Introduction
Bladder cancer is the 9th leading cause of
cancer-related deaths with ~430,000 new cases (3.1% of total) and
accounts for ~16,500 deaths worldwide annually (1,2).
Notably, it is notorious for the high rate of local recurrence
(~70%); thus, additional surgical resection is repeatedly required
for patients even during their entire life (3). In fact, it is reported that bladder
cancer is the most expensive human cancer to treat based on the
cumulative/patient cost from diagnosis until death (4). For this reason, there is an urgent
need to develop novel treatment strategies to counteract such a
tenacious disease.
Histone acetylases and histone deacetylases (HDACs)
play opposing activities in the acetylated level of histones
resulting in various degrees of gene expression. Remarkably, the
HDAC-induced extensive deacetylated level of histones has been
linked to carcinogenesis by suppressing the expression of tumor
regulatory genes, such as p21(WAF/CIP1) (5). In contrast, HDAC inhibitors may
reverse this process by blocking HDAC activity leading to the
re-expression of silenced regulatory genes (6,7),
thereby inducing cytotoxicity in cancer cells and acting as a
potential new class of anticancer agents (7,8). In
addition, HDAC inhibitors have recently been noted for their
ability to activate not only histones, but also non-histone
substrates in diverse cellular responses, including cell cycle
arrest, differentiation, apoptosis and altering metastasis in
numerous cancer cell types (9). A
plethora of structurally diverse HDAC inhibitors have been
identified (10) and some of them
have exhibited demonstrable antitumor activity and have a favorable
safety profile in clinical studies (11). To date, four HDAC inhibitors are
approved by the United States Food and Drug Administration (US FDA)
(12), including suberoylanilide
hydroxamic acid (SAHA), romidepsin (FK-228), belinostat (PXD-101)
and panobinostat (LBH-589) for the treatment of lymphoma or
multiple myeloma.
Trichostatin A (TSA), a hydroxamic acid-derived
phytochemical that originally serves as an antifungal antibiotic,
is a classic pan-HDAC inhibitor selectively repressing the class I
and II HDAC families of enzymes at nanomolar concentrations
(13). Despite the expensive
production and toxicity in clinical trials, TSA is now mainly
regarded as a prototype compound with great potency to be a useful
reference tool for further investigation of new HDAC inhibitors
(14). TSA has been widely reported
to induce cell cycle arrest (15,16),
promote apoptosis (17,18), and suppress angiogenesis (19,20) or
metastasis (21,22) in various types of tumors. However,
the precise molecular mechanisms underlying the antitumor actions
in urothelial carcinoma (UC) have not been fully delineated.
Therefore, in the present study, we aimed to elucidate how TSA
regulates the related apoptotic pathways by targeting 5,637 bladder
cancer cells. Furthermore, the presented implications found between
TSA and UC may provide essential evidence not only in the
identification of therapeutic targets, but also further for
effective antitumor drug development.
Materials and methods
Cell culture
The 5,637 urothelial cell line, a grade II
carcinoma, was purchased from the Bioresource Collection and
Research Center (Hsinchu, Taiwan). The 5,637 cells were maintained
in RPMI-1640 medium (Gibco Life Technologies, Grand Island, NY,
USA) supplemented with 10% fetal bovine serum (FBS; Biological
Industries, M.P. Ashrat, Israel), 1.5 g/l sodium bicarbonate, 4.5
g/l D-glucose, 1% penicillin-streptomycin (Gibco Life
Technologies), 1 mM sodium pyruvate and 10 mM HEPES. Cells were
plated on 100-mm plastic dishes and incubated in a CO2
incubator at 37°C, with 5% CO2 and 95% filtered air.
Cell viability assay
Cell viability was determined using a colorimetric
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. The cells were seeded in 96-well plates at a density of
4×103 cells/well for 24 h, and then were incubated with
various concentrations of test agents for another 12–36 h. MTT was
added into the medium at 37°C for 2 h, and the medium was then
discarded and dimethyl sulfoxide (DMSO) was added to dissolve the
formazan product. Each well was measured by light absorbance at 540
nm. The result was expressed as the percentage of the normal
saline-treated control group.
Cell cycle analysis
Firstly, 1×106 cells were seeded in
100-mm dishes for a 24-h incubation. Afterwards, TSA or sterile
alcohol was added for another 12 and 24 h of treatment. The cells
were then harvested, centrifuged at 800 × g for 5 min and fixed
with ice-cold 75% ethanol overnight at 4°C. Following removal of
the ethanol, the cells were stained with a DNA staining solution
[containing 1 mg/ml propidium iodide (PI) and 10 mg/ml RNase A
dissolved in phosphate-buffered saline (PBS)] for 30 min at room
temperature. The relative DNA content of the stained cells was
measured using a FACScan flow cytometer (BD Biosciences, San Jose,
CA, USA). The cell doublets were removed by gating the left area of
the FL2-W vs. FL2-A dot plot for analysis. The cell cycle data from
flow cytometry were analyzed using ModFit LT™ software (Verity
Inc., Sunnyvale, CA, USA).
Annexin V-FITC and PI staining
Apoptotic cells were also detected using Annexin V
labeling. Cells were treated as above and harvested, and then
stained with 2 µl Annexin V-FITC and 2 µl PI staining solution
(Bio-Genesis Technologies, Inc., Taipei, Taiwan) in the dark at
room temperature for 15 min. The cell samples were immediately
analyzed by the same flow cytometry and software program as
previously mentioned.
Measurement of mitochondrial membrane
potential (MMP, ΔΨm)
The fluorescence intensity of Rhodamine 123
(Sigma-Aldrich, St. Louis, MO, USA), which is permeable to the
mitochondrial membrane then specifically quenched in the
mitochondria due to the MMP, was used as a measure of membrane
damage. In brief, 5×105 cells were incubated with 5 µM
Rhodamine 123 for 10 min at 37°C. The cells were then centrifuged
at 200 × g for 6 min, resuspended in PBS, and kept on ice in the
dark. The staining intensity was determined using the FACS analysis
(Ex/Em = 485/535 nm) as previously mentioned. Negative staining for
Rhodamine 123 represented the loss of ΔΨm.
Western blot analysis
Briefly, the TSA-treated and control cells were
washed with PBS and incubated for 20 min in 150 µl of lysis buffer
containing PRO-PREP™ protein extraction solution (iNtRON
Biotechnology, Gyeonggi-do, Korea), 0.1 M NaF and 0.5 M vanadate.
The mixture was centrifuged at 12,000 × g for 5 min, and then the
total protein extract in the supernatant fluid was collected and
aliquoted for 30 µg for further analysis. Protein concentrations
were determined using the Pierce BCA assay kit (Thermo Scientific,
Rockford, IL, USA). For immunoblotting, proteins on the analytical
10% SDS-PAGE gels were transferred to a polyvinylidene difluoride
membrane using a trans-blot apparatus. Antibodies against cleaved
caspase-3, caspase-9, PARP, survivin (Cell Signaling Technology,
Inc., Danvers, USA), α-tubulin, GAPDH, β-actin (GeneTex, Irvine,
CA, USA), p-Akt, cyclin D1, p21 (Santa Cruz Biotechnology, Paso
Robles, CA, USA) and Sp1 (Upstate Biotechnology, Lake Placid, NY,
USA) were used as the primary antibodies. Mouse, rabbit or goat IgG
antibodies conjugated to horseradish peroxidase were used as the
secondary antibodies. An enhanced chemiluminescence kit and
Chemi-Smart 3000 system (Vilber Lourmat Corporation, Torcy, France)
were used for detection, and the quantity of each band was
determined using MultiGauge software (Fujifilm, Tokyo, Japan).
