Introduction
Liver cancer is the most common type of cancer and
the third leading cause of cancer-related deaths worldwide due to
its poor prognosis (1). Treatment
of liver cancer includes surgery, intervention therapy,
chemotherapy and liver transplantation, however the therapeutic
effect is not satisfactory. As a non-invasive treatment,
chemotherapy is still a major method for advanced tumor at present.
However, no systemic chemotherapy has been demonstrated to be
consistently efficacious and acquisition of drug-resistant
phenotypes is often associated with liver cancer chemotherapy.
Reducing the drug resistance of chemotherapy is a major clinical
problem (2).
Escape from chemotherapy is positively correlated
with the unfolded protein response (UPR) and autophagy (3–5).
Autophagy plays a significant role in energy homeostasis by
degradation and recycling of damaged or harmful intracellular
components. Overload of misfolded or unfolded proteins in the
endoplasmic reticulum (ER) leads to a failure of protein
degradation by proteasomes, causing the upregulation of autophagy
(6–8). It has been revealed that autophagy may
function as a cytoprotective mechanism, since the signaling
pathways of intrinsic and extrinsic apoptosis are blocked after
autophagy activation and result in adaptation and survival of tumor
cells under hypoxic and inflammatory conditions (9–11). In
addition, an increasing number of studies have revealed that
autophagy may also contribute to resistance to chemotherapy for
cancer (12–14). Although autophagy is a potential
therapeutic target in adjuvant chemotherapy, its exact role is
still unclear.
Adenosine (Ado) is an endogenous purine nucleoside
composed of an adenine attached to a ribose sugar molecule moiety,
and is present ubiquitously in all the organs, tissues and cells.
Accumulating evidence has revealed that Ado induces apoptosis in a
variety of cancer cells (15–17).
ER stress and the mitochondrial pathway may be involved in the
Ado-induced apoptosis process (18–20).
Cellular adaptation to ER stress is achieved by the activation of a
highly conserved signal transduction pathway known as the UPR,
which alleviates stress at an early stage and triggers apoptosis if
homeostasis fails over a prolonged time frame. UPR involves an ER
molecular chaperone (GRP78), ER stress sensor proteins (PERK, IRE-1
and ATF-6) and their downstream signaling pathways (21). During ER stress, PERK dissociates
from GRP78 and activates ATF4, which promotes transcription of
genes related to cell survival and pro-apoptotic factors such as
CHOP. CHOP can alter the permeability of the mitochondrial membrane
and lead to the subsequent release of pro-apoptotic molecules
cytochrome c (Cyt C), which further activates caspases to
promote cell apoptosis (22). In
our previous studies, we demonstrated that Ado-induced apoptosis
was associated with activation of ER stress (19,23).
However, whether Ado affects autophagy, or whether autophagy plays
a protective role on cells is unclear. Therefore, it is necessary
to further investigate the relationship between autophagy and
apoptosis.
Materials and methods
Cell culture and experimental
groups
The human hepatoblastoma HepG2 cell line (Institute
of Cell Biology at the Chinese Academy of Sciences, Shanghai,
China) were cultured in Dulbecco's modified Eagle's medium (DMEM)
containing 10% (v/v) fetal bovine serum, penicillin (final
concentration, 100 U/ml), and streptomycin (final concentration,
100 µg/ml) (all from Thermo Fisher Scientific, Inc., Waltham, MA,
USA), under a humidified atmosphere of 5% CO2 and 95%
air at 37°C. This growth medium was changed every two or three
days, and cells were passaged at ~80% confluence. To validate that
autophagy participates in Ado-induced apoptosis, the autophagy
inhibitor LY294002 (LY; Calbiochem, San Diego, CA, USA) and the
autophagy inducer rapamycin (Rapa) were pre-treated and 1% dimethyl
sulfoxide (DMSO) was used as a control (Control).
Transient transfection
For RNAi experiments, the plasmid encoding a small
interference RNA (siRNA) targeted against AMP-activated protein
kinase (AMPK) (si-AMPK) or an empty plasmid vector only expressing
GFP (control siRNA) was constructed. We first constructed four
si-AMPK sequences and these interference plasmids were named
si-AMPK1, si-AMPK-2, si-AMPK-3 and si-AMPK-4, respectively. The
plasmid which had the highest inhibition efficiency (78%) was
selected for the next experiments (data not shown). The best
sequence of si-AMPK, 5′-CUGAGUUGCAUAUACUGUA-3′ and control-siRNA,
5′-GACGAGCGGCACGUGCACA-3′ were synthesized by GenePharma Co., Ltd.
(Shanghai, China). For transfection, cells were trypsinized and
seeded in 6-well plates at a density of 4×105
cells/well. Two days after reaching confluence, HepG2 cells were
cultured in a serum-free medium for 1 h and transfected with 20 µM
of the target gene or control siRNA using Lipofectamine 2000
(Thermo Fisher Scientific, Inc.) according to a method described in
our previous study (19). Following
a change of fresh medium 6 h later, the transfected cells were
incubated with or without 2.0 mM Ado in complete medium for a
further 24 h, then the cells were collected and named: Adenosine
treatment group (Ado), Ado + si-AMPK or control siRNA group. These
transfected cells were processed for western blot analysis and
measurement of mitochondrial membrane potential.
MTT assay to detect the cell
viability
HepG2 cells were seeded in a 96-well plate
(5×103 cells/well) in a humidified atmosphere with 5%
CO2 at 37°C and treated with Ado alone (0, 1.0, 2.0, 3.0
and 4.0 mM) for 12, 24 and 48 h; or 2.0 mM Ado alone, 10 µM LY
alone or 2.0 mM Ado in combination with 10 µM LY for 12, 24, 36 and
48 h. Subsequently, 10 µl MTT (5 mg/ml) was added to each well and
cells were incubated for an additional 4 h. Following removal of
the supernatant, DMSO (100 µl/well) was added to dissolve the blue
formazan crystals converted from MTT by HepG2 cells. Cell viability
was assessed using a microplate reader at an optical density of 560
nm (Wellscan K3; KHB Labsystems, Helsinki, Finland). The experiment
was repeated three times.
Cell cycle analysis
HepG2 cells were seeded in a 96-well culture plate
and incubated with 2.0 mM Ado at 37°C for different time-points (6,
12 and 24 h). Cells were collected after trypsin treatment, washed
with phosphate-buffered saline (PBS), and then cell pellets were
resuspended in 1 ml of 50 µg/ml solution of propidium iodide (PI)
in buffer, and incubated in the dark at room temperature for 15
min. The PI fluorescence was assessed on a FACScan flow cytometer
(FACSCalibur; BD Biosciences, San Jose, CA, USA) and the cell cycle
distribution was analyzed using ModFit 3.2 software (Verity
Software House, Inc., Topsham, ME, USA).
Hoechst staining and observation of
nuclear structure
HepG2 cells were seeded in a 96-well plate with
3×103 cells/well overnight, and then incubated with Ado
alone, Ado + Rapa or Ado + LY respectively for an additional 12 h.
Then the cells were washed with PBS and stained by adding 0.5
ml/well Hoechst 33258(10 mg/l) (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) followed by washing in PBS. Condensed or
fragmented nuclei, characteristic of apoptotic cells, were assessed
by fluorescence microscopy. The fraction of apoptotic cells was
calculated as the number of apoptotic cells divided by the number
of total cells. Apoptosis scores were determined, from five
randomly selected fields, by counting 500 cells directly in each
sample using a blinded method.
