Introduction
Hepatocellular carcinoma (HCC) is the most common
primary malignancy of the liver, the sixth most common cancer
overall and the third most common cause of cancer-related death
worldwide (1). Despite the
decreasing incidence of HCC in Asia, western countries are
continuously encountering new cases. HCC has one of the worst
prognoses of any cancer, with a 5-year survival rate of
approximately 15–25% (2,3). Diagnosis at advanced stages and
metastasis are associated with a poor prognosis. In clinical
practice, HCC is characterized by hypervascularity which is
required for tumor growth, invasion and metastasis. Tumor
angiogenesis has been reported to be a significant predictor of
death and is correlated with the tumor stage (4).
Angiotensin II type 1 (AT1) receptor blocker (ARB)
is an antihypertensive drug with established feasibility and safety
in clinical use. In addition to HCC cells, various cancer cells
have recently been reported to express AT1 receptors (5–7).
Several ARBs have been shown to inhibit angiogenesis in various
cancer cells expressing AT1 receptor. Angiotensin II promotes cell
proliferation during cancer development, and ARBs may suppress
proliferation by antagonizing the AT1 receptor (8–10).
ARBs inhibit the growth of breast (11), endometrial (12) and gastric cancer cells (13) in several studies. According to some
epidemiological studies, the use of ARBs may increase the risk of
cancer (14). In contrast, the use
of an ARB treatment in hypertensive patients is associated with
lower cancer incidence and mortality rates in other studies
(15,16).
Among these agents, telmisartan is a widely
prescribed drug for the treatment of hypertension, diabetic
nephropathy and heart failure. Telmisartan has been shown to
inhibit cell proliferation by inducing apoptosis in various cancer
cell lines including endometrial (12), prostate 17), renal (18) and colon (19) cancer cell lines. Although
telmisartan induces cell cycle arrest in hematological malignancies
(20), few studies have examined
the main antitumor effects of telmisartan, other than apoptosis, on
cell cycle arrest in non-hematological malignancies. As shown in
our previous study, telmisartan inhibits human esophageal
adenocarcinoma cell proliferation and tumor growth by inducing cell
cycle arrest through the regulation of cell cycle-related proteins
via the AMP-activated protein kinase (AMPK)/mammalian target of
rapamycin (mTOR) pathway (21).
AMPK is a cellular energy sensor that is expressed in almost all
eukaryotic cells (22) and
regulates cell growth and proliferation by modulating the mTOR
signaling pathway (23,24). In HCC cells, we assumed that
telmisartan regulates the proliferation of cancer cells though AMPK
activation and focused on AMPK/mTOR signaling.
The present study evaluated the effects of
telmisartan on the growth of HCC cell lines and its mechanism of
action. Furthermore, possible mechanisms associated with the
antitumor effects of telmisartan, including apoptosis, receptor
tyrosine kinases (RTKs), angiogenesis and microRNAs (miRNAs), were
also explored.
Materials and methods
Chemicals
Telmisartan and valsartan were purchased from Tokyo
Chemical Industry Co. (Tokyo, Japan). Losartan and irbesartan were
purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
We prepared solutions of telmisartan, valsartan and irbesartan by
diluting with dimethyl sulfoxide (DMSO). Losartan was prepared by
diluting with H2O. The stock solutions were stored at
−20°C.
Cell lines and culture
We used five human HCC cell lines, the HLF, HLE,
HuH-7 and PLC/PRF/5 cells were obtained from the Japanese Cancer
Research Bank (Osaka, Japan), and HepG2 cells were supplied by
Riken Cell Bank (Tsukuba, Japan). HLF cells were cultured in
Dulbeccos modified Eagles medium (DMEM) supplemented with 5% fetal
bovine serum (FBS) and penicillin-streptomycin (100 µg/ml) in a
humidified atmosphere containing 5% CO2. HLE, HuH-7 and
PLC/PRF/5 cells were cultured in DMEM supplemented with 10% FBS and
penicillin-streptomycin. HepG2 cells were cultured in MEM
supplemented with 10% FBS and penicillin-streptomycin.
Cell proliferation assay
Cell proliferation assays were conducted using the
Cell Counting kit-8 (Dojindo Laboratories, Kumamoto, Japan)
according to the manufacturers instructions. Each of the five cell
lines was seeded onto 96-well plates (5.0×103
cells/well) and were cultured in 100 µl of the corresponding
medium. After 24 h, the seeded cells were treated with telmisartan,
valsartan, losartan or irbesartan (0, 10, 50 or 100 µM) and were
cultured for an additional 48 h. At the indicated time-points, the
medium in each well was replaced with 100 µl of medium containing
the CCK-8 reagent, and the cells were incubated for 3 h. The
absorbance was measured at a wavelength of 450 nm using an
automated microplate reader.
Flow cytometric analyses of the cell
cycle and apoptosis
We conducted a flow cytometric analysis using the
Cycle Phase Determination kit (Cayman Chemical Company, Ann Arbor,
MI, USA) to assess the growth inhibition mechanism. HLF cells
(1.0×106 cells/100 mm-diameter dish) were treated with
100 µM telmisartan or DMSO control for 12–48 h. Fixed cells were
washed with phosphate-buffered saline (PBS) and then stored at
−20°C until analysis by flow cytometry. On the day of analysis, the
cells were washed with cold PBS, suspended in 100 µl of PBS plus 10
µl of RNase A (250 µg/ml) and incubated for 30 min. Then, 110 µl of
propidium iodide (PI) stain (100 µg/ml) was added to each
suspension, and the cells were incubated at 4°C for at least 30 min
prior to analysis.
Apoptosis was analyzed after the telmisartan
treatment using flow cytometry and an Annexin V-FITC Early
Apoptosis Detection kit (Cell Signaling Technology, Boston, MA,
USA). HLF cells (1.0×106 cells/100 mm-dimeter dish) were
treated with 100 µM telmisartan or DMSO control for 12 h. Apoptotic
and necrotic cell death were analyzed by double staining with
FITC-conjugated Annexin V and PI; this staining method is based on
the binding of Annexin V to apoptotic cells with exposed
phosphatidylserines and the labeling of late apoptotic/necrotic
cells with membrane damage by PI. Staining was performed according
to the manufacturers instructions. We repeated the same experiment
three times to compare the proportion of apoptotic cells in the
telmisartan-treated group and the non-treated group.
Flow cytometry was conducted using a Cytomics FC 500
flow cytometer (Beckman Coulter, Indianapolis, IN, USA) with an
argon laser (488 nm). The percentages of cells were analyzed using
Kaluza software (Beckman Coulter).
Western blot analysis
Cell lysates were prepared at 4°C as previously
described (25). HLF cells
(1.0×106 cells/100 mm-dimeter dish) were seeded and
cultured for 24 and 48 h after treatment with 100 µM of telmisartan
or DMSO control. The cells were lysed with a protease inhibitor
cocktail: PRO-PREP complete protease inhibitor mixture (iNtRON
Biotechnology, Seongnam, Korea). Supernatants containing the
soluble cellular proteins were collected and stored at −80°C until
use. Protein concentrations were measured using a NanoDrop 2000
spectrofluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
Protein aliquots (1–10 µg) were resuspended in sample buffer and
separated on 10% Tris-glycine gradient gels via sodium dodecyl
sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (26). After blocking, the membranes were
incubated with primary antibodies followed by horseradish
peroxidase (HRP)-conjugated secondary antibodies (27).
The antibodies used for western blot analyses were
obtained from the following sources: antibodies against cyclin D1
and cyclin E were obtained from Thermo Fisher Scientific; CDK4,
CDK6 and CDK2 were obtained from Santa Cruz Biotechnology (Santa
Cruz, CA, USA); AMPKα, p-AMPKα (thr172), mTOR, p-mTOR (thr2448),
p70S6K and p-p70S6K (thr389) antibodies were purchased from Cell
Signaling Technology; and anti-β-actin was purchased from
Sigma-Aldrich (St. Louis, MO, USA). The secondary antibodies
included HRP-conjugated anti-mouse and anti-rabbit IgG (Cell
Signaling Technology).
Enzyme-linked immunosorbent assay
(ELISA) for measuring apoptosis
Caspase-cleaved cytokeratin 18 (cCK18) levels were
measured using the M30 Apoptosense ELISA kit obtained from Peviva
Ab (Bromma, Sweden) to evaluate whether telmisartan exerted a
pro-apoptotic effect (28). HLF
cells (5.0×103 cells/well) were seeded on a 96-well
plate and cultured for 6, 24 and 48 h following treatment with 100
µM of telmisartan. The cells were lysed in polyoxyethylene octyl
phenyl ether (NP-40) (Wako Pure Chemical Industries). The
subsequent ELISA procedures were performed according to the
manufacturers instructions. The amounts of antigen in the control
and unknown samples were calculated using a standard curve.
Antibody arrays of apoptosis-related
protein profiles
HLF cells were cultured for 12 h after treatment
with 100 µM of telmisartan or DMSO control and then lysed in
PRO-PREP. The human apoptosis antibody array kit (R&D Systems,
Minneapolis, MN, USA) was used to assess the levels of
apoptosis-related proteins. Briefly, proteins were captured by
antibodies spotted on a nitrocellulose membrane. Then, the levels
of apoptosis-related proteins were assessed using an HRP-conjugated
antibody followed by detection via chemiluminescence, and each
array membrane was exposed to X-ray film using a chemiluminescence
detection system (Perkin-Elmer, Waltham, MA, USA). The
immunoreactive band density obtained from this array was analyzed
by densitometric scanning (TIc scanner; Shimazu Co., Ltd., Kyoto,
Japan). We repeated the same experiment three times to compare the
telmisartan-treated group with the non-treated group.