Statistical analysis
Numerical data are expressed as the mean ± standard
error from triplicate experiments. Statistical differences were
analyzed using one-way analysis of variance analysis followed by
Student's t-test. All statistics were calculated using SigmaPlot
version 12.5 (Systat Software, Inc., San Jose, CA, USA).
Results
TSA alters cell morphology and
significantly reduces cell viability
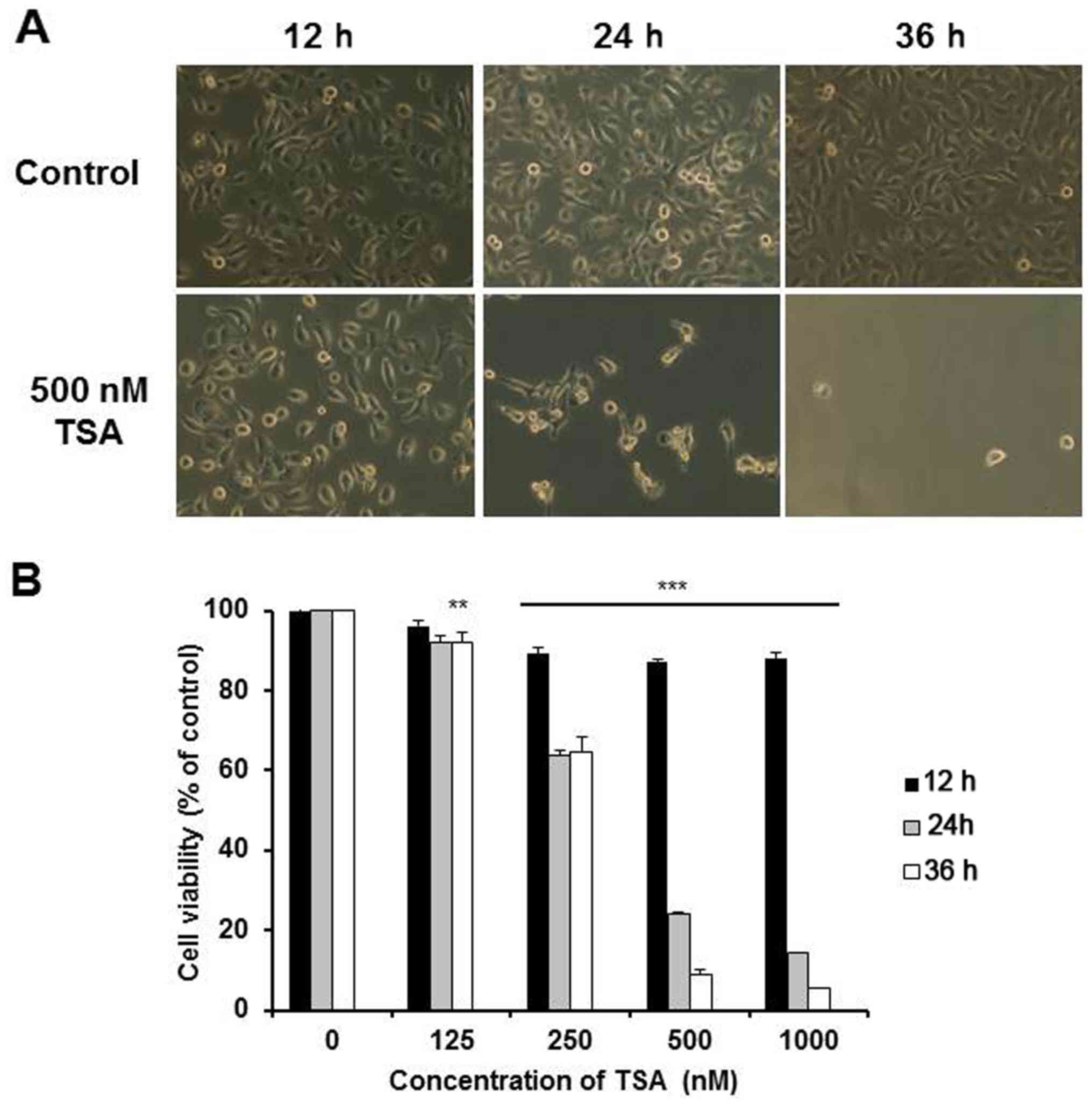
TSA is a very toxic chemical in 5,637 cells. As
shown in Fig. 1A, the 5,637 cells
in the normal culture exhibited a spindle-shaped morphology. After
exposure to 500 nM TSA for 12 h, some cells rounded up. After 24 h,
>70% of the cells rounded up and the attached cells showed
marked change to an elongated shape with filamentous protrusions.
The viability of the 5,637 cells was then assessed using MTT assay.
The 5,637 cells treated with TSA displayed decreased cell viability
in a dose-dependent manner after 24 h (Fig. 1B). Furthermore, it was found that
most of the cells died after co-culture with 500 nM TSA for 36 h,
which was also well reflected in the markedly low cell viability
presented here. Since the IC50 value of TSA was
determined between 250 and 500 nM after a 24-h treatment, these
concentrations were used in the following experiments.