Fluorescence microscope and flow
cytometric analysis of apoptosis
Apoptotic cells were quantified with an Annexin V
and PI detection kit (Nanjing KeyGen Biotechnology Co., Ltd.,
Nanjing, China), and by flow cytometric analysis. Briefly, cells
were treated with 2.0 mM Ado for 24 h, washed twice with PBS,
resuspended in 1 ml of binding buffer, stained for 15 min at room
temperature with Annexin V and PI, and sorted on a FACSCalibur
instrument with CellQuest software (BD Biosciences).
MDC staining and observation of
autophagy
HepG2 cells in 24-well plates were treated with or
without Ado for 24 h. Subsequently, the cells were incubated with
50 µmol/l monodansylcadaverine (MDC) (Sigma-Aldrich; Merck KGaA) in
medium at 37°C for 1 h. The cells were then fixed with 4%
paraformaldehyde for 15 min. Subsequent to rinsing twice with cold
PBS and photographed by fluorescence photometry.
Protein extraction and western
blotting
Western blot analysis was performed as previously
described (19). Briefly, protein
(50 µg) was subjected to electrophoresis on 12.5% SDS-PAGE and then
transferred onto nitrocellulose membranes. The membranes were
blocked with 5% non-fat dry milk in Tris-buffered saline containing
0.5% Tween-20 at room temperature for 60 min. Then the transferred
membranes were incubated with primary antibodies in TBS at 4°C
overnight. The primary antibodies LC3-I/II (D3U4C; cat. no. 12741),
Beclin-1 (D40C5; cat. no. 3495), p62 (D5E2; cat. no. 8025), cleaved
caspase-3 (Asp175; cat. no. 8025) were obtained from Cell Signaling
Technology, Inc. (Beverly, MA, USA). ATF4 (cat. no. ab23760), GRP78
(cat. no. ab21685), PERK (cat. no. ab65142), p-AMPK (cat. no.
ab133448), AMPK (cat. no. ab80039), p-ULK1 (cat. no. ab229540),
ULK1 (cat. no. ab167139), p-mTOR (cat. no. ab84400), mTOR (cat. no.
ab32028), CHOP (cat. no. ab11419), cytochrome c (cat. no.
ab110325) and β-actin (cat. no. ab8226) were purchased from Abcam
(Cambridge, MA, USA). The dilution of all primary antibodies was
1:1,000. After washing with Tris-buffered saline with Tween-20
(TBST3) times for 8 min, the membranes were incubated with
peroxidase-conjugated secondary antibodies (goat anti-mouse IgG,
A32723 or goat anti-rabbit IgG; A32732; both 1:5,000, Invitrogen;
Thermo Fisher Scientific, Inc.) for 1 h at room temperature. Bands
were visualized with an ECL detection system (Thermo Fisher
Scientific, Inc.). Protein expression was analyzed by the Quantity
One software (National Institutes of Health, Bethesda, MD, USA) and
normalized to that of β-actin. In addition, a cell Mitochondria
Isolation kit (Beyotime Institute of Biotechnology, Jiangsu, China)
was used to extract proteins in mitochondria in order to analyze
cytochrome c.
RNA preparation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
assay
Total RNA was extracted from cells using the total
RNA kit (Thermo Fisher Scientific, Inc.) and cDNAs were synthesized
using a Revert Aid First Strand cDNA Synthesis kit (Fermentas;
Thermo Fisher Scientific, Inc.) in accordance with the
manufacturer's instructions. The primer sequences used for RT-PCR
analysis are listed in Table I.
Real-time RT-PCR was performed with an ABI PRISM 7900 Sequence
Detection System (Applied Biosystems, Inc., Foster City, CA, USA)
using SYBR-Green PCR Master Mix (Roche Diagnostics GmbH, Mannheim,
Germany). The thermal cycling conditions were as follows: 95°C for
10 min; 95°C for 15 sec, and then 58°C for 30 sec for 40 cycles.
All reactions were run in triplicate. Amplified samples were
subjected to agarose electrophoresis with ethidium bromide as the
fluorescent dye. Analysis of the relative gene expression data was
performed by the 2−ΔΔCq method (24).
 | Table I.Primer sequences for RT-PCR. |
Table I.
Primer sequences for RT-PCR.
| Genes | Sequence |
|---|
| 18s | F:
5′-AAACGGCTACCACATCCAAG-3′ |
|
| R:
5′-CAATTACAGGGCCTCGAAAG-3′ |
| Caspase-3 | F:
5′-AGAGCTGGACTGCGGTATTGGAG-3′ |
|
| R:
5′-GAACCATGACCCGTCCCTTG-3′ |
| Cytochrome
c | F:
5′-CTTACACAGCCGCCAATA-3′ |
|
| R:
5′-CTTCTTCTTAATGCCGACAA-3′ |
| GRP78 | F:
5′-CCTGGTACTGCTTGATGTAT-3′ |
|
| R:
5′-TTCTGCTGTATCCTCTTCAC-3′ |
| CHOP | F:
5′-CTTCATACATCACCACACCT-3′ |
|
| R:
5′-GTAGTCAGTAGCCACTTCT-3′ |
| PERK | F:
5′-TGTCGCCAATGGGATAGTGACGAA-3′ |
|
| R: 5′
AATCCGGCTCTCGTTTCCATGTCT-3′ |
| ATF4 | F:
5′-CTGACCACGTTGGATGACAC-3′ |
|
| R:
5′-GGGCTCATACAGATGCCTCT-3′ |
| Beclin-1 | F:
5′-GAGTTTCAAGATCCTGGACCGTGTCA-3′ |
|
| R:
5′-CTGTTGGCACTTTCTGTGGACATCA-3′ |
| AMPK | F:
5′-AAACCCACAGAAATCCAAACAC-3′ |
|
| R:
5′-CCTTCCATTCATAGTCCAACTG-3′ |
| ULK1 | F:
5′-ACCGCATTCACAGCATCACT −3′ |
|
| R:
5′-ACCGCATTCACAGCATCACT −3′ |
| mTOR | F:
5′-CTGGGACTCAAATGTGTGCAGTTC-3′ |
|
| R:
5′-GAACAATAGGGTGAATGATCCGGG-3′ |
| p62 | F:
5′-GGCCGCCCTGTTCCCCG-3′ |
|
| R:
5′-GCCGGCACTCTTTTTTCTCTT-3′ |
Measurement of mitochondrial membrane
potential (ΔΨm)
HepG2 cells were seeded at 4×105
cells/well into 6-well plates. After 24 h of incubation, the cells
were incubated Ado, LY or Rapa for 24 h. Then the cells were washed
twice with ice-cold PBS, centrifuged at 600 × g for 5 min.
Subsequently, the cell pellets were resuspended in culture medium
with 5 µmol/l Rhodamine-123 and incubated at 37°C in the dark for
30 min, and then washed and resuspended in culture medium. The mean
fluorescence intensity of Rhodamine-123 was detected using a flow
cytometer (FACSCalibur; BD Biosciences) at an excitation of 488 nm
and an emission of 585 nm.
Statistical analysis
Sample capacity for each experiment was adjusted
according to the variance obtained. Data were expressed as the mean
± SD. All statistics were calculated using the SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA). All experiments described were
performed at least in triplicate. The statistical significance of
differences was analyzed by Student's t-test between two groups or
one-way analysis of variance (ANOVA) followed by Dunnett's post hoc
test for multiple comparisons. P<0.05 was considered to indicate
a statistically significant difference.