Antibody arrays of p-RTKs
HLF cells were cultured for 48 h after treatment
with 100 µM of telmisartan or DMSO control and then lysed in
PRO-PREP. Human p-RTK array kits (R&D Systems) were used
according to the manufacturers instructions. The levels of
phosphoproteins were assessed using an HRP-conjugated antibody.
Each array was exposed to X-ray film and analyzed using the same
method as described for the other antibody arrays. We repeated the
same experiment three times to compare the telmisartan-treated
group with the non-treated group.
Antibody arrays of angiogenetic
protein profiles
HLF cells were cultured for 48 h after treatment
with 100 µM of telmisartan or DMSO control and then lysed in
PRO-PREP. The RayBio Human Angiogenesis Antibody array (RayBiotech,
Norcross, GA, USA) was used according to the manufacturers
protocol. This method is a dot-based assay that enables the
detection and comparison of 20 angiogenesis-specific cytokines.
Each array was exposed to X-ray film and analyzed using the same
method as described the other antibody arrays. We repeated the same
experiment three times to compare the telmisartan-treated group
with the non-treated group.
Microarray analysis of miRNAs
After treatment with 100 µM telmisartan or the DMSO
control for 48 h, total RNA was extracted from HLF cells using the
miRNeasy Mini kit (Qiagen, Hilden, Germany) according to the
manufacturers instructions. Each RNA sample typically exhibited
A260/280 ratios between 1.9 and 2.1, which were
evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies,
Santa Clara, CA, USA). After measuring RNA quantity using the RNA
6000 Nano kit (Agilent Technologies), the samples were labeled
using a miRCURY Hy3 Power Labeling kit (Exiqon A/S, Vedbaek,
Denmark) and hybridized to a human miRNA Oligo Chip (v.21; Toray
Industries, Inc., Tokyo, Japan). Scanning was conducted using the
3D-Gene Scanner 3000 (Toray Industries). The 3D-Gene extraction
version 1.2 software (Toray Industries) was used to calculate the
raw signal intensity of the images. The raw data were analyzed
using the GeneSpring GX 10.0 software (Agilent Technologies) to
assess the differences in miRNA expression between the
telmisartan-treated and control samples. Global normalization was
performed on raw data that were above the background level.
Differentially expressed miRNAs were determined using Welchs
t-test. The false discovery rate was computed using the
Benjamini-Hochberg method (29).
Hierarchical clustering was performed using the farthest neighbor
method with the absolute uncentered Pearsons correlation
coefficient as a metric. A heatmap was produced with the relative
expression intensity of each miRNA, in which the base-2 logarithm
of intensity was median-centered for each row.
Real-time quantitative polymerase
chain reaction (qPCR) analysis of miRNAs
We compared the expression levels obtained from the
miRNA arrays with real-time qPCR measurements to validate the data
for miR-3651 and miR-7-5p which exhibited significantly greater
changes in expression in the microarray analysis. Total RNA was
extracted as described above and was diluted to 2.0 ng/µl. TaqMan
microRNA assays (Applied Biosystems, Waltham, MA, USA) were adopted
to determine the expression levels of miRNAs using U6 small nuclear
RNA (RNU6B) as an internal control (Assay ID: 464410mat for
miR-3651; 000268 for miR-7-5p; and 001093 for U6). miRNAs were
reverse transcribed using the TaqMan microRNA Reverse Transcription
kit (Applied Biosystems). Reverse transcription was prepared in 15
µl reaction volumes consisting of 5 µl of RNA, 3 µl of 5X RT primer
and 12 µl of reverse transcription Master Mix. PCRs were performed
in the MicroAmp Fast Optical 96-Well Reaction Plate (Applied
Biosystems). Each well contained a 20 µl reaction consisting of 2
µl of cDNA, 1 µl of 20X qPCR assay, 7 µl of nuclease-free water and
10 µl of TaqMan Fast Advanced Master Mix (Applied Biosystems).
Using the ViiA7 real-time PCR system (Applied Biosystems),
reactions were denatured by an incubation at 95°C for 20 sec
followed by 40 cycles of 1 sec at 95°C and 20 sec at 60°C. The
relative expression levels of miR-3651 and miR-7-5p were calculated
using the comparative Ct method according to the following formula:
2-ΔCt (ΔCt = miRCt - U6Ct).
Statistical analyses
All statistical analyses were performed using the
GraphPad Prism software version 6.0 (GraphPad Software, Inc., San
Diego, CA, USA). Unpaired t-tests were conducted to compare the
data between groups. A P-value of <0.05 was considered
significant.
Results
Telmisartan inhibits the proliferation
of human HCC cells
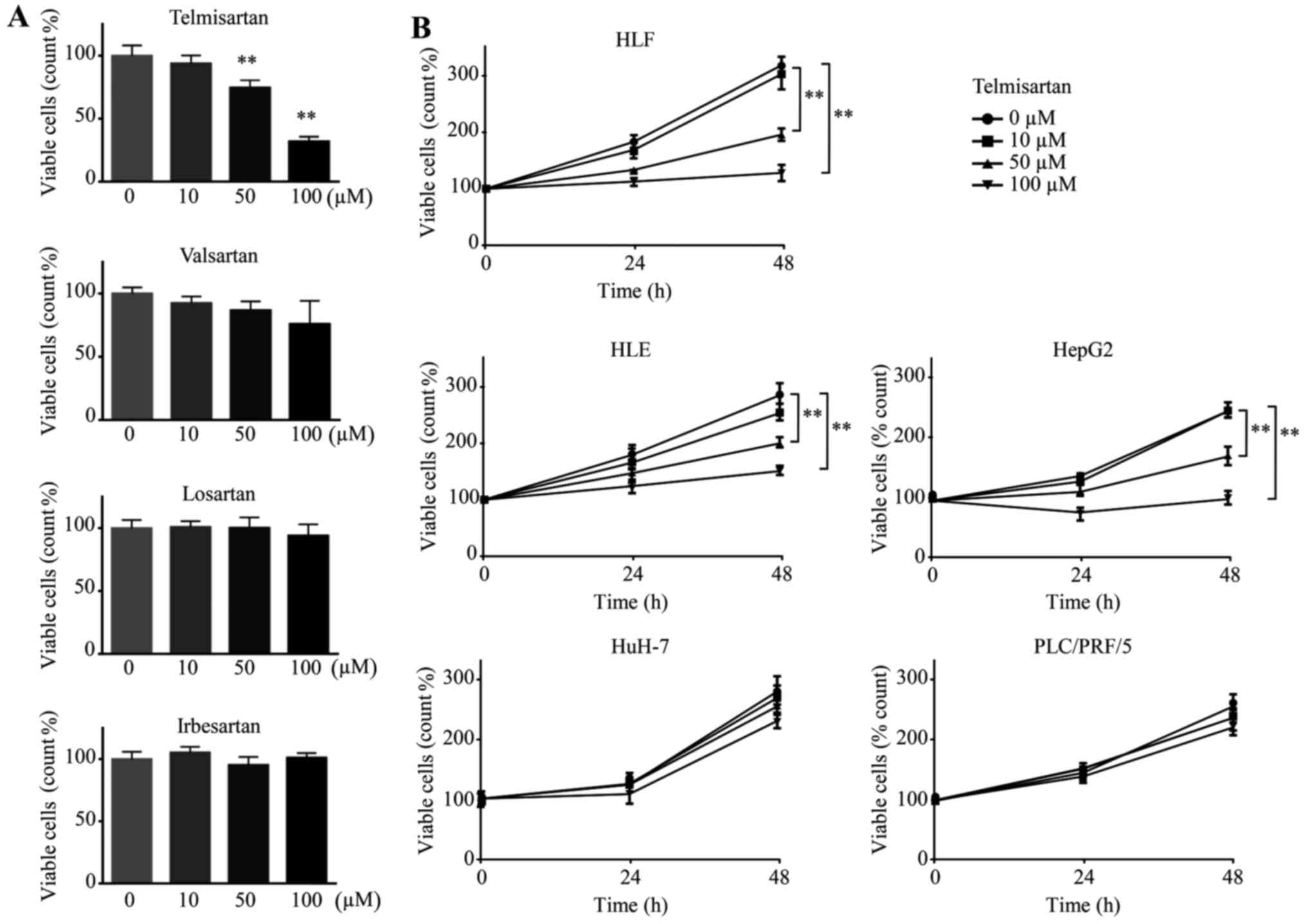
We conducted a cell proliferation assay and examined
the antitumor effects of four ARBs (telmisartan, valsartan,
losartan and irbesartan) on the HLF cell line after treatment for
48 h. Telmisartan reduced the proliferation of HLF cells, whereas
none of the other ARBs (valsartan, losartan and irbesartan)
affected the proliferation of the HCC cell lines (Fig. 1A). Additionally, five HCC cell lines
(HLF, HLE, HepG2, HuH-7 and PLC/PRF/5) were treated with
telmisartan, which reduced the proliferation of three HCC cell
lines (HLF, HLE and HepG2) but not that of HuH-7 and PLC/PRF/5
cells (Fig. 1B). HLF, HLE and HepG2
cells are classified as poorly differentiated types of HCC cells,
and HuH-7 and PLC/PRF/5 cells are classified as well differentiated
types of HCC cells (30). Thus,
telmisartan inhibits the proliferation of three HCC cell lines in a
dose-dependent manner.