TSA causes cell cycle arrest of the
5,637 cells
HDAC inhibitors have been reported to cause growth
arrest at the G1 and G2/M phases in a wide variety of tumor cells
(23). To elucidate whether TSA
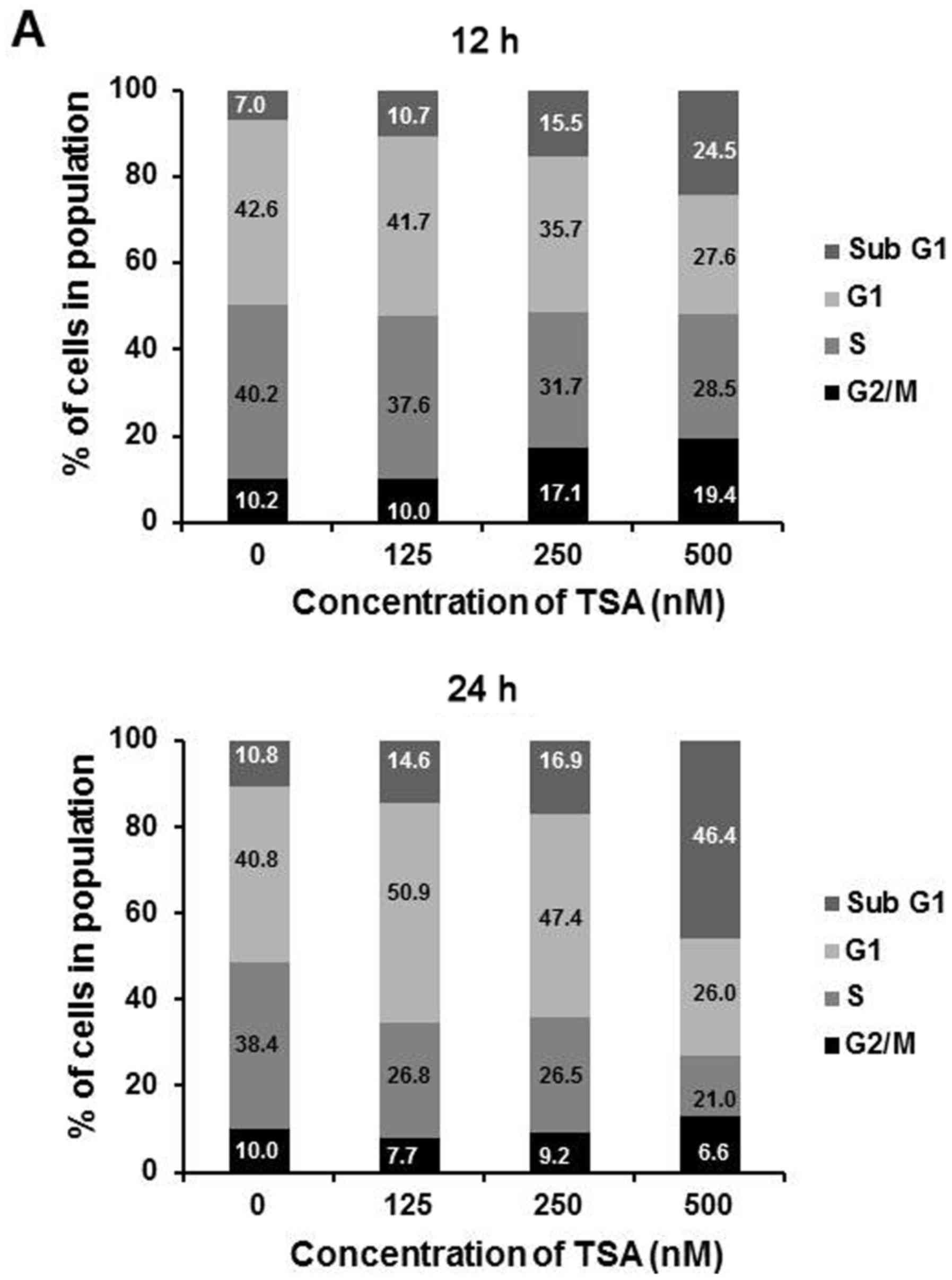
induces growth arrest in 5,637 cells, flow cytometric analysis was
used. We measured cell cycle distribution at 12 and 24 h,
respectively, after treatment with varying concentrations of TSA
(125, 250 and 500 nM). The results showed that G2/M arrest and the
sub-G1 portion were formatted along with the rising concentration
of TSA at 12 h. At 24 h, there was a moderate increase in G1 phase
portion at a lower dosage (125 and 250 nM), while a large degree of
cell death was correlated with enhanced sub-G1 phase cells observed
under a high dosage (500 nM) of TSA (Fig. 2A). Since p21 is the principal
cyclin-dependent kinase inhibitor (CDKI) involved in cell cycle
arrest upon DNA damage (24), we
analyzed p21 expression by western blotting following TSA
treatments. As shown in Fig. 2B,
p21 expression was significantly increased in a dose-dependent
manner following a 12-h treatment. In contrast, cyclin D1 was
correspondingly found downregulated following both a 12- and 24-h
treatment (Fig. 2B). These findings
demonstrated that TSA may regulate G2/M and G1 phase arrest by
modulating both p21 and cyclin D1 expression in 5,637 cells. Due to
the rationale that HDAC inhibitors may regulate gene expression,
the p21 mRNA expression level was then detected to verify whether
TSA has an effect on p21 mRNA. However, as shown in Fig. 2C, the amount of p21 mRNA did not
change following TSA treatment, suggesting that an alternative
pathway was attributable to the enhanced p21 protein accumulation
rather than the direct transcriptional activation of TSA.
TSA induces cell death via apoptosis
in 5,637 cells
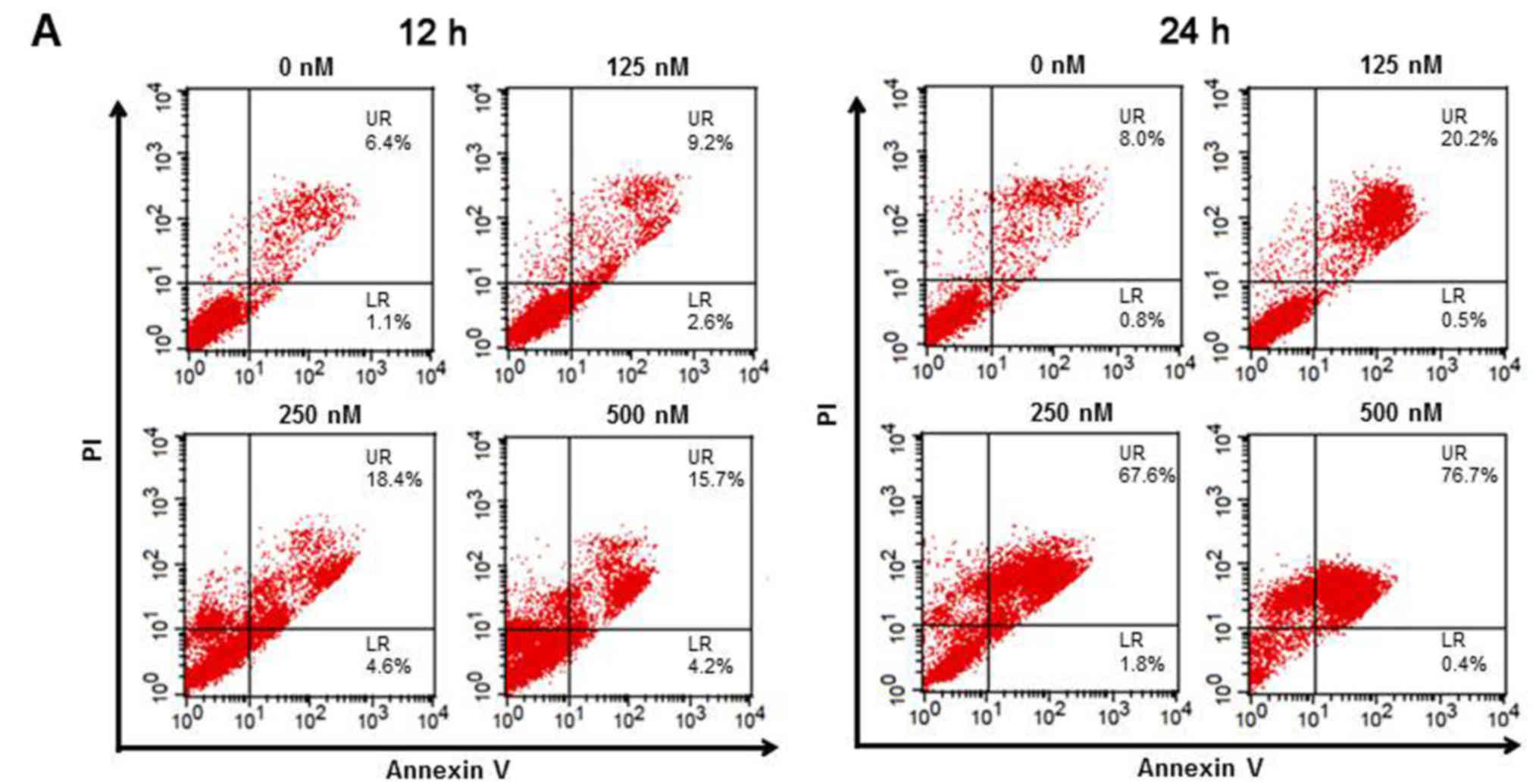
Based on the evidence from the cell cycle assay that
TSA induced severe death of 5,637 cells, we next examined the
apoptosis status using Annexin V-PI staining in cells exposed again
to four varying concentrations of TSA for 12 and 24 h. As shown in
Fig. 3A, TSA caused dose-dependent
apoptosis based on an increase in the number of Annexin V-positive
cells at 12 h and at 24 h (UR, late apoptosis). In addition,
apoptosis was further confirmed by the evidence of cleaved PARP and
cleaved caspase-3 as detected by western blotting (Fig. 3B). In essence, the cell viability
was restored when the pan-caspase inhibitor Z-VAD-FMK was applied
(Fig. 3C), suggesting that
TSA-induced cell death was mediated by the caspase-dependent
apoptotic pathway.