Results
Ado inhibits cell viability, induces
ER stress and G0/G1 cell cycle arrest
Cell viability was investigated using an MTT assay.
HepG2 cells were treated with different concentrations of Ado
(1.0–4.0 mM) for 12, 24 and 48 h. As revealed in Fig. 1A, 1.0 mM Ado did not significantly
affect cell viability from 12 to 48 h. However, Ado treatment at
2.0–4.0 mM significantly decreased viability as time increased from
12 to 48 h, revealing that Ado inhibited the growth of HepG2 cells
in a time- and concentration-dependent manner. In addition,
Ado-induced suppression of viability was due to the arrest of cell
cycle progression, which was confirmed by the flow cytometric
assay. Our data demonstrated that Ado significantly increased the
percentage of cells in the G0/G1 stage and decreased the percentage
of cells in the S and G2/M phases in HepG2 cells, compared with the
control (Fig. 1B). Statistical
results revealed that Ado also increased the percentage of sub-G1
phase (an apoptotic peak), which was consistent with our results by
fluorescence microscopy observation and Annexin V/PI assay
(Figs. 3B and 4A).
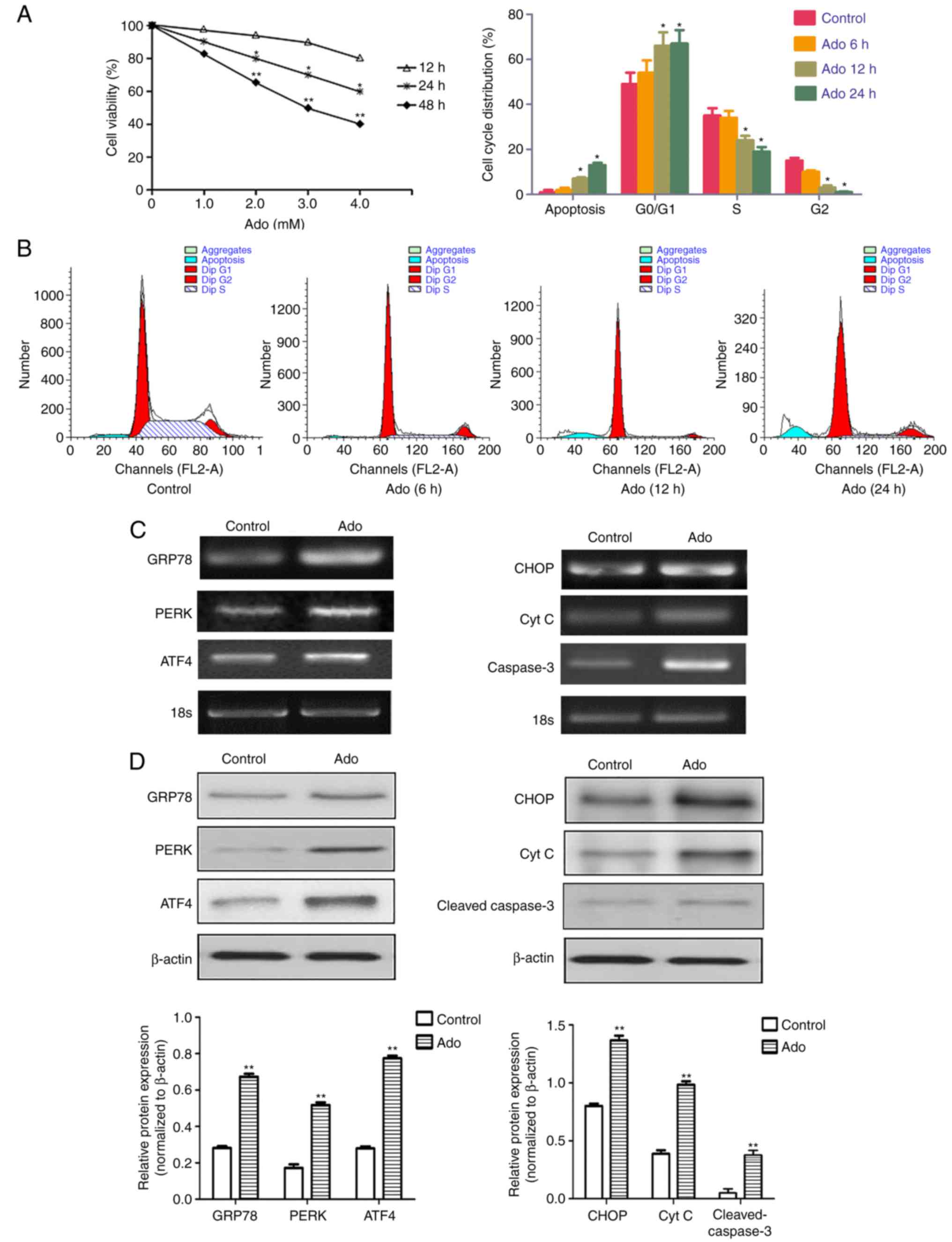 | Figure 1.Ado inhibits cell viability and
triggers ER stress in HepG2 cells. (A) Time- and dose-dependent
cytotoxic effects of Ado on HepG2 cells. Cells were treated with
different concentrations of Ado (1.0–4.0 mM) for 12, 24, or 48 h.
Cell viability was determined by MTT assay. Results are expressed
as percentages of cell growth relative to initial number of viable
cells. (B) Cells were treated with 2.0 mM for 6, 12, or 24 h. The
cell cycle distribution was evaluated using flow cytometric assay
by PI staining. (C) HepG2 cells were treated with 2.0 mM Ado for 24
h. Cells were collected and subjected to total RNA extraction. The
mRNA levels of ER stress-related genes GRP78, PERK, ATF4, CHOP, Cyt
C and caspase-3 were assessed by RT-PCR. (D) HepG2 cells underwent
the aforementioned treatment and were collected and subjected to
western blot analyses with specific antibodies directed against
GRP78, PERK, ATF4, CHOP, Cyt C and cleaved caspase-3, or β-actin.