Telmisartan induces cell cycle arrest
in G0/G1 phase and regulates cell
cycle-related proteins
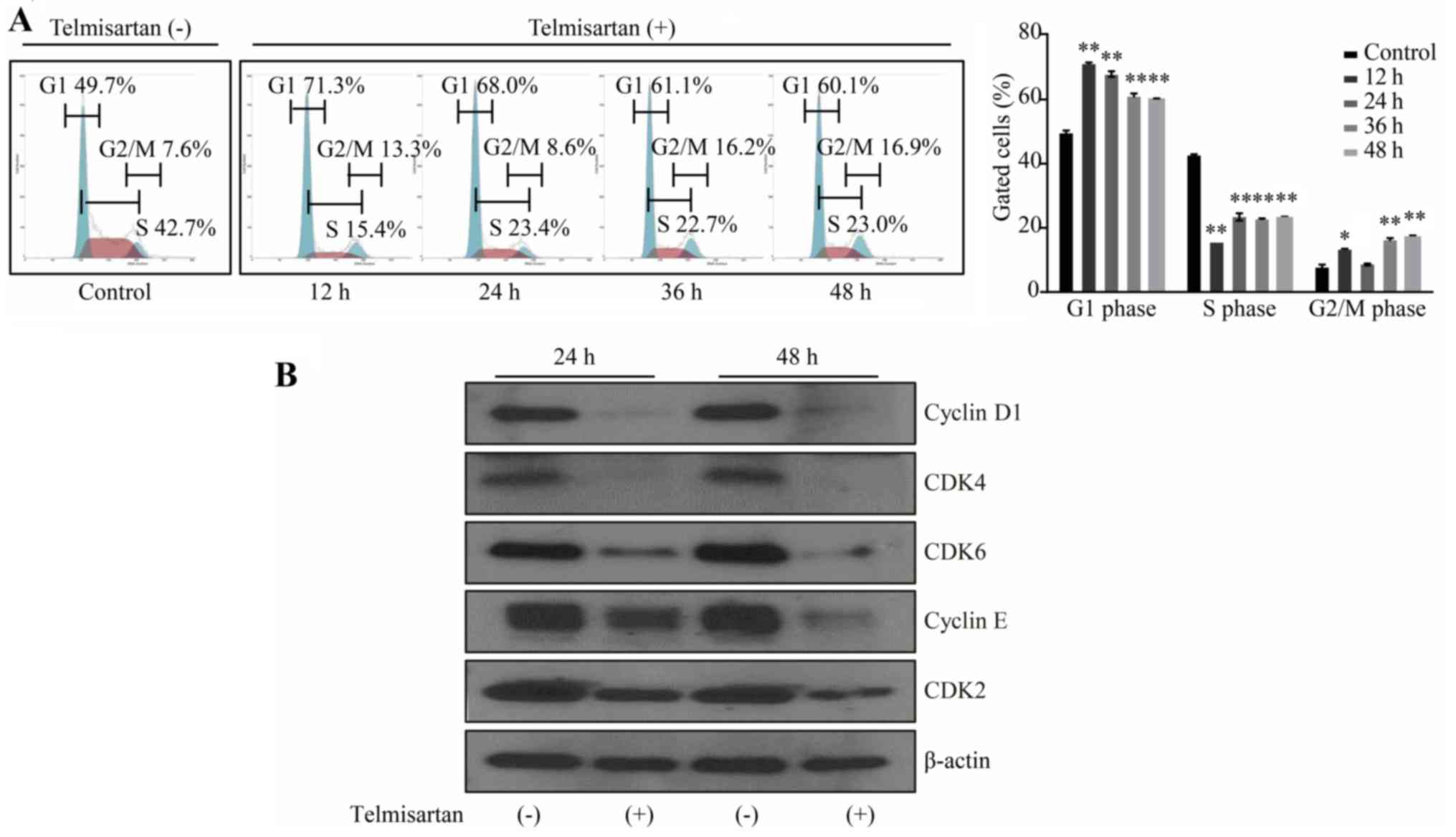
We examined cell cycle progression using flow
cytometry to further investigate the effects of telmisartan on the
HCC cell lines. Treatment with 100 µM telmisartan significantly
increased the population of cells in G1 phase and
reduced the population of cells in S phase at 12, 24, 36 and 48 h
after treatment (Fig. 2A). The
effect peaked in 12 h after treatment and lasted up to 48 h.
The effects of telmisartan on the expression of
various cell cycle-related proteins in HLF cells were evaluated by
western blotting. Cells were treated with 0 or 100 µM telmisartan
for 24 or 48 h. Cyclin D1 and cyclin E, which are key proteins
involved in the transition from the G0 to G1
phase, exhibited the greatest reduction in expression; telmisartan
inhibited cyclin D1 and cyclin E expression in a time-dependent
manner (Fig. 2B). Additionally,
analysis of other proteins associated with the G0 to
G1 transition indicated that the levels of CDK4, CDK6
(the catalytic subunits of cyclin D1) and CDK2 (the catalytic
subunit of cyclin E) were decreased in HLF cells after the addition
of telmisartan.
Telmisartan induces AMPK
phosphorylation and inhibits the mTOR pathway and p70S6K
phosphorylation in HLF cells
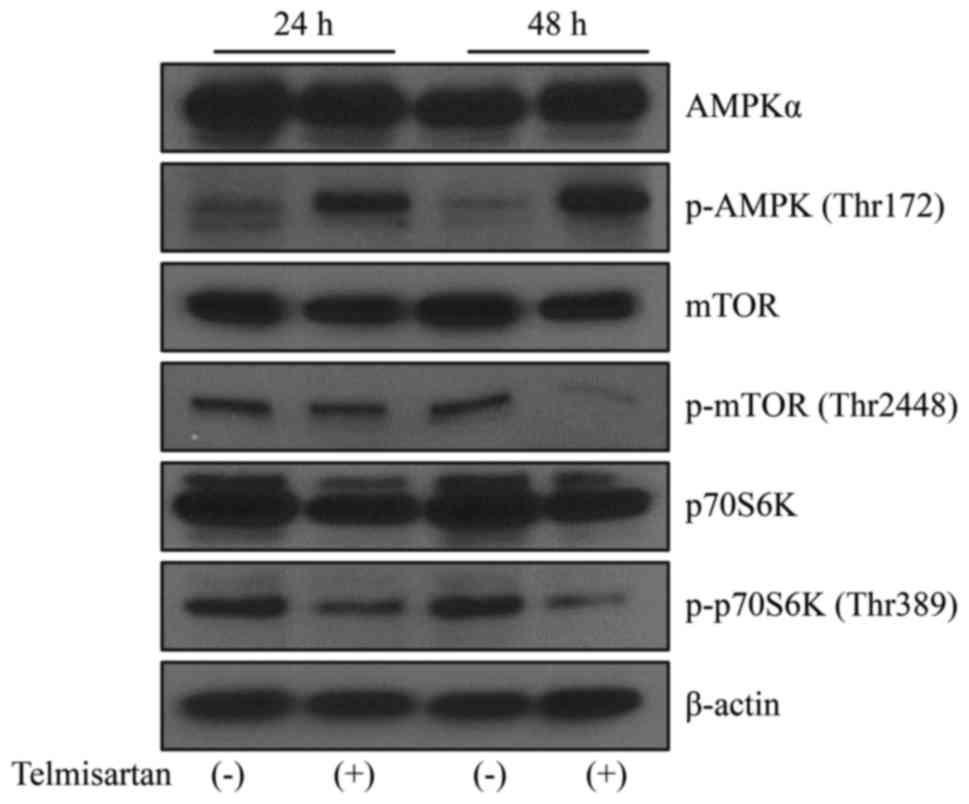
We focused on AMPK/mTOR signaling to identify the
mechanism by which telmisartan induced cell cycle arrest.
Telmisartan induced the phosphorylation of AMPK (thr172) in HLF
cells, and this effect persisted for at least 48 h (Fig. 3). The levels of the p-mTOR and
p-p70S6K proteins decreased in HLF cells following the telmisartan
treatment. Thus, telmisartan inhibits cancer cell proliferation by
inducing AMPK/mTOR signaling in HCC cells.