TSA induces apoptosis in 5,637 cells
via the mitochondrial pathway by causing MMP dissipation and
caspase-9 activation
The progression of apoptosis has been closely
related to the injury suffered from MMP collapse (25). Since the TSA-induced apoptotic clues
come from the activation of intrinsic pathway, subsequently we
measured the change in MMP to assess whether the apoptosis process
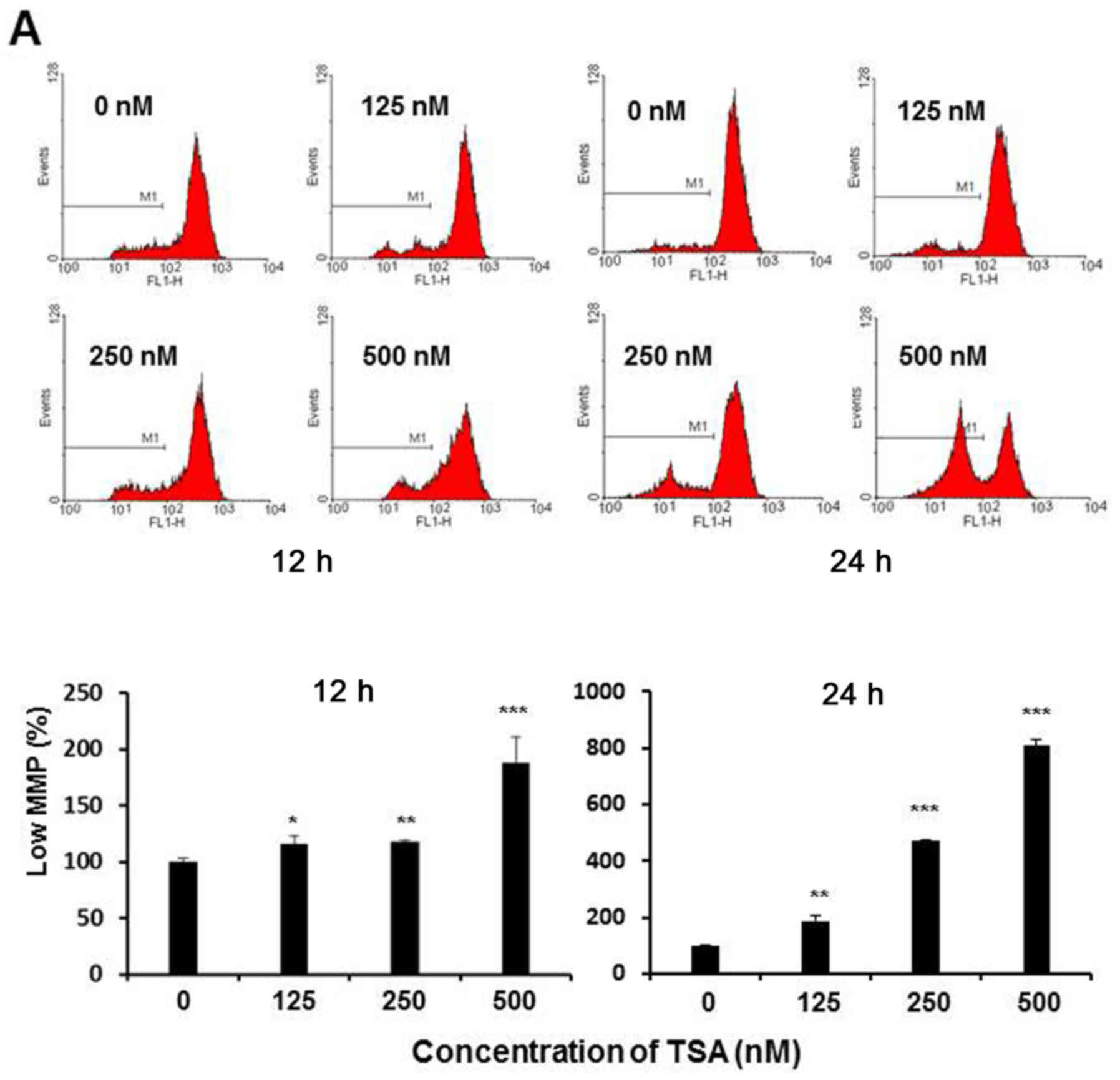
was associated with irreversible mitochondrial depolarization. In
the present study, the loss of MMP in TSA-treated 5,637 cells was
determined by use of Rhodamine 123 dye via flow cytometric assay.
As shown in Fig. 4A, the loss of
MMP was noted in the histograms, in which a clear peak shift was
shown along with the increasing TSA concentration, indicating that
TSA could lead to depolarization of the inner mitochondrial
membrane in a dose-dependent manner. The percentage of the cells
with mitochondrial depolarization was 13.6, 14.9 and 16.1% in
response to 125, 250 and 500 nM of TSA treatment at 12 h, and 27.4,
50.8 and 77.8% at 24 h, respectively. Furthermore, the downstream
apoptotic effector caspase-9 was also demonstrated to be activated
accompanied by the MMP loss (Fig.
4B). Collectively, these observations strongly support the
notion that TSA-induced apoptosis in 5,637 cells occurred through
the intrinsic mitochondrial pathway.