The density of the corresponding bands was assessed quantitatively
using image analysis software and corrected by reference to the
value of β-actin. Bar graphs represent the mean fluorescence
intensity. *P<0.05, **P<0.01 denotes significant difference
from the normal control HepG2 cells; Ado, adenosine; ER,
endoplasmic reticulum; Cyt C, cytochrome c; PI, propidium
iodide. |
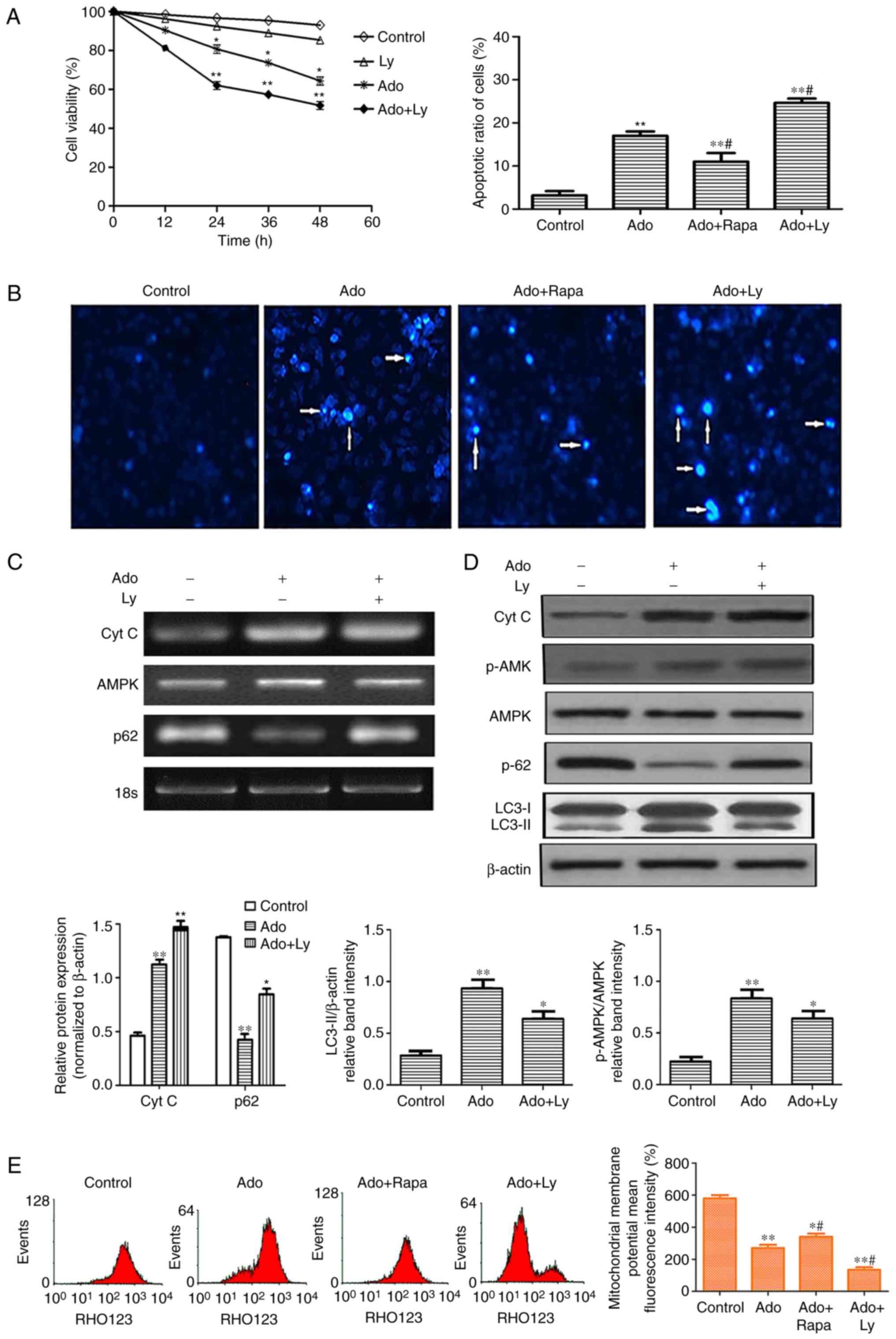 | Figure 3.Inhibition of autophagy enhances ER
stress-mediated apoptotic cell death. (A) HepG2 cells were treated
with 2.0 mM Ado or 10 µM LY alone. In the combination treatment
group, the cells were pre-treated with 10 µM LY for 2 h, followed
by 2.0 mM Ado treatment. Cell viability was determined by MTT assay
at the indicated time-points. (B) Cell apoptosis was detected by
Hoechst staining. Images were captured under a fluorescence
microscope (original magnification, ×200). Arrows indicate
apoptotic cells. (C) The mRNA levels of Cyt C, AMPK and p62 were
assessed by RT-PCR. (D) HepG2 cells underwent the aforementioned
treatment and were collected and subjected to western blot analyses
with specific antibodies directed against Cyt C, p-AMPK, AMPK, p62,
LC3-I and LC3-II, or β-actin. Bar graphs represent the mean protein
band intensity. Relative quantity of the aforementioned proteins
was computed. (E) Mitochondrial membrane potential (ΔΨm) indicated
by Rhodamine-123 was detected by flow cytometry. Bar graphs
represent the mean fluorescence intensity. *P<0.05, **P<0.01
vs. the control; #P<0.05 vs. the Ado group. ER,
endoplasmic reticulum; Ado, adenosine; Cyt C, cytochrome c;
AMPK, AMP-activated protein kinase; LC3-II, microtubule-associated
protein1 light chain 3-II. |
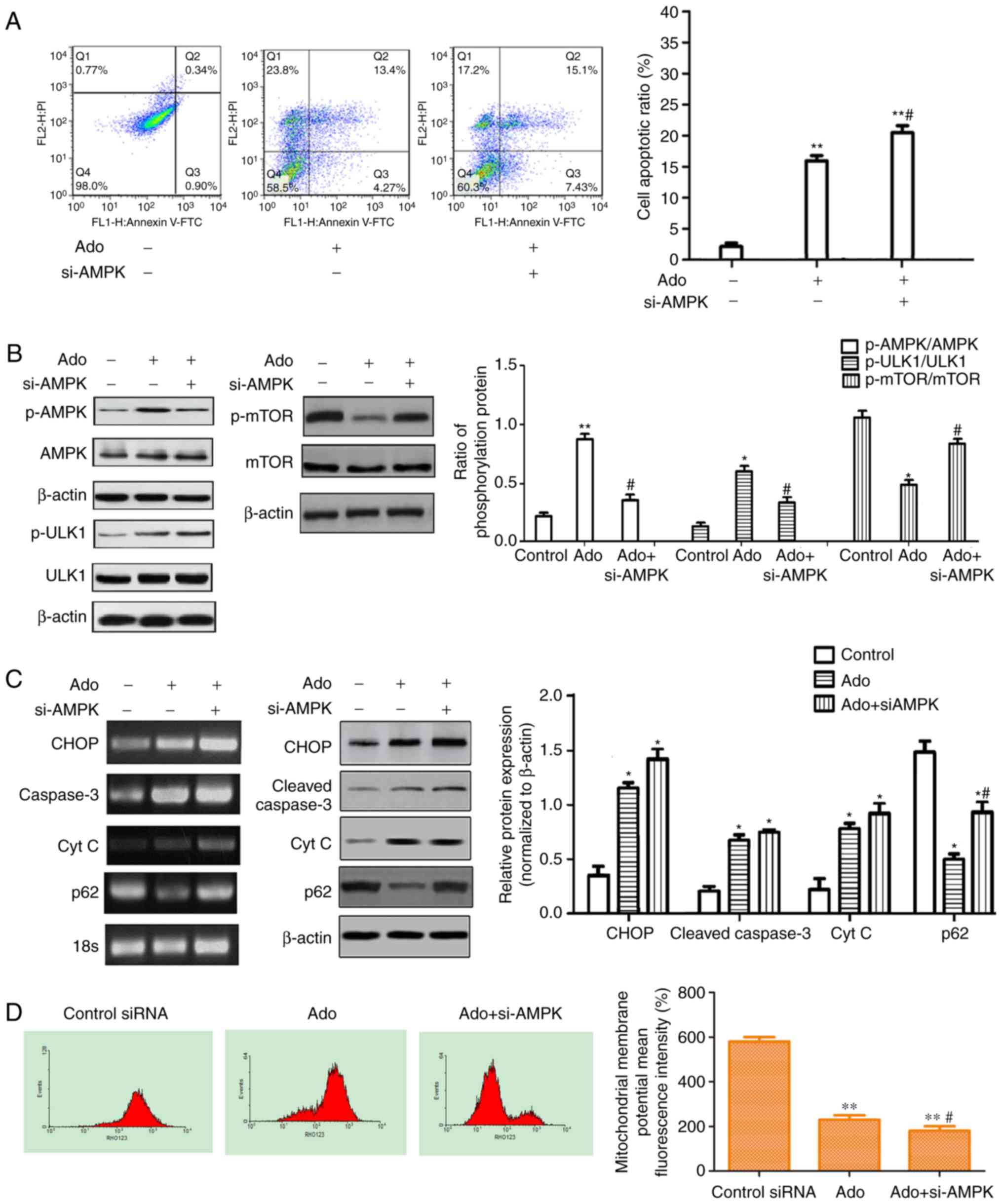 | Figure 4.Inhibition of autophagy by AMPK siRNA
enhances Ado-induced apoptosis and inhibits the AMPK/mTOR/ULK1
pathway. (A) HepG2 cells were transfected with AMPK siRNA (20 µM)
or control siRNA (transfected with empty vector, 20 µM), then
exposed to 2.0 mM Ado or not for 24 h and then the cell apoptotic
ratio was determined by flow cytometry (FACS) with Annexin V-FITC
and PI double staining. (B) HepG2 cells underwent the
aforementioned treatment and were collected and the protein levels
of the AMPK/mTOR/ULK1 pathway were assessed by western blotting.