Telmisartan partially contributes to
the induction of apoptosis in HLF cells
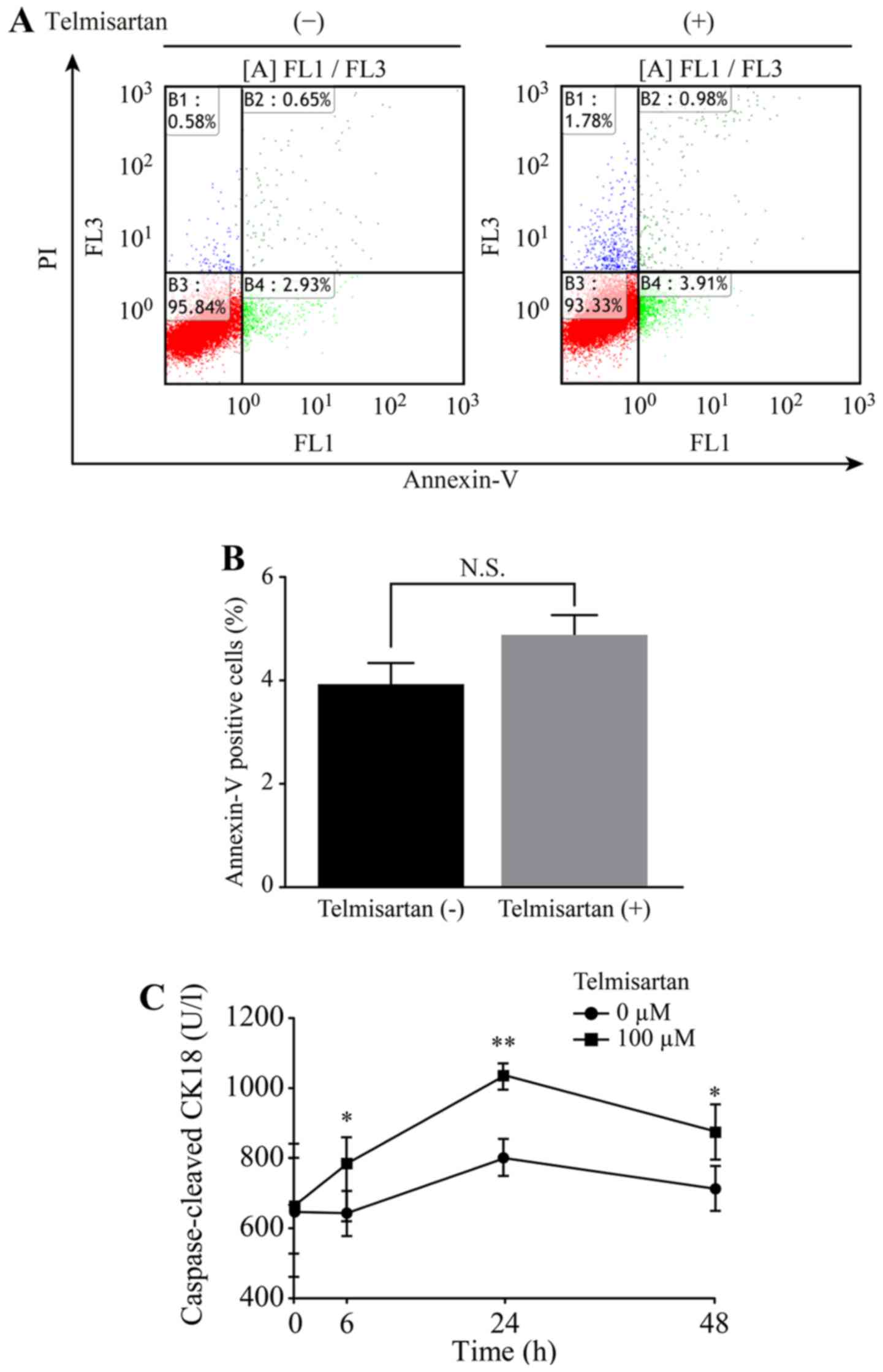
We detected apoptotic cells after telmisartan
treatment using flow cytometry. The different quadrants presented
in Fig. 4A represent living cells
(lower left quadrant), early apoptotic cells (lower right quadrant)
and late apoptotic cells (upper right quadrant). A comparison of
the percentage of Annexin V positive cells revealed that the
average of the triplicate assessment was 4.9% in the
telmisartan-treated group and 3.9% in the non-treated group
(Fig. 4B). Telmisartan slightly
increased the proportion of apoptotic HLF cells 12 h after
treatment, but this change was not statistically significant.
HLF cells were subsequently treated with 100 µM
telmisartan and the levels of cCK18 following treatment were
measured using an M30 ELISA kit to determine whether telmisartan
induced apoptosis. Telmisartan significantly increased the cCK18
levels at 6, 24 and 48 h after treatment (Fig. 4C).
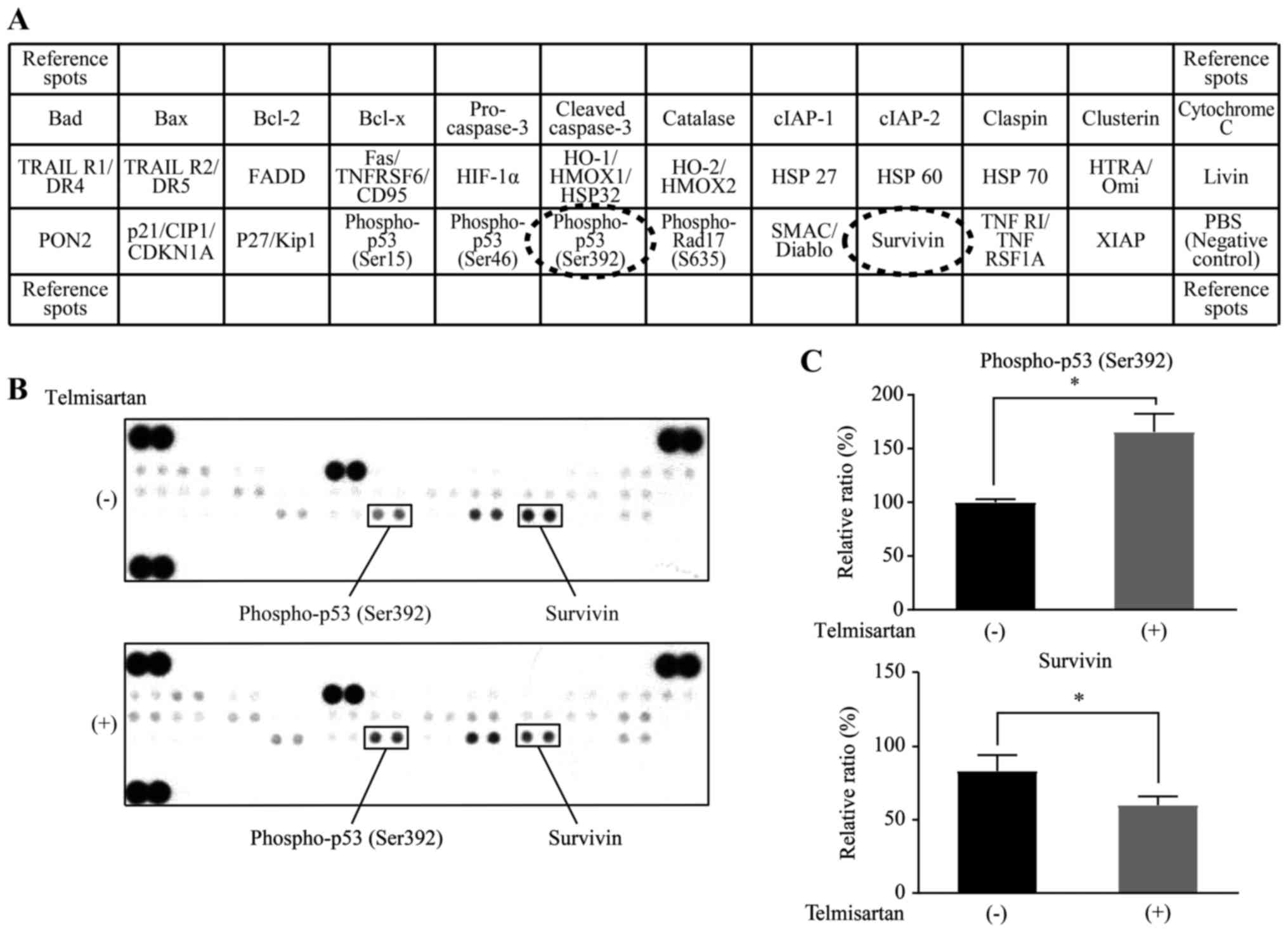
Additionally, we used an apoptosis array system to
identify the apoptosis-associated proteins that were involved in
the antitumor effects of telmisartan. The use of an antibody array
enabled us to screen 35 apoptosis-associated proteins in HLF cells
treated with or without telmisartan (Fig. 5A). Telmisartan increased the levels
of phospho-p53 (ser392) and decreased the levels of survivin in HLF
cells (Fig. 5B). Based on
densito-metry, the intensities of the phospho-p53 (ser392) and
survivin spots from the telmisartan-treated HLF cells were 165.6
and 71.6% of the intensity of the untreated HLF cells, respectively
(Fig. 5C). Based on these results,
telmisartan may partially inhibit HCC cell proliferation by
inducing apoptosis.