TSA suppresses the PI3K-Akt signaling
pathway in 5,637 cells
Previously published data explored in various cancer
cell studies have indicated a role for PI3K/Akt in the cytotoxocity
of TSA (26,27). We next examined whether the PI3k-Akt
signaling pathway may also contribute to TSA-induced apoptosis in
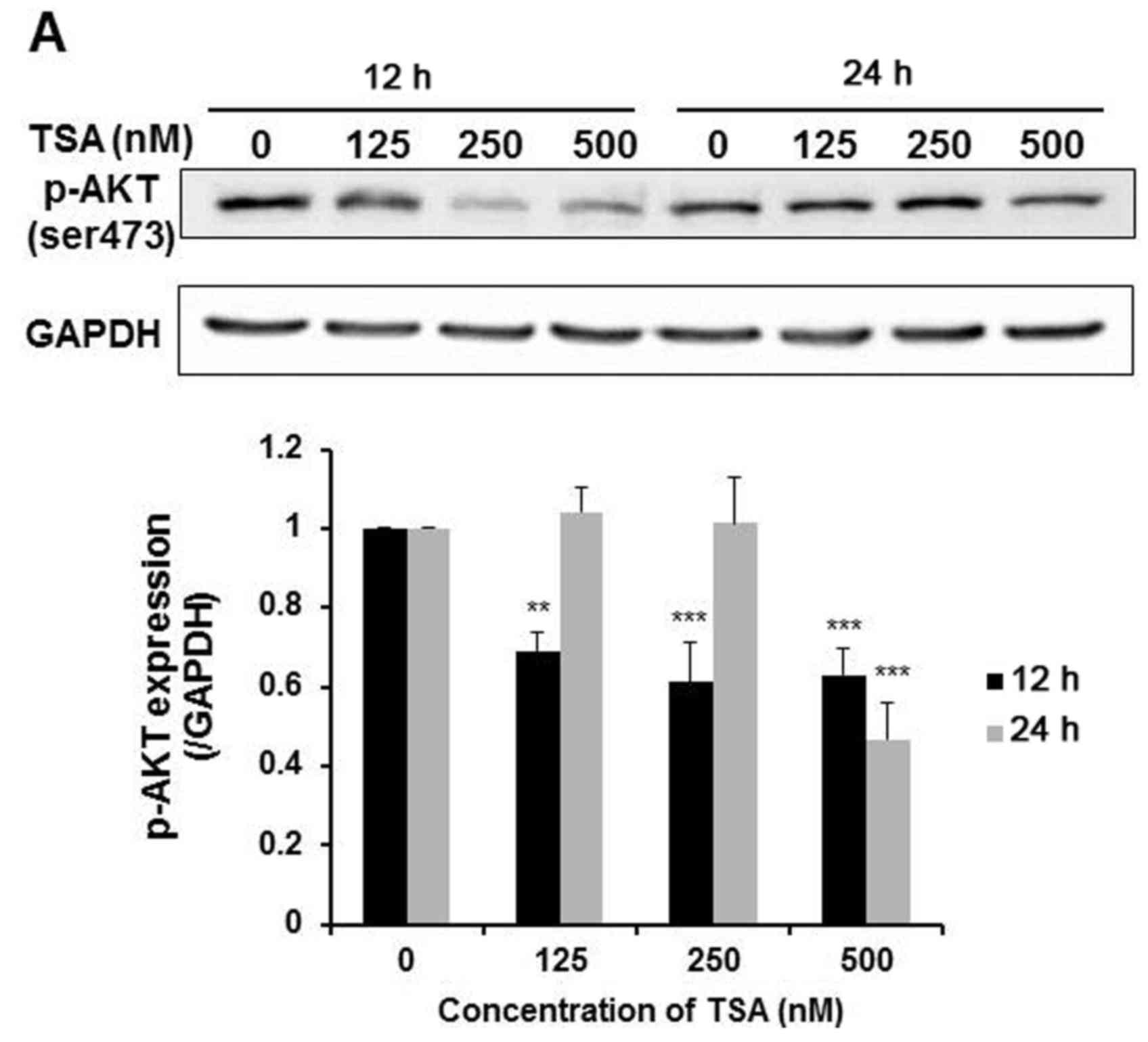
5,637 cells. As shown in Fig. 5A,
TSA dose-dependently reduced Akt phosphorylation (ser473) after 12
h of treatment. While at 24 h, only 500 nM TSA suppressed Akt
phosphorylation. This evidence suggested that TSA-induced
suppression of the PI3K/Akt pathway was more effective at early
time points. Moreover, when cells were treated with PI3K inhibitor
LY294002 (10 µM) alone, pro-apoptotic molecule caspase-9 was also
subsequently activated (Fig. 5B),
which was consistent with a previous study showing that LY294022
attenuates cell viability in 5,637 cells (28). Given these observations, we suggest
that the enhanced apoptosis in 5,637 cells induced by TSA is
mediated, at least in part, through the PI3K-Akt pathway.
TSA causes Sp1 downregulation and
suppresses survivin expression in 5,637 cells
Reports have previously noted that TSA suppresses
Sp1 binding to the promoter of survivin (BIRC5), which belongs to a
member of the inhibitor of apoptosis (IAP) family that inhibits
caspase activation thereby leading to induction of apoptosis
(29,30). Thus, in the present study, we
examined whether TSA affects Sp1 and survivin expression in 5,637
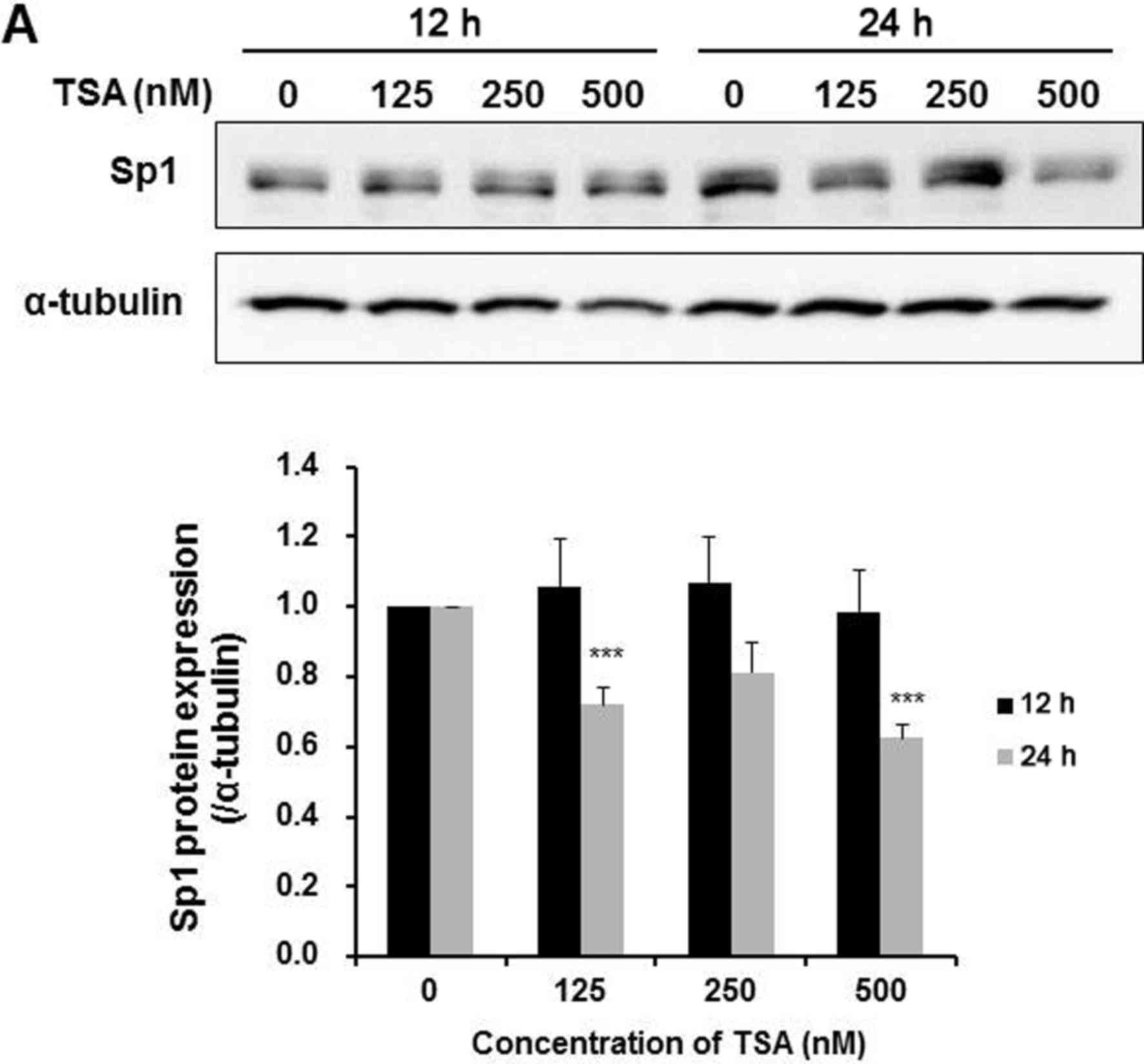
cells. Results from Fig. 6A
demonstrated that TSA dose-dependently downregulated Sp1 expression
after a 24-h treatment, accompanied by a decrease in the survivin
protein level (Fig. 6B). However,
to further make clear the essential effect exerted by Sp1, we used
mithramycin A, which is a prototypic Sp1 inhibitor possessing
capacity to displace Sp1 from its transcription sites (31), to block Sp1 activity. As shown in
Fig. 6C, the cell growth was
markedly suppressed by treatment of mithramycin A. However,
pre-treatment of pan-caspase inhibitor for 1 h antagonized the
influence of mithramycin A and effectively restored the cell
viability (Fig. 6D). This evidence
indicated that TSA counteracted Sp1 transcriptional activity, which
at least led to downregulation of survivin protein expression
following apoptosis at 24 h in 5,637 cells.