The phosphorylation ratios of p-AMPK/AMPK, p-ULK1/ULK1 and
p-mTOR/mTOR were computed (C) The mRNA and protein levels of ER
stress-related genes were assessed by RT-PCR and western blotting.
HepG2 cells underwent the aforementioned treatment and were
collected and subjected to western blot analyses with specific
antibodies directed against CHOP, cleaved caspase-3, p62 and Cyt C
or β-actin. Bar graphs represent the mean protein band intensity.
(D) Mitochondrial membrane potential (ΔΨm) indicated by
Rhodamine-123 was detected by flow cytometry. Bar graphs represent
the mean fluorescence intensity. *P<0.05, **P<0.01 vs.
control; #P<0.05 vs. Ado group. Ado, adenosine; AMPK,
AMP-activated protein kinase; PI, propidium iodide; ER, endoplasmic
reticulum. |
The endoplasmic reticulum (ER) is a highly dynamic
organelle in eukaryotic cells. Many studies have indicated that
once UPRs (PERK, ATF6 and/or IRE1) are activated, they initiate an
early adaptive response to inhibit transcription and translation,
and to increase the expression of GRP78 (4,19).
However, with prolonged stress, additional responses are initiated,
including caspase-12/-9/-3 and ERK/ATF4/CHOP, which can promote
cell apoptosis (25). In our
previous study, it was demonstrated that the caspase-12/-9/-3
pathway played an important role in Ado-mediated apoptosis
(19). To investigate whether other
ER stress pathways following Ado treatment were activated in HepG2
cells, RT-PCR and western blot analyses were performed to detect
the expression of GRP78, PERK, ATF4, CHOP, Cyt C and cleaved
caspase-3. The results revealed that the mRNA and protein
expression levels of the aforementioned relative genes were
significantly increased after Ado treatment, indicating that the
ATF4/CHOP pathway also participated in Ado-induced apoptosis
(Fig. 1C and D), which enriched our
understanding of Ado-induced apoptosis through ER stress in HepG2
cells. Collectively, these data indicated that the effect of Ado on
the inhibition of cell growth was associated with G0/G1 cell cycle
arrest and ER stress-related apoptosis.
Ado induces autophagy in HepG2
cells
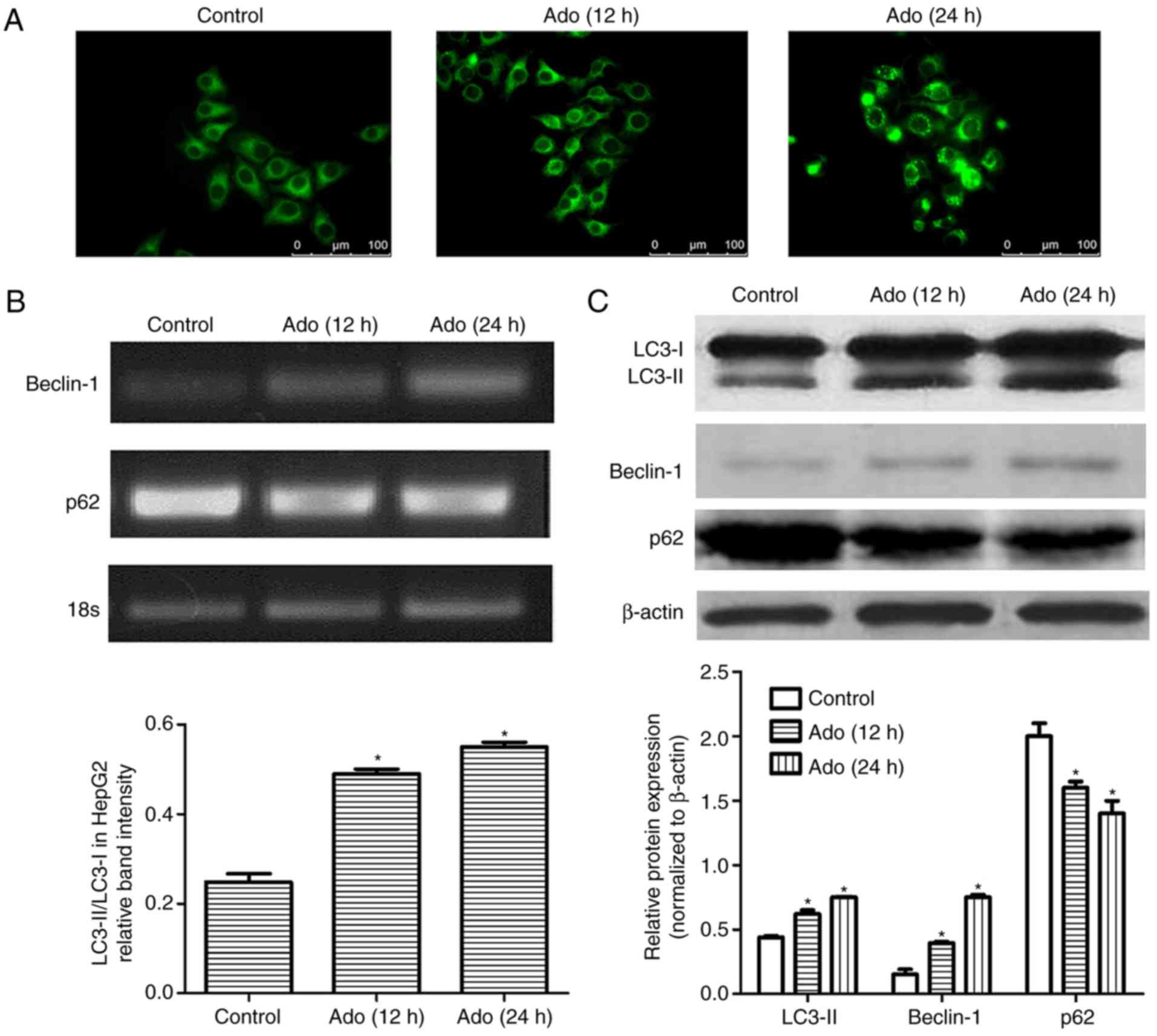
In the present study, three different methods were
applied to explore autophagic flux. Firstly, fluorescence
microscopic evaluation revealed an increased amount of
autophagosomes in the cells treated with Ado from 12 to 24 h
(Fig. 2A). Secondly, we assessed
the expression of autophagy-related proteins, Beclin-1 and p62.