Telmisartan reduces the p-ErbB3 levels
in HLF cells
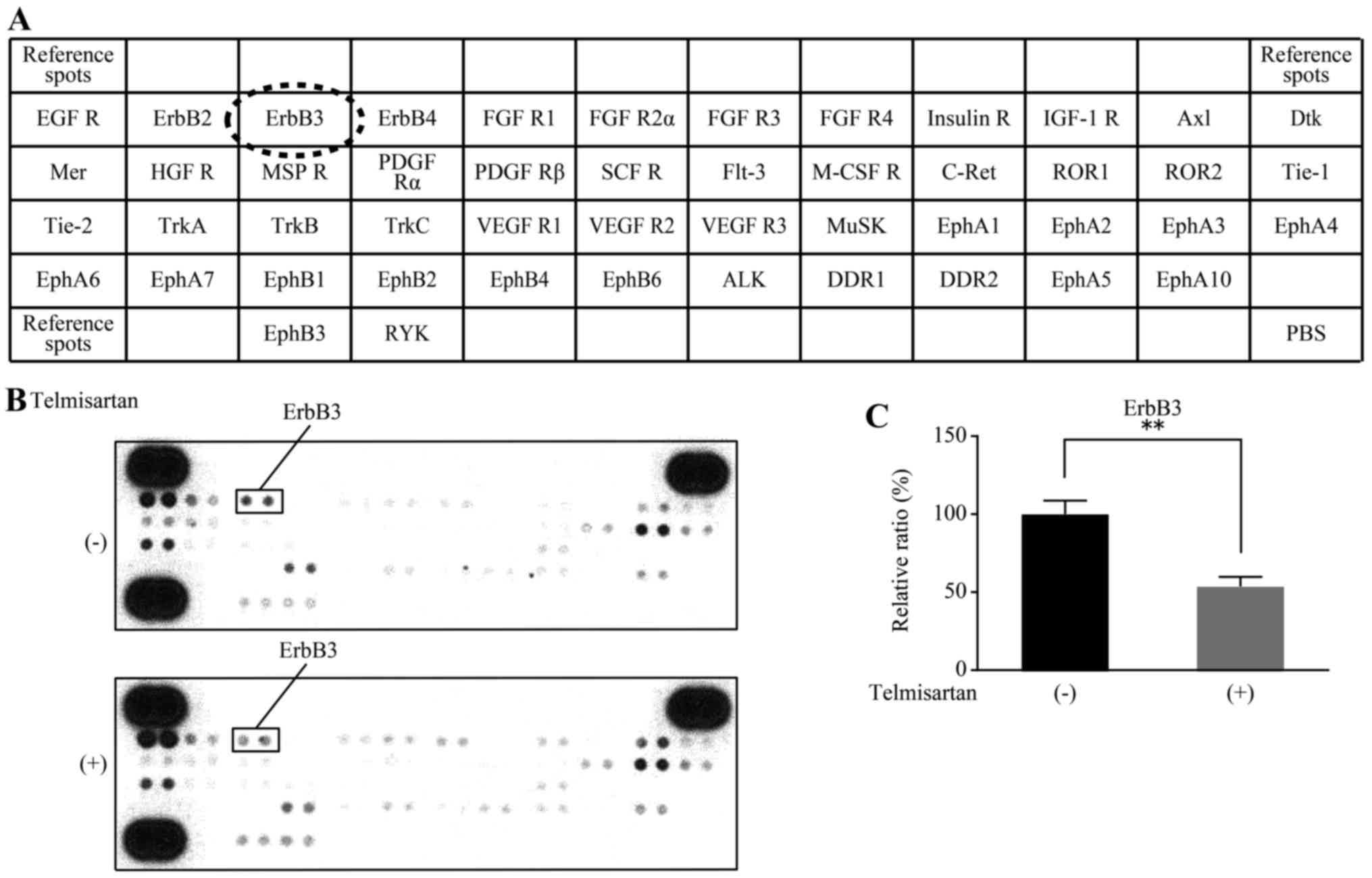
We used a p-RTK array to identify the key RTKs
associated with the antitumor effects of telmisartan. We
simultaneously analyzed the expression of 46 differentially
activated RTKs in HLF cells 48 h after telmisartan administration
using an antibody array (Fig. 6A).
Telmisartan reduced the levels of p-ErbB3 in HLF cells (Fig. 6B). The densitometric analyses of
p-ErbB3 showed a decrease of 53.7% (Fig. 6C). Thus, telmisartan may decrease
the expression of cell cycle regulatory proteins by inhibiting
ErbB3 activation in HCC cells.
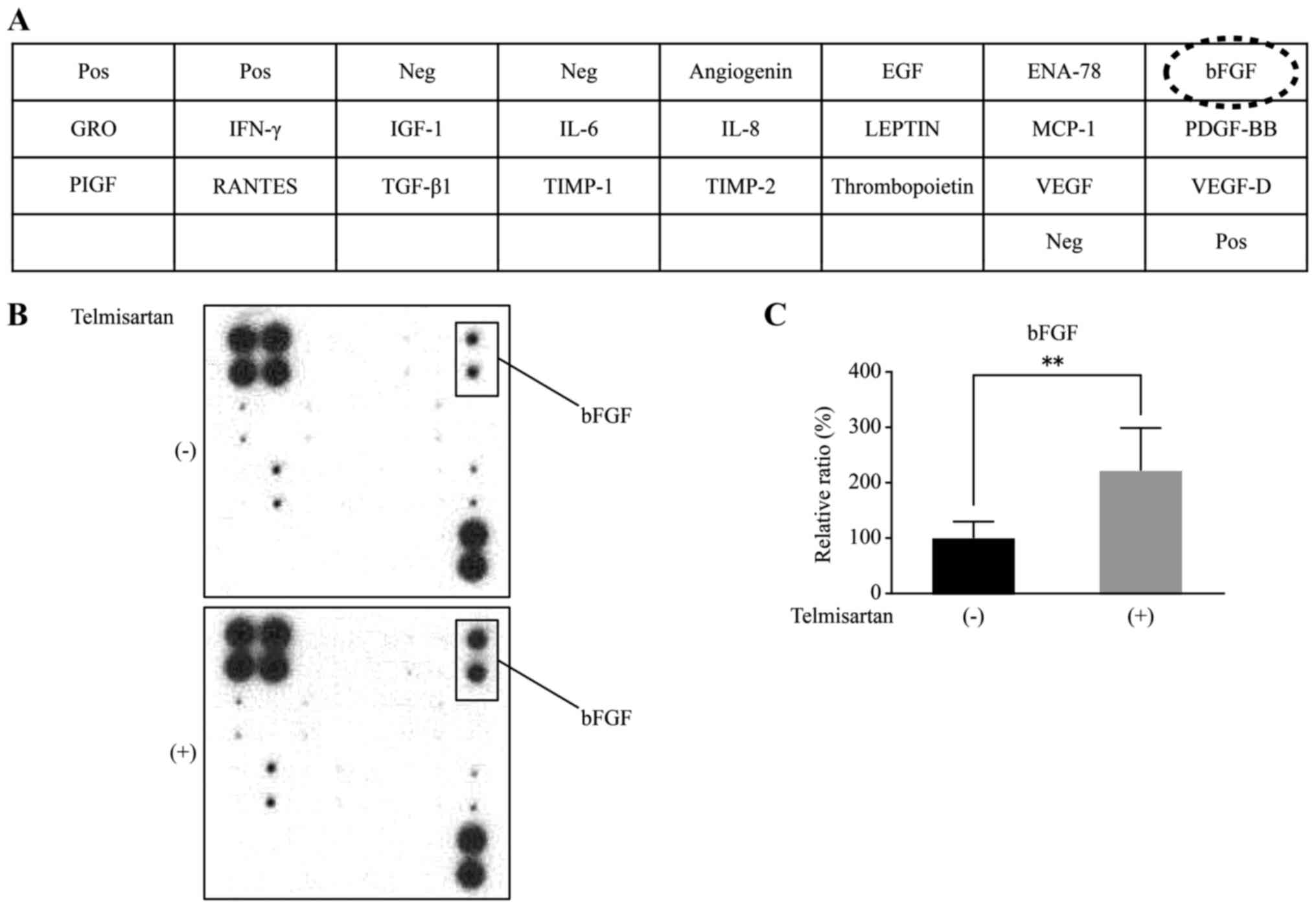
Telmisartan affects angiogenesis in
HLF cells
An antibody-based analysis of angiogenesis was
conducted to investigate the antitumor effects of telmisartan and
to examine the relationship between angiogenesis and telmisartan
(Fig. 7A). Using the antibody
array, we simultaneously screened the expression levels of 20
different angiogenesis-related proteins in HLF cells treated with
or without telmisartan. The telmisartan treatment increased the
b-fibroblast growth factor (bFGF) levels in HLF cells as detected
by the protein array (Fig. 7B).
According to the densitometric analyses, the ratio of the intensity
of the bFGF spots obtained from the telmisartan-treated cells to
the spots obtained from the untreated cells was 221.7% (Fig. 7C).
