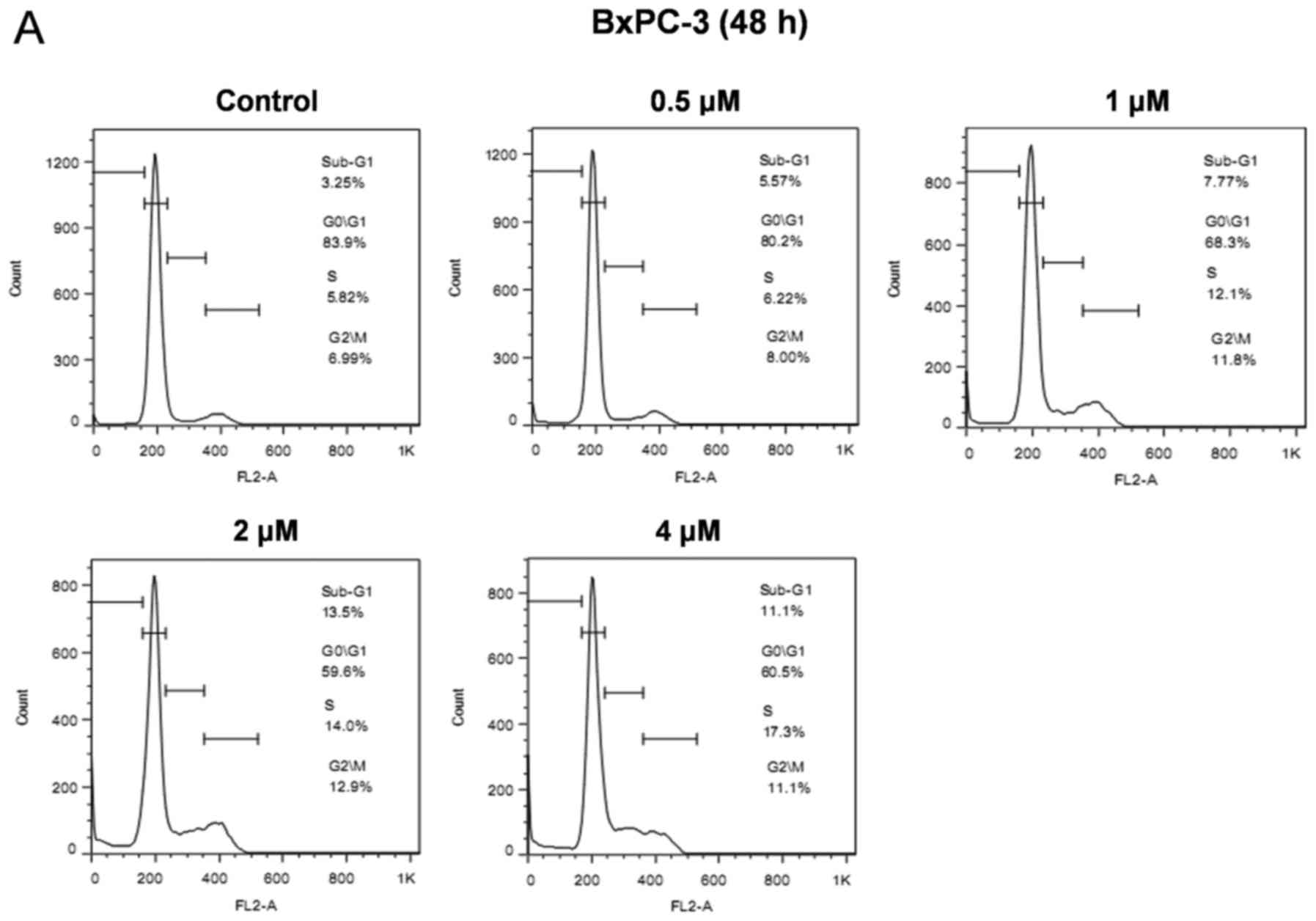 | Figure 2.AZ20 induces cell cycle arrest,
inhibits the activity of CHK1, and increases DNA damage in
pancreatic cancer cells. (A and B) BxPC-3 and HPAC cells were
treated with variable concentrations of AZ20 for 48 h. The cells
were fixed with 80% ethanol, stained with PI, and subjected to flow
cytometric analysis to determine cell cycle distribution.
Representative histograms are shown. (C and D) BxPC-3 and HPAC
cells were treated with variable concentrations of AZ20 for 48 h.
Whole cell lysates were subjected to western blotting and probed
with anti-PARP-1, γH2AX, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1, -CDK1,
-p-CDK2, -CDK2, -RRM1, -RRM2 or -GAPDH antibody. |
AZ20 treatment induces cell cycle
arrest and DNA damage in pancreatic cancer cells
To investigate the effect of AZ20 on cell cycle
progression in pancreatic cancer cells, we treated BxPC-3 and HPAC
cells with the indicated concentrations of AZ20, and then analyzed
cell cycle distribution in the cells by PI staining and flow
cytometric analyses. AZ20 treatment induced S and G2/M arrest in a
dose-dependent manner in the BxPC-3 cells (Fig. 2A). Similar results were also
obtained in the HPAC cells (Fig.
2B). These results showed that the effect of AZ20 treatment on
cell cycle checkpoints is activation rather than abrogation,
suggesting that AZ20 treatment caused DNA damage in the cells.
To test this possibility, we investigated the
effects of AZ20 treatment on the expression of γH2AX, a biomarker
of DNA double-strand breaks (27),
and the downstream signaling of ATR. In both BxPC-3 and HPAC cells,
AZ20 treatment increased the expression of γH2AX, indicative of DNA
damage. AZ20 also caused obviously decreased phosphorylation of
CHK1 at serine-345, CDC25C at serine-216 and CDK1 at tyrosine-15,
demonstrating suppression of the CHK1/CDC25C/CDK1 pathway resulting
from ATR inhibition. In contrast, AZ20 treatment increased the
phosphorylation of CDK2 at tyrosine-15 (Fig. 2C and D). It has recently been
reported that inhibition of ATR causes decreased expression of RRM2
in cancer cells (12). Although
AZ20 treatment caused downregulation of RRM2 in BxPC-3 cells, it
had no effect on RRM2 expression in HPAC cells. Notably, AZ20
treatment substantially decreased the expression of RRM1 in the
BxPC-3 and in HPAC cells, although to a much lesser extent
(Fig. 2C and D). These data suggest
that the induction of DNA damage by AZ20 was partially due to its
negative effect on RR expression.
AZ20 enhances GEM-induced DNA damage and cell death.
To examine the effect of AZ20 on GEM-induced cell death and cell
cycle progression, BxPC-3 and HPAC cells were treated with 1 µM
AZ20 and 80 nM GEM alone or in combination for 48 h. Compared to
the single drug treatment, AZ20 and GEM combination caused
significantly increased cell death accompanied by substantially
increased PARP-1 cleavage and γH2AX expression in both cell lines
(Fig. 3A, F and G). The presence of
AZ20 also significantly decreased GEM-induced S phase arrest in
both cell lines, and G2/M phase arrest in BxPC-3 cells (Fig. 3B-E). AZ20 suppressed GEM-induced
CHK1 phosphorylation at serine-345, indicating a negative effect on
GEM-induced checkpoint activation. Although GEM treatment had
differential effects on the phosphorylation of CDC25C (serine-216)
in BxPC-3 and HPAC cells (induction in BxPC-3 cells, while
suppression in HPAC cells), it increased tyrosine-15
phosphorylation of CDK1/2 in both cell lines, which was almost
completely abolished by AZ20 (Fig. 3F
and G). These data indicate that AZ20 increases GEM-induced
cell death through abrogation of cell cycle checkpoints and
enhancement of DNA damage.
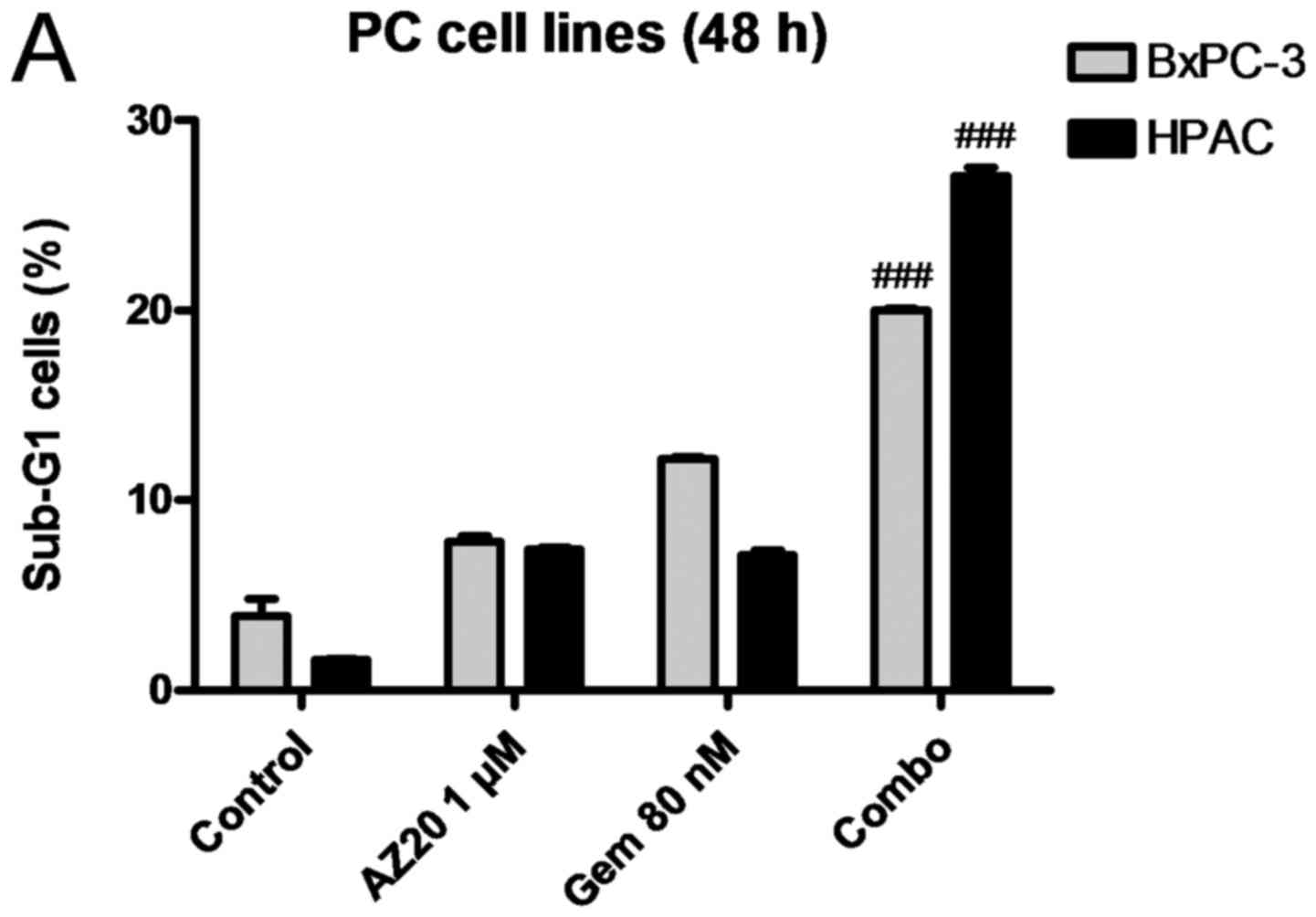 | Figure 3.AZ20 enhances GEM-induced cell death
in pancreatic cancer cells. (A) BxPC-3 and HPAC cells were treated
with 1 µM AZ20 and 80 nM GEM alone or simultaneously for 48 h. The
cells were fixed with 80% ethanol, stained with PI and subjected to
flow cytometric analysis to determine cell death (sub-G1) and cell
cycle distribution. The data are presented as means of triplicates
± standard errors from 1 representative experiment. (B-E) Cell
cycle results particularly the percentage of cells in the S phase
in BxPC-3 and HPAC cell lines and G2/M phase in the BxPC-3 cells
are graphed as means of triplicates ± standard errors from 1
representative experiment. (F and G) BxPC-3 and HPAC cells were
treated with 1 µM AZ20 and 80 nM GEM alone or simultaneously for 48
h. Whole cell lysates were subjected to western blotting and probed
with anti-PARP-1, -γH2AX, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1,
-CDK1, -p-CDK2, -CDK2, -RRM1, -RRM2 or -GAPDH antibody; *p<0.05
(GEM vs. control), **p<0.005 (GEM vs. control), ***p<0.001
(GEM vs. control), ##p<0.005 (combo vs. GEM),
###p<0.001 (combo vs. GEM). (H) Relative RRM2 protein
levels are expressed as means ± standard errors from 3 independent
experiments; *p<0.05 (GEM vs. control), **p<0.005 (GEM vs.
control), ***p<0.001 (GEM vs. control), ##p<0.005
(combo vs. GEM), ###p<0.001 (combo vs. GEM). |
Consistent with previous studies (7,8), GEM
treatment resulted in increased protein levels of RRM1 in the HPAC
cells and RRM2 in both BxPC-3 and HPAC cell lines. Notably,
addition of AZ20 almost completely abolished GEM-induced RR
expression in both cell lines (Fig.