Beclin-1 is an initiative regulator of autophagy and p62 is a
marker of autophagosome degradation. p62 decreases when autophagy
is triggered and accumulates when autophagy is inhibited. The
results demonstrated that Ado significantly reduced p62 levels in a
time-dependent manner. Conversely, Ado increased Beclin-1 and
LC3-II expression (Fig. 2B and C).
Thirdly, the expression of LC3-II and the ratio of LC3-II/LC3-I was
significantly increased after Ado treatment in HepG2 cells
(Fig. 2C). Collectively, all these
data demonstrated that Ado induced autophagy in HepG2 cells.
Ado induces cell apoptosis and LY
enhances Ado-induced apoptosis
Since cell growth inhibition is always associated
with cell apoptosis, we further detected whether Ado induced
apoptosis by Hoechst staining. Following Ado treatment for 24 h,
cell nuclei became condensed and shrunk. In addition, typical
apoptotic bodies appeared and the cell apoptotic ratio increased
(Fig. 3B). Apoptotic cell death,
besides characteristics such as chromatin condensation and
apoptotic body formation, was also accompanied by typical
mitochondrial changes. The latter includes enhanced membrane
permeability, a decrease in mitochondrial membrane potential (ΔΨm)
and release of cytochrome c into the cytosol. To investigate
the role of Ado-induced autophagy on apoptosis in HepG2 cells,
cells were pretreated with autophagy inhibitor LY294002 (LY, 10 µM)
or autophagy activator rapamycin (Rapa, 20 µM) for 1 h, followed by
treatment with Ado for an additional 24 h. As revealed in Fig. 3A, 10 µM LY only slightly reduced the
viability of HepG2 cells at different time-points, but there was no
significant difference when compared with the control group.
However, 2.0 mM Ado in combination with 10 µM LY significantly
reduced the viability with the increase of time from 24 to 48 h.
Furthermore, the pharmacological inhibition of autophagy by LY
significantly increased the mRNA and protein expression levels of
cytochrome c and p62, decreased the expression level of
LC3-II, and enhanced the loss of the ΔΨm. Conversely, combination
treatment of Ado with Rapa, significantly increased the ΔΨm and
decreased the number of apoptotic cells, compared with the Ado
alone group (Fig. 3B and E). These
results revealed that the inhibition of autophagy increased
Ado-induced cytotoxic effects and the activation of autophagy
alleviated Ado-induced apoptosis, indicating that autophagy plays a
protective role in Ado-induced apoptosis in HepG2 cells.
Ado induces autophagy by activating
AMPK and downregulating the mTOR signaling pathway
As aforementioned, it was determined that HepG2
cells treated with Ado exhibited increased apoptosis and autophagy.
It is well-known that the AMPK/mTOR pathway functions as an
autophagy regulator under starvation or other cellular stress
conditions (26). Ado may increase
the ratio of AMP/ATP in cells and thus lead to the activation of
AMPK, which may promote autophagosome formation (27). In the present study, RT-PCR and
western blot analysis demonstrated that Ado increased the mRNA and
phosphorylated protein expression of AMPK (Figs. 3D and 4B). Therefore, the possible role of the
AMPK-mediated autophagy signaling pathway was further investigated.
The AMPK-siRNA plasmids were used to achieve a specific knockdown
of AMPK in HepG2 cells. Ado increased the ratio of p-AMPK/AMPK,
p-ULK1/ULK1, and decreased the ratio of p-mTOR/mTOR and p62
expression level. Knockdown of AMPK abrogated Ado-induced
activation of p-ULK1 and mTOR inhibition, increased p62 expression
and the cell apoptosis ratio (Fig.
4A-C). Furthermore, we observed that knockdown of AMPK
significantly increased ER stress-related apoptosis as evidenced by
increased induction of CHOP, cleaved caspase-3, Cyt C (Fig. 4C) and the loss of mitochondrial
membrane potential (ΔΨm) (Fig. 4D).
Collectively, in HepG2 cell lines, Ado-induced AMPK/mTOR pathway
activation partially blocked ER stress and decreased apoptotic cell
death.
Discussion
Ado is a common metabolite of ATP, which exhibits
cytotoxic effects at high concentrations. Numerous in vitro
experiments have demonstrated that Ado can induce apoptotic cell
death in various types of cancer cells via several mechanisms,
including promotion of cell cycle arrest, apoptosis and suppression
of signal transduction (15–17).
However, the mechanisms underlying the cytotoxic effects of Ado are
not totally understood. It was demonstrated that ER stress was
involved in Ado-induced apoptosis in a previous study (19). In the early stage of ER stress, UPR
plays an important role to maintain cell homeostasis and there is
cross-talk between UPR and autophagy (18). We hypothesized that autophagy
probably participated in this process. In the present study, we
demonstrated that Ado induced both apoptosis and autophagy
concurrently in HepG2 cells. The main mechanism of autophagy was
found to be via AMPK activation of intracellular energy sensor and
subsequent inhibition of the main autophagy suppressor mTOR
(Fig. 4).
Clinically, it has been suggested that the cell
cycle is a primary target for cancer treatment (28). In the present study, Ado was
observed to cause a time- and concentration-dependent inhibition of
cell proliferation in HepG2 cells (Fig.
1A). The results of cell cycle analysis revealed that Ado
arrested cells in the G0/G1 phase and prevented them from
transitioning to the S phase (Fig.
1B). G0 is the resting phase in which cells stop dividing and
leave the cell cycle, while cells prepare energy and material for
DNA replication in the G1 phase (29). Therefore, arrest of cells in the
G0/G1 phase results in the obstruction of mitosis and cellular DNA
synthesis. Thus, Ado-induced proliferation inhibition was related
to cell cycle arrest in HepG2 cells.
Cell apoptosis is a programmed cell death process
triggered. There are three different pathways, including the cell
death receptor apoptotic pathway, ER stress and the mitochondrial
apoptotic pathway (30). The ER
stress apoptotic pathway involves UPR and loss of ΔΨm and the
subsequent release of cytochrome c from mitochondria to the
cytoplasm (21,22). In the present study, Hoechst
staining revealed that Ado treatment caused obvious nuclear
condensation, which is a typical characteristic of apoptosis
(Fig. 3B). Flow cytometric
assessment of Annexin V/PI staining further confirmed that Ado
increased the proportion of apoptotic cells (Fig. 4A). The results of RT-PCR and western
blotting demonstrated that Ado significantly increased the mRNA and
protein expressions of GRP78, PERK, ATF4, CHOP, cleaved caspase-3
and cytochrome c (Fig. 1C and
D). Concurrently, Ado also caused the loss of ΔΨm (Fig. 3E), revealing that Ado treatment
impaired the mitochondria and caused the aberrant release of
cytochrome c from mitochondria into cytoplasm. Collectively,
these findings indicated that Ado induced ER-related apoptosis in
HepG2 cells.
Autophagy is a dynamic process comprised of two
consecutive stages. The first step is autophagosome formation. The
second step is autophagosome clearance, which involves autolysosome
formation via autophagosome-lysosome fusion (31). Inhibiting autophagic flux can
sensitize cells to stimulus-induced damage and cell death (3–5). In
the present study, in addition to the induction of apoptosis, we
also determined for the first time that Ado induced autophagy in
HepG2 cells, as demonstrated by the increased autophagic vacuoles
(Fig. 2A), accompanied with the
increased levels of Beclin-1, the conversion of the molecular form
of LC3 (LC3-I) to LC3-II, and the decreased expression of p62
(Fig. 2B and C). These are commonly
used as markers for detecting autophagy (32).