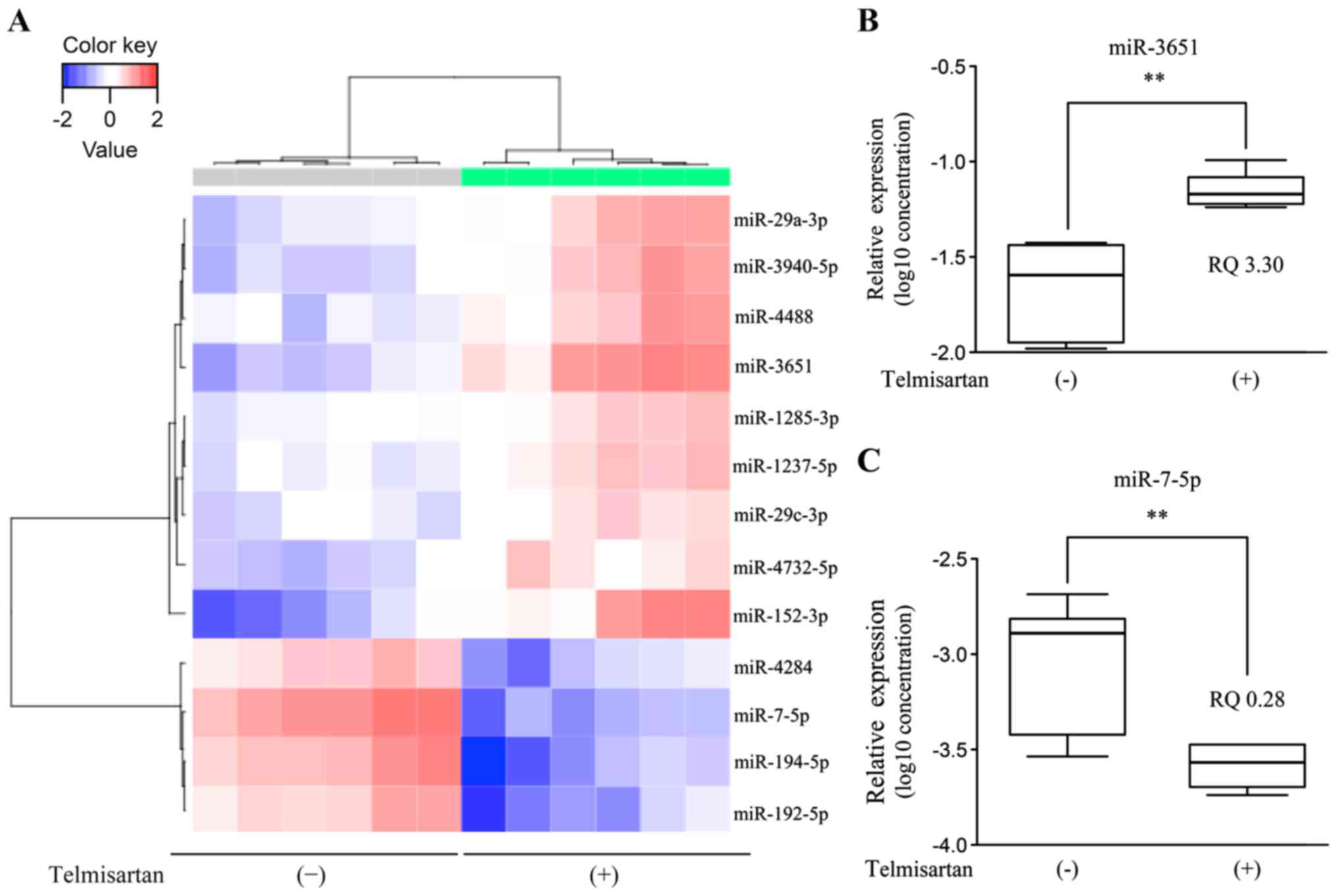
Telmisartan affects miRNA
expression
Using a custom microarray platform, we analyzed the
expression levels of 2,555 miRNA probes in the cell lines in the
presence or absence of the telmisartan treatment. Treatment with
100 µM telmisartan for 48 h upregulated the expression of 108
miRNAs and downregulated the expression of 55 miRNAs in HLF cells.
miR-3651 was significantly upregulated and miR-7-5p was
significantly downregulated (Table
I). An unsupervised hierarchical clustering analysis was
conducted using Pearsons correlation, and the results indicated
that HLF cells treated with telmisartan in vitro clustered
together and were separate from the untreated cell lines (Fig. 8A).
 | Table I.Statistical results and chromosomal
locations of miRNAs evaluated in HLF cells treated with or without
telmisartan that exhibited a fold change (FC)>1.5, FC<0.67,
or a P-value <0.005. |
Table I.
Statistical results and chromosomal
locations of miRNAs evaluated in HLF cells treated with or without
telmisartan that exhibited a fold change (FC)>1.5, FC<0.67,
or a P-value <0.005.
| miRNA | Fold change
(Treated/Untreated) | P-value | Chromosomal
location |
|---|
| Upregulated |
|
|
|
|
miR-4294 | 3.19 | 0.0010 | 10 |
|
miR-152-3p | 2.65 | 0.0005 | 17q21.32 |
|
miR-6088 | 2.62 | 0.0034 | 19 |
|
miR-3651 | 2.45 | <0.0001 | 9 |
|
miR-3940-5p | 2.06 | 0.0002 | 19 |
|
miR-7150 | 2.02 | 0.0040 | 9 |
|
miR-642b-3p | 2.00 | 0.0011 | 19 |
|
miR-6795-5p | 1.95 | 0.0017 | 19 |
|
miR-3191-3p | 1.91 | 0.0031 | 19 |
|
miR-1469 | 1.89 | 0.0015 | 15 |
|
miR-29a-3p | 1.84 | 0.0005 | 7q32.3 |
|
miR-4488 | 1.84 | 0.0006 | 11 |
|
miR-6887-5p | 1.81 | 0.0048 | 19 |
|
miR-6808-5p | 1.77 | 0.0027 | 1 |
|
miR-6774-5p | 1.75 | 0.0027 | 16 |
|
miR-4732-5p | 1.73 | 0.0003 | 17 |
|
miR-1246 | 1.72 | 0.0019 | 2 |
|
miR-4442 | 1.70 | 0.0017 | 3 |
|
miR-1237-5p | 1.63 | 0.0004 | 11 |
|
miR-362-5p | 1.62 | 0.0022 | X |
|
miR-1343-5p | 1.61 | 0.0039 | 11 |
|
miR-193b-5p | 1.57 | 0.0040 | 16 |
|
miR-29c-3p | 1.54 | 0.0004 | 1q32.2 |
|
miR-6769a-5p | 1.53 | 0.0018 | 16 |
|
miR-8059 | 1.52 | 0.0015 | 17 |
|
miR-1285-3p | 1.52 | 0.0008 | 7 |
|
miR-29b-3p | 1.51 | 0.0039 | 7 |
|
miR-8064 | 1.51 | 0.0046 | 3 |
|
miR-23c | 1.51 | 0.0030 | X |
| Downregulated |
|
|
|
|
miR-7-5p | 0.29 | <0.0001 | 9 |
|
miR-194-5p | 0.31 | 0.0005 | 1 |
|
miR-192-5p | 0.37 | 0.0004 | 11q13.1 |
|
miR-4284 | 0.46 | 0.0004 | 7 |
|
miR-1303 | 0.56 | 0.0038 | 5 |
|
miR-425-5p | 0.58 | 0.0042 | 3 |
|
miR-6807-5p | 0.59 | 0.0033 | 19 |
|
miR-503-5p | 0.60 | 0.0046 | X |
|
miR-15b-3p | 0.60 | 0.0015 | 3 |
|
miR-377-3p | 0.60 | 0.0028 | 14 |
|
miR-542-3p | 0.64 | 0.0026 | X |
|
miR-3918 | 0.65 | 0.0012 | 6 |
|
miR-200a-3p | 0.65 | 0.0019 | 1 |
The results of the qPCR assay revealed that the mean
miR-3651 level was obviously increased in the telmisartan-treated
HLF cells compared with the controls, whereas miR-7-5p expression
was significantly decreased in the telmisartan-treated cells
compared to the untreated cells (Fig.
8B).
Discussion
ARBs are widely prescribed drugs for the treatment
of hypertension. Despite their widespread use, the use of ARBs is
associated with an 8% increased risk of cancer (14). However, telmisartan is not
associated with an increased risk of cancer (31). Among the ARBs tested in the present
study, only telmisartan inhibited HLF cell proliferation.
Additionally, telmisartan dose-dependently inhibited the
proliferation of three HCC cell lines (HLF, HLE and HepG2) but not
the HuH-7 and PLC/PRF/5 cell lines. HLF, HLE and HepG2 cells are
classified as poorly differentiated types of HCC cells, and HuH-7
and PLC/PRF/5 cells are classified as well differentiated types of
HCC cells (30). Accordingly,
poorly differentiated HCC cells were more sensitive to the
telmisartan treatment than well to moderately differentiated HCC
cells.
Many previous studies have shown that telmisartan
exerts an antitumor effect through the induction of apoptosis, as
has been demonstrated in endometrial (12), prostate (17), renal (18) and colon cancer (19). However, few reports, including our
data in esophageal adenocarcinoma (21), have shown that cell cycle arrest is
the major mechanism underlying the antitumor effect of telmisartan.
Similarly, the present study showed that telmisartan induced cell
cycle arrest at the G0/G1 phase by modulating
the expression of cell cycle regulatory proteins in HCC cells.
According to our flow cytometric analyses, telmisartan induced
significant cell cycle arrest in HLF cells. These findings were
further corroborated by an analysis of cell cycle-related proteins.
The levels of the cell cycle regulatory proteins cyclin D1, cyclin
E and CDKs were substantially reduced. Specific cyclin/CDK
complexes are activated at different times during cell cycle
progression. Complexes of CDK4 and CDK6 with cyclin D1 are required
for G1 phase progression, whereas complexes of CDK2 with
cyclin E are required for the G1-S transition (32). The expression of various cell
cycle-related proteins is related to cancer prognosis (32,33).