3F-H). These results suggest that AZ20 enhances GEM sensitivity
in pancreatic cancer cells, at least partially through abrogation
of GEM-induced RR expression.
AZ20 increases GEM-induced DNA damage
at an early time point
Previous studies have demonstrated that DNA
fragmentation at the late stage of cell apoptosis also induces
γH2AX expression (27). To provide
direct evidence that the AZ20 and GEM combination indeed cooperate
in inducing DNA damage, we determined the expression of γH2AX and
other relevant proteins in BxPC-3 and HPAC pancreatic cancer cells
post 8 h drug treatments. Consistent with the results obtained in
the cells post 48 h drug treatments, the combination treatment
resulted in substantially increased expression of γH2AX in the
cells compared to individual drug treatment. Neither single nor
combination drug treatment had obvious effects on cell morphology
and PARP-1 cleavage, demonstrating that the drug treatments did not
induce cell death under these conditions. AZ20 treatment also
abolished GEM-induced RRM2 expression in the cells. In contrast,
there was no obvious effect of the drug treatments on the
CHK1/CDC25C/CDK1/2 pathway proteins (Fig. 4A-C). These results suggest that AZ20
enhances the antitumor activity of GEM mainly by increasing
GEM-induced DNA damage and decreasing GEM-induced RRM2 expression
rather than abrogating GEM-induced checkpoint activation.
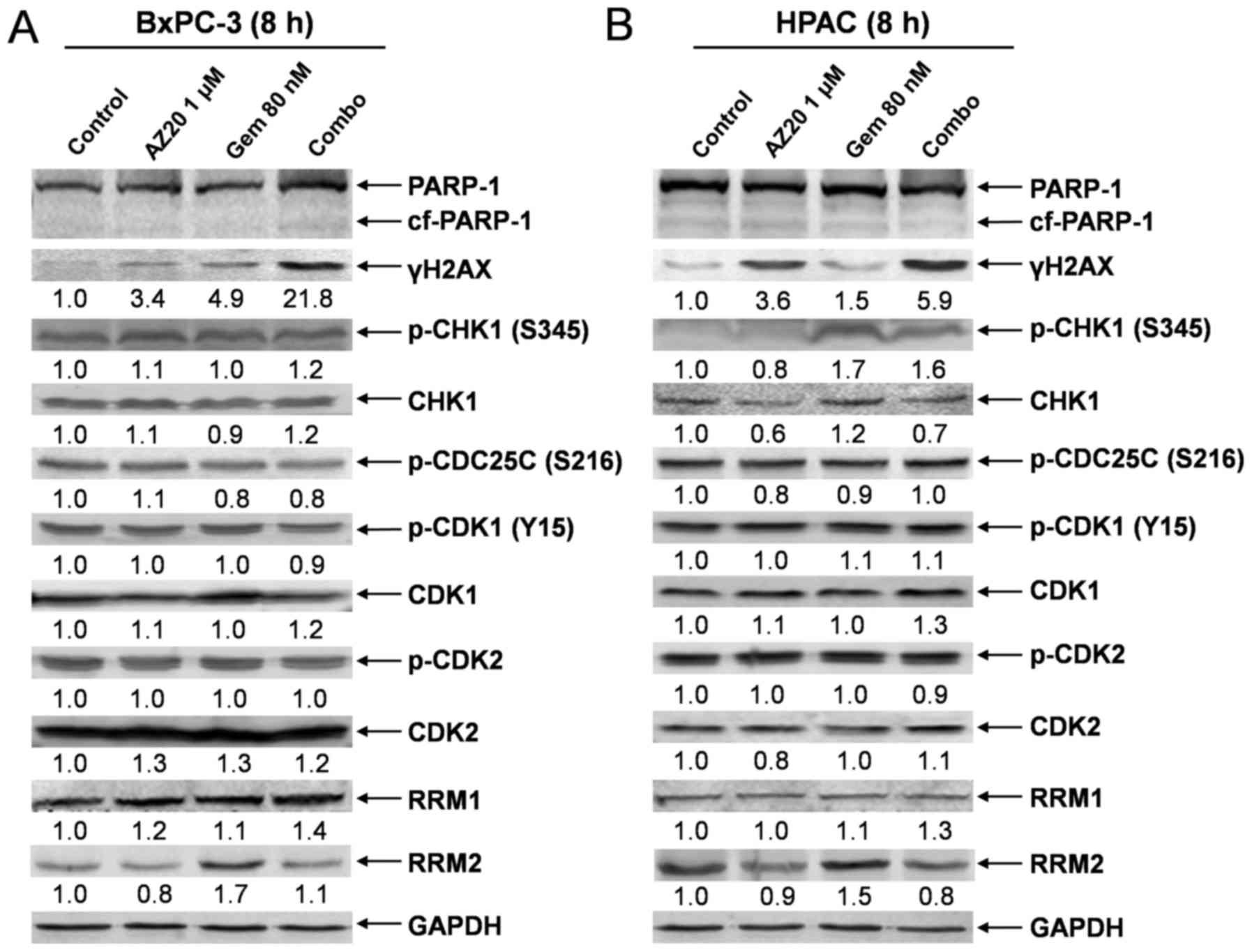 | Figure 4.AZ20 enhances GEM-induced DNA damage
in pancreatic cancer cells. (A and B) BxPC-3 and HPAC cells were
treated with 1 µM AZ20 and 80 nM GEM alone or simultaneously for 8
h. Whole cell lysates were subjected to western blotting and probed
with anti-PARP-1, -γH2AX, -p-CHK1, -CHK1, -p-CDC25C, -p-CDK1,
-CDK1, -p-CDK2, -CDK2, -RRM1, -RRM2 or -GAPDH antibody. (C)
Relative RRM2 protein levels are graphed as means ± standard errors
from 3 independent experiments. (D and E) BxPC-3 and HPAC cells
were treated with 1 µM AZ20 and 80 nM GEM alone or simultaneously
for 8 h, and then the cells were subjected to alkaline comet assay.
Representative comets are shown. (F and G) Comet assay results are
graphed as the median percentage of DNA in the tail from 3
replicate gels ± SEM; ***p<0.001 (GEM vs. control),
##p<0.005 (combo vs. GEM), ###p<0.001
(combo vs. GEM). |
To further confirm the effect of AZ20 on GEM-induced
DNA damage, comet assay was performed after 8 h drug treatment. As
shown in Fig. 4D-G, both AZ20 and
GEM induced DNA strand breaks, as assessed by the percentage of DNA
content in the comet tail compared to the no drug treatment
control. Furthermore, combined treatment with AZ20 with GEM
significantly increased DNA strand breaks compared to the single
drug treatment in both BxPC-3 and HPAC cell lines. These results
provide compelling evidence that AZ20 enhances GEM-induced DNA
damage prior to cell death in pancreatic cancer cells.
AZ20 increases the antitumor activity
of GEM in a synergistic manner
Finally, we determined the direction and extent of
antitumor interactions between AZ20 and GEM in the 5 pancreatic
cancer cells by MTT assays and calculating CI values using CalcuSyn
software. As shown in Table I,
addition of AZ20 significantly decreased the IC50 values
of GEM in each of the pancreatic cancer cell lines tested. By
calculating CI values using the CalcuSyn software, we demonstrated
that the antitumor interactions between the 2 agents were
synergistic (CI < 0.9). These results demonstrated that
combination of GEM with AZ20 had synergistic antitumor activities
in pancreatic cancer cells.
 | Table I.AZ20 enhances GEM sensitivity in a
synergistic manner in pancreatic cancer cell lines. |
Table I.
AZ20 enhances GEM sensitivity in a
synergistic manner in pancreatic cancer cell lines.
|
|
| IC50 of
gemcitabine (nM) in the absence or presence of AZ20 (µM) |
|
|---|
|
|
|
|
|
|---|
| Cell line | IC50 of
AZ20 (µM) | 0 | 0.25 | 0.5 | 1 | aP-value |
|---|
| ASPC-1 | 2.4±0.3 | 657.5±59.7 | 255.8±29.3
(0.49) | 141.0±23.3
(0.42) | 109.6±27.7
(0.58) |
<0.001 |
| BxPC-3 | 2.3±0.5 | 26.6±5.0 | 13.7±2.0
(0.63) | 8.6±0.7
(0.55) | 4.6±0.2
(0.62) | <0.05 |
| CFPAC-1 | 2.4±0.2 |
6.5±0.8 | 3.8±0.5
(0.69) | 2.7±0.2
(0.62) | 2.0±0.3
(0.72) | <0.05 |
| HPAC |
0.8±0.04 | 14.7±1.9 | 3.8±0.3
(0.56) | 1.9±0.1
(0.73) | ND |
<0.005 |
| MIAPaCa-2 | 1.4±0.09 | 14.7±1.7 | 5.1±0.5
(0.52) | 3.3±0.4
(0.59) | 2.6±0.5
(0.90) |
<0.005 |
Discussion
Previous studies have mainly focused on the role of
ATR in cell cycle checkpoints, and suggest that abrogation of cell
cycle checkpoints is the main mechanism by which ATR inhibitors
synergize with DNA damaging agents (18,19).
In contrast to these previous studies, in the present study we
found that the novel ATR inhibitor AZ20 enhanced the antitumor
activity of GEM mainly by enhancing GEM-induced DNA double-strand
breaks and by abolishing GEM-induced RRM2 expression rather than
abrogating GEM-induced checkpoint activation.
Similar to studies using DNA damaging agents in
combination with ATR inhibitors (18,19,28),
we found that AZ20 synergized with GEM to induce apoptosis in
pancreatic cancer cells. AZ20 abrogated GEM-induced p-CHK1 and RRM2
expression, while enhancing GEM-induced DNA damage. Although we
found that AZ20 treatment reduced the percentages of GEM-induced S
and G2/M phase cells, the amount of reduction was not very
substantial at 48 h post-treatment. At 8 h post combined AZ20 and
GEM treatment, we did not detect changes in CDK1/2 phosphorylation,
although we did detect a significant increase in DNA damage. Based
on these results, we believe that the effects of AZ20 on cell cycle
distribution were due to death of cells in the S and G2/M phases
rather than abrogation of the cell cycle checkpoints. Fokas et
al used the selective ATR inhibitor VE-822 in pancreatic cancer
cells and found that it caused G1 arrest, although they did not
determine levels of phosphorylated CDK1 and CDK2 (19). However, they found that VE-822 had
no effect on GEM-induced S phase arrest (19). Their results are similar to our
results, further supporting our findings that ATR inhibition
enhances GEM through means other than cell cycle checkpoint
abrogation.
ATR inhibitors were developed based on the evidence
that ATR mediates S and G2/M cell cycle arrest. Thus, in cells with
deficient G1 checkpoints, DNA damage repair may rely on the S and
G2/M checkpoints for repair. Inhibition of ATR in G1-deficient
cells may result in accumulation of DNA damage and cause cell death
(17,29). Several studies have since supported
this hypothesis and have demonstrated that ATR inhibitors abrogate
the S and G2/M cell cycle checkpoints (reviewed in ref. 17). However, in the present study we used
p53 wild-type HPAC cells and found that the ATR inhibitor AZ20
enhanced GEM-induced cell death. Our finding suggests that AZ20 did
not enhance GEM by abrogating the S and G2/M cell cycle checkpoints
rather that AZ20 enhanced GEM-induced DNA damage in pancreatic
cancer cells.
ATR inhibition has been demonstrated to cause
downregulation of RRM2 (12,30).
Consistent with that study, our results showed increased RRM2 after
treatment with GEM, which was decreased by the addition of AZ20.
Moreover, GEM-induced DNA damage was significantly enhanced by
AZ20. In addition to RRM2, RRM1 modulation has been shown to
directly influence GEM efficacy (31). Zhou et al performed a kinome
screen to identify sensitizers for RRM1-dependent GEM efficacy
(32). They identified CHK1 as a
major therapeutic target capable of overcoming GEM resistance in
non-small cell lung cancer. Our results are similar in that
targeting ATR, directly upstream of CHK1, enhanced GEM-induced
apoptosis in pancreatic cancer cells. In BxPC-3 cells, GEM
treatment alone caused downregulation of RRM1, which was further
downregulated when combined with GEM (48 h treatment). While in
HPAC cells, AZ20 treatment alone did not have an effect on RRM1 and
RRM2, though it did abrogate induction of RRM1 and RRM2 by GEM
treatment. It has been reported that knockdown of RRM2 could lead
to apoptosis in cancer cells (33).
Thus, abrogation of GEM-induced RRM2 expression by AZ20 may enhance
GEM-induced cell death in pancreatic cancer cells. However, changes
in RRM2 were detected after treatment for only 8 h, while the
effects on RRM1 were not detected until later, suggesting that the
effects on RRM2 contributed to the enhanced cell killing, whereas
contribution from RRM1 may have been consequential rather than
causal.
Taken together, our data showed that targeting ATR
using AZ20 can enhance the antitumor activity of GEM through
induction of DNA damage and decrease in RRM2 expression in
pancreatic cancer cells. Accordingly, combination of AZ20 with GEM
may represent a potential chemotherapeutic regimen for treating
pancreatic cancer. Although our studies were limited to in
vitro models, our findings support the further development of
AZ20 in combination with GEM for the treatment of pancreatic
cancer.
References
|
1
|
Cid-Arregui A and Juarez V: Perspectives
in the treatment of pancreatic adenocarcinoma. World J
Gastroenterol. 21:9297–9316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Majumder S, Chari ST and Ahlquist DA:
Molecular detection of pancreatic neoplasia: Current status and
future promise. World J Gastroenterol. 21:11387–11395. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gresham GK, Wells GA, Gill S, Cameron C
and Jonker DJ: Chemotherapy regimens for advanced pancreatic
cancer: A systematic review and network meta-analysis. BMC Cancer.
14:4712014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Sousa Cavalcante L and Monteiro G:
Gemcitabine: Metabolism and molecular mechanisms of action,
sensitivity and chemoresistance in pancreatic cancer. Eur J
Pharmacol. 741:8–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Karnitz LM, Flatten KS, Wagner JM,
Loegering D, Hackbarth JS, Arlander SJ, Vroman BT, Thomas MB, Baek
YU, Hopkins KM, et al: Gemcitabine-induced activation of checkpoint
signaling pathways that affect tumor cell survival. Mol Pharmacol.
68:1636–1644. 2005.PubMed/NCBI
|
|
7
|
Voutsadakis IA: Molecular predictors of
gemcitabine response in pancreatic cancer. World J Gastrointest
Oncol. 3:153–164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qin L, Dong Z and Zhang JT: 14-3-3σ
regulation of and interaction with YAP1 in acquired gemcitabine
resistance via promoting ribonucleotide reductase expression.
Oncotarget. 7:17726–17736. 2016.PubMed/NCBI
|
|
9
|
Maréchal A and Zou L: DNA damage sensing
by the ATM and ATR kinases. Cold Spring Harb Perspect Biol.
5:52013. View Article : Google Scholar
|
|
10
|
Couch FB, Bansbach CE, Driscoll R, Luzwick
JW, Glick GG, Bétous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et
al: ATR phosphorylates SMARCAL1 to prevent replication fork
collapse. Genes Dev. 27:1610–1623. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sørensen CS and Syljuåsen RG: Safeguarding
genome integrity: The checkpoint kinases ATR CHK1 and WEE1 restrain
CDK activity during normal DNA replication. Nucleic Acids Res.
40:477–486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Buisson R, Boisvert JL, Benes CH and Zou
L: Distinct but concerted roles of ATR DNA-PK, and Chk1 in
countering replication stress during S phase. Mol Cell.
59:1011–1024. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kwok M, Davies N, Agathanggelou A, Smith
E, Petermann E, Yates E, Brown J, Lau A and Stankovic T: Synthetic
lethality in chronic lymphocytic leukaemia with DNA damage response
defects by targeting the ATR pathway. Lancet. 385 Suppl 1:S582015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fokas E, Prevo R, Hammond EM, Brunner TB,
McKenna WG and Muschel RJ: Targeting ATR in DNA damage response and
cancer therapeutics. Cancer Treat Rev. 40:109–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weber AM and Ryan AJ: ATM and ATR as
therapeutic targets in cancer. Pharmacol Ther. 149:124–138. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Prevo R, Fokas E, Reaper PM, Charlton PA,
Pollard JR, McKenna WG, Muschel RJ and Brunner TB: The novel ATR
inhibitor VE-821 increases sensitivity of pancreatic cancer cells
to radiation and chemotherapy. Cancer Biol Ther. 13:1072–1081.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fokas E, Prevo R, Pollard JR, Reaper PM,
Charlton PA, Cornelissen B, Vallis KA, Hammond EM, Olcina MM,
McKenna W Gillies, et al: Targeting ATR in vivo using the novel
inhibitor VE-822 results in selective sensitization of pancreatic
tumors to radiation. Cell Death Dis. 3:e4412012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Foote KM, Blades K, Cronin A, Fillery S,
Guichard SS, Hassall L, Hickson I, Jacq X, Jewsbury PJ, McGuire TM,
et al: Discovery of
4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole
(AZ20): A potent and selective inhibitor of ATR protein kinase with
monotherapy in vivo antitumor activity. J Med Chem. 56:2125–2138.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nghiem P, Park PK, Kim Y, Vaziri C and
Schreiber SL: ATR inhibition selectively sensitizes G1
checkpoint-deficient cells to lethal premature chromatin
condensation. Proc Natl Acad Sci USA. 98:pp. 9092–9097. 2001;
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mukhopadhyay UK, Senderowicz AM and
Ferbeyre G: RNA silencing of checkpoint regulators sensitizes
p53-defective prostate cancer cells to chemotherapy while sparing
normal cells. Cancer Res. 65:2872–2881. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Toledo LI, Murga M, Zur R, Soria R,
Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR and
Fernandez-Capetillo O: A cell-based screen identifies ATR
inhibitors with synthetic lethal properties for cancer-associated
mutations. Nat Struct Mol Biol. 18:721–727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ruzankina Y, Schoppy DW, Asare A, Clark
CE, Vonderheide RH and Brown EJ: Tissue regenerative delays and
synthetic lethality in adult mice after combined deletion of Atr
and Trp53. Nat Genet. 41:1144–1149. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang G, Niu X, Zhang W, Caldwell JT,
Edwards H, Chen W, Taub JW, Zhao L and Ge Y: Synergistic antitumor
interactions between MK-1775 and panobinostat in preclinical models
of pancreatic cancer. Cancer Lett. 356:656–668. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie C, Drenberg C, Edwards H, Caldwell JT,
Chen W, Inaba H, Xu X, Buck SA, Taub JW, Baker SD, et al:
Panobinostat enhances cytarabine and daunorubicin sensitivities in
AML cells through suppressing the expression of BRCA1, CHK1, and
Rad51. PLoS One. 8:e791062013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Redon CE, Nakamura AJ, Zhang YW, Ji JJ,
Bonner WM, Kinders RJ, Parchment RE, Doroshow JH and Pommier Y:
Histone gammaH2AX and poly(ADP-ribose) as clinical pharmacodynamic
biomarkers. Clin Cancer Res. 16:4532–4542. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jossé R, Martin SE, Guha R, Ormanoglu P,
Pfister TD, Reaper PM, Barnes CS, Jones J, Charlton P, Pollard JR,
et al: ATR inhibitors VE-821 and VX-970 sensitize cancer cells to
topoisomerase i inhibitors by disabling DNA replication initiation
and fork elongation responses. Cancer Res. 74:6968–6979. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kastan MB, Zhan Q, el-Deiry WS, Carrier F,
Jacks T, Walsh WV, Plunkett BS, Vogelstein B and Fornace AJ Jr: A
mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is
defective in ataxia-telangiectasia. Cell. 71:587–597. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
D'Angiolella V, Donato V, Forrester FM,
Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP and
Pagano M: Cyclin F-mediated degradation of ribonucleotide reductase
M2 controls genome integrity and DNA repair. Cell. 149:1023–1034.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jordheim LP, Sève P, Trédan O and Dumontet
C: The ribonucleotide reductase large subunit (RRM1) as a
predictive factor in patients with cancer. Lancet Oncol.
12:693–702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou J, Chen Z, Malysa A, Li X, Oliveira
P, Zhang Y and Bepler G: A kinome screen identifies checkpoint
kinase 1 (CHK1) as a sensitizer for RRM1-dependent gemcitabine
efficacy. PLoS One. 8:e580912013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rahman MA, Amin ARMR, Wang D, Koenig L,
Nannapaneni S, Chen Z, Wang Z, Sica G, Deng X, Chen ZG, et al: RRM2
regulates Bcl-2 in head and neck and lung cancers: A potential
target for cancer therapy. Clin Cancer Res. 19:3416–3428. 2013.
View Article : Google Scholar : PubMed/NCBI
|