Both apoptosis and autophagy are important in normal
physiology and in a wide range of diseases and cross-talk also
occurs between them. Studies have revealed that after treatment
with antitumor drugs, some cancer cells undergo autophagy as a
temporary survival mechanism, and the suppression of autophagy
leads to apoptosis and enhances antitumor effects (3). However, some treatments also result in
the induction of autophagic cell death or both apoptosis and
autophagy (12). To further
investigate the interplay between autophagy and apoptotic death,
HepG2 cells were pretreated with autophagy inhibitors (LY) or an
autophagy inducer (Rapa). LY has been revealed to inactivate
Akt/PKB, decrease the expression of phosphorylated Akt (Ser473),
inhibit cancer cell growth and induce apoptosis. LY has also been
revealed to act on HepG2 cells, induce significant nuclear pyknosis
and reduce cytoplasmic volume (33). However, the anticancer effect of LY
is closely related to its concentration. As revealed in Fig. 3A, 10 µM LY only slightly reduced the
viability of HepG2 cells at different time-points. When the dose of
LY increased to 30 µM, MTT assays revealed that after 12, 24, 36
and 48 h of treatment, the rates of viability were 91.41±9.33,
83.82±9.14, 75.14±8.14 and 64.71±6.78%, respectively (data not
shown). The viability of HepG2 cells significantly decreased,
compared with the control group (P<0.05). This was in line with
a previous study (34). Thus, the
dose of 10 µM LY, which did not significantly affect the viability
of HepG2 cells, was used in subsequent experiments in order to
observe the cytotoxic effect of Ado and the role of inhibition of
autophagy. The results revealed that LY, the autophagy inhibitor,
increased the expression level of p62 and decreased the expression
level of LC3-II, enhancing Ado-induced apoptosis and proliferation
inhibition in HepG2 cells (Fig.
3A-D). In contrast, pretreatment with Rapa significantly
decreased the apoptotic ratio and increased the mitochondrial
membrane potential (Fig. 3B and E).
These results indicated that autophagy regulated Ado-induced
apoptosis and played a protective role in HepG2 cells.
Cell autophagy is affected by AMPK phosphorylation
and activity of Mtor (35). AMPK is
a major regulator of cellular energy homeostasis, and it regulates
carbohydrate and fat metabolism in order to maintain the cellular
energy balance (36,37). AMPK is activated by metabolic
stressors that deplete ATP and increase AMP, and by upstream
kinases that induce its phosphorylation at Thr172. Several avenues
of evidence have pointed to Ado as an activator of AMPK by
increasing intracellular AMP concentrations or by binding to the
Ado receptors on the cell membrane (38,39).
The mechanism by which AMPK activates autophagic response
presumably involves downregulation of mTOR and activation of ULK1
(40). As physiological stresses
result in both AMPK activation and mTOR inhibition, ULK1 initiates
the autophagy process (41). In the
present study, our results demonstrated that Ado activated the
AMPK/mTOR/ULK1 pathway, as evidenced by the increased ratio of
p-AMPK/AMPK, p-ULK1/ULK1 and the decreased ratio of p-mTOR/mTOR.
Knockdown of AMPK by si-AMPK abolished Ado-induced mTOR inhibition
and ULK1 activation, thereby downregulating autophagy, which was
ascertained by the increased p62 expression (Fig. 4C). These data revealed that Ado
activated AMPK and triggered autophagy in a double-pronged
mechanism of directly activating ULK1 and inhibiting mTOR protein
expression, which was consistent with the increased autophagy
(Fig. 2A-C). Concurrently, we also
observed that knockdown of AMPK further enhanced CHOP pathway
activation, as evidenced by increased expression levels of CHOP,
cleaved caspase-3, the cell apoptosis ratio and decreased
mitochondrial membrane potential (ΔΨm) (Fig. 4A, C and D). These results indicated
that the AMPK/mTOR/ULK1 pathway was involved in Ado-induced
autophagy and autophagy played a protective effect in Ado-induced
apoptosis. Therefore, our study demonstrated that AMPK may act as
an important factor governing the cross talk between apoptosis and
autophagy in HepG2 cells. Whether Ado alters the ratio of
intracellular AMP/ATP or directly activates its receptors on the
cell membrane and leads to AMPK activation remains to be further
explored.
In conclusion, our present study revealed that
Ado-induced ER stress resulted in apoptosis and autophagy
concurrently. Autophagy may regulate Ado-induced cytotoxicity via
the activation of the AMPK/mTOR/ULK1 signaling pathway and
autophagy may play a protective role in the apoptotic process.
Inhibition of autophagy may effectively enhance the anticancer
effect of Ado in human hepatoblastoma HepG2 cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Guangdong
Natural Science Foundation in China (no. 2014A030313470) and the
Guangdong Science and Technology Planning Project (no.
2014A020212284). This study was also supported by the Department of
Education, Guangdong Government under the Top-tier University
Development Scheme for Research and Control of Infectious
Diseases.
Availability of data and materials
The datasets generated/analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LFW designed and wrote the study. HCZ contributed
significantly to the data analysis and the manuscript preparation.
XTZ, ZJP, LXL and GPL performed the experiments and wrote the
manuscript. JLF analyzed the data and approved the final version of
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mokdad AA, Singal AG and Yopp AC: Advances
in local and systemic therapies for hepatocellular cancer. Curr
Oncol Rep. 18:92016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin J, Wu L, Bai X, Xie Y, Wang A, Zhang
H, Yang X, Wan X, Lu X, Sang X, et al: Combination treatment
including targeted therapy for advanced hepatocellular carcinoma.
Oncotarget. 7:71036–71051. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou J, Li G, Zheng Y, Shen HM, Hu X, Ming
QL, Huang C, Li P and Gao N: A novel autophagy/mitophagy inhibitor
liensinine sensitizes breast cancer cells to chemotherapy through
DNM1L-mediated mitochondrial fission. Autophagy. 11:1259–1279.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xie WY, Zhou XD, Li Q, Chen LX and Ran DH:
Acid-induced autophagy protects human lung cancer cells from
apoptosis by activating ER stress. Exp Cell Res. 339:270–279. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chi KH, Wang YS, Huang YC, Chiang HC, Chi
MS, Chi CH, Wang HE and Kao SJ: Simultaneous activation and
inhibition of autophagy sensitizes cancer cells to chemotherapy.