Therefore, the major cell cycle regulators (cyclin D1 and cyclin E)
may be intracellular targets of telmisartan in human HCC cell
lines.
More recently, telmisartan was shown to contribute
to the activation of AMPK in vascular endothelial cells (34,35).
AMPK is a serine/threonine protein kinase that plays many important
roles in cellular energy homeostasis (36). Recent studies have documented that
the pharmacological activation of AMPK can block cancer cell growth
in various human cancers (37). In
HCC, we also assumed that telmisartan regulates the proliferation
of cancer cells through AMPK activation. In the present study,
telmisartan exerted its antitumor effects by inducing AMPK
phosphorylation in HCC cells, suggesting that activation of the
AMPK/mTOR pathway inhibits the expression of cell cycle regulatory
proteins. AMPK activation has been shown to inhibit the mTOR
pathway and p70S6K phosphorylation, which are involved in protein
synthesis, suggesting that this pathway may regulate cell
proliferation in various cancer cells (38,39).
These reports support our finding that AMPK/mTOR signaling is a
pivotal pathway underlying the antiproliferative effect of
telmisartan on HCC cells.
As described above, telmisartan inhibits cell
proliferation by inducing apoptosis in various cancer cell lines,
including urological cancer cell lines (17,18).
It also induces apoptosis and DNA fragmentation in human urological
cancer (40). The mechanism by
which telmisartan induces apoptosis in endometrial cancer was
recently elucidated and involves the downregulation of Bcl-2 and
Bcl-xL, as well as the upregulation of cleaved caspase-3 and PARP
(12). HLF cells were treated with
or without 100 µM telmisartan for 12 h and analyzed using flow
cytometry to determine whether telmisartan induced apoptosis.
Telmisartan did not significantly increase the proportion of
apoptotic cells, but it increased the cCK18 levels in HLF cells
after 6, 24 and 48 h of treatment. Additionally, telmisartan
increased the levels of phospho-p53 (ser392) and decreased survivin
expression in HLF cells. The p53 protein is activated by
phosphorylation to induce tumor cell apoptosis (41), and survivin is a member of a family
of apoptosis inhibitors that play key roles in regulating cell
division and inhibiting apoptosis by blocking caspase activation.
However, telmisartan might inhibit HCC cell proliferation partially
by inducing apoptosis and regulating the levels of
apoptosis-related proteins. Some experimental results, including
flow cytometry and western blotting, suggest that the antitumor
effect of telmisartan in hepatocellular carcinoma is due to cell
cycle arrest rather than apoptosis.
Several signaling pathways, such as the
Ras/Raf/mitogen-activated protein kinase (MAPK), epidermal growth
factor receptor (EGFR), insulin-like growth factor receptor,
Wnt/β-catenin and Akt/mTOR signaling pathways, have been implicated
in hepatic carcinogenesis. In particular, the Ras/Raf/MAPK pathway
is typically activated in HCC as a result of increased signaling
from upstream growth factors and the inactivation of tumor
suppressor genes (42). Telmisartan
reduced the phosphorylation of ErbB3 in HCC cells, as measured
using p-RTK arrays. In the present study, telmisartan decreased
mTOR expression. mTOR, a major downstream target of the EGFR
family, regulates p70S6K. Thus, telmisartan might inhibit cell
cycle regulatory proteins by decreasing ErbB3 phosphorylation to
regulate HCC cell proliferation.
Candesartan, another ARB, was recently shown to
significantly reduce transforming growth factor β1 (TGF-β1)
expression and suppress tumor proliferation and stromal fibrosis
(13). Candesartan also
significantly inhibits the growth of tumor xenografts and
angiogenesis in mice (13). In the
present study, telmisartan increased the bFGF levels in HLF cells.
Thus, HLF cells that escape the antitumor effects of telmisartan
may express angiogenesis-related proteins.
miRNAs are small non-coding RNA molecules that can
regulate the development and progression of various cancers
(43). The expression of several
miRNAs was significantly altered in vitro following
telmisartan treatment. We identified 163 differentially expressed
miRNAs (108 upregulated and 55 downregulated) in HLF cells in
response to telmisartan treatment using a microarray analysis.
Several miRNAs that were upregulated upon telmisartan treatment
have been reported to be tumor suppressors associated with
decreased expression of cyclin/CDK complexes and anti-apoptotic
proteins. For instance, the miR-29 family targets Bcl-2 (44), miR-29c-3p modulates cyclin E
expression (45), and miR-29b-3p
represses CDK2 expression (46). In
addition, numerous studies have examined the target molecules of
miRNAs associated with cancer progression: miR-126-5p directly
regulates a disintegrin and metalloprotease domain 9 (ADAM9) and
metalloproteinase 7 (MMP7) expression (47), and miR-152-3p represses DNA
methyltransferase 1 (DNMT1) expression (48). Notably, several miRNAs that were
down-regulated upon telmisartan treatment have been reported to be
oncomiRNAs associated with increased expression of CDK inhibitors:
miR-7 inhibits p21-activated kinase 1 (PAC1) (49) and miR-194 directly targets p27kip1
(50). It is possible that these
miRNAs interact in a complicated manner and contribute to the
antitumor effect of telmisartan, but the suppression of tumor
growth via miRNAs has not been fully elucidated. Despites these
limitations, our findings have important implications.
In conclusion, telmisartan inhibits human HCC cell
proliferation by inducing cell cycle arrest via the regulation of
cell cycle-related proteins.
Acknowledgements
We thank Ms. Kayo Hirose, Ms. Kana Ogawa, Ms. Keiko
Fujikawa, Ms. Miwako Watanabe, Ms. Megumi Okamura and Ms. Fuyuko
Kokado for their skillful technical assistance.
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
AT1
|
angiotensin II type 1
|
|
ARBs
|
angiotensin II type 1 receptor
blockers
|
|
AMPK
|
AMP-activated protein kinase
|
|
mTOR
|
mammalian target of rapamycin
|
|
cCK18
|
caspase-cleaved cytokeratin 18
|
|
RTKs
|
receptor tyrosine kinases
|
|
CDK
|
cyclin-dependent kinase
|
|
bFGF
|
b-fibroblast growth factor
|
|
EGFR
|
epidermal growth factor receptor
|
References
|
1
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Altekruse SF, Henley SJ, Cucinelli JE and
McGlynn KA: Changing hepatocellular carcinoma incidence and liver
cancer mortality rates in the United States. Am J Gastroenterol.
109:542–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim SR, Kudo M, Hino O, Han KH, Chung YH
and Lee HS: Organizing Committee of Japan-Korea Liver Symposium:
Epidemiology of hepatocellular carcinoma in Japan and Korea. A
review. Oncology. 75 Suppl 1:13–16. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Muto J, Shirabe K, Sugimachi K and Maehara
Y: Review of angiogenesis in hepatocellular carcinoma. Hepatol Res.
45:1–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Itabashi H, Maesawa C, Oikawa H, Kotani K,
Sakurai E, Kato K, Komatsu H, Nitta H, Kawamura H, Wakabayashi G,
et al: Angiotensin II and epidermal growth factor receptor
cross-talk mediated by a disintegrin and metalloprotease
accelerates tumor cell proliferation of hepatocellular carcinoma
cell lines. Hepatol Res. 38:601–613. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wolgien MC Molina, da Silva ID Guerreiro,
Nazário AC Pinto, Nakaie CR, Correa-Noronha SA, de Noronha SM
Ribeiro and Facina G: Genetic association study of angiotensin II
receptor types 1 (A168G) and 2 (T1247G and A5235G) polymorphisms in
breast carcinoma among Brazilian Women. Breast Care (Basel).
9:176–181. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miyajima A, Kosaka T, Asano T, Asano T,
Seta K, Kawai T and Hayakawa M: Angiotensin II type I antagonist
prevents pulmonary metastasis of murine renal cancer by inhibiting
tumor angiogenesis. Cancer Res. 62(15s): 1–4179. 2002.
|
|
8
|
Kinoshita J, Fushida S, Harada S, Yagi Y,
Fujita H, Kinami S, Ninomiya I, Fujimura T, Kayahara M, Yashiro M,
et al: Local angiotensin II-generation in human gastric cancer:
Correlation with tumor progression through the activation of
ERK1/2, NF-kappaB and survivin. Int J Oncol. 34:1573–1582. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okamoto K, Tajima H, Ohta T, Nakanuma S,
Hayashi H, Nakagawara H, Onishi I, Takamura H, Ninomiya I, Kitagawa
H, et al: Angiotensin II induces tumor progression and fibrosis in
intrahepatic cholangiocarcinoma through an interaction with hepatic
stellate cells. Int J Oncol. 37:1251–1259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du N, Feng J, Hu LJ, Sun X, Sun HB, Zhao
Y, Yang YP and Ren H: Angiotensin II receptor type 1 blockers
suppress the cell proliferation effects of angiotensin II in breast
cancer cells by inhibiting AT1R signaling. Oncol Rep. 27:1893–1903.
2012.PubMed/NCBI
|
|
11
|
Chen X, Meng Q, Zhao Y, Liu M, Li D, Yang
Y, Sun L, Sui G, Cai L and Dong X: Angiotensin II type 1 receptor
antagonists inhibit cell proliferation and angiogenesis in breast
cancer. Cancer Lett. 328:318–324. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koyama N, Nishida Y, Ishii T, Yoshida T,
Furukawa Y and Narahara H: Telmisartan induces growth inhibition,
DNA double-strand breaks and apoptosis in human endometrial cancer
cells. PLoS One. 9:e930502014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okazaki M, Fushida S, Harada S, Tsukada T,
Kinoshita J, Oyama K, Tajima H, Ninomiya I, Fujimura T and Ohta T:
The angiotensin II type 1 receptor blocker candesartan suppresses
proliferation and fibrosis in gastric cancer. Cancer Lett.
355:46–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sipahi I, Debanne SM, Rowland DY, Simon DI
and Fang JC: Angiotensin-receptor blockade and risk of cancer:
Meta-analysis of randomised controlled trials. Lancet Oncol.
11:627–636. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bhaskaran K, Douglas I, Evans S, van Staa
T and Smeeth L: Angiotensin receptor blockers and risk of cancer:
Cohort study among people receiving antihypertensive drugs in UK
General Practice Research Database. BMJ. 344:e26972012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Makar GA, Holmes JH and Yang YX:
Angiotensin-converting enzyme inhibitor therapy and colorectal
cancer risk. J Natl Cancer Inst. 106:djt3742014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Funao K, Matsuyama M, Kawahito Y, Sano H,
Chargui J, Touraine JL, Nakatani T and Yoshimura R: Telmisartan is
a potent target for prevention and treatment in human prostate
cancer. Oncol Rep. 20:295–300. 2008.PubMed/NCBI
|
|
18
|
Funao K, Matsuyama M, Kawahito Y, Sano H,
Chargui J, Touraine JL, Nakatani T and Yoshimura R: Telmisartan as
a peroxisome proliferator-activated receptor-γ ligand is a new
target in the treatment of human renal cell carcinoma. Mol Med Rep.
2:193–198. 2009.PubMed/NCBI
|
|
19
|
Lee LD, Mafura B, Lauscher JC, Seeliger H,
Kreis ME and Gröne J: Antiproliferative and apoptotic effects of
telmisartan in human colon cancer cells. Oncol Lett. 8:2681–2686.
2014.PubMed/NCBI
|
|
20
|
Kozako T, Soeda S, Yoshimitsu M, Arima N,
Kuroki A, Hirata S, Tanaka H, Imakyure O, Tone N, Honda S, et al:
Angiotensin II type 1 receptor blocker telmisartan induces
apoptosis and autophagy in adult T-cell leukemia cells. FEBS Open
Bio. 6:442–460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fujihara S, Morishita A, Ogawa K, Tadokoro
T, Chiyo T, Kato K, Kobara H, Mori H, Iwama H and Masaki T: The
angiotensin II type 1 receptor antagonist telmisartan inhibits cell
proliferation and tumor growth of esophageal adenocarcinoma via the
AMPKα/mTOR pathway in vitro and in vivo. Oncotarget. 8:8536–8549.
2017.PubMed/NCBI
|
|
22
|
Rehman G, Shehzad A, Khan AL and Hamayun
M: Role of AMP-activated protein kinase in cancer therapy. Arch
Pharm (Weinheim). 347:457–468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dann SG and Thomas G: The amino acid
sensitive TOR pathway from yeast to mammals. FEBS Lett.
580:2821–2829. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fingar DC and Blenis J: Target of
rapamycin (TOR): An integrator of nutrient and growth factor
signals and coordinator of cell growth and cell cycle progression.
Oncogene. 23:3151–3171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Masaki T, Tokuda M, Yoshida S, Nakai S,
Morishita A, Uchida N, Funaki T, Kita Y, Funakoshi F, Nonomura T,
et al: Comparison study of the expressions of myristoylated
alanine-rich C kinase substrate in hepatocellular carcinoma, liver
cirrhosis, chronic hepatitis, and normal liver. Int J Oncol.
26:661–671. 2005.PubMed/NCBI
|
|
26
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Towbin H, Staehelin T and Gordon J:
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: Procedure and some applications. Proc Natl
Acad Sci USA. 76:pp. 4350–4354. 1979; View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schutte B, Henfling M, Kölgen W, Bouman M,
Meex S, Leers MP, Nap M, Björklund V, Björklund P, Björklund B, et
al: Keratin 8/18 breakdown and reorganization during apoptosis. Exp
Cell Res. 297:11–26. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J R Stat Soc Series B Stat Methodol. 57:289–300.
1995.
|
|
30
|
Morishita A, Iwama H, Fujihara S, Sakamoto
T, Fujita K, Tani J, Miyoshi H, Yoneyama H, Himoto T and Masaki T:
MicroRNA profiles in various hepatocellular carcinoma cell lines.
Oncol Lett. 12:1687–1692. 2016.PubMed/NCBI
|
|
31
|
Tascilar K, Azoulay L, DellAniello S,
Bartels DB and Suissa S: The use of telmisartan and the incidence
of cancer. Am J Hypertens. 29:1358–1365. 2016.PubMed/NCBI
|
|
32
|
Masaki T, Shiratori Y, Rengifo W, Igarashi
K, Yamagata M, Kurokohchi K, Uchida N, Miyauchi Y, Yoshiji H,
Watanabe S, et al: Cyclins and cyclin-dependent kinases:
Comparative study of hepatocellular carcinoma versus cirrhosis.
Hepatology. 37:534–543. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matsuda Y: Molecular mechanism underlying
the functional loss of cyclindependent kinase inhibitors p16 and
p27 in hepatocellular carcinoma. World J Gastroenterol.
14:1734–1740. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kurokawa H, Sugiyama S, Nozaki T, Sugamura
K, Toyama K, Matsubara J, Fujisue K, Ohba K, Maeda H, Konishi M, et
al: Telmisartan enhances mitochondrial activity and alters cellular
functions in human coronary artery endothelial cells via
AMP-activated protein kinase pathway. Atherosclerosis. 239:375–385.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Myojo M, Nagata D, Fujita D, Kiyosue A,
Takahashi M, Satonaka H, Morishita Y, Akimoto T, Nagai R, Komuro I,
et al: Telmisartan activates endothelial nitric oxide synthase via
Ser1177 phosphorylation in vascular endothelial cells. PLoS One.
9:e969482014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kahn BB, Alquier T, Carling D and Hardie
DG: AMP-activated protein kinase: Ancient energy gauge provides
clues to modern understanding of metabolism. Cell Metab. 1:15–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: Metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kato K, Gong J, Iwama H, Kitanaka A, Tani
J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, et al:
The antidiabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Matsuyama M, Funao K, Kuratsukuri K,
Tanaka T, Kawahito Y, Sano H, Chargui J, Touraine JL, Yoshimura N
and Yoshimura R: Telmisartan inhibits human urological cancer cell
growth through early apoptosis. Exp Ther Med. 1:301–306. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hussain SP, Schwank J, Staib F, Wang XW
and Harris CC: TP53 mutations and hepatocellular carcinoma:
Insights into the etiology and pathogenesis of liver cancer.
Oncogene. 26:2166–2176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Galuppo R, Ramaiah D, Ponte OM and Gedaly
R: Molecular therapies in hepatocellular carcinoma: What can we
target? Dig Dis Sci. 59:1688–1697. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Morishita A and Masaki T: miRNA in
hepatocellular carcinoma. Hepatol Res. 45:128–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xiong Y, Fang JH, Yun JP, Yang J, Zhang Y,
Jia WH and Zhuang SM: Effects of microRNA-29 on apoptosis,
tumorigenicity, and prognosis of hepatocellular carcinoma.
Hepatology. 51:836–845. 2010.PubMed/NCBI
|
|
45
|
Ding DP, Chen ZL, Zhao XH, Wang JW, Sun J,
Wang Z, Tan FW, Tan XG, Li BZ, Zhou F, et al: miR-29c induces cell
cycle arrest in esophageal squamous cell carcinoma by modulating
cyclin E expression. Carcinogenesis. 32:1025–1032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xiao L, Rao JN, Zou T, Liu L, Cao S,
Martindale JL, Su W, Chung HK, Gorospe M and Wang JY: miR-29b
represses intestinal mucosal growth by inhibiting translation of
cyclin-dependent kinase 2. Mol Biol Cell. 24:3038–3046. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Felli N, Felicetti F, Lustri AM, Errico
MC, Bottero L, Cannistraci A, De Feo A, Petrini M, Pedini F,
Biffoni M, et al: miR-126&126* restored expressions play a
tumor suppressor role by directly regulating ADAM9 and MMP7 in
melanoma. PLoS One. 8:e568242013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Braconi C, Huang N and Patel T:
MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor
suppressor gene expression by interleukin-6 in human malignant
cholangiocytes. Hepatology. 51:881–890. 2010.PubMed/NCBI
|
|
49
|
Reddy SD, Ohshiro K, Rayala SK and Kumar
R: MicroRNA-7, a homeobox D10 target, inhibits p21-activated kinase
1 and regulates its functions. Cancer Res. 68:8195–8200. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wu X, Liu T, Fang O, Leach LJ, Hu X and
Luo Z: miR-194 suppresses metastasis of non-small cell lung cancer
through regulating expression of BMP1 and p27kip1.
Oncogene. 33:1506–1514. 2014. View Article : Google Scholar : PubMed/NCBI
|