Discussion
During the past several years, there is growing
evidence to suggest that HDAC inhibitors may be applied with
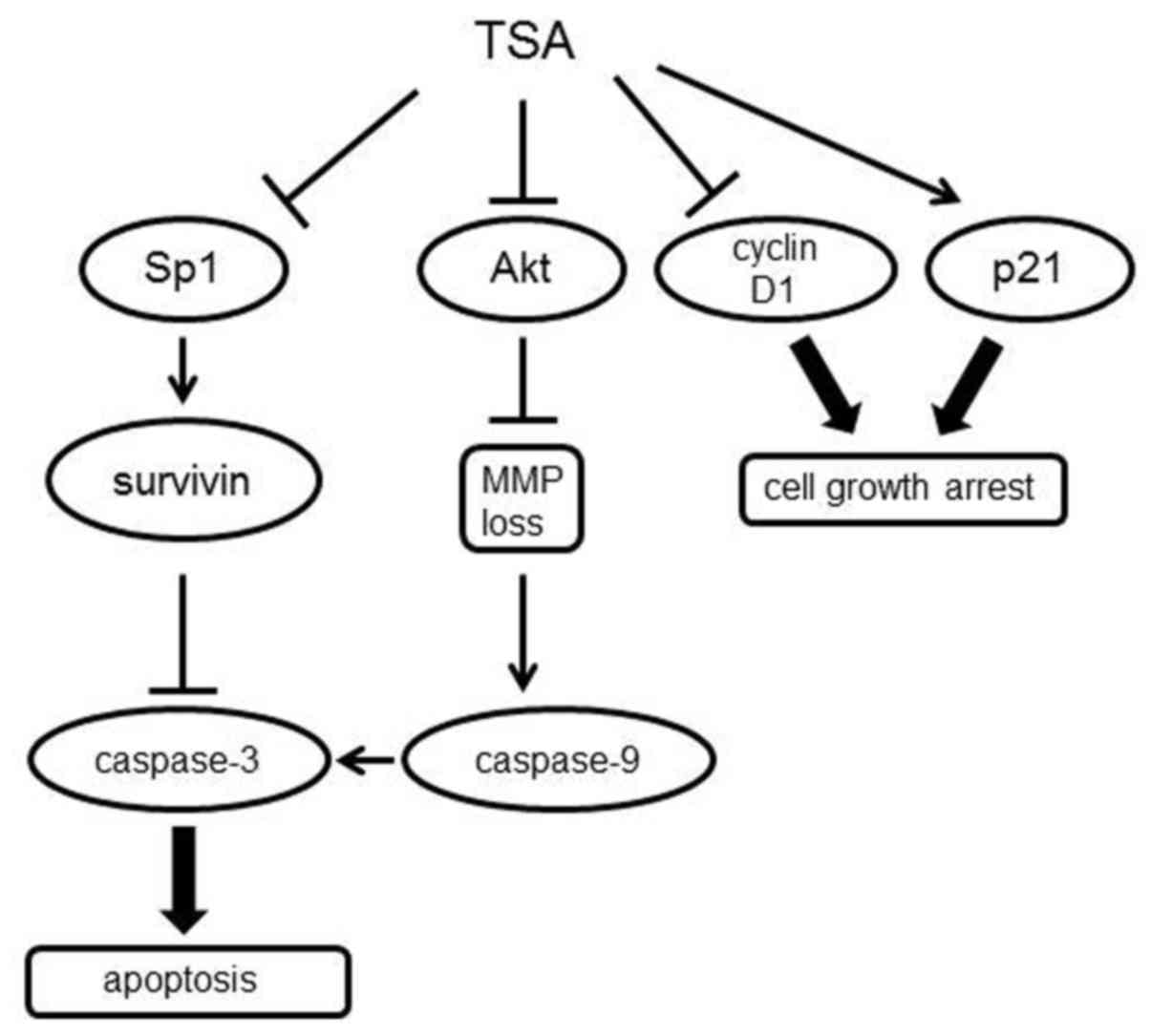
favorable outcome in cancer treatment (32). In the present study, we found that
HDAC inhibitor TSA caused 5,637 urinary bladder cell death via cell
cycle arrest and intrinsic apoptosis (Fig. 7). TSA reduced cell viability
(Fig. 1) and caused cell cycle
arrest at the G2/M and G1 phases (Fig.
2A), which may result from the increased expression of CDK
inhibitor p21 at 12 h and decreased expression of cyclin D1 that
play a pivotal role in cell cycle progression (Fig. 2B). In the classical route, TP53 gene
activation increases transcription of p21 mRNA, leading to cell
cycle arrest at G1/S and/or G2/M transition (33). However, in 5,637 cells, TP53 was
found to bear point mutations at the core domain to affect the
ability of p53 to bind DNA (34).
Therefore, TSA was likely to induce a p53-independent mechanism to
trigger p21 accumulation, similar to a previous study on
gemcitabine-induced p21 expression in 5,637 cells (35). Moreover, our current data showed
that the p21 expression change was originated by an altered protein
level instead of transcriptional regulation since its mRNA amount
was unchanged after TSA treatment (Fig.
2C). A wide variety of evidence suggests that p21 induction
could be affected by some forms of regulation (36). In the present study, TSA treatment
did not alter p21 mRNA, while it increased p21 protein at 12 h
followed by repressed expression at 24 h, which was most likely due
to the change in translation and/or protein degradation. However,
the kinetics underlying the TSA-induced expression regulation of
the p21 protein level in 5,637 cells remains obscure; thus, further
study is required to elucidate this issue. Different from p21, TSA
was found to reduce cyclin D1 protein expression at both 12 and 24
h (Fig. 2B). One study provided
relevant evidence that TSA induced cyclin D1 degradation in a
ubiquitin-dependent 26S proteasome pathway (37), which was most likely responsible for
cyclin D1 protein reduction in our results. Notably, although
cyclin D1 protein decreased both at 12 and 24 h, G1 arrest was
observed only at 24 h (Fig. 2A).
This circumstance may be attributed to cyclin E which is involved
in G1 to S phase progression to compensate for cyclin D1 loss at
the early time point (38). In
addition to cell cycle arrest, TSA caused apoptotic cell death
characterized by upregulated levels of pro-apoptotic markers such
as caspase-3 and cleaved PARP (Fig.
3B), indicating that factors other than p53 signaling could
control apoptotic induction in 5,637 cells.
Mitochondria play a crucial role in the intrinsic
pathway of apoptosis owing to the release of pro-apoptotic
intermediates from the intermembrane space, such as cytochrome
c, Smac/Diablo and AIF, which in turn amplify the following
caspase cascade, leading to the final damage to the cell (39). In this respect, the sustained
opening of the mitochondrial permeability transition pore (MPTP) in
the mitochondrial inner membrane has been associated with rupture
of the outer membrane causing subsequent loss of MMP and release of
pro-apoptotic factors (40). In our
results, we demonstrated that TSA-induced apoptosis was
commensurate with altered MMP (Fig.
4A) as well as subsequent caspase-9 cleavage (Fig. 4B), indicating that TSA induced
apoptosis via the mitochondrial pathway in 5,637 cells. However,
our results also showed that the induction of 5,637 cell death was
closely linked with PI3K/Akt pathway inhibition particularly at 12
h. Akt is known to enhance cell survival through the
phosphorylation-dependent inhibition of certain pro-apoptotic
pathways. Furthermore, activated Akt has been documented to
localize in mitochondria and play important functions concerning
energy metabolism and cell survival (41). TSA is known to induce Akt
dephosphorylation by disrupting HDAC-protein phosphatase 1 (PP1)
complex, consequently leading to inactivation of this kinase route
in a PP1-dependent manner (42). In
contrast, deregulated MMP has a causative linkage with Akt
signaling since dephosphorylated Akt may lose the ability to
protect mitochondria from MPTP opening (43), which is well concordant with our
current results that TSA caused significant MMP loss (Fig. 4A) and pAKT de-phosphorylation
(Fig. 5A), and LY294002 treatment
also induced caspase-9 activation (Fig.
5B). These results supported that the signaling of TSA-mediated
apoptosis at the early phase in 5,637 cells was correlated to the
blockage of the PI3K-Akt pathway.
Our data demonstrated that treatment with the
specific Sp1 inhibitor, mithramycin A, led to a marked decrease in
cell viability (Fig. 6C), which was
restored following pre-treatment with Z-VAD-FMK (Fig. 6D). These features clearly
demonstrated that Sp1 plays a pivotal role in the anti-apoptotic
regulation of 5,637 cells. Survivin is a small member of the IAP
family and is highly expressed in malignant lesions due to the
close association with cell cycle transition as well as
anti-apoptotic activity commonly coupling to the poor outcomes of
cancer therapies (44). Currently,
various clinical trials targeting the overexpression of survivin or
activation of its related signaling pathways may pave a promising
way for cancer intervention (45,46).
Published data indicate a role for Sp1 in regulating survivin gene
transcription since numerous Sp1 binding sites exist in the
survivin promoter region (47). It
is important to note that in our present results, both Sp1 and
survivin protein levels were downregulated at later time points
(Fig. 6), indicating that
impairment of the Sp1-survivin pathway contributed to, at least in
part, the TSA-induced apoptosis in 5,637 cells. It is known that
TSA reduces cell viability by recruiting p53 or p63 to counteract
the Sp1-survivin cascade (29,30).
However, mutated p53 and intrinsically anti-apoptotic isoform of
p63 (ΔNp63α) retained in 5,637 cells (48) could not repress survivin expression
and induce apoptosis. Alternatively, given that TSA-mediated Akt
dephosphorylation has been reported to activate GSK3β (42) and GSK3β-mediated phosphorylation
facilitates Sp1 degradation (49),
there is a plausible association between deactivated Akt with
downstream Sp1 protein degradation as well as correspondingly
decreased expression of survivin in 5,637 cells. Meanwhile, the
compromised DNA binding ability of Sp1 resulting from the direct
TSA-induced acetylation presumably also aided in reduced survivin
expression to some extent (50).
Collectively, we conclude that TSA exerts multifaceted effects
including the regulation of Sp1-survivin expression and then
contributes to the tumor-suppressive behavior observed in 5,637
cells.
Acknowledgements
The present study was supported by grants from the
Ministry of Science and Technology MOST104-2320-B-415-001-MY3 of
the Republic of China, Taiwan.
Glossary
Abbreviations
Abbreviations:
|
TSA
|
trichostatin A
|
|
CDKI
|
cyclin-dependent kinase inhibitor
|
|
MMP
|
mitochondrial membrane potential
|
|
MPTP
|
mitochondrial permeability transition
pore
|
|
HDAC
|
histone deacetylase
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
Sp1
|
specificity protein 1
|
|
IAP
|
inhibitor of apoptosis
|
References
|
1
|
Stewart BW and Wild C: International
Agency for Research on Cancer and World Health Organization: World
Cancer Report. 2014.
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaufman DS, Shipley WU and Feldman AS:
Bladder cancer. Lancet. 374:239–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Avritscher EB, Cooksley CD, Grossman HB,
Sabichi AL, Hamblin L, Dinney CP and Elting LS: Clinical model of
lifetime cost of treating bladder cancer and associated
complications. Urology. 68:549–553. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ocker M and Schneider-Stock R: Histone
deacetylase inhibitors: Signalling towards p21cip1/waf1.
Int J Biochem Cell Biol. 39:1367–1374. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hitomi T, Matsuzaki Y, Yokota T, Takaoka Y
and Sakai T: p15INK4b in HDAC inhibitor-induced growth
arrest. FEBS Lett. 554:347–350. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rosato RR and Grant S: Histone deacetylase
inhibitors: Insights into mechanisms of lethality. Expert Opin Ther
Targets. 9:809–824. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Batty N, Malouf GG and Issa JP: Histone
deacetylase inhibitors as anti-neoplastic agents. Cancer Lett.
280:192–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang J and Zhong Q: Histone deacetylase
inhibitors and cell death. Cell Mol Life Sci. 71:3885–3901. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mottamal M, Zheng S, Huang TL and Wang G:
Histone deacetylase inhibitors in clinical studies as templates for
new anticancer agents. Molecules. 20:3898–3941. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoon S and Eom GH: HDAC and HDAC
inhibitor: From cancer to cardiovascular diseases. Chonnam Med J.
52:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vanhaecke T, Papeleu P, Elaut G and
Rogiers V: Trichostatin A-like hydroxamate histone deacetylase
inhibitors as therapeutic agents: Toxicological point of view. Curr
Med Chem. 11:1629–1643. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Van Beneden K, Mannaerts I, Pauwels M, Van
den Branden C and van Grunsven LA: HDAC inhibitors in experimental
liver and kidney fibrosis. Fibrogenesis Tissue Repair. 6:12013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yamashita Y, Shimada M, Harimoto N,
Rikimaru T, Shirabe K, Tanaka S and Sugimachi K: Histone
deacetylase inhibitor trichostatin A induces cell-cycle
arrest/apoptosis and hepatocyte differentiation in human hepatoma
cells. Int J Cancer. 103:572–576. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Noh EJ, Lim DS, Jeong G and Lee JS: An
HDAC inhibitor, trichostatin A, induces a delay at G2/M
transition, slippage of spindle checkpoint, and cell death in a
transcription-dependent manner. Biochem Biophys Res Commun.
378:326–331. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cohen HY, Lavu S, Bitterman KJ, Hekking B,
Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM and Sinclair
DA: Acetylation of the C terminus of Ku70 by CBP and PCAF controls
Bax-mediated apoptosis. Mol Cell. 13:627–638. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Medina V, Edmonds B, Young GP, James R,
Appleton S and Zalewski PD: Induction of caspase-3 protease
activity and apoptosis by butyrate and trichostatin A (inhibitors
of histone deacetylase): Dependence on protein synthesis and
synergy with a mitochondrial/cytochrome c-dependent pathway.
Cancer Res. 57:3697–3707. 1997.PubMed/NCBI
|
|
19
|
Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE,
Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, et al: Histone
deacetylases induce angiogenesis by negative regulation of tumor
suppressor genes. Nat Med. 7:437–443. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deroanne CF, Bonjean K, Servotte S, Devy
L, Colige A, Clausse N, Blacher S, Verdin E, Foidart JM, Nusgens
BV, et al: Histone deacetylases inhibitors as anti-angiogenic
agents altering vascular endothelial growth factor signaling.
Oncogene. 21:427–436. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoshikawa M, Hishikawa K, Marumo T and
Fujita T: Inhibition of histone deacetylase activity suppresses
epithelial-to-mesenchymal transition induced by TGF-beta1 in human
renal epithelial cells. J Am Soc Nephrol. 18:58–65. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu LT, Chang HC, Chiang LC and Hung WC:
Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2
activation and cancer cell invasion. Cancer Res. 63:3069–3072.
2003.PubMed/NCBI
|
|
23
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park SJ, Kim MJ, Kim HB, Sohn HY, Bae JH,
Kang CD and Kim SH: Trichostatin A sensitizes human ovarian cancer
cells to TRAIL-induced apoptosis by down-regulation of
c-FLIPL via inhibition of EGFR pathway. Biochem
Pharmacol. 77:1328–1336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen CS, Weng SC, Tseng PH, Lin HP and
Chen CS: Histone acetylation-independent effect of histone
deacetylase inhibitors on Akt through the reshuffling of protein
phosphatase 1 complexes. J Biol Chem. 280:38879–38887. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu JY, Tsai KW, Li YZ, Chang YS, Lai YC,
Laio YH, Wu JD and Liu YW: Anti-bladder-tumor effect of baicalein
from Scutellaria baicalensis Georgi and its application in
vivo. Evid Based Complement Alternat Med.
2013:5797512013.PubMed/NCBI
|
|
29
|
Hsu YF, Sheu JR, Lin CH, Yang DS, Hsiao G,
Ou G, Chiu PT, Huang YH, Kuo WH and Hsu MJ: Trichostatin A and
sirtinol suppressed survivin expression through AMPK and p38MAPK in
HT29 colon cancer cells. Biochim Biophys Acta. 1820:104–115. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hsu YF, Sheu JR, Hsiao G, Lin CH, Chang
TH, Chiu PT, Wang CY and Hsu MJ: p53 in trichostatin A induced C6
glioma cell death. Biochim Biophys Acta. 1810:504–513. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sleiman SF, Langley BC, Basso M, Berlin J,
Xia L, Payappilly JB, Kharel MK, Guo H, Marsh JL, Thompson LM, et
al: Mithramycin is a gene-selective Sp1 inhibitor that identifies a
biological intersection between cancer and neurodegeneration. J
Neurosci. 31:6858–6870. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Newbold A, Falkenberg KJ, Prince HM and
Johnstone RW: How do tumor cells respond to HDAC inhibition? FEBS
J. 283:4032–4046. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jänicke RU, Sohn D, Essmann F and
Schulze-Osthoff K: The multiple battles fought by anti-apoptotic
p21. Cell Cycle. 6:407–413. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vinall RL, Ripoll AZ, Wang S, Pan CX and
deVere White RW: MiR-34a chemosensitizes bladder cancer cells to
cisplatin treatment regardless of p53-Rb pathway status. Int J
Cancer. 130:2526–2538. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
da Silva GN, de Camargo EA and Salvadori
DM: Toxicogenomic activity of gemcitabine in two
TP53-mutated bladder cancer cell lines: Special focus on
cell cycle-related genes. Mol Biol Rep. 39:10373–10382. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Warfel NA and El-Deiry WS: p21WAF1 and
tumourigenesis: 20 years after. Curr Opin Oncol. 25:52–58. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Alao JP, Stavropoulou AV, Lam EW, Coombes
RC and Vigushin DM: Histone deacetylase inhibitor, trichostatin A
induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast
cancer cells. Mol Cancer. 5:82006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Keenan SM, Lents NH and Baldassare JJ:
Expression of cyclin E renders cyclin D-CDK4 dispensable for
inactivation of the retinoblastoma tumor suppressor protein,
activation of E2F, and G1-S phase progression. J Biol
Chem. 279:5387–5396. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rasola A and Bernardi P: The mitochondrial
permeability transition pore and its involvement in cell death and
in disease pathogenesis. Apoptosis. 12:815–833. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lim S, Smith KR, Lim ST, Tian R, Lu J and
Tan M: Regulation of mitochondrial functions by protein
phosphorylation and dephosphorylation. Cell Biosci. 6:252016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Alao JP, Stavropoulou AV, Lam EW and
Coombes RC: Role of glycogen synthase kinase 3 beta (GSK3beta) in
mediating the cytotoxic effects of the histone deacetylase
inhibitor trichostatin A (TSA) in MCF-7 breast cancer cells. Mol
Cancer. 5:402006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Miyamoto S, Murphy AN and Brown JH: Akt
mediates mitochondrial protection in cardiomyocytes through
phosphorylation of mitochondrial hexokinase-II. Cell Death Differ.
15:521–529. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ambrosini G, Adida C and Altieri DC: A
novel anti-apoptosis gene, survivin, expressed in cancer and
lymphoma. Nat Med. 3:917–921. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cheng Q, Ling X, Haller A, Nakahara T,
Yamanaka K, Kita A, Koutoku H, Takeuchi M, Brattain MG and Li F:
Suppression of survivin promoter activity by YM155 involves
disruption of Sp1-DNA interaction in the survivin core promoter.
Int J Biochem Mol Biol. 3:179–197. 2012.PubMed/NCBI
|
|
46
|
Glaros TG, Stockwin LH, Mullendore ME,
Smith B, Morrison BL and Newton DL: The ‘survivin suppressants’ NSC
80467 and YM155 induce a DNA damage response. Cancer Chemother
Pharmacol. 70:207–212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li F and Altieri DC: Transcriptional
analysis of human survivin gene expression. Biochem J. 344:305–311.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee HO, Lee JH, Choi E, Seol JY, Yun Y and
Lee H: A dominant negative form of p63 inhibits apoptosis in a
p53-independent manner. Biochem Biophys Res Commun. 344:166–172.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wei S, Chuang HC, Tsai WC, Yang HC, Ho SR,
Paterson AJ, Kulp SK and Chen CS: Thiazolidinediones mimic glucose
starvation in facilitating Sp1 degradation through the
up-regulation of beta-transducin repeat-containing protein. Mol
Pharmacol. 76:47–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Duan H, Heckman CA and Boxer LM: Histone
deacetylase inhibitors down-regulate bcl-2 expression and
induce apoptosis in t(14;18) lymphomas. Mol Cell Biol.
25:1608–1619. 2005. View Article : Google Scholar : PubMed/NCBI
|