Oncotarget. 7:58075–58088. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rubiolo JA, López-Alonso H, Martínez P,
Millán A, Cagide E, Vieytes MR, Vega FV and Botana LM: Yessotoxin
induces ER-stress followed by autophagic cell death in glioma cells
mediated by mTOR and BNIP3. Cell Signal. 26:419–432. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Artero-Castro A, Perez-Alea M, Feliciano
A, Leal JA, Genestar M, Castellvi J, Peg V, Ramón Y Cajal S and
Lleonart ME: Disruption of the ribosomal P complex leads to
stress-induced autophagy. Autophagy. 11:1499–1519. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Netea-Maier RT, Klück V, Plantinga TS and
Smit JW: Autophagy in thyroid cancer: Present knowledge and future
perspectives. Front Endocrinol. 6:222015. View Article : Google Scholar
|
|
9
|
Kovaleva V, Mora R, Park YJ, Plass C,
Chiramel AI, Bartenschlager R, Döhner H, Stilgenbauer S, Pscherer
A, Lichter P, et al: miRNA-130a targets ATG2BDICER1 to
inhibit autophagy and trigger killing of chronic lymphocytic
leukemia cells. Cancer Res. 72:1763–1772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jing Z, Sui X, Yao J, Xie J, Jiang L, Zhou
Y, Pan H and Han W: SKF-96365 activates cytoprotective autophagy to
delay apoptosis in colorectal cancer cells through inhibition of
the calcium/CaMKIIγ/AKT-mediated pathway. Cancer Lett. 372:226–238.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Y, Gong W, Yang ZY, Zhou XS, Gong C,
Zhang TR, Wei X, Ma D, Ye F and Gao QL: Quercetin induces
protective autophagy and apoptosis through ER stress via the
p-STAT3/Bcl-2 axis in ovarian cancer. Apoptosis. 22:544–557. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu JL, Hu XL, Guo AY, Wang CJ, Wen YY and
Cang SD: Endoplasmic reticulum stress promotes autophagy and
apoptosis and reverses chemoresistance in human ovarian cancer
cells. Oncotarget. 8:49380–49394. 2017.PubMed/NCBI
|
|
13
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo XL, Hu F, Zhang SS, Zhao QD, Zong C,
Ye F, Guo SW, Zhang JW, Li R, Wu MC, et al: Inhibition of p53
increases chemosensitivity to 5-FU in nutrient-deprived
hepatocarcinoma cells by suppressing autophagy. Cancer Lett.
346:278–284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu LX, Deng W, Zhou XT, Chen RP, Xiang
MQ, Guo YT, Pu ZJ, Li R, Wang GF and Wu LF: The mechanism of
adenosine-mediated activation of lncRNA MEG3 and its antitumor
effects in human hepatoma cells. Int J Oncol. 48:421–429. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hajiahmadi S, Panjehpour M, Aghaei M and
Shabani M: Activation of A2b adenosine receptor regulates ovarian
cancer cell growth: Involvement of Bax/Bcl-2 and caspase-3. Biochem
Cell Biol. 93:321–319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu S, Hou D, Chen P, Zhang Q, Lv B, Ma Y,
Liu F, Liu H, Song EJ, Yang D, et al: Adenosine induces apoptosis
through TNFR1/RIPK1/P38 axis in colon cancer cells. Biochem Biophys
Res Commun. 460:759–765. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hirsch C, Gauss R, Horn SC, Neuber O and
Sommer T: The ubiquitylation machinery of the endoplasmic
reticulum. Nature. 458:453–460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu LF, Guo YT, Zhang QH, Xiang MQ, Deng W,
Ye YQ, Pu ZJ, Feng JL and Huang GY: Enhanced antitumor effects of
adenoviral-mediated siRNA against GRP78 gene on adenosine-induced
apoptosis in human hepatoma HepG2 cells. Int J Mol Sci. 15:525–544.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yasuda Y, Saito M, Yamamura T, Yaguchi T
and Nishizaki T: Extracellular adenosine induces apoptosis in
Caco-2 human colonic cancer cells by activating caspase-9/-3 via
A2a adenosine receptors. J Gastroenterol. 44:56–65.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gorman AM, Healy SJ, Jäger R and Samali A:
Stress management at the ER: Regulators of ER stress-induced
apoptosis. Pharmacol Ther. 134:306–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Singh N, Robinson-Taylor KS,
Dorsett-Martin WA, Morris MW Jr, Earl TM and Anderson CD:
Hepatocyte autophagy is linked to C/EBP-homologous protein,
Bcl2-interacting mediator of cell death, and BH3-interacting domain
death agonist gene expression. J Surg Res. 195:588–595. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu LF, Wei BL, Guo YT, Ye YQ, Li GP, Pu ZJ
and Feng JL: Apoptosis induced by adenosine involves endoplasmic
reticulum stress in EC109 cells. Int J Mol Med. 30:797–804. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bromati CR, Lellis-Santos C, Yamanaka TS,
Nogueira TC, Leonelli M, Caperuto LC, Gorjão R, Leite AR, Anhê GF
and Bordin S: UPR induces transient burst of apoptosis in islets of
early lactating rats through reduced AKT phosphorylation via
ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr
Comp Physiol. 300:R92–R100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Varshney R, Varshney R, Mishra R, Gupta S,
Sircar D and Roy P: Kaempferol alleviates palmitic acid-induced
lipid stores, endoplasmic reticulum stress and pancreatic β-cell
dysfunction through AMPK/mTOR-mediated lipophagy. J Nutr Biochem.
57:212–227. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Imamura K, Ogura T, Kishimoto A, Kaminishi
M and Esumi H: Cell cycle regulation via p53 phosphorylation by a
5′-AMP activated protein kinase activator,
5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human
hepatocellular carcinoma cell line. Biochem Biophys Res Commun.
287:562–567. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oura K, Tadokoro T, Fujihara S, Morishita
A, Chiyo T, Samukawa E, Yamana Y, Fujita K, Sakamoto T, Nomura T,
et al: Telmisartan inhibits hepatocellular carcinoma cell
proliferation control by inducing cell cycle arrest. Oncol Rep.
38:2825–2835. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kang S, Kang MS, Ryu E and Myung K:
Eukaryotic DNA replication: Orchestrated action of multi-subunit
protein complexes. Mutat Res. 809:58–69. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kenific CM and Debnath J: Cellular and
metabolic functions for autophagy in cancer cells. Trends Cell
Biol. 25:37–45. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park JH, Lee JE, Shin IC and Koh HC:
Autophagy regulates chlorpyrifos-induced apoptosis in SH-SY5Y
cells. Toxicol Appl Pharmacol. 268:55–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Buontempo F, Ersahin T, Missiroli S,
Senturk S, Etro D, Ozturk M, Capitani S, Cetin-Atalay R and Neri
ML: Inhibition of Akt signaling in hepatoma cells induces apoptotic
cell death independent of Akt activation status. Invest New Drugs.
29:1303–1313. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xing CG, Zhu BS, Liu HH, Lin F, Yao HH,
Liang ZQ and Qin ZH: LY294002 induces p53-dependent apoptosis of
SGC7901 gastric cancer cells. Acta Pharmacol Sini. 29:489–498.
2008. View Article : Google Scholar
|
|
35
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ojuka EO, Jones TE, Nolte LA, Chen M,
Wamhoff BR, Sturek M and Holloszy JO: Regulation of GLUT4
biogenesis in muscle: Evidence for involvement of AMPK and
Ca2+. Am J Physiol Endocrinol Metab. 282:E1008–E1013.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth and metabolism. Nat Cell
Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Galardo MN, Riera MF, Pellizzari EH,
Sobarzo C, Scarcelli R, Denduchis B, Lustig L, Cigorraga SB and
Meroni SB: Adenosine regulates Sertoli cell function by activating
AMPK. Mol Cell Endocrinol. 330:49–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aymerich I, Foufelle F, Ferré P, Casado FJ
and Pastor-Anglada M: Extracellular adenosine activates
AMP-dependent protein kinase (AMPK). J Cell Sci. 119:1612–1621.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bolster DR, Crozier SJ, Kimball SR and
Jefferson LS: AMP-activated protein kinase suppresses protein
synthesis in rat skeletal muscle through down-regulated mammalian
target of rapamycin (mTOR) signaling. J Biol Chem. 277:23977–23980